

Summary
Glycoprotein processing along a complex highly compartmentalized pathway is a hallmark of eukaryotic cells. We followed the kinetics of intracellular, site-specific glycan processing of a model protein with five distinct N-glycosylation sites and deduced a mathematical model of the secretory pathway that describes a complex set of processing reactions localized in defined intracellular compartments such as the endoplasmic reticulum the Golgi, or the lysosome. The model was able to accommodate site-specific N-glycan processing and we identified phosphorylated glycan structures of the mannose-6-phosphate pathway responsible for the lysosomal sorting of the glycoprotein. Importantly, our model protein can take different routes of the cellular secretory pathway, resulting in an increased glycan complexity of the secreted protein.
Subject areas: Cell biology, Integrative aspects of cell biology, Mathematical biosciences
Graphical abstract
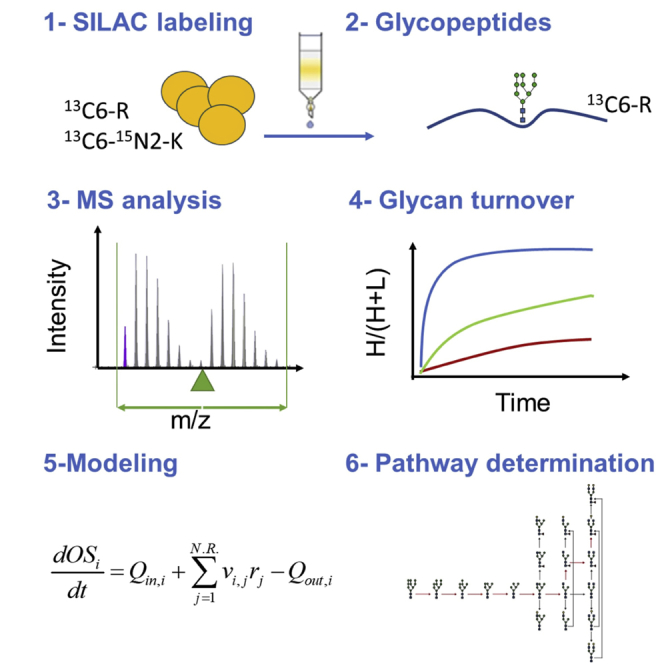
Highlights
-
•
The intracellular processing of an N-glycoprotein was analyzed in CHO cells
-
•
Kinetic data were obtained by MS/MS analysis in combination with SILAC
-
•
Mathematical model of the processing and the secretory pathway was deduced
-
•
Testing of the model revealed the predicted phosphorylated glycan species
Cell biology; Integrative aspects of cell biology; Mathematical biosciences.
Introduction
Post-translational modifications are essential for the function and trafficking of proteins, at the cellular and organism level (van Kooyk and Rabinovich, 2008). N-glycosylation is one of the major modifications of secretory proteins that affects more than one-fifth of the proteins of mammalian cells (Zielinska et al., 2012). Contrary to other post-translational modifications, N-glycosylation is a multi-step process, requiring a complex set of enzymes and donor-substrates, precisely localized across multiple compartments of the secretory pathway (Colley et al., 2015). A trademark of N-glycosylation is the diversity and heterogeneity of the glycan structures generated from a clearly defined Glc3Man9GlcNAc2 oligosaccharide that is added in the ER to nascent polypeptides by an oligosaccharyltransferase (OST) (Breitling and Aebi, 2013; Colley et al., 2015; Ruiz-Canada et al., 2009; Wild et al., 2018). After maturation of the glycan along the secretion pathway, highly diverse, species-specific structures are observed in eukaryotic cells (North et al., 2010).This species specificity is because of a defined set of processing enzymes. In multicellular organisms, differential expression of these enzymes can result in cell-type specific glycan structures. In addition, N-glycans of mature proteins are heterogeneous: (1) Different glycosites of the same protein can carry different glycostructures and (2) different glycan structures can be found on the same site of glycosylation (site-specific heterogeneity) (Losfeld et al., 2017; Varki, 1998). It is well established that glycan processing can be modulated by many biophysical parameters such as: enzyme and substrate concentrations, enzyme-substrate exposure time, substrate affinities, accessibility of the N-glycan to the processing machinery, and interaction of the glycan with the glycoprotein itself (Del Val et al., 2016; Dell et al., 2010; Varki, 1998). In addition, the segregation of the processing machinery into different reaction compartments (e.g., ER and Golgi) along the pathway results in a sequential modification of the glycans (Bard and Chia, 2016; Moremen et al., 2012; Rose, 2012). This organization of glycoprotein processing in different intracellular compartments is a hallmark of the eukaryotic cell. The identification and characterization of the biochemical properties of the processing enzymes as well as the assignment of their localization in the compartmentalized pathway has been a central aspect of glycobiology research over the years (Moremen et al., 2012; Stanley et al., 2015).
It is noteworthy that the site-specific heterogeneity does not reflect the output of a “random” process; in contrast, it is highly reproducible. Numerous, variable, but clearly defined parameters modulate this glycan heterogeneity. The activity and substrate specificity of the glycoenzymes, their biochemical properties, as well as the localization of the processing enzymes along the secretory pathway have been identified as essential drivers of heterogeneity (Colley et al., 2015). The site-specific processing is directed by the protein itself (Arigoni-Affolter et al., 2019; Moremen et al., 2012; Varki and Kornfeld, 2015). Steric hindrance and interaction of the glycan with the protein surface (Hang et al., 2015; Nagae and Yamaguchi, 2018; Narimatsu et al., 2019; Wu et al., 2017) have been identified as a major modulatory factor (Losfeld et al., 2017).
Concomitantly, site-specific glycan heterogeneity as the output of glycoprotein biogenesis renders the characterization of the process extremely complex: a quantitative analysis of site-specific structural heterogeneity for a given glycoprotein is required to describe the output of the pathway. The latest innovation in mass spectrometry has allowed for an improvement of glycosylation profiling, in particular the study of site-specific glycosylation (Chi et al., 2020; Xiao et al., 2019; Zhu and Desaire, 2015). The rapid development of highly sensitive glycoproteomic methods made it even possible to characterize intracellular processing intermediates (Arigoni-Affolter et al., 2019). We developed a new methodology combining stable isotope labeling by amino acid in cell culture (SILAC) with parallel reaction monitoring (PRM) targeted mass spectrometry that allows for the analysis of turnover for intracellular glycoprotein intermediates in vivo and the subsequent mathematical modeling of glycoprotein biogenesis along the secretory pathway. Using IgG biogenesis in Chinese Hamster Ovary (CHO) cells as a model system, the methods were first used on the glycosylation of a single glycosite, allowing for the description of the IgG biogenesis along the secretory pathway and provided a basis to quantify the glycoprotein machinery (Arigoni-Affolter et al., 2019).
Here, we extended our analytical approach on the model protein disulphide isomerase (PDI) with 5 N-glycosylation sites that are processed site-specifically when expressed in CHO cells. In contrast to IgG, this model protein revealed a much more complex intracellular processing that involved not only the classical ER to Golgi secretory pathway but also autophagy and a mannose-6-phosphate regulated lysosomal route. Our experimental data allow for a mathematical modeling of a complex secretory pathway that includes multiple connective pathways between organelles that are defined by their specific biosynthetic activities.
Results
Comparison of site-specific intracellular and secreted glycoprofiles
To establish a sensitive analytical system of intracellular glycoprotein processing, we generated a CHO cell line that stably expressed the yeast PDI model protein. An N-terminal (His) 10 tag that directly followed the signal sequence (Losfeld et al., 2017) allowed for an efficient enrichment of the protein irrespective of its intracellular processing stage. Special focus was given on identical culturing time of the cells for the experimental replicates. Intracellular PDI (from cell lysates) and secreted PDI (from cell supernatants) were isolated, and glycopeptides were analyzed by PRM-based mass spectrometry targeting the 5 peptides containing a glycosylation sequon. For each candidate peptide, the entire set of potential glycan structures of the N-glycosylation pathway, were monitored. Site-specific glycosylation profiles were obtained for intracellular (Figure 1A) and secreted (Figure 1B) PDI. As expected, we observed significant differences: intracellular PDI presented mostly oligomannosidic structures, whereas the secreted protein was predominantly modified with complex glycan structures. This extensive remodeling of intracellular and secreted N-glycans is in agreement with the previously described site-specific glycosylation pattern of transiently secreted PDI (Losfeld et al., 2017). Though only low levels of complex N-glycan processing intermediates were detected on intracellular PDI, the sensitive PRM mass spectrometry allowed for the reliable detection of very low abundance intermediates (less than 0.5% of the total pool of intracellular PDI). With respect to site-specific processing, and in agreement with previous results (Losfeld et al., 2017), we noted on the secreted protein that processing site 2 and 3 was much more elaborate than on site 4. This higher site-specific diversity was also observed on the intracellular protein, but the high mannose structures remained the most abundant N-glycans at all sites.
Figure 1.

Site-specific relative glycan abundance of intracellular and secreted PDI
Each panel represents respectively the relative abundance of the neutral glycans on each glycosite of intracellular (Panel A) and secreted PDI (Panel B). In Panel A, the offset sub-panel is zooming on data for which the abundance was very low, with a Yaxis maximum set to 5%. Data represent the average of three biological replicates.
Glycan turnover is site-specific
We combined the analysis of site-specific glycosylation by LC-MS/MS in PRM mode with a pulse regime of SILAC labeling to monitor the turnover of site-specific N-glycan structures. To do so, CHO cells expressing PDI were first cultivated in regular (light) Cellvento CHO-220 medium supplemented with arginine and lysine, and then shifted to (heavy) medium containing labeled 13C6-L-Arginine and 13C6-15N2-L-Lysine. After the shift, cells were sampled every 20 min for 2 h and every 30 min for the following 3 h. Intracellular PDI was purified from cell extracts and prepared for targeted PRM-based mass spectrometric analysis. A total of 43 glycopeptides with neutral glycan structures were targeted for each glycosite. It is relevant to note that the assignment of glycan structures was based on MS1 and MS2 data. Detailed knowledge of N-glycoprotein biogenesis (Colley et al., 2015; Stanley et al., 2015) and the observed MS2 fragmentation patterns allowed for a clear definition of the structure. For instance, the Hex12HexcNAc2, Hex11HexNac2 or the Hex10HexNAc2 structures were assigned to Glc3Man9GlcNAc2, Glc2Man9GlcNAc2 and GlcMan9GlcNAc2, respectively, in CHO cells. Both the light and the heavy labeled glycopeptide counterparts were measured simultaneously by adapting the method from Arigoni-Affolter et al. (2019). In brief, an extra step of purification by HILIC was used to enrich for glycopeptides, the simultaneous monitoring of multiple glycopeptides was rendered possible by a time scheduling with a set time-window per glycosite, the collision energy was lowered for glycosidic bond breakage to 11 or 16 depending on the glycopeptide charge, the isolation window was enlarged to 7Da and centered to the average +1Da mass of the light and heavy mass to monitor simultaneously the light and heavy counterpart of a given glycopeptide. The MS2 quantification was performed on the second peak of the isotopic distribution to avoid cross contamination between the light and heavy signals of larger glycopeptides. Relative quantification of the labeled glycopeptide was based on a manually expanded library of Skyline for automated peptide analysis (Table S1) using the MS2 signals. Intracellular glycan turnover was determined by calculating the ratio between the neo-synthesized glycopeptide (heavy signal) and the total glycopeptide pool (sum of heavy and light signals) for each time point (Figures 2 and S1). For all figures, data were plotted only for detectable structures intracellularly as summarized in Figure 1A. The obtained ratios were plotted over time for each glycostructures on the different sites. The turnover of the specific glycans was structure as well as site specific. For instance, the turnover rate of Hex9HexNac2 was slower on site 4 as compared to the other sites but still faster than the Hex5HexNAc2 turnover of all sites.
Figure 2.

Site-specific intracellular glycan turnovers and modeled kinetics
The site-specific glycan turnovers are represented by the plotting of the fractional labeling (heavy signal/(heavy + light signal)) of each glycan intermediates over time. A representation of the glycan structure deducted from the monosaccharide composition was added on the left of its corresponding plot, potential isoforms are not represented. The different sites are represented by shapes color coded as follows: site3 in purple, site4 in red and site5 in yellow. The modeled turnover kinetics are shown as solid line curves of the corresponding colors. Each data point represents the average of three biological replicates. Error bars represent the calculated standard deviations.
Our experimental set-up was based on the enrichment of PDI polypeptides via an N-terminal (His) 10 tag and therefore allowed for the characterization of very early glycoprocessing events that can take place on nascent polypeptides: indeed, low but quantifiable levels of Hex12GlcNAc2-glycopeptides were detected for all sites except for site 4 (Figure 3). Of interest, these reaction products of the oligosaccharyltransferase were turned over with site-specific rates, but, surprisingly, for site 3 and 5, Hex12HexNac2 presented a slower rate than its glucosidase I product Hex11HexNac2 or the glucosidase II product Hex10HexNAc2 (Figure 3B). This slow turnover of a preceding processing intermediate was best explained by the hypothesis that the measured turnover represented the average behavior of different intracellular pools of a given glycopeptide (Arigoni-Affolter et al., 2019) (Figure 3A). Therefore, differential processing of identical, site-specific N-glycan structures in separate intracellular pools was taken into account in the further evaluation of the experimental data. Figure 3B shows the calculated single pool model (gray line) and compares it to the experimental data and double pool model (colored data points and lines, respectively) for the individual sites.
Figure 3.

Modeling of glucosylated species and Comparison of Sec-Y178A-PDI glycan turnovers
(A) Chain reactions used for the modeling using a single (left) or a double pool (right) of Hex12HexNac2.
(B) Kinetic data (colored shapes) and modeled kinetics (solid lines) of glucosylated species. The site-specific glycan turnovers are represented by the plotting of the fractional labeling of each glycan intermediates over time. The different sites are color coded as follows: site1 green, site2 blue, site3 in purple, site4 in red and site5 in yellow. The gray line represents the modeled kinetics assuming single pools of the respective processing intermediates. Each data point represents the average of three biological replicates. Error bars represent the calculated standard deviations.
(C) The Y178A mutation affects site 4 but not site 2 glycan processing. The site-specific glycan turnovers of Sec-PDI and Sec-Y178A-PDI are represented by the plotting of the fractional labeling of each glycan intermediates over time. A representation of the glycan structure was added on the left of its corresponding plot. The different sites are color coded as follows: site2 Sec-PDI in dark blue, site2 Sec-Y178A-PDI in light blue, site 4 Sec-PDI in dark red and site 4 Sec-Y178A-PDI in light red. Each data point represents the average of three biological replicates. Error bars represent the calculated standard deviations.
To validate our analytical system, we took advantage of a cis-acting PDI mutation that affected site-specific N-glycan processing. Previously, we showed that the mutation of the tyrosine178 in the close vicinity of site 4 into an alanine residue led to a strong and highly selective change in glycan processing at this site and we proposed that an altered kinetic of intracellular glycan processing was responsible for this alteration (Losfeld et al., 2017). Our analytical approach now enabled an evaluation of this hypothesis and, at the same time, allowed for a stringent test of the experimental approach. We investigated how the intracellular glycan turnover of Sec-PDI-Y178A site 4 compared to the wildtype site 4. N-glycan processing at the other sites served as an internal control (Figure 3C). As expected, the intracellular processing kinetics of site 1, 2, 3 and 5 glycans (shown for site 2 in Figure 3C) was not affected by the Y178A mutation, whereas turnover of site 4 glycans was altered. Of interest, not all processing reactions of the site 4 glycan were affected to the same extend. Hex10HexNAc2 turnover was the same for wild-type and mutant PDI whereas Hex8HexNAc2 processing improved significantly. Although independent experiments were used to obtain wild-type and mutant PDI expressing cell lines and turnover data, the only difference was observed for site 4 glycan processing. We concluded that our experimental system produced highly reproducible results and was ideally suited to describe intracellular N-glycan processing at a site-specific level.
The canonical ER to Golgi secretory pathway is unable to describe glycan turnover rates
Next, we tried to fit the experimental data (turnover of site-specific individual glycan structures) to a mathematical model developed for the processing of IgG (Arigoni-Affolter et al., 2019). This model consisted of a given network of compartmentalized processing and transport reactions as well as site-specific processing rates. In particular, all processing events are restricted to the ER-Golgi secretory system, with the exception of quality control pathways that include oligomannose species in the ER being subjected to ERAD (cytosol) and ERLAD (lysosome) degradation events. This modeling approach did correctly reproduce the turnover rates observed for a subset of oligosaccharides, but we noted severe inconsistencies of the modeled and experimentally determined turnover rates for many N-glycan species. In particular, complex type N-glycan structures generated in the Golgi revealed very different kinetics and low turnover rates: for some complex type glycans such as Fuc (HexNAc2Hex3) HexNac3Hex on site2 or Fuc (HexNAc2Hex3) HexNAc on all sites, only 20% were turned over after 300 min. The observation of complex type glycans being turned over with kinetics comparable to cell division (∼24h) was not consistent with a model where all complex glycans resided solely in the Golgi apparatus, because protein traveling through the Golgi cisternae were expected to be secreted within 35 min (Juan-Colas et al., 2018). Furthermore, we noted that complex glycans without terminal galactose (e.g., FucHexNAc2 and FucHexNac3) of sites 3 and 5 presented a slower intracellular turnover than the galactosylated structures with equivalent number of branches of the same site (e.g., FucHexNac2Hex). In addition, we observed typical “lysosomal” degradation structures such as FucHex3HexNac2, Hex3HexNac2 and Hex4HexNac2, suggesting the potential targeting of PDI from the Golgi to a degradation pathway. To account for these phenomena, we implemented the additional transport of the PDI protein from the Golgi to the maturing endosomes (Scott et al., 2014). In this model, a fraction of PDI was directed to the lysosome by a trans-Golgi localized sorting event (Varki and Kornfeld, 2015), whereas the other fraction followed the canonical secretory pathway to the plasma membrane. This bifurcation of the pathway generated distinct PDI populations that were processed by a defined set of hydrolases known to be localized in the endosomal pathway (Figure 4) (Braulke and Bonifacino, 2009; Ni et al., 2006; Varki and Kornfeld, 2015). We describe the endosomal processing system with a series of hydrolase-defining equations equivalent to the system used to model the Golgi apparatus, where transport and processing occur simultaneously (as detailed in the STAR methods). In this model we mathematically represented the activity of the enzymes with logistic functions (i.e., reaches an asymptotic level with respect to the position in the endosome as shown in Figures 5A and 5B) to represent the well-known pH-dependent activity of these enzymes as well as the pH gradient along the Golgi to lysosome pathway (Scott et al., 2014; Varki and Kornfeld, 2015). Subsequently, PDI molecules that exit this pathway were either degraded (in the lysosome) or secreted to explain the FucHex3HexNac2, Hex3HexNac2 and Hex4HexNac2 glycans detected on secreted PDI protein.
Figure 4.

Glycan synthesis and degradation network
The network is represented such as the different plans represent the different compartments, inside each compartments the potential structures are ordered according to the processing events and the different glycosyltransferases and glycosylhydrolases activity are schematized by black arrows. The brown arrows represent the transport events.
Figure 5.

Enzymatic activity and PDI localization along the processing pathway
(A) Normalized enzymatic activities in the Golgi. The values of enzymatic activity toward PDI obtained by the model were normalized to 1 arbitrary unit and plotted in function of the total Golgi length set to 1.
(B) Normalized enzymatic activities in the endosomes. The values of enzymatic activity toward PDI obtained by the modeled were normalized to 1 arbitrary unit and plotted in function of the total endosomes’ length set to 1.
(C) Intracellular localization of PDI. The pie chart presents the relative amount in % of PDI calculated to be localized in the ER, Golgi, endosomes, and lysosomes by the model.
Within this approach, the turnover rate of each glycan species was ultimately determined by the time they spend in the different compartments: species that exhibit a fast turnover were assigned to the Golgi and the early part of the endosome, whereas species that showed slow turnover rates (>3600 min) were present in all the compartments up to the lysosome. Using this extended network, we obtained the model curves presented as solid lines in Figures 2 and S1 with an excellent fit to the experimental data. In addition, we noted that the abundance of the different site-specific PDI glycoforms reflected the experimentally determined relative values (Figure S2). Besides, the model allowed for the calculation of the kinetic parameters k of each enzyme at the protein and site-specific level, respectively, as presented in Tables S3A and S3B and Figure S3.
Although the ER-Golgi-Endosome-Lysosome framework was capable of correctly reproducing the processing kinetics of “late” glycan structures at all sites of PDI, it failed to correctly model the high abundance (75–95%) and the slow turnover of intracellular high mannose structures that, at the same time, represented minor species (5–15%) of glycans on secreted PDI. We concluded that a pool of high mannose PDI glycoprotein with slow turnover was present in the CHO cells. This pool was different from the rapidly turning over high mannose pool in the ER that fed into the Golgi compartment. To accommodate these experimental observations, we included a direct pathway of ER resident PDI glycoprotein to the lysosome. This pathway that can be interpreted as autophagy of the ER (Smith and Wilkinson, 2017) affected a major fraction of intracellular PDI. This population of PDI constituted about 70% of the total intracellular PDI (Figure 5C). As was the case for PDI glycoprotein transported via the Golgi to the lysosome, degradation of PDI reaching this compartment via the autophagy pathway was slow, reflected by the measured slow turnover rates. We concluded that the extension of the mathematical model with a “Golgi to lysosome” pathway and an “ER to lysosome” was able to accommodate both the very diverse N-glycan structures of PDI as well as their experimentally determined turnover rates. Importantly, this mathematical model of processing pathways was able to accommodate the experimental data of differential processing of N-glycans at all the five sites of PDI. It varied the kinetic parameters of individual, site specific processing reactions but the five independent (site-specific) calculations predicted a similar distribution of PDI protein among the different pathways.
PDI is targeted to endosomes by phosphorylated glycans
Our mathematical model predicted two ways of targeting PDI to the lysosome, ER autophagy and an endosomal pathway that affected Golgi-derived glycoprotein. The second pathway resembles the well-known mannose-6-phosphate pathway (Gabel and Kornfeld, 1982; Goldberg and Kornfeld, 1981; Kornfeld et al., 1982) that depends on the phosphorylation of oligomannosidic glycans (Couso et al., 1986; Varki et al., 1983) (Figure 6). These modified glycans serve as a signal for a directed transport of the glycoprotein to the lysosome (Varki and Kornfeld, 2015). Therefore, this protein-specific glycan modification allowed for an experimental evaluation of our model. Based on the detailed knowledge of the Man6P processing (Varki and Kornfeld, 2015) (Varki et al., 1983) (Dahms et al., 1987; Lobel et al., 1987) (Dahms et al., 1989), we developed a Man6P processing pathway (Figure 6) and experimentally addressed the site-specific modification of PDI by PRM mass spectrometry. Indeed, we detected phosphorylated glycans with different, site-specific abundance (Figure S4). Of interest we observed phosphorylated glycans containing up to 10 and 11 hexoses, suggesting that glucosylated species could be processed in the Man6P pathway.
Figure 6.
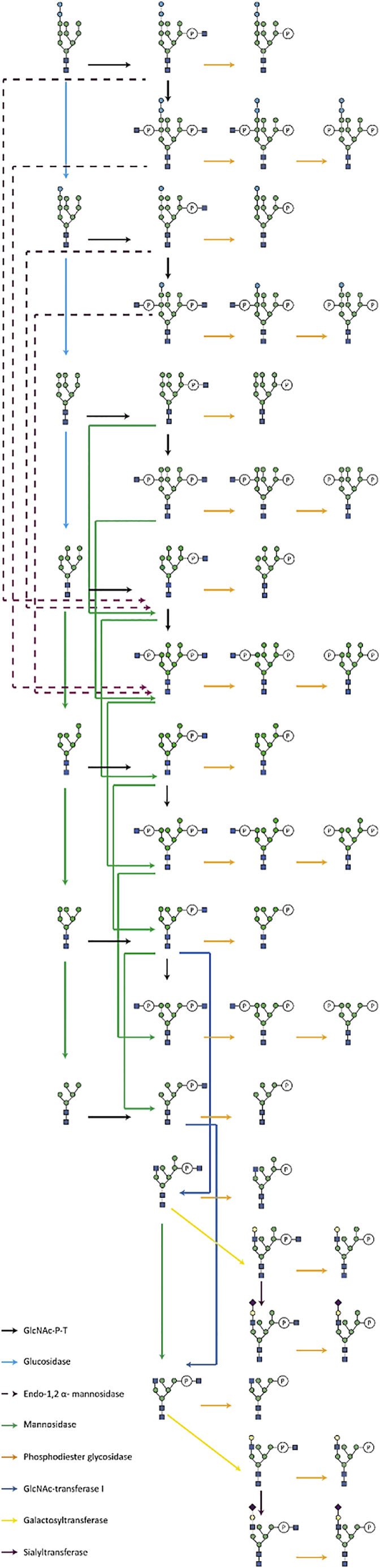
Mannose-6-phosphate pathway
The scheme represents the different processing events of the mannose-6-phosphate pathway. The different enzymatic events are color coded according to the processing enzyme. Potential isoforms of the glycan structures are not represented.
The turnover rate of the phosphorylated glycans was measured in a SILAC-PRM experiment and the results obtained for sites 2 and 5 are presented in Figure 7 (colored shapes). We noted that the turnover rates of the different phosphorylated N-glycans of PDI were very similar, but they differed in their time of the first detection. We also noted a site-specific distribution of phosphorylated N-glycan structures, but their processing rates were not site-specific. Processing of phosphoglycans was faster than that of neutral glycans and followed the kinetics of Golgi and early endosome. We therefore used the same modeling approach as for neutral N-glycans, in the network presented in Figure 6, obtaining model curves (solid lines in Figure 7) that correctly overlay to the experimental observations.
Figure 7.

Phosphorylated glycan turnover
The site-specific phosphorylated glycan turnovers are represented by the plotting of the fractional labeling of each glycan intermediates over time. A representation of the glycan structure was added on the left of its corresponding plot, potential isoforms are not represented. The different sites are color coded as follows: site2 in blue and site5 in yellow and their modeled turnover kinetics are shown as solid line curves of the corresponding colors. Each data point represents the average of three biological replicates. Error bars represent the calculated standard deviations.
Discussion
Processing of N-linked glycans occurs along the secretory pathway and the differential localization of the processing enzymes in defined compartments such as the ER or the Golgi is a hallmark of this eukaryote-specific process. Specific N-glycan processing steps have long been used as a functional definition of the compartments (Dmitrieff and Sens, 2012; Reynders et al., 2011; Zhong, 2011) and defined processing enzymes are markers for these compartments (Dunphy et al., 1985; Rabouille et al., 1995; Velasco et al., 1993). Numerous studies have analyzed the secretory pathway by monitoring N-glycan processing of model proteins or enzyme localization in combination with genetic or pharmacological approaches to characterize the secretory pathway in detail (Kenworthy et al., 2004; Lippincott-Schwartz et al., 1991; Presley et al., 1998; Runge and Robbins, 1986; Stevens et al., 1982; Ziman et al., 1996). However, comprehensive analyses of N-glycan processing remain rare (Chen et al., 2020; Gilgunn et al., 2016; Liu et al., 2020; Venkatakrishnan et al., 2020). This is primarily due to the fact that a precise description of site-specific N-glycan structures has only become available because of recent advances in MS-based glycoproteomics techniques. In combination with time-resolved SILAC approaches, it became possible to characterize the secretory routes of a model glycoprotein and to define the corresponding kinetics of the intracellular N-glycan processing pathways (Arigoni-Affolter et al., 2019). We now extended our experimental approach and followed the processing of the model protein yeast PDI that is N-glycosylated at five sites. N-glycan processing of this protein has been studied extensively using heterologous expression in insect and CHO cells (Hang et al., 2015; Losfeld et al., 2017), the biochemical characterization of individual processing reaction and the structural characterization of its N-glycans (Mathew et al., 2021) as well as the role of N-glycans on the protein structure (Weiß et al., 2021).
Our experimental approach revealed information about two distinct components of the processing pathway of PDI along the secretory pathway: (1) The route taken by the model protein along the pathway and 2) the site-specific processing of the N-glycans in the different compartments.
The modeling of the processing pathway of PDI was based on the analysis of the processing of the 5 different N-glycans of PDI resulting in five independent datasets. Though the processing of the N-glycans is site-specific, the underlying routing of PDI along the pathway must be the same for the corresponding 5 different models. Because of possible bifurcations in the pathway (exit from the ER; exit from the Golgi) several intracellular routes of the protein were possible. We noted that this intracellular pathway of PDI differed significantly from the route defined previously for IgG (neither ERAD nor ERLAD; significant levels of the protein directed to the lysosome), but the relative proportions of these intracellular routes were very similar when independently calculated from the 5 individual glycan processing datasets. This observation not only strongly supported our experimental approach but also confirmed the hypothesis that different proteins take different routes within the cell. Importantly, intracellular processing of a given protein has to be viewed as a composite of the different routes resulting in a heterogeneous pathway that is protein specific. Taking into account the mass balance in our mathematical model, we calculated that 86% of the synthesized PDI was secreted directly from the Golgi, 6% from the endosome and 8% was degraded in the lysosome.
We noted that important routes of intracellular PDI led to the lysosome (as indicated by lysosome-specific processing). We experimentally confirmed the route between the Golgi and the endosomes/lysosomes, with the involvement of the mannose-6-phosphate modification. Indeed, PDI was modified by the P-GlcNAc-transferase and targeted to the lysosome. Although surprising for a protein that is not a lysosomal enzyme, a few other non-lysosomal resident proteins were previously described as modified by the mannose-6-phosphate, such as the DNAse I, uteroferrin or TGF-β proteins (Varki and Kornfeld, 2015). A possible explanation of this modification can be structural as PDI contains many lysine residues, shown to be a recognition motif for the P-GlcNAc-transferase in the case of lysosomal enzymes (Cuozzo and Sahagian, 1994). As an alternative route to the lysosome, we postulated a direct transport of PDI from the ER. Based on the calculated kinetic parameters, this pathway was significantly different/slower than the previously proposed ERLAD pathway (Arigoni-Affolter et al., 2019; Bernasconi and Molinari, 2011) and we tentatively assigned this pathway to the autophagy process of the ER. It is important to note that the kinetics of the different intracellular sections of the secretory pathway differ significantly in PDI processing. Although the exit from the ER to the Golgi results in rapid passage through the Golgi and in rapid secretion or slow processing in the endosomal pathway, the autophagy pathway is characterized by slow N-glycan processing. Therefore, the intracellular pools of N-glycan processing intermediates differ significantly from the secreted products.
The intracellular processing reactions rarely go to completion. Accordingly, site-specific N-glycan heterogeneity observed on secretory proteins is the result of the different reactivity of the N-glycans toward the different mostly incomplete processing reactions, but also of the different intracellular routes that are taken by a given protein.
Yeast PDI heterologously expressed in CHO cells turned out to be an excellent model to analyze this site-specific processing of the N-glycans. Our studies shed light on the route between the Golgi and the endosomes/lysosomes, with the involvement of the mannose-6-phosphate modification. Indeed, PDI was modified by the P-GlcNAc-transferase and targeted to the lysosome. Though it is not clear why PDI glycans are modified by the Man-6-P pathway, we were able to define an elaborate Man-6-P modification pathway that is based on the kinetic data of processing intermediates. Surprisingly, we detected early ER processing structures such as Glc2Man9GlcNAc2 that were phosphorylated by the Golgi localized P-GlcNAc-transferase. Subsequent ER processing intermediates were also found to enter the Man-6-P pathway. Further processing of these structures was possibly linked to the enzymatic activity of the endo-α1,2-mannosidase described to be able to cut out a mannose linked to glucoses residues from the A-branch of glycans (Kukushkin et al., 2012) (Schroder et al., 2018). Furthermore, the kinetics of phosphorylated glycans indicated that once the P-GlcNAc group added, there was no longer a site-specific effect on the kinetic, because all phosphorylated had the same kinetic behavior (Figure 7). The difference of distribution in the phosphorylated structures between the sites could thus be attributed to the distribution of the precursor glycans on each site before phosphorylation.
With respect to processing of N-glycans in the ER, we were surprised to reliably detect the Glc3Man9GlcNAc2 N-glycan, the product of oligosaccharyltransferase, and the subsequent glucosylated processing products at different N-glycosylation sites of PDI. Of interest, our results showed that the Glc3Man9GlcNac2 was turned over much slower than the subsequent Glc2Man9GlcNAc2 structure. This kinetic behavior can only be explained by the coexistence of two separate Glc3Man9GlcNac2 pools that have different accessibility for the glucosidase I processing enzyme. We can only speculate about the nature of these two pools. What comes to mind are the separate and differentially localized action of the two OST complexes, containing STT3A and STT3B (Shrimal et al., 2015, 2019), or two distinct folding pathways of PDI that result in differential accessibility. Irrespective of the interpretation, our experimental approach made it possible to study the kinetics of the very first processing steps of N-glycoprotein biogenesis with unprecedented site-specific and temporal resolution.
Our results add further support to the model that site-specific N-glycan heterogeneity is because of differential kinetic rates of the processing reactions. Evidently, a major determinant of these kinetic rates is the protein context at a given site (Hang et al., 2015; Losfeld et al., 2017; Moremen et al., 2012; Varki, 1998; Weiß et al., 2021). The analysis of the mutant Tyr 178 that affects only site 4 processing now reveals what processing steps are affected by the mutation in vivo. Our data confirmed the in vitro results obtained with purified substrate and enzymes (Mathew et al., 2021).
Of interest, not all the processing reactions in the pathway displayed site-specificity (Figure S4A). For example, we observed that FucT was the most active on site 2 and 5, whereas GnTIV and V was most active on site1 and barely active on other sites. Notably, the order of preferential activity for enzymes like Golgi ManI and GnTI was in agreement with in vitro results published in Mathew et al. (2021). The endosomal hydrolases presented a site-specific activity as well, with the N-acetylglucosaminidase and the fucosidase being most active on site 4.
Our studies on the kinetics of intracellular N-glycoprotein processing revealed a protein-specific sorting among different intracellular routes of the secretory system. This mixed exposure to very different processing pathways increased the heterogeneity of final processing products at a structural and a site-specific level. In addition, the kinetics of N-glycan processing in the individual compartments of the secretory pathway differ significantly: rapid processing, short residence time in and rapid secretion from the Golgi, slow turnover, long residence time and slow secretion/degradation in the endolysosomal compartment. Therefore, intracellularly, our model protein PDI was predominantly present in the endolysosomal compartment but the major portion of the synthesized PDI was secreted via the Golgi pathway. We envisage that this complex secretory machinery combines protein-specific information (via the protein structure) with regulated properties of the processing pathways (enzyme levels, portioning) to yield a highly specific and divers glycoproteome essential for differentiation processes in multicellular eukaryotes. Following the kinetics of intracellular glycoprotein processing via quantitative glycoproteomics will be an essential tool to characterize these regulatory mechanisms. We foresee that the quantitative analysis of the intracellular glycoforms of a given model protein(s) in combination with our mathematical model can describe the “secretory status” of a cell, similar to a “gene activity status” displayed by a transcriptome analysis.
Limitations of the study
Our results highlight the complexity of N-glycoprotein processing in mammalian cells and the necessity for a quantitative analysis of intracellular processing intermediates to understand the site-specific heterogeneity of glycostructures. However, our analytical approach is too complex for a standardized procedure available in non-specialized laboratories. A multitude of detailed quantitative studies using different model proteins will be required for the formulation of a comprehensive mathematical model of the secretory pathway that allows for the prediction of the intracellular processing pathways based on a quantitative analysis of a secreted and mature glycoprotein.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Iodoacetamide | Sigma | I6125 |
| Trypsin | Promega | V5113 |
| Cellvento™ CHO-220 | Merck Millipore | 102577 |
| RPMI | Gibco | 21875034 |
| Penicillin/streptomycin | Corning | 1406-05-9 |
| Ultraglutamine | Lonza | BE17-605E/U1 |
| Recombinant insulin | Sigma | I3536 |
| Lysine | Sigma | L5626 |
| Arginine | Sigma | A5131 |
| 13C6-L-Arginine | Silantes | 201203902 |
| 13C6-15N2-L-Lysine | Silantes | 211603902 |
| Protino Ni-NTA beads | Macherey Nagel | 745400.100 |
| Deposited data | ||
| Mass spectrometry raw data | B-Fabric ETH | Upon request |
| Experimental models: Cell lines | ||
| Freestyle CHO-S | Thermofisher scientific | R80007 |
| Sec-Pdi-CHO | ETH Zurich | Upon request |
| Sec-Y178APdi-CHO | ETH Zurich | Upon request |
| Recombinant DNA | ||
| pcDNA3.1-His-Sec-PDI | ETH Zurich | Upon request |
| pcDNA3.1-His-SecY178A-PDI | ETH Zurich | Upon request |
| Software and algorithms | ||
| Skyline | MacCoss Lab Software | Start Page: /home/software/Skyline |
| Matlab R2020a | Mathworks | https://ch.mathworks.com/products/matlab.html |
| Microsoft Visual Studio 2013 | Microsoft | https://visualstudio.microsoft.com/ |
Resource availability
Lead contact
-
•
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Markus Aebi markus.aebi@micro.biol.ethz.ch
Materials availability
-
•
Cell lines generated in this study have been stored in ETH Microbiology institute repository.
Experimental model and subject details
Cells
Previously cloned vectors pcDNA3.1-His-Sec-PDI and pcDNA3.1-His-SecY178A-PDI were individually transfected into Freestyle CHO-S cells (Thermofisher scientific) as previously described (Losfeld et al., 2017). After 24h, a geneticin selection pressure was applied to transfected cells. To do so, cells were transferred into RPMI supplemented with 10% FBS, 1% Gln, 1% penicillin/streptomycin and 100 μg/ml of geneticin. Adherent surviving cells were passaged 10 times to obtain heterogeneous stably expressing CHO cell lines. The heterogeneous cell lines were then submitted to a sub-cloning by limited dilution to obtain homogeneous cell lines respectively called Sec-PDI-CHO and Sec-Y178APDI-CHO.
Method details
Cell culture
The established stable CHO cell lines were cultured in suspension at 37°C and 350rpm in spin tubes (TPP) using a customized cell culture medium without lysine and arginine, derived from Cellvento™ CHO-220 (Merck-Millipore). Powdered medium was reconstituted in water at 19.54 g/L, added with 2g/L sodium carbonate and supplemented by 0.4% of recombinant insulin (Sigma), 1% ultraglutamine (Lonza), 1% Penicillin/streptomycin (Corning), and by 40 mg/L of lysine (Sigma) and 100 mg/L arginine (Sigma) later called light medium. For SILAC experiments, light arginine and lysine were replaced by heavy labeled 13C6-L-Arginine and 13C6-15N2-L-Lysine (Silantes), later called heavy medium. Cell density was maintained at 1∗106 cells/ml every other day.
Kinetic experiments
Two days before the kinetic analysis, 2 tubes per time point are seeded with 7∗107 cells at a density of 2∗106 cells/ml in light medium. On the day of the experiment, cells are spun down 3minat 100g, and the cell pellet is resuspended in 35ml of heavy medium at 37°C. Cells are then incubated at 37°C and sampled every 20 minutes for 2 hours, and every 30 minutes for the following 3 hours. For the kinetic analysis of the mannose-6-phosphate pathway, cells were sampled every 60 minutes for 5 hours. After incubation, the cells are centrifuged 1minat 300g the supernatant and the cells are flash frozen separately and stored at −80°C until purification.
Denaturing purification
For denaturing purification, the binding, washing, and elution buffer used were containing 6M urea. Intracellular PDI proteins were obtained by lysing the cells in a Tris/Cl 62.5mM buffer at pH 6.8, with 3% Triton-100, then sonicated for 1min30 on ice and centrifuged at 3500 g for 3 min. For the analysis of the mannose-6-phosphate samples, the lysis buffer was supplemented with phosphatase inhibitors: 400 mM tartrate, 100 mM sodium fluoride, 115 mM sodium molybdate. After centrifugation, the soluble fraction was diluted 4 times in PBS/Urea. The cell lysate was then incubated with Protino Ni-NTA beads (Macherey-Nagel) on a rotating wheel for 3hat 4°C. After binding, the beads were washed by 15 column volumes of 25mM imidazole in PBS/Urea, and proteins were eluted by 4 column volumes of 250 mM imidazole in PBS/Urea. Eluted proteins were concentrated with Amicon YM-30 filtering device (Millipore-Sigma), and buffer was exchanged to PBS. The purified proteins were then stored at −20°C before further analyses.
Mass spectrometry samples preparation
Samples were prepared following the FASP protocol used previously (Losfeld et al., 2017; Wisniewski et al., 2009). Briefly, proteins were applied onto Amicon YM-30 filter device (Millipore), reduced for 1h at 37°C by 50 mM dithiothreitol in 50 mM ammonium bicarbonate, alkylated by 65 mM iodoacetamide in 50 mM ammonium bicarbonate for 1hat 37°C. Samples were then washed 3 times by 50 mM ammonium bicarbonate and digested by trypsin (Promega) overnight at 37°Cat a 1/80 μg of protein ratio. Digested peptides were collected by centrifugation and dried.
The samples were then submitted to a batch ZIC-HILIC (Sequant) chromatography (Hagglund et al., 2004). In brief, beads were first washed by 20 volumes of 0.5% FA and spun down 1.5minat 2000g. Beads were then equilibrated 2 times in 20 volumes of 80% ACN, 0.5% FA. 100ul of beads slurry were added to the samples after re-suspension by 50ul of 80% ACN, 0.5% FA. The beads and samples were incubated at room temperature for 30min under agitation. The beads were washed three times with 250ul 80% ACN, 0.5% FA. The peptides were eluted 3 times by 250ul 0.5% FA. The pooled elution was then dried by speedvac.
Peptides were then desalted twice by zip-tip C18 (Millipore). For each sample 3 separate biological replicates were analyzed.
Mass spectrometry method and data acquisition
Samples were analyzed thanks to a Q Exactive HF mass spectrometer from Thermo Fischer Scientific with a PicoviewTM nanospray source 500 model (New Objective) following a method adapted from (Arigoni-Affolter et al., 2019). In brief, the samples were loaded onto a nanoACQUITY UPLC 2G C18 trap column (180 μm × 20 mm, 100 Å, 5 μm particle size) and separated on a nanoACQUITY UPLC BEH130C18 column (75 μm × 250 mm, 130 Å, 1.7 μm particle size), at a constant flow rate of 300 nL/min, with a column temperature of 50°C and a linear gradient of 1−35% acetonitrile/0.1% formic acid in 42 min, followed by a sharp increase to 98% acetonitrile in 2 min and then held isocratically for another 10 min.
The mass spectrometry acquisition was composed of a full scan MS spectrum, followed by 10 sequential PRM scans based on the inclusion list containing the glycopeptides m/z for all potential glycan structures for each glycosites. All scans were acquired with a resolution of 60000 at 100 m/z. Full-scan MS spectra were acquired between 800 and 2000 m/z. For HCD MS/MS runs, the normalized collision energy (NCE) was setup at either 16% for precursor ions with a z = 2 and 11% for precursors with a z = 3 or 4, and an AGC target value of 5 × 105. The quadrupole isolation window for each precursor was 7 m/z. The m/z included in the inclusion list were calculated as the average of (light and heavy peptides) +1 m/z (Tables S1 and S2). The different glycosite containing peptides were acquired thanks to a scheduled run with a retention window per glycopeptide backbone.
Quantification and statistical analysis
Mass spectrometry data analysis using Skyline
MS2-level quantification was performed with the software Skyline (Arigoni-Affolter et al., 2019) by inserting manually the chemical formula for each transition into the software as specific ions. All transitions were chosen among fragment ions generated by the breakage of glycosidic bonds only. In addition, the presence of glycan oxonium ions was also used to confirm the glycopeptides identities. The peak height of the second peak of the isotopic distribution was used for quantification of each transition (Tables S1 and S2).
Mathematical modeling
The mechanistic model used to describe the time and space-dependent formation of the different oligosaccharide structures is based on a previous work developed for humanized monoclonal antibodies produced in CHO cells (Arigoni-Affolter et al., 2019), extended to reproduce the behavior exhibited by PDI throughout the entire pathway. In short, the model comprises four separate compartments (ER, Golgi apparatus, Endosomal system, Lysosome) connected according to Figure 4. Each compartment is characterized by its own set of differential equations to account for glycan processing and protein transport, folding or degradation, when present. Within this framework, each site is uniquely described by the set of parameters that describes the site-specific glycan processing, but shares protein-related parameters (folding, transport, and degradation rates) with all other sites.
Modeling of the N-Glycan pathway in the ER
The first species considered in the model is Glc3Man9. This species is considered as the sum of two different populations, Glc3Man9A and Glc3Man9B, which differ in term of which domain of the OST is responsible for their transfer to the nascent protein. For both species the input term is the cell-specific productivity of heavy-labelled PDI, which in turn depends on the kinetic of incorporation of heavy amino acids in the cytoplasm during RNA translation. To account for these mechanisms, the cell-specific productivity of heavy-labeled PDI was modelled as first order kinetic according to the following equation:
Where is the maximum PDI productivity at steady state (normalized to be equal to 1), and is the time constant that accounts for the intracellular incorporation of heavy amino acids, which was set equal to 10 minutes according to a 13C flux analysis estimation on CHO cells (Duarte et al., 2014).
For simplicity, OST-catalyzed transfer of the Glc3Man9 glycan to the heavy-labeled peptide was assumed to occur co-translationally with peptide synthesis. Partitioning between the A and B pools is dictated by a single factor , such that:
with representing the input terms in the mass balance for Glc3Man9A and Glc3Man9B and .
After glucose trimming, both pools converge to a single Man9 glycan population. At this stage, the glycan can be further trimmed down to Man5 with the action of the ER mannosidases in the quality control machinery of the ER, or it can fold and become a possible substrate for translocation to the Golgi apparatus. Folding was assumed to be a single-step process, characterized by a single kinetic constant , which is the same for all 5 sites (folding is assumed to be completed once all domains are folded). Transport to the Golgi was assumed to be fast (no accumulation of folded species in the ER) and efficient (no misfolded species are transported).
In order to better represent the experimentally observed data and kinetic trends for PDI, an autophagy pathway was implemented in the model to account for ER-to-Lysosome transport of PDI. The reaction rate for this step is represented with a single kinetic constant and results in the generation of an additional pool of Man9-5 glycans in a separate compartment. These glycans can be further trimmed down to oligomannose species with less than 2 mannoses by the action of lysosomal mannosidases or they can be removed from the system due to degradation of the entire protein (represented by the kinetic term .
The mass balances for singe oligosaccharides in the ER assume homogeneous distribution of enzymes and substrates (perfect mixing hypothesis). For a given oligosaccharide , the mass balance can be written as follow:
The term is the input term and is zero for all species except for Glc3Man9. The term in the summation accounts for all possible reactions in the ER network where is either a reactant or a product. The notation uses the coefficients and for substrates and reactions respectively, whereas and represent the stoichiometric coefficient and the reaction rate for the reaction considered. The term represents outward transport to Golgi and Lysosome due to correct folding or autophagy and assumes the form:
Where is the appropriate kinetic constant for transport.
Modeling of the N-Glycan pathway in the Golgi apparatus
Correctly folded PDI structures travel across the Golgi and are modified by the sequential action of compartmentalized enzymes. Transport across the compartment occurs at constant linear velocity, with residence time in the compartment set at 20 min.
To represent enzyme segregation across the Golgi the axial coordinate, the activity of Golgi-resident enzymes was modelled as Gaussian functions:
Where is the activity of enzyme at position and ,, , represent the peak height, the peak position and the width of the enzymatic window of activity, respectively.
Similar to the ER, the reaction rates for enzyme-catalyzed reactions in the Golgi follow first order kinetic with respect to enzyme concentration. At each position the reaction rate for a reaction catalyzed by a generic enzyme can be written in the form:
Golgi-to-endosome transport was modelled by including a sorting enzyme with similar characteristics (first-order kinetic, Gaussian distribution for activity). This sorting enzyme acts equally on all oligosaccharides without restrictions, removing them from the Golgi and transferring an equal amount to the Endosomal system.
The mass balance for oligosaccharides traveling the Golgi accounts for concentrations that vary both in time and space (where space is the axial position in the Golgi) according to the equation below:
The terms and represent the length and residence time of the Golgi apparatus, respectively. The length of the Golgi was arbitrarily set to 1. The initial condition (at ) is given by the amount has been transported from the ER during the integration interval . Species that reach are secreted in the extracellular environment.
Modeling of the N-Glycan pathway in the endosomal compartment
The endosomal system is mathematically similar to the Golgi apparatus: the PDI pool that was transported from the Golgi apparatus travels in space along an axis (starting from the initial position 0) at constant speed while it is being modified by specific enzymes.
The distribution of enzyme activity along the apparatus was assumed to follow a logistic behavior according to the following equation:
With representing a different pH optimum for the different enzymes (pH is assumed to vary uniformly along ) and is arbitrarily set to 15.
The oligosaccharides that reach the end of the endosomal compartment (normalized to ) are either secreted (and add to the pool of secreted species from the Golgi) or transported to the lysosomal compartment for final degradation (and add to the pool from the ER). The best representation of the experimental data was achieved by assuming 45% of the total endosomal pool being secreted and 55% being transported to the Lysosome for degradation.
The structure of the mass balance equation is the same as the one used in the Golgi apparatus but acts upon a different stoichiometric network.
Modeling of the N-Glycan pathway in the lysosome
In the model, the lysosome receives material from ER and Endosome and provides a way to close the intracellular mass balance by accounting for the degradation of PDI and attached oligosaccharides. Mathematically, the lysosome behaves in the same way as the ER, with uniform distribution of enzymes and substrate. For each oligosaccharide, two reactions are possible: the first one involves trimming of the accessible monosaccharides, according to the same stoichiometric network and kinetic parameters of the endosomal compartments (only peak activity is considered for the enzymes, since it is assumed that enzymes in the lysosome work at optimal pH), while the second one involves degradation of the entire protein (degradation of the protein also removes the glycan from the system). The latter is characterized by a single parameter, , which is the same for all sites and all structures.
The structure of the mass balance equation is the same as the one used in the ER.
Model construction and parameter estimation
The systems of ordinary differential equations (ODEs) for ER and Lysosome were solved in Matlab R2020a (Mathworks, MA). The system of partial differential equations of Golgi and Endosome were solved in FORTRAN using the solver DLSODES from ODEPACK. The solution was discretized using first-order central finite difference and 50 grid points. The entire system was simulated for 360 min for the Golgi and 1800 min for the Endosome, with a resolution of 50 and 60 minutes respectively. Parameter optimization was performed using the GDE3 algorithm variant of the genetic algorithm. The objective function was defined as the sum of least square errors between experimental data and model predictions for heavy-to-light ratios, intracellular and extracellular fractions. The computations were run separately for each site, until common parameters converged to a single set that could fit all experimental data. Parameter estimation was performed on a computing cluster. The limit of the genetic algorithm was set to 250 generations and it took approximately 2 days to compute all generations.
Acknowledgments
We thank the Functional Genomics Center Zurich for help with the mass spectrometry analyses. We thank Dr. Ilaria Arigoni-Affolter for the fruitful discussions. This work was supported by ETH Zurich and the Swiss National Science Foundation grant 310030_162636 to M.A.
Author contributions
Conceptualization, M.A. and M.E.L.; Methodology, M.E.L., E.S., and C.W.T.L.; Investigation, M.E.L. and E.S.; Validation, M.E.L. and C.W.T.L.; Formal Analysis, E.S. and M.E.L., Writing – original draft, M.E.L., E.S. and M.A., Visualization, M.E.L.; Supervision, M.A. and M.E.L., Funding acquisition, M.A.
Declaration of interests
The authors declare no competing interest.
Published: November 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105417.
Supplemental information
Data and code availability
-
•
Mass spectrometry raw data reported in this paper were deposited on B-Fabric data management from the FGCZ.
-
•
Code reported in this paper will be shared by the lead contact upon request.
-
•
All data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Arigoni-Affolter I., Scibona E., Lin C.W., Bruhlmann D., Souquet J., Broly H., Aebi M. Mechanistic reconstruction of glycoprotein secretion through monitoring of intracellular N-glycan processing. Sci. Adv. 2019;5:eaax8930. doi: 10.1126/sciadv.aax8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F., Chia J. Cracking the glycome encoder: signaling, trafficking, and glycosylation. Trends Cell Biol. 2016;26:379–388. doi: 10.1016/j.tcb.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Bernasconi R., Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 2011;23:176–183. doi: 10.1016/j.ceb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braulke T., Bonifacino J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta. 2009;1793:605–614. doi: 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Breitling J., Aebi M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harbor Perspect. Biol. 2013;5:a013359. doi: 10.1101/cshperspect.a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Wang R., Li D., Zuo C., Wen P., Liu H., Chen Y., Fujita M., Wu Z., Yang G. Comprehensive analysis of the glycome and glycoproteome of bovine milk-derived exosomes. J. Agric. Food Chem. 2020;68:12692–12701. doi: 10.1021/acs.jafc.0c04605. [DOI] [PubMed] [Google Scholar]
- Chi B., Veyssier C., Kasali T., Uddin F., Sellick C.A. At-line high throughput site-specific glycan profiling using targeted mass spectrometry. Biotechnol. Rep. (Amst) 2020;25:e00424. doi: 10.1016/j.btre.2020.e00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley K.J., Varki A., Kinoshita T. In: Essentials of Glycobiology. Varki A., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., Darvill A.G., Kinoshita T., Packer N.H., et al., editors. 2015. Cellular organization of glycosylation; pp. 41–49. [DOI] [Google Scholar]
- Couso R., Lang L., Roberts R.M., Kornfeld S. Phosphorylation of the oligosaccharide of uteroferrin by UDP-GlcNAc:glycoprotein N-acetylglucosamine-1-phosphotransferases from rat liver, Acanthamoeba castellani, and Dictyostelium discoideum requires alpha 1,2-linked mannose residues. J. Biol. Chem. 1986;261:6326–6331. [PubMed] [Google Scholar]
- Cuozzo J.W., Sahagian G.G. Lysine is a common determinant for mannose phosphorylation of lysosomal proteins. J. Biol. Chem. 1994;269:14490–14496. [PubMed] [Google Scholar]
- Dahms N.M., Lobel P., Breitmeyer J., Chirgwin J.M., Kornfeld S. 46 kd mannose 6-phosphate receptor: cloning, expression, and homology to the 215 kd mannose 6-phosphate receptor. Cell. 1987;50:181–192. doi: 10.1016/0092-8674(87)90214-5. [DOI] [PubMed] [Google Scholar]
- Dahms N.M., Lobel P., Kornfeld S. Mannose 6-phosphate receptors and lysosomal enzyme targeting. J. Biol. Chem. 1989;264:12115–12118. [PubMed] [Google Scholar]
- Del Val I.J., Polizzi K.M., Kontoravdi C. A theoretical estimate for nucleotide sugar demand towards Chinese Hamster Ovary cellular glycosylation. Sci. Rep. 2016;6:28547. doi: 10.1038/srep28547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell A., Galadari A., Sastre F., Hitchen P. Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int. J. Microbiol. 2010;2010:148178. doi: 10.1155/2010/148178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitrieff S., Sens P. Golgi apparatus: homotypic fusion maintains biochemical gradients within the Golgi and improves the accuracy of protein maturation. Int. J. Biochem. Cell Biol. 2012;44:718–721. doi: 10.1016/j.biocel.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Duarte T.M., Carinhas N., Barreiro L.C., Carrondo M.J., Alves P.M., Teixeira A.P. Metabolic responses of CHO cells to limitation of key amino acids. Biotechnol. Bioeng. 2014;111:2095–2106. doi: 10.1002/bit.25266. [DOI] [PubMed] [Google Scholar]
- Dunphy W.G., Brands R., Rothman J.E. Attachment of terminal N-acetylglucosamine to asparagine-linked oligosaccharides occurs in central cisternae of the Golgi stack. Cell. 1985;40:463–472. doi: 10.1016/0092-8674(85)90161-8. [DOI] [PubMed] [Google Scholar]
- Gabel C.A., Kornfeld S. Lysosomal enzyme phosphorylation in mouse lymphoma cell lines with altered asparagine-linked oligosaccharides. J. Biol. Chem. 1982;257:10605–10612. [PubMed] [Google Scholar]
- Gilgunn S., Millán Martín S., Wormald M.R., Zapatero-Rodríguez J., Conroy P.J., O'Kennedy R.J., Rudd P.M., Saldova R. Comprehensive N-glycan profiling of avian immunoglobulin Y. PLoS One. 2016;11:e0159859. doi: 10.1371/journal.pone.0159859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg D.E., Kornfeld S. The phosphorylation of beta-glucuronidase oligosaccharides in mouse P388D1 cells. J. Biol. Chem. 1981;256:13060–13067. [PubMed] [Google Scholar]
- Hagglund P., Bunkenborg J., Elortza F., Jensen O.N., Roepstorff P. A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J. Proteome Res. 2004;3:556–566. doi: 10.1021/pr034112b. [DOI] [PubMed] [Google Scholar]
- Hang I., Lin C.W., Grant O.C., Fleurkens S., Villiger T.K., Soos M., Morbidelli M., Woods R.J., Gauss R., Aebi M. Analysis of site-specific N-glycan remodeling in the endoplasmic reticulum and the Golgi. Glycobiology. 2015;25:1335–1349. doi: 10.1093/glycob/cwv058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan-Colas J., Hitchcock I.S., Coles M., Johnson S., Krauss T.F. Quantifying single-cell secretion in real time using resonant hyperspectral imaging. Proc. Natl. Acad. Sci. USA. 2018;115:13204–13209. doi: 10.1073/pnas.1814977115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy A.K., Nichols B.J., Remmert C.L., Hendrix G.M., Kumar M., Zimmerberg J., Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J. Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld S., Reitman M.L., Varki A., Goldberg D., Gabel C.A. Steps in the phosphorylation of the high mannose oligosaccharides of lysosomal enzymes. Ciba Found. Symp. 1982:138–156. doi: 10.1002/9780470720745.ch8. [DOI] [PubMed] [Google Scholar]
- Kukushkin N.V., Easthope I.S., Alonzi D.S., Butters T.D. Restricted processing of glycans by endomannosidase in mammalian cells. Glycobiology. 2012;22:1282–1288. doi: 10.1093/glycob/cws088. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Glickman J., Donaldson J.G., Robbins J., Kreis T.E., Seamon K.B., Sheetz M.P., Klausner R.D. Forskolin inhibits and reverses the effects of brefeldin A on Golgi morphology by a cAMP-independent mechanism. J. Cell Biol. 1991;112:567–577. doi: 10.1083/jcb.112.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Fu Y., Huang Z., Liu Y., Liu B.F., Cheng L., Liu X. A comprehensive analysis of subclass-specific IgG glycosylation in colorectal cancer progression by nanoLC-MS/MS. Analyst. 2020;145:3136–3147. doi: 10.1039/d0an00369g. [DOI] [PubMed] [Google Scholar]
- Lobel P., Dahms N.M., Breitmeyer J., Chirgwin J.M., Kornfeld S. Cloning of the bovine 215-kDa cation-independent mannose 6-phosphate receptor. Proc. Natl. Acad. Sci. USA. 1987;84:2233–2237. doi: 10.1073/pnas.84.8.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losfeld M.E., Scibona E., Lin C.W., Villiger T.K., Gauss R., Morbidelli M., Aebi M. Influence of protein/glycan interaction on site-specific glycan heterogeneity. Faseb. J. 2017;31:4623–4635. doi: 10.1096/fj.201700403R. [DOI] [PubMed] [Google Scholar]
- Mathew C., Weiß R.G., Giese C., Lin C.W., Losfeld M.E., Glockshuber R., Riniker S., Aebi M. Glycan-protein interactions determine kinetics of N-glycan remodeling. RSC Chem. Biol. 2021;2:917–931. doi: 10.1039/d1cb00019e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen K.W., Tiemeyer M., Nairn A.V. Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae M., Yamaguchi Y. Biophysical analyses for probing glycan-protein interactions. Adv. Exp. Med. Biol. 2018;1104:119–147. doi: 10.1007/978-981-13-2158-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narimatsu Y., Joshi H.J., Nason R., Van Coillie J., Karlsson R., Sun L., Ye Z., Chen Y.H., Schjoldager K.T., Steentoft C., et al. An atlas of human glycosylation pathways enables display of the human glycome by gene engineered cells. Mol. Cell. 2019;75:394–407.e5. doi: 10.1016/j.molcel.2019.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X., Canuel M., Morales C.R. The sorting and trafficking of lysosomal proteins. Histol. Histopathol. 2006;21:899–913. doi: 10.14670/HH-21.899. [DOI] [PubMed] [Google Scholar]
- North S.J., Huang H.H., Sundaram S., Jang-Lee J., Etienne A.T., Trollope A., Chalabi S., Dell A., Stanley P., Haslam S.M. Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J. Biol. Chem. 2010;285:5759–5775. doi: 10.1074/jbc.M109.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley J.F., Smith C., Hirschberg K., Miller C., Cole N.B., Zaal K.J., Lippincott-Schwartz J. Golgi membrane dynamics. Mol. Biol. Cell. 1998;9:1617–1626. doi: 10.1091/mbc.9.7.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C., Hui N., Hunte F., Kieckbusch R., Berger E.G., Warren G., Nilsson T. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J. Cell Sci. 1995;108:1617–1627. doi: 10.1242/jcs.108.4.1617. [DOI] [PubMed] [Google Scholar]
- Reynders E., Foulquier F., Annaert W., Matthijs G. How Golgi glycosylation meets and needs trafficking: the case of the COG complex. Glycobiology. 2011;21:853–863. doi: 10.1093/glycob/cwq179. [DOI] [PubMed] [Google Scholar]
- Rose D.R. Structure, mechanism and inhibition of Golgi alpha-mannosidase II. Curr. Opin. Struct. Biol. 2012;22:558–562. doi: 10.1016/j.sbi.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Ruiz-Canada C., Kelleher D.J., Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell. 2009;136:272–283. doi: 10.1016/j.cell.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge K.W., Robbins P.W. A new yeast mutation in the glucosylation steps of the asparagine-linked glycosylation pathway. Formation of a novel asparagine-linked oligosaccharide containing two glucose residues. J. Biol. Chem. 1986;261:15582–15590. [PubMed] [Google Scholar]
- Schroder S.P., Kallemeijn W.W., Debets M.F., Hansen T., Sobala L.F., Hakki Z., Williams S.J., Beenakker T.J.M., Aerts J., van der Marel G.A., et al. Spiro-epoxyglycosides as activity-based probes for glycoside hydrolase family 99 endomannosidase/endomannanase. Chemistry. 2018;24:9983–9992. doi: 10.1002/chem.201801902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott C.C., Vacca F., Gruenberg J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014;31:2–10. doi: 10.1016/j.semcdb.2014.03.034. [DOI] [PubMed] [Google Scholar]
- Shrimal S., Cherepanova N.A., Gilmore R. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin. Cell Dev. Biol. 2015;41:71–78. doi: 10.1016/j.semcdb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimal S., Cherepanova N.A., Mandon E.C., Venev S.V., Gilmore R. Asparagine-linked glycosylation is not directly coupled to protein translocation across the endoplasmic reticulum in Saccharomyces cerevisiae. Mol. Biol. Cell. 2019;30:2626–2638. doi: 10.1091/mbc.E19-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M., Wilkinson S. ER homeostasis and autophagy. Essays Biochem. 2017;61:625–635. doi: 10.1042/EBC20170092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P., Taniguchi N., Aebi M. In: Essentials of Glycobiology. Varki A., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., Darvill A.G., Kinoshita T., Packer N.H., et al., editors. 2015. N-Glycans; pp. 99–111. [DOI] [Google Scholar]
- Stevens T., Esmon B., Schekman R. Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell. 1982;30:439–448. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y., Rabinovich G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008;9:593–601. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- Varki A. Factors controlling the glycosylation potential of the Golgi apparatus. Trends Cell Biol. 1998;8:34–40. doi: 10.1016/s0962-8924(97)01198-7. [DOI] [PubMed] [Google Scholar]
- Varki A., Kornfeld S. In: Essentials of Glycobiology. Varki A., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., Darvill A.G., Kinoshita T., Packer N.H., et al., editors. 2015. P-type lectins; pp. 423–433. [DOI] [PubMed] [Google Scholar]
- Varki A., Sherman W., Kornfeld S. Demonstration of the enzymatic mechanisms of alpha-N-acetyl-D-glucosamine-1-phosphodiester N-acetylglucosaminidase (formerly called alpha-N-acetylglucosaminylphosphodiesterase) and lysosomal alpha-N-acetylglucosaminidase. Arch. Biochem. Biophys. 1983;222:145–149. doi: 10.1016/0003-9861(83)90511-8. [DOI] [PubMed] [Google Scholar]
- Velasco A., Hendricks L., Moremen K.W., Tulsiani D.R., Touster O., Farquhar M.G. Cell type-dependent variations in the subcellular distribution of alpha-mannosidase I and II. J. Cell Biol. 1993;122:39–51. doi: 10.1083/jcb.122.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan V., Dieckmann R., Loke I., Tjondro H.C., Chatterjee S., Bylund J., Thaysen-Andersen M., Karlsson N.G., Karlsson-Bengtsson A. Glycan analysis of human neutrophil granules implicates a maturation-dependent glycosylation machinery. J. Biol. Chem. 2020;295:12648–12660. doi: 10.1074/jbc.RA120.014011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiß R.G., Losfeld M.E., Aebi M., Riniker S. N-glycosylation enhances conformational flexibility of protein disulfide isomerase revealed by microsecond molecular dynamics and Markov state modeling. J. Phys. Chem. B. 2021;125:9467–9479. doi: 10.1021/acs.jpcb.1c04279. [DOI] [PubMed] [Google Scholar]
- Wild R., Kowal J., Eyring J., Ngwa E.M., Aebi M., Locher K.P. Structure of the yeast oligosaccharyltransferase complex gives insight into eukaryotic N-glycosylation. Science. 2018;359:545–550. doi: 10.1126/science.aar5140. [DOI] [PubMed] [Google Scholar]
- Wisniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wu Z., Liu Y., Li L., Wan X.F., Zhu H., Guo Y., Wei M., Guan W., Wang P.G. Decoding glycan protein interactions by a new class of asymmetric N-glycans. Org. Biomol. Chem. 2017;15:8946–8951. doi: 10.1039/c7ob02303k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H., Sun F., Suttapitugsakul S., Wu R. Global and site-specific analysis of protein glycosylation in complex biological systems with Mass Spectrometry. Mass Spectrom. Rev. 2019;38:356–379. doi: 10.1002/mas.21586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W. Golgi during development. Cold Spring Harbor Perspect. Biol. 2011;3:a005363. doi: 10.1101/cshperspect.a005363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Desaire H. Carbohydrates on proteins: site-specific glycosylation analysis by mass spectrometry. Annu. Rev. Anal. Chem. 2015;8:463–483. doi: 10.1146/annurev-anchem-071114-040240. [DOI] [PubMed] [Google Scholar]
- Zielinska D.F., Gnad F., Schropp K., Wisniewski J.R., Mann M. Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol. Cell. 2012;46:542–548. doi: 10.1016/j.molcel.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Ziman M., Chuang J.S., Schekman R.W. Chs1p and Chs3p, two proteins involved in chitin synthesis, populate a compartment of the Saccharomyces cerevisiae endocytic pathway. Mol. Biol. Cell. 1996;7:1909–1919. doi: 10.1091/mbc.7.12.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Mass spectrometry raw data reported in this paper were deposited on B-Fabric data management from the FGCZ.
-
•
Code reported in this paper will be shared by the lead contact upon request.
-
•
All data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.