

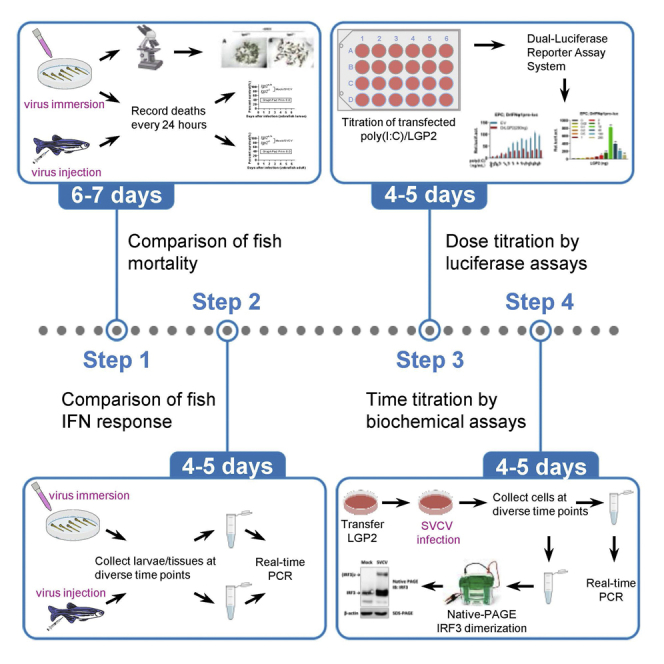
Summary
Here, we present a protocol to characterize zebrafish LGP2 as a dual regulator of interferon (IFN) response. We detail in vivo assays using time-lapse comparison of IFN response between wild-type and lgp2 knockout zebrafish following spring viraemia of carp virus (SVCV) infection. We also describe in vitro assays including titration of infection duration in SVCV-infected fish cells to determine changes in IFN response. This protocol is effective to illuminate a regulatory switch of LGP2 in fish cells toward virus infection.
For complete details on the use and execution of this protocol, please refer to Gong et al. (2022).1
Subject areas: Immunology, Model Organisms, Molecular Biology, Signal Transduction, Protein Biochemistry
Graphical abstract
Highlights
-
•
Protocol to identify a dual regulation of zebrafish LGP2 following viral infection
-
•
In vivo assays to compare fish mortality and IFN responses following viral infection
-
•
In vitro assays including dose titration and time titration in SVCV-infected fish cells
-
•
Applicable to a variety of fish cell types having IFN response to stimuli
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we present a protocol to characterize zebrafish LGP2 as a dual regulator of interferon (IFN) response. We detail in vivo assays using time-lapse comparison of IFN response between wild-type and lgp2 knockout zebrafish following spring viraemia of carp virus (SVCV) infection. We also describe in vitro assays including titration of infection duration in SVCV-infected fish cells to determine changes in IFN response. This protocol is effective to illuminate a regulatory switch of LGP2 in fish cells toward virus infection.
Before you begin
LGP2 (Laboratory of genetics and physiology 2) lacks the N-terminal CARDs (caspase activation and recruitment domains) that are required for signaling transduction in RIG-I (Retinoic acid inducible gene-I) and MDA5 (melanoma differentiation associated gene 5). In mammals, LGP2 is initially identified as a feedback inhibitor of RIG-I/MDA5 signaling but subsequently as an activator of MDA5 signaling and an inhibitor of RIG-I signaling.2 We previously established that zebrafish LGP2 is a dual regulator of fish IFN response toward viral infection.1,3 The protocol below describes the detailed steps in zebrafish larvae and adults as well as in cultured fish cells to determine LGP2’s dual regulation of fish IFN response. Before proceeding, it is imperative to generate lgp2-deficient zebrafish and validate a normal IFN response of cultured fish cells when infected with virus. The key point is to preestablish titration methods, including intensive titration of viral titer and infection duration, the doses of diverse stimuli, such as poly(I:C) and even LGP2 itself. Given the conservation and disparities of zebrafish and human LGP2, this protocol can be used in cultured mammalian cells, including HEK293T and COS7 cells, with a minor modification.4
Institutional permissions
Animal experiments and treatments were performed according to the Guide for Animal Care and Use Committee of Institute of Hydrobiology, Chinese Academy of Sciences (IHB, CAS, Protocol No. 2016–018).
Culture fish cell lines
Timing: 1 day
-
1.
Grow epithelioma papulosum cyprini cells (EPC) in medium 199 (Gibco) supplemented with 10% fetal bovine serum (FBS) at 28°C in a humidified incubator containing 5% CO2.
CRITICAL: Cell culture must be undertaken in microbiological safety cabinet using aseptic technique to ensure sterility. EPC cells are used here due to relatively high transfection efficiency and the fact that RLR-triggered IFN response can be easily detected when EPC cells respond to SVCV infection. EPC cells can be replaced by other fish cell lines with similar characteristics, such as grass carp C. idellus ovary cells (CO), and crucian carp (C. auratus) blastulae embryonic cells (CAB) in our lab.
Prepare SVCV solution
-
2.Propagate SVCV.
-
a.Seed EPC cells in 25 cm2 cell culture flasks to reach monolayers after overnight (8–14 h) culture.
-
b.Pour off medium carefully and wash cells with prewarmed serum-free M199 medium or PBS.
-
c.Inoculate virus by pouring 0.5 mL 1×103 TCID50/mL (tissue culture infective dose 50% per mL) SVCV to the culture flask for 20–30 min.
-
d.Roll flask gently to ensure that all cells contact SVCV during inoculation.
-
e.Pipette out SVCV and wash cells again with prewarmed serum-free M199 medium.
-
f.Add 4–5 mL prewarmed M199 medium containing 0.5%–2% FBS, and place flasks in a 28°C incubator for at least 2–3 days.
-
g.Freeze and thaw cells three times to thoroughly release virus when > 80% cytopathic effects (CPEs) appear.
-
h.Move all lysates to 50 mL aseptic centrifuge tubes after mixing well by pipettes.
-
i.Centrifugate at 13,000 × g for 15 min.
-
j.Sterilize supernatants through 0.22 μm membrane filter and store at −80°C.
-
a.
-
3.Titrate SVCV virus.
-
a.Put EPC cells in 96-well plates (100 μL/well) overnight (8–14 h) to reach a monolayer.
-
b.10-fold serially dilute SVCV virus in 96-well plates or microcentrifuge tubes. Make sure you resuspend the dilutions very well before going to the next dilution.
-
c.Pour off medium carefully, and wash cells with prewarmed serum-free M199 medium or PBS.
-
d.Transfer virus dilutions to 96-well plates, with 8 wells each gradient and 100 μL each well.
-
e.Leave the plates in a 28°C incubator for 5–7 days to ensure viral propagation.
-
f.Check daily under an inverted microscope.
-
g.Stain cells by dropping 1% crystal violet solution for 1–2 h.
-
h.Discard the dye solution and calculate TCID50/mL.
-
a.
Breed WT (lgp2+/+) or lgp2−/− zebrafish
-
4.Keep zebrafish in a circulation system, which continuously filters and aerates the system water.
-
a.Store aeration water in the large water tank to supplement the loss of system water.
-
b.Maintain the room temperature (RT) to be between 27°C–28°C.
-
c.Set the system water temperature to be between 28°C–28.5°C, and the lighting conditions at 14:10 h (light:dark).
-
a.
-
5.
Transfer mature WT and lgp2−/− zebrafish into two 3-L tanks for breeding at the night before embryo collection. The ratio of females to males is 2:1 or 1:1.
-
6.
Turn on the lights in the next morning, and you can see that females are chased by males for breeding under the stimulation of light.
-
7.
Collect and mix all embryos from similar clutches within 3 h post mating.
-
8.
Wash embryos with system water to remove debris.
-
9.
Transfer about 200 embryos to a 10-cm dish filled with fresh system water in a 28°C incubator. The incubator provides physiological light/dark cycle to ensure normal development.
-
10.
Remove dead embryos daily to keep the system water clean. The embryos develop into larvae 3 days post fertilization (dpf).
-
11.
Perform viral-infection assays with zebrafish larvae or adults.
Prepare buffers
-
12.
Make 1% crystal violet solution.
-
13.
Make 1× Tris-Gly working buffer (inside and outside inner modules), TBST, transfer buffer, 5% milk according to the recipe provided below.
-
14.
Make 8% native-PAGE gels according to the recipe provided below.
Prepare sampling tools
-
15.
Bake animal dissection forceps, animal anatomical scissors and ceramic beads in an oven at 240°C for 4 h to inactivate the residue RNase.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CaIRF3 Rabbit mAb (1:5000) | Sun et al.6 | N/A |
| Zebrafish LGP2 Rabbit mAb (1:5000) | Gong et al.1 | N/A |
| β-Actin Rabbit mAb (1:5000) | Cell Signaling Technology | Cat# 8457; RRID: AB_10950489 |
| HRP Goat Anti-Rabbit IgG (H+L) (secondary antibody) (1:5000) | ABclonal, China | Cat# AS014, RID: AB_2769854 |
| Bacterial and virus strains | ||
| Spring viraemia of carp virus (SVCV) | Gong et al.1 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Protease inhibitor cocktail | Bimake | Cat# B14002 |
| Phosphatase inhibitor cocktail | Bimake | Cat# B15001 |
| poly(I:C) | SIGMA | Cat# P0913; CAS: 42424-50-0 |
| RNAlater RNA stabilization reagent | QIAGEN | Cat# 76106 |
| Methylene blue | Solarbio, China | Cat# M8030 |
| MS-222 | Alfa | Cat# 1809123 |
| Crystal violet stain solution (1%) | Solarbio, China | Cat# G1062 |
| Penicillin-streptomycin (100×), sterile | MedChemExpress | Cat# HY-K1006 |
| RIPA lysis buffer (weak) | Beyotime, China | Cat# P0013D |
| Non-denaturing PAGE loading buffer (5×) | Beyotime, China | Cat# P0016 |
| Sodium deoxycholate | Macklin, China | Cat# D6128; CAS: 302-95-4 |
| Ammonium persulfate | Macklin, China | Cat# A801034; CAS: 7727-54-0 |
| 1 M Tris-HCl (pH=8.8) | Biosharp, China | Cat# BL515B |
| 1 M Tris-HCl (pH=6.8) | Biosharp, China | Cat# BL514A |
| 30% Acrylamide-bisacrylamide (29:1) | Biosharp, China | Cat# BL513B |
| N, N, N′, N′-Tetramethylethylenediamine (TEMED) | Beyotime, China | Cat# ST728 |
| Glycin | Genview | Cat# FG149 |
| Tris-base | Biofroxx | Cat# 1115KG001; CAS: 77-86-1 |
| Polyethylenimine, linear (MW 25,000) (PEI) | Sigma-Aldrich | Cat# 23966-1 |
| PBS | Monad | Cat# CR10601S |
| Tween-20 | DING GUO, China | Cat# DH358-4 |
| Clarity Western ECL substrate | BIO-RAD | Cat# 1705061 |
| Methanol | SCR, China | Cat# 40064260; CAS: 67-56-1 |
| Non-fat powdered milk | BBI, China | Cat# A600669 |
| medium 199 (Gibco) | HyClone | Cat# SH30253.01 |
| Fetal bovine serum (FBS) | WISENT | Cat# 086-150 |
| Opti-mem | Invitrogen | Cat# 31985-070 |
| Critical commercial assays | ||
| Endo-free Plasmid Mini Kit II | OMEGA | Cat# D6950-02 |
| SV Total RNA Isolation | Promega | Cat# Z3100 |
| TRIzol Plus RNA Purification kit | Invitrogen | Cat#12183555 |
| GoScript™ Reverse Transcription System | Promega | Cat# A5001 |
| Universal Blue qPCR SYBR Green Master Mix | YEASEN, China | Cat# 11201ES08 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat# P0913 |
| Deposited data | ||
| Raw and analyzed data | This paper | Lead contact, Yibing Zhang (ybzhang@ihb.ac.cn) |
| Experimental models: Cell lines | ||
| Epithelioma papulosum cyprini cells (EPC) | ATCC (CRL-2872) | N/A |
| Ovary cells of grass carp (CO) | Kept in IHB, CAS | N/A |
| Crucian carp (C. auratus L.) blastulae embryonic cells (CAB) | Chen et al.7 | N/A |
| Experimental models: Organisms/strains | ||
| Zebrafish (Danio rerio) strain AB: larvae (6 pdf) and adults (60 dpf, male: female =1:1) | China Zebrafish Resource Center | N/A |
| Recombinant DNA | ||
| pcDNA3.1(+) | Invitrogen | Cat# V79020 |
| DrIFNφ1pro-luc (expression plasmid) | Sun et al.8 | N/A |
| DrLGP2 (expression plasmid) | Gong et al.1 | N/A |
| pRL-TK (expression plasmid) | Promega | Cat# E2241 |
| Software and algorithms | ||
| GraphPad Prism 8.0 | Dotmatics | https://www.graphpad.com/ |
| Other | ||
| Mini-PROTEAN® tetra system | BIO-RAD | Cat# 1658004 |
| Mini Trans-Blot® Electrophoretic Transfer Cell | BIO-RAD | Cat# 170-3930 |
| Mini-PROTEAN® system casting stand | BIO-RAD | Cat# 1658050 |
| LUMINESCENT IMAGE ANALYZER | LISTED | Model: ImageQuant LAS 4000 mini |
| 24-well plate | LABSELECT, China | Cat# 11320 |
| RNase-free microcentrifuge tubes | BBI, China | Cat# F611541-0001 |
| Animal dissection forceps | Fine Science Tools | Cat# 11295-10 |
| Animal anatomical scissors | Easybio, China | CAT#BE6621 |
| Ceramic beads, 1.4 mm (325 g) | QIAGEN | CAT#13113-325 |
Materials and equipment
10× tris-glycine (Tris-Gly) stock buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-base | 250 mM | 30.2 g |
| Glycine | 2 M | 144 g |
| ddH2O | N/A | Add to 1 L |
| Total | N/A | 1 L |
Store at room temperature (20°C–25°C) for at least half year.
1× Tris-Gly working buffer (inside tank)
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× Tris-Gly stock buffer | 1× | 100 mL |
| Sodium deoxycholate | 0.2% | 2 g |
| ddH2O | N/A | Add to 1 L |
| Total | N/A | 1 L |
Store at room temperature for at least half year.
1× Tris-Gly working buffer (outside tank)
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× Tris-Gly stock buffer | 1× | 100 mL |
| ddH2O | N/A | Add to 1 L |
| Total | N/A | 1 L |
Store at room temperature for at least half year.
Native-PAGE gel (8% separation gel)
| Reagent | Final concentration | Amount |
|---|---|---|
| ddH2O | N/A | 2.3 mL |
| 30% acrylamide-bisacrylamide (29:1) | 8.1% | 1.35 mL |
| 1 M Tris-HCl (pH=8.8) | 250 mM | 1.25 mL |
| 10% Ammonium Persulfate (APS)a | 0.1% | 50 μL |
| TEMED | 0.06% | 3 μL |
| Total | N/A | About 5 mL |
Mix well, pour into the rubber plate quickly and avoid bubbles. Add ddH2O slowly for liquid sealing. Store in fume hood for 30–60 min. Carry out the entire process in fume hood. a10% APS: 1 g Ammonium persulfate in 10 mL ddH2O. Store 10% APS at −20°C for three months, or at 4°C for a week.
Native-PAGE gel (5% stacking gel)
| Reagent | Final concentration | Amount |
|---|---|---|
| ddH2O | N/A | 1.35 mL |
| 30% acrylamide-bisacrylamide(29:1) | 5% | 335 μL |
| 1 M Tris-HCl (pH=6.8) | 125 mM | 250 μL |
| 10% APS | 0.1% | 20 μL |
| TEMED | 0.1% | 2 μL |
| Total | N/A | About 2 mL |
Mix well, pour into the rubber plate quickly and avoid bubbles. Insert the comb quickly. Store in fume hood for 30–60 min. Carry out the entire process in fume hood.
Transfer buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-base | 26 mM | 3.13 g |
| Glycine | 200 mM | 14.4 g |
| Methanol | 20% | 200 mL |
| ddH2O | N/A | Add to 1 L |
| Total | N/A | 1 L |
Mix well, store at room temperature for at least half year.
5% milk
| Reagent | Final concentration | Amount |
|---|---|---|
| Non-fat powdered milk | 5% | 5 g |
| TBST | N/A | Add to 100 mL |
| Total | N/A | 100 mL |
Mix well, store at 4°C for <12 h.
TBST buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-base | 25 mM | 3.03 g |
| NaCl | 137 mM | 8 g |
| KCl | 2.7 mM | 0.2 g |
| Tween-20 | 0.1% | 1 mL |
| ddH2O | N/A | Add to 1 L |
| Total | N/A | 1 L |
Mix well, store at room temperature for <12 h.
Step-by-step method details
Susceptible comparison of WT and lgp2−/− zebrafish larvae to SVCV infection
To determine the biological role of LGP2 in zebrafish survival against virus infection, WT (lgp2+/+) and lgp2−/− zebrafish larvae are directly immersed with SVCV in two 10-cm dishes, respectively. Dead larvae are easily distinguished from live larvae due to severe body curling, which is beneficial to illustrate by taking pictures (Figure 1).
-
1.
Sterilize the aerated water (system water) by 0.22 μm filter unit.
-
2.Take lgp2+/+ or lgp2−/− zebrafish larvae into two containers with system water.
-
a.Absorb methylene blue-containing water as far as possible.
-
b.Wash zebrafish larvae with sterilized system water for 2–3 times to remove residual methylene blue.
-
a.
-
3.
Transfer 50 lgp2+/+ larvae and 50 lgp2−/− larvae to two 10-cm dishes, respectively. Each dish contains 20 mL of system water supplemented with 200 μL penicillin-streptomycin solution (final concentrations: 100 U/mL of penicillin, and 100 mg/mL of streptomycin sulfate) to prevent the growth of bacteria.
-
4.
Mix SVCV solution to reach a viral titer of 2 × 106 TCID50/mL.
-
5.
Place dishes at 28°C for 2 days through checking every few hours.
-
6.
Add MS-222 (10 g/100 mL) at a ratio of 1/1,000 to anesthetize lgp2+/+ and lgp2−/− larvae, which will stop fish swimming and keep stationary within 1 min after anesthetization.
-
7.
Take pictures as soon as possible.
Figure 1.

Susceptible comparison of lgp2+/+ and lgp2−/− zebrafish larvae to SVCV infection
(A and B) Representative imaging of lgp2+/+ (A) and lgp2−/− (B) zebrafish larvae (6 dpf, n=50) immersed with SVCV (2×106 TCID50/mL) for 2 d. Dead larvae indicated by red arrows are easily discriminated from live larvae due to body curling.
Death curve of lgp2+/+ and lgp2−/− zebrafish larvae in response to SVCV infection
This section describes how to draw a death curve of zebrafish larvae by survival comparison of WT and lgp2−/− zebrafish larvae immersed with SVCV.
-
8.
Transfer 100 lgp2+/+ and lgp2−/− larvae to two 10-cm dishes, respectively. Each dish contains 20 mL of system water, supplemented with 200 μL penicillin-streptomycin (final concentrations: 100 U/mL of penicillin, and 100 mg/mL of streptomycin sulfate).
-
9.
Mix SVCV solution to reach a viral titer of 5 × 105 TCID50/mL.
-
10.
Put dishes at 28°C. Check every few hours and pick out dead larvae immediately.
-
11.
Record deaths in the two groups every 24 h until one group of larvae is completely dead.
-
12.
Draw the death curve by data processing tools such as Prism 8.0.
IFN response comparison of lgp2+/+ and lgp2−/− zebrafish larvae to SVCV infection
In response to SVCV infection, IFN response is significantly induced in both lgp2+/+ and lgp2−/− zebrafish larvae by upregulation of ifn and ISGs. The subtle differences can be detected by time lapse comparison of mRNA levels of ifn and ISGs between lgp2+/+ and lgp2−/− zebrafish larvae (6dpf) following SVCV infection.
-
13.
Take 300 lgp2+/+ or lgp2−/− zebrafish larvae to two 10-cm dishes, respectively. Each dish contains 20 mL of sterilized system water, supplemented with 200 μL penicillin-streptomycin solution (final concentrations: 100 U/mL of penicillin, and 100 mg/mL of streptomycin sulfate).
-
14.
Add SVCV solution into two dishes up to a viral concentration of 5 × 105 TCID50/mL, and place dishes at a 28°C incubator.
-
15.
Collect 30 lgp2+/+ larvae and 30 lgp2−/− larvae, at 0, 3, 6, 12, 24, 36, 48, 72 h post infection, to two 1.5 mL RNase-free microcentrifuge tubes, respectively.
-
16.
Remove the residual virus solution by washing with PBS twice.
-
17.
Add 200 μL of RNAlater RNA Stabilization Reagent (Qiagen) into each tube, and store at −80°C.
-
18.Thaw all samples after sampling is finished. For each sample at a time point:
-
a.Take all larvae out with dissection forceps.
-
b.Absorb residual solution on a filter paper.
-
c.Put the sample into a fresh 1.5 mL RNase-free microcentrifuge tube.
-
a.
-
19.Lyse samples:
-
a.Add 1 mL of tissue lysis solution (according to different Kits. e.g., Trizol-based lysis buffer provided in TRIzol Plus RNA Purification kit) together with several baked ceramic beads.
-
b.Homogenize fully on a homogenizer. Or homogenize samples with other available methods.
-
a.
-
20.
Centrifuge at 15,000 × g for 10 min at 4°C.
-
21.
Transfer supernatants carefully to fresh 1.5 mL microcentrifuge tubes.
-
22.
Extract total RNA and remove cellular DNA by RNase-free DNase.
-
23.
Synthesize first-strand cDNA by random primers or Oligo(dT) primers.
-
24.
Detect mRNA levels of ifn and ISGs by real-time PCR.
-
25.
Analyze data with data processing software such as Prism 8.0.
Death curve of lgp2+/+ and lgp2−/− zebrafish adults in response to SVCV infection
This section describes the steps to compare mortality rates between WT and lgp2−/− zebrafish adults in response to SVCV infection. Unlike lgp2+/+ and lgp2−/− zebrafish larvae which are easily immersed with SVCV solution in 10-cm dishes, lgp2+/+ and lgp2−/− zebrafish adults (60 dpf) are housed in two tanks at 28°C and infected with SVCV by intraperitoneal injection.
-
26.
Take 50 lgp2+/+ zebrafish adults and 50 lgp2−/− zebrafish adults in two tanks at 28°C, respectively.
-
27.
Infect fish by intraperitoneal injection with SVCV (108 TCID50/mL), 20 μL each.
-
28.Place tanks at 28°C.
-
a.Check fish every few hours.
-
b.Pick out dead fish immediately.
-
a.
-
29.
Record deaths every 24 h until one group is dead.
-
30.
Draw the death curve by data processing software such as Prism 8.0.
IFN response comparison of lgp2+/+ and lgp2−/− zebrafish adults to SVCV infection
IFN response is significantly induced by SVCV infection in zebrafish tissues by upregulation of ifn and ISGs. The subtle difference can be detected by time lapse comparison of mRNA levels of ifn and ISGs in different tissues of lgp2+/+ and lgp2−/− zebrafish adults (Figure 2).
-
31.
Repeat steps 26–28.
-
32.
Sample at 0, 3, 6, 12, 24, 36, 48, 72 h post infection, with 4 lgp2+/+ adults or 4 lgp2−/− zebrafish adults (male: female =1:1) at a given time point.
-
33.
Add MS-222 (10 g/100 mL) at a ratio of 1/1,000 to anesthetize fish, and snip the tail for bloodletting.
-
34.
For each time point, collect gill, liver, spleen, head kidney, trunk kidney and other tissues of WT or lgp2−/− zebrafish adults (n=4) to a 200 μL RNase-free microcentrifuge tube, respectively.
-
35.
Add 200 μL of RNAlater RNA Stabilization Reagent (Qiagen), and store at −80°C.
-
36.
Repeat steps 18–25.
Figure 2.

IFN response comparison of lgp2+/+ and lgp2−/− zebrafish adults to SVCV infection
(A) Time course comparison of mRNA expression of ifnφ1 gene in trunk kidney between lgp2+/+ and lgp2−/− zebrafish adults (60 dpf) following SVCV infection.
(B) The induction ratio of ifnφ1 gene in (A) was calculated based on the fold induction at the same time point, followed by normalization to the ratio at 0 h post infection, which was set to 1, indicating no change.
Luciferase assays to determine IFN response by titration of poly(I:C)
Transfection of fish cells with poly(I:C), a synthetic analogue of virus dsRNA, is believed to mimic virus infection. The concentrations of poly(I:C) correspond to cellular concentrations of virus dsRNA produced during virus infection: low concentrations of transfected poly(I:C) might be analogue to the intracellular concentrations of viral dsRNA at the early stage of virus infection and high concentrations correspond to the late stage of virus infection4 (Figure 3).
-
37.
Seed EPC cells in 24-wells plates. Each well contain 0.5 mL M199 supplemented with 10% FBS, to reach monolayers after overnight (8–14 h) culture at 28°C.
-
38.Prepare transfection mixture.
-
a.Mix three kinds of plasmids at a ratio of 10:10:1, DrIFNφ1pro-luc (200 ng), DrLGP2 (200 ng) and pRL-TK (20 ng).
-
b.Dilute the indicated plasmids in 50 μL Opti-mem, which contain Polyethylenimine (PEI, MW25000, 1 μg/μL of storage concentration) with a ratio of 1:3 plasmid/PEI (μg/μL).
-
a.
-
39.
Add the prepared transfection mixtures into EPC cells, each well with 50 μL. Each concentration of poly(I:C) is performed in triplicate.
-
40.
24 h later, transfect cells again by PEI with poly(I:C) at increasing concentrations (0, 0.25, 0.5, 1, 1.5, 2, 4, 8, 10, 20, 40, 80 ng/mL).
-
41.
Another 24 h later, collect cells for luciferase assays according to the Dual-Luciferase Reporter Assay System (Promega, USA).
Figure 3.

Luciferase assays to determine IFN promoter activation by titration of poly(I:C)
EPC cells seeded in 24-well plates were transfected with DrIFNφ1pro-luc (200 ng), pRL-TK (20 ng) and DrLGP2 (200 ng). 24 h later, cells were transfected again with poly(I:C) at increasing concentrations (from 0 to 80 ng/mL) for another 24 h, followed by luciferase assays.
Luciferase assays to determine IFN response by titration of LGP2 itself
Overexpression of zebrafish LGP2 in fish cells provokes a strong IFN response at low doses but a weak one at high doses, indicating that the function switch of zebrafish LGP2 can be determined by luciferase assays through titration of zebrafish LGP2 itself (Figure 4). Similar dual regulation of human LGP2 can be detected in mammalian cells only when low concentration of poly(I:C) (such as 4 ng/mL) is present,4 based on below protocol with a minor modification by transfection again with low concentration of poly(I:C).
-
42.
Seed EPC cells (each well containing 0.5 mL M199 supplemented with 10% FBS) in 24-wells plates to reach monolayers after overnight (8–14 h) culture at 28°C.
-
43.Prepare transfection mixture similar to step 38:
-
a.Mix three kinds of plasmids: DrIFNφ1pro-luc (200 ng), pRL-TK (20 ng), together with DrLGP2 at increasing doses (0, 0.02, 0.1, 0.2, 0.5,1, 2, 5, 10, 40, 100, 200 ng).
-
b.Dilute these plasmids with 50 μL Opti-mem, which contains PEI with a ratio of 1:3 plasmid (μg)/PEI (μL).
-
a.
-
44.
Add the prepared transfection mixture into EPC cells, each well with 50 μL. Each dose of LGP2 is performed in triplicate.
-
45.
24–48 h later, collect cells for luciferase assays according to the Dual-Luciferase Reporter Assay System (Promega, USA).
Figure 4.

Luciferase assays to determine IFN promoter activation by titration of LGP2 itself
EPC cells seeded in 24-well plates were co-transfected DrIFNφ1pro-luc (200 ng), pRL-TK (20 ng) and DrLGP2 at increasing doses (from 0 to 200 ng) for 48 h, followed by luciferase assays.
Biochemical assays to determine LGP2’s dual regulation on IFN response
SVCV infection triggers IRF3 dimerization in fish cells (Figure 5). In SVCV-infected EPC cells that are overexpressed with zebrafish LGP2, the dual regulation of zebrafish LGP2 on cellular IFN response can be determined by time course analyses of the change in IRF3 dimerization by native PAGE, and the change in cellular IFN and ISG expression by real-time PCR.
-
46.
Seed EPC cells in 3.5-cm dishes to reach monolayers after overnight (8–14 h) culture at 28°C, each dish with 2 mL M199 supplemented with 10% FBS.
-
47.Prepare transfection mixture similar to step 38:
-
a.Set DrLGP2 plasmids at increasing doses (0, 40, 1,000 ng).
-
b.Dilute plasmids of each dose with 200 μL Opti-mem, which contains PEI with a ratio of 1:3 plasmid (μg)/PEI (μL).
-
a.
-
48.
Add the prepared transfection mixture into dishes, each dish with 200 μL.
-
49.Infect cells with SVCV at 24 h post transfection.
-
a.Add SVCV solution to dishes to a final concentration at 5 × 105 TCID50/mL and mix gently.
-
b.Put dishes at a 28°C incubator.
-
a.
-
50.
Sample at 0, 6, 12, 24, 36, 72 h post infection. Remove culture medium, and wash cells gently with cold PBS 2–3 times.
-
51.For real-time PCR analyses.
-
a.Add cellular lysis buffer to dishes, 500 μL each.
-
b.Transfer cell lysates to a 1.5 mL RNase-free microcentrifuge tube, and store at −80°C.
-
c.Thaw all samples for RNA extraction according to SV Total RNA Isolation System Protocol (https://www.promega.com.cn/resources/protocols/technical-manuals/0/sv-total-rna-isolation-system-protocol/), after sampling is finished.
-
d.Remove cellular DNA by treatment with RNase-free DNase.
-
e.Synthesize first-strand cDNA by reverse transcription with random primers or Oligo(dT) primers.
-
f.Detect mRNA levels of ifn and ISGs by real-time PCR.
-
g.Analyze data with data processing software such as Prism 8.0.
-
a.
-
52.For Native-PAGE analysis.
-
a.Add 200 μL RIPA lysis buffer (weak, Beyotime) on ice. RIPA is supplemented with EDTA-free cocktail and phosphatase inhibitor before use.
-
b.Lyse cells on a shaker for 30 min at 4°C.
-
c.Transfer cells lysates to 1.5 mL microcentrifuge tubes on ice, respectively.
-
d.Centrifuge at 15,000 × g for 10 min at 4°C.
-
e.Transfer supernatants to 4 fresh 200 μL microcentrifuge tubes on ice, each tube with 50 μL.
-
f.Do western blots to analyze IRF3 dimerization by Native-PAGE electrophoresis.
-
a.
-
53.Detect IRF3 dimerization by native-PAGE electrophoresis.
-
a.Prepare 8% native-PAGE gel according to the recipe provided above.
-
b.Add 1× Tris-Gly working buffer (inside and outside inner modular), and run for 30 min at 40 V before loading samples.
-
c.Add 12.5 μL 5× non-denaturing PAGE loading buffer to each sample (50 μL lysates) on ice, and mix well.
-
d.Load samples and run for 3 h at 80 V on ice.CRITICAL: Running electrophoresis at low temperature: place the electrophoresis tank in ice, and put an ice box inside the electrophoresis tank at the same time. If needed, replace with fresh ice boxes to sustain low temperature environment during electrophoresis.
-
e.Using Bio-Rad transfer system, arrange sponge > filter paper > gel > 0.45 μm PVDF membrane > filter paper > sponge in transfer buffer orderly and thoroughly, and run for 50–80 min at 95 V on ice, to transfer proteins from gel to membrane.Alternatives: Besides wet transfer above, other transfer methods, such as dry transfer and semi-dry transfer, might be option.CRITICAL: Before transfer, put PVDF membrane into 100% methanol solution for 15 s, wash PVDF membrane with transfer buffer 1–2 times, and keep membrane wet in transfer buffer.
-
f.Wash the membrane 3 × 10 min with TBST after transfer.
-
g.Block the membrane in TBST containing 5% milk for 30–60 min at RT (20°C–25°C) or 12 h at 4°C.
-
h.Wash the membrane 3 × 10 min with TBST.
-
i.Incubate the membrane with IRF3 antibody solution (1:5000 dilution in TBST supplemented with 1% milk, for 12 h at 4°C or 2 h at RT.
-
j.Remove antibody solution, wash the membrane 3 × 10 min using TBST.
-
k.Incubate the membrane in secondary antibody solution (1:5000 in TBST supplemented with 1% milk) at RT for 1–2 h.
-
l.Remove antibody solution, wash the membrane 3 × 10 min using TBST.
-
m.Do chemiluminescent visualization of protein bands by ImageQuant LAS 4000 mini system
-
i.Make a solution by mixing equal volumes of enhancer solution and peroxide solution (Clarity Western ECL Substrate, BIO-RAD).
-
ii.Drip the solution frequently on the top of membrane for 2 min at RT.
-
iii.Capture image with a chemiluminescent imaging system, or other methods.CRITICAL: Antibody specific to crucian carp IRF3 is described previously.6 Antibody dilution is dependent on the storage titer of a given antibody.
-
i.
-
a.
Figure 5.

Native-PAGE analysis of IRF3 dimerization by SVCV infection
EPC cells seeded in 3.5 cm dishes were infected with SVCV (final concentration at 5 × 105 TCID50/mL). 36 h later, IRF3-specific antibody was used to determine IRF3 dimerization by Native-PAGE.
Expected outcomes
This protocol is designed to characterize the dual regulatory roles of zebrafish LGP2 in IFN response.1,4 Comparison of WT and knockout zebrafish in response to SVCV infection demonstrates that LGP2 is essential for zebrafish survival against viral infection (Figure 1). Real-time PCR analyses of ifn and ISG expression indicate that LGP2 acts as an activator of IFN response in the early phase of SVCV infection (<24 h post infection) and an inhibitor in the late phase of SVCV infection (>24 h post infection) (Figure 2). The function switch of zebrafish LGP2 can be observed in fish cells transfected with poly(I:C) at increasing concentrations by luciferase assays (Figure 3).
Given that overexpression of zebrafish LGP2 alone can stimulate IFN promoter activation, titration of LGP2 expression levels shows that low doses of LGP2 function as an activator, and high doses of LGP2 as an inhibitor (Figure 4). As a typical ISG, the change of LGP2 expression is tightly related to the different stages of virus infection; therefore, the assays regarding titration of LGP2 expression levels still indicate the dual regulation of LGP2 on fish IFN response. Finally, the dual regulation of zebrafish LGP2 can be confirmed by biochemical assays, such as time course analyses of the change in IRF3 dimerization by native PAGE, and the change in cellular IFN and ISG expression by real-time PCR.1
Limitations
Zebrafish ifn and several ISGs are modulated in comparison of IFN response between WT and knockout zebrafish in this protocol. Transcriptome and proteome might be further performed at the same time points after SVCV infection to assist the understanding of the changes in ifn and ISG expression. In vitro assays are performed by overexpression of zebrafish LGP2 in fish cells. To avoid the interference from endogenous LGP2, these overexpression assays might be carried out in LGP2-dificient fish cells to further clarify LGP2 function.
Troubleshooting
Problem 1
lgp2+/+ and lgp2−/− zebrafish adults do not lay eggs on the same day, or zebrafish embryos are not enough for follow-up infection assays (Refer to: Breed WT or lgp2−/− zebrafish).
Potential solution
Before feeding fish more frequently up to 3–4 times daily for 2–3 days, facilitating females to empty dead eggs by setting up breeding tanks and pre-breeding is helpful to improve breeding efficiency. To avoid female dystocia due to over-feeding, feeding frequency and feeding duration can be adjusted in time. Breeding females are characterized by abdominal bulge, with yellow and translucent abdomen near the fecal orifice.
Problem 2
Infection assays fail to observe the differential mortality between lgp2+/+ and lgp2−/− zebrafish due to dying too quickly (all dead <24 h post infection) or too slowly (starting to die >72 h post infection) (Refer to step-by-step method details: 8–12).
Potential solution
A pretest is recommended to obtain the optimized viral titer for infection assays under same conditions.
Problem 3
During viral infection assays, system water in dishes or tanks is contaminated by other microbes (Refer to step-by-step method details: 3–5, 8–10, 13–14).
Potential solution
System water is sterilized through 0.22 μm filter units before use. In addition, a supplement with penicillin and streptomycin is helpful to inhibit bacteria growth. Importantly, check frequently and pick up dead larvae immediately.
Problem 4
Real-time PCR or luciferase assays does not show a positive regulation of LGP2 on IFN response at the early infection and a negative regulation at the later infection (Refer to step-by-step method details: 23–24, 36, 41, 45).
Potential solution
The key point of this protocol is to utilize titration methods, including dense titration of viral titer and infection duration, the doses of diverse stimuli, such as poly(I:C) and even LGP2 itself. It is recommended to set viral titers, infection duration and stimuli doses more densely to optimize experiment parameters.
Problem 5
Overloading during plasmid transfection causes imperfect results (Refer to step-by-step method details: 43 and 47).
Potential solution
During transfection, adequate empty vector pcDNA3.1(+) is added to guarantee equivalent amounts of total plasmid DNA transfected in all wells. Consistent results can be obtained when total doses of transfected plasmids are used at less than a threshold, such as <600 ng in 0.5 mL/well in 24-well plates. The total doses of transfected plasmids are proportional to the number of fish cells seeded in different plates or dishes. For example, 4-fold doses of the plasmids are transfected in 6-well plates compared to that in 24-well plates, and a 3.5-cm dish corresponds to a well of 6-well plates.
Problem 6
Fish IRF3 dimerization is not shown by Native-PAGE assays (Refer to step-by-step method details: 52–53).
Potential solution
Collecting cells, lysing cells, loading samples and running electrophoresis are performed at 4°C. Avoid freezing and thawing of cellular lysates frequently, and use antibodies specific to fish IRF3.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yi-Bing Zhang (ybzhang@ihb.ac.cn).
Materials availability
The expression plasmids generated in this protocol are available from the lead contact.
Acknowledgments
This work was supported by the grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA24010308), the National Key R&D Program of China (2018YFD0900302), the National Natural Science Foundation of China (31972826, 32102838), and the Freshwater Ecology and Biotechnology Laboratory (2022FBZ03).
Author contributions
Y.-B.Z. conceived the project, and Y.-B.Z. and X.-Y.G. designed the experiments. X.-Y.G. performed the experiments. Y.-B.Z. and X.-Y.G. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Xiu-Ying Gong, Email: 13018024014@163.com.
Yi-Bing Zhang, Email: ybzhang@ihb.ac.cn.
Data and code availability
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Gong X.Y., Zhang Q.M., Zhao X., Li Y.L., Qu Z.L., Li Z., Dan C., Gui J.F., Zhang Y.B. LGP2 is essential for zebrafish survival through dual regulation of IFN antiviral response. iScience. 2022;25:104821. doi: 10.1016/j.isci.2022.104821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rehwinkel J., Gack M.U. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020;20:537–551. doi: 10.1038/s41577-020-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Q.M., Zhao X., Li Z., Wu M., Gui J.F., Zhang Y.B. Alternative splicing transcripts of zebrafish LGP2 gene differentially contribute to IFN antiviral response. J. Immunol. 2018;200:688–703. doi: 10.4049/jimmunol.1701388. [DOI] [PubMed] [Google Scholar]
- 4.Gong X.Y., Qu Z.L., Li Y.L., Sun H.Y., Zhao X., Dan C., Gui J.F., Zhang Y.B. Function conservation and disparities of zebrafish and human LGP2 genes in fish and mammalian cells responsive to poly(I:C) Front. Immunol. 2022;13:985792. doi: 10.3389/fimmu.2022.985792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawakami K., Patton E.E., Orger M. Second Edition. Volume 1451. MIMB; 2016. Zebrafish: methods and protocols. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 6.Sun F., Zhang Y.B., Liu T.K., Gan L., Yu F.F., Liu Y., Gui J.F. Characterization of fish IRF3 as an IFN-inducible protein reveals evolving regulation of IFN response in vertebrates. J. Immunol. 2010;185:7573–7582. doi: 10.4049/jimmunol.1002401. [DOI] [PubMed] [Google Scholar]
- 7.Chen M.R., Chen H.X., Yi Y.L. The establishment of a heteroploid line from crucian carp and its biological characterisitics. J. Fish. China. 1985;9:121–130. [Google Scholar]
- 8.Sun F., Zhang Y.B., Liu T.K., Shi J., Wang B., Gui J.F. Fish MITA serves as a mediator for distinct fish IFN gene activation dependent on IRF3 or IRF7. J. Immunol. 2011;187:2531–2539. doi: 10.4049/jimmunol.1100642. [DOI] [PubMed] [Google Scholar]
- 9.Kinkel M.D., Eames S.C., Philipson L.H., Prince V.E. Intraperitoneal injection into adult zebrafish. J. Vis. Exp. 2010 doi: 10.3791/2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.