
Figure 1.
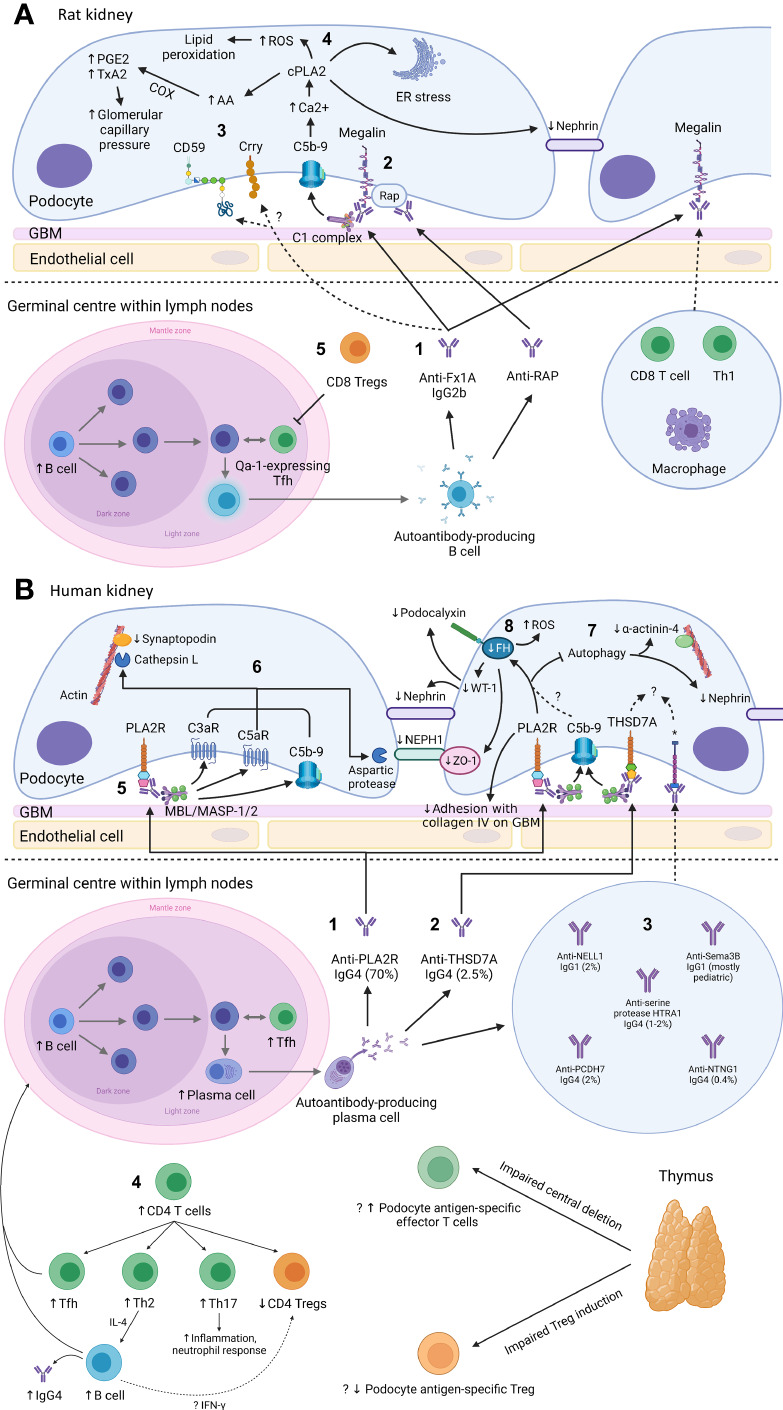
Mechanism of podocyte injury in Heymann nephritis (A) and primary membranous nephropathy (B) Created with BioRender.com. GBM, glomerular basement membrane; ROS, reactive oxygen species; cPLA2, cytosolic phospholipase A2; PGE2, prostaglandin E2; TxA2, thromboxane A2; COX, cyclo-oxygenase; AA, arachidonic acid; ER, endoplasmic reticulum; RAP, receptor associated protein; Treg, regulatory T cell; Th1, CD4+ T helper-1 cell; Tfh, CD4+ T follicular helper cell; FH, fumarate hydratase; WT-1, Wilms tumor-1; PLA2R, M-type phospholipase A2 receptor 1; C3aR, C3a receptor; C5aR, C5a receptor; MBL, mannan-binding lectin; MASP, mannan-binding lectin serine protease; ZO-1, zonula occludens-1; THSD7A, thrombospondin type-1 domain-containing protein 7A; NELL1, neural epidermal growth factor-like 1 protein; Sema3B, semaphorin 3B; PCDH7, protocadherin 7; HTRA1, high temperature requirement A serine peptidase 1; NTNG1, netrin G1; Th2, CD4+ T helper-2 cell; Th17, CD4+ T helper-17 cell; IFN-γ, interferon-γ; *cognate antigen for anti-NELL1, anti-Sema3B, anti-PCDH7, anti-serine protease HTRA1, and anti-NTNG1 autoantibodies respectively. (A): 1. In Heymann nephritis, anti-Fx1A antibodies bind primarily to megalin on podocytes and activate the classical complement pathway (via the IgG2b subclass). 2. Anti-RAP antibodies are also detected and may represent epitope spreading due to the association between megalin and RAP, which assists the transport of megalin from ER to cell surface. 3. Complement activation may be potentially exacerbated by binding of anti-Fx1A antibodies to complement regulatory proteins Crry and CD59. 4. Sublethal C5b-9 injury causes intracellular calcium influx, activating cPLA2, which hydrolyzes membrane phospholipids of the podocyte, ER and nuclear envelope. This causes ER stress, generation of ROS, disruption of the slit diaphragm protein nephrin, and release of AA with subsequent COX-mediated generation of prostanoids (PGE2, TxA2) that increase glomerular filtration pressure, exacerbating proteinuria. Glomerular IgG and complement deposition attract glomerular infiltrates of CD8+ T cells, Th1 cells, and macrophages, potentially via the IgG Fc receptor and anaphylatoxins C3a/C5a. 5. Autoantibody production in Heymann nephritis is at least in part dependent on Qa-1-expressing Tfh cells, which are inhibited by CD8+ Tregs. (B): 1. In primary membranous nephropathy (PMN), autoantibodies are predominantly directed against PLA2R, and 2. less commonly against THSD7A, 3. NELL1, PCDH7, serine protease HTRA1 and NTNG1, except in pediatric PMN where Sema3B is the most common podocyte antigen targeted by autoantibodies. 4. Increased B cells and Tfh cells are observed, which interact in the germinal center of lymph nodes to induce differentiation of B cells into high-affinity antibody-producing plasma cells. Autoantibodies in PMN are primarily of the IgG4 subclass, potentially due to Th2-mediated IL-4 production. Loss of tolerance to podocyte antigens such as PLA2R may be due to loss of thymus-derived podocyte antigen-specific Tregs. 5. Glycosylated anti-PLA2R IgG4 bind the MBL/MASP-1/2 complex to activate the lectin complement pathway. 6. This causes activation of C3aR and C5aR as well as deposition of C5b-9, which activates cathepsin L- and an aspartic protease-mediated proteolysis of the actin cytoskeleton protein synaptopodin and slit diaphragm protein NEPH1 respectively. Anti-PLA2R antibody-containing serum is also associated with reduced FH, impaired autophagy and reduced adhesion to the GBM, though it is unclear whether these effects are mediated via complement activation. 7. Impairment in autophagy results in internalization of nephrin and the actin cytoskeleton protein α-actinin-4. 8. Reduced FH leads to intracellular fumarate accumulation, which is associated with increased generation of ROS, reduced slit diaphragm protein ZO-1 and reduced transcription of WT-1, which normally activates the expression of podocyte proteins nephrin and podocalyxin. The mechanisms of podocyte injury of other autoantibodies associated with PMN remain incompletely understood.