

Abstract
Mitochondrial dysfunction is known to contribute to a range of diseases, and primary mitochondrial defects strongly impact high-energy organs such as the heart. Platforms for high-throughput and human-relevant assessment of mitochondrial diseases are currently lacking, hindering the development of targeted therapies. In the past decade, human-induced pluripotent stem cells (iPSCs) have become a promising technology for drug discovery in basic and clinical research. In particular, human iPSC-derived cardiomyocytes (iPSC-CMs) offer a unique tool to study a wide range of mitochondrial functions and possess the potential to become a key translational asset for mitochondrial drug development. This review summarizes mitochondrial functions and recent therapeutic discoveries, advancements and limitations of using iPSC-CMs to study mitochondrial diseases of the heart with an emphasis on cardiac applications.
Keywords: cardiomyocyte, cardiovascular disease, heart, iPSC, mitochondria, stem cell
Beyond being the colloquial cellular ‘powerhouse’, mitochondria play multifaceted roles in physiology, including the regulation of biosynthetic growth, waste removal, fission, fusion, stress signalling and the balance of cellular life and death [1–3]. Mitochondrial functions are crucial for nearly all cells and tissues, from highly proliferative tumours to highly aerobic, post-mitotic tissues. Therefore, mitochondrial dysfunction presents heterogeneously across many cell types and is implicated in a wide range of human diseases [4,5]. Primary mitochondrial defects are the most common inherited metabolic disorders, affecting nearly 1 : 5000 people with a clinical presentation at any age and in any organ [4,5]. Primary defects can lead to devastating clinical manifestations, including lactic acidosis, skeletal myopathy, deafness, blindness, subacute neurodegeneration, intestinal dysmotility and peripheral neuropathy [4]. Secondary mitochondrial dysfunction is a well-accepted maladaptive mechanism associated with the progression and potentiation of diabetes, cancer, obesity, neuromuscular and cardiovascular diseases [6].
Currently, there are no viable mitochondrial therapies. Drug development of mitochondrial-targeted therapies is greatly hindered by broad clinical manifestations, genetic heterogeneity and incomplete understanding of basic mitochondrial phenotype–genotype relationships [7,8]. While the number of promising clinical trials for mitochondrial-targeted therapies has substantially improved in recent years, they still lag behind the speed of diagnosis [9]. New investigational platforms, cutting-edge technologies and treatment approaches are urgently needed [10]. This review will focus on how cardiac-specific mechanisms modelled by human-induced pluripotent stem cell (iPSC) technology have led to new therapeutic insights into mitochondrial diseases.
Mitochondrial modelling: past and present
The heart is a high-energy consuming organ and thus contains more mitochondria than any other tissue in the body [11]. Mitochondrial dysfunction is well-observed in the progression of various etiologies of heart failure [12–14]. The failing heart was first described as being an ‘engine out of fuel’ in 1939 [15], and the energy-starvation hypothesis has since been heavily pursued [16–18]. Today, myocardial energetics, or energy metabolism in the heart, remains a topic of interest because energy-sparing drugs, such as angiotensin-converting enzyme (ACE) inhibitors and beta-receptor blockers [19–21], were shown to improve patient outcomes. Increasing energy demand, achieved by inotropes, worsens clinical outcomes [22]. Identifying and targeting mitochondrial mechanisms pertinent to heart function holds potential for new cardiac therapies [23].
Although cardiomyocytes provide an opportune system for mitochondrial research, they have been highly challenging to study in cell culture due to their post-mitotic nature. Murine models have been extensively utilized to study cardiac mitochondria for the past three decades [24]. While animal modelling is indispensable for the preclinical evaluation of therapeutic agents, this platform is largely incompatible with high-throughput drug discovery and personalized medicine [25,26]. Furthermore, inherent species differences in heart rate, arrhythmic responses, temporal disease progression and the lack of comorbidities in rodent models lead to an incomplete human pathogenesis model [27,28].
The advent of iPSC technology provides a unique tool for investigating human diseases. Reprogramming donor cells using a non-integrating virus can yield pluripotent iPSCs capable of differentiating into diverse cell types [25]. This platform features two notable advantages over existing traditional models. First, they provide an unlimited source of human cardiac cell types, which can be expanded, matured and maintained in cell culture for over 200 days [29–31]. The establishment of robust differentiation protocols has resulted in low-cost, high purity induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) for mass production [32,33] and broad applications in regenerative medicine, diagnostics, drug toxicity and efficacy testing [25]. Second, iPSCs are genetically matched to their donors and recapitulate clinical phenotypes in a dish, providing disease- and patient-specific platforms for therapeutic interrogation. Disease-specific platforms developed so far include long QT syndrome [34,35], hypertrophy [36], and dilated cardiomyopathy [37,38] applications. Thus, the iPSC-CM platform has emerged as a powerful tool for translational medicine that circumvents some existing limitations of historical models.
Cell-based assays and multi-omics molecular techniques have significantly increased detection sensitivities in parallel to advancements in iPSC technologies, thus providing cellular and mitochondrial resolution unimaginable only a few years ago (Fig. 2). Thus, the combination of iPSC advancement with high-throughput cell-based assays paves an exciting trajectory for studying diverse mitochondrial functions.
Fig. 2.
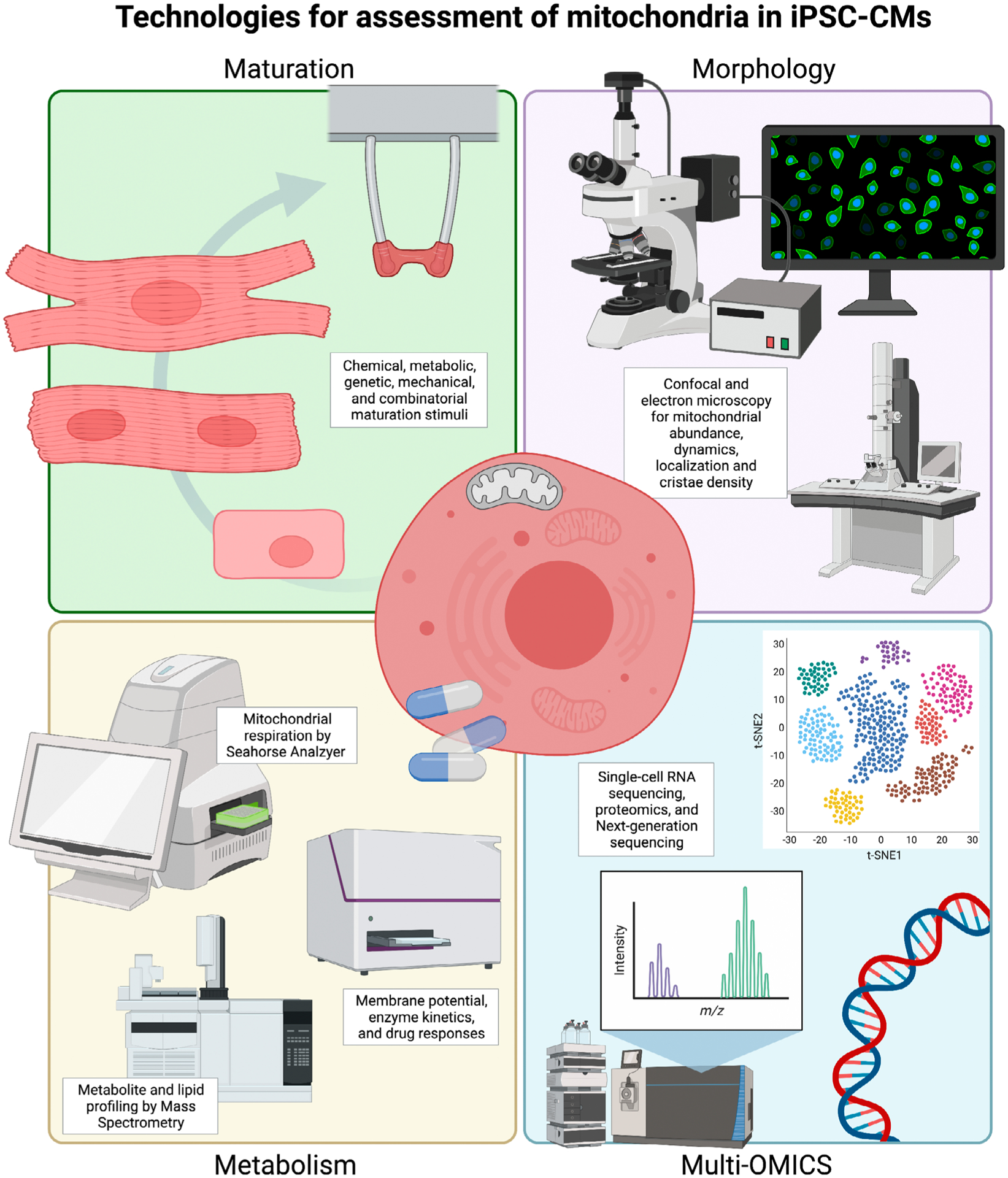
Summary of iPSC-CM technology compatible with mitochondrial and metabolic assessments. Consistent with advancements in iPSC-CM maturation strategies, new optimizations to study mitochondrial function on cellular platforms have emerged to include new methods for measuring metabolic responses, morphology, gene, transcript and protein expression. Created with BioRender.com.
Here, we summarize distinctive functional features of mitochondria, including their genome, membrane architecture, flexible energy metabolism and oxidative phosphorylation (OXPHOS) machinery. We review the current literature for each function wherein human-iPSC-CM models have led to new therapeutic insights relevant to mitochondrial diseases (Table 1, Fig. 1). We then discuss technological advancements, current limitations and future recommendations for mitochondrial studies using iPSC-CM platforms.
Table 1.
Summary of mitochondrial studies using iPSC-CMs.
| Target | Product | Pathway | Disease | Reference |
|---|---|---|---|---|
| mtDNA | mtDNA deletions | mtDNA homeostasis | Kearns-Sayre syndrome | Sequiera et al., Cells (2021) [40] |
| mt-RNR2 | 16S mt-rRNA | DNA replication | Hypertrophic cardiomyopathy | Li et al., Stem CellReports (2018) [41] |
| POLG | POLG2 | DNA replication | Leigh syndrome | Liang et al., EMBO Medicine (2020) [42] |
| mt-TL1 | mt-tRNA | Translation | MELAS | Yokota et al., Cell Death and Disease (2017) [46] |
| mtDNA | mtDNA mutations | mtDNA homeostasis | MELAS | Perales-Clemente et al., EMBO (2016) [47] |
| TAZ | Tafazzin 1 | Cardiolipin production | Barth syndrome | Wang et al., Nature Medicine (2014) [53] |
| DNAJC19 | TIM44 | Protein import | Dilated cardiomyopathy | Rohani et al., Canadian Journal of Cardiology (2020) [56] |
| HADHA | ECHA | Fatty acid oxidation | Sudden infant death syndrome | Miklas et al., Nature Communications (2019) [69] |
| FXN | Frataxin | Iron-sulfur cluster production | Friedreich’s ataxia | Hick et al., Disease Models and Mechanisms (2013) [74] |
| SCO2 | SCO2 | Complex IV Assembly | Cardiomyopathy | Hallas et al., J Mol. Cell Medicine (2018) [76] |
Fig. 1.
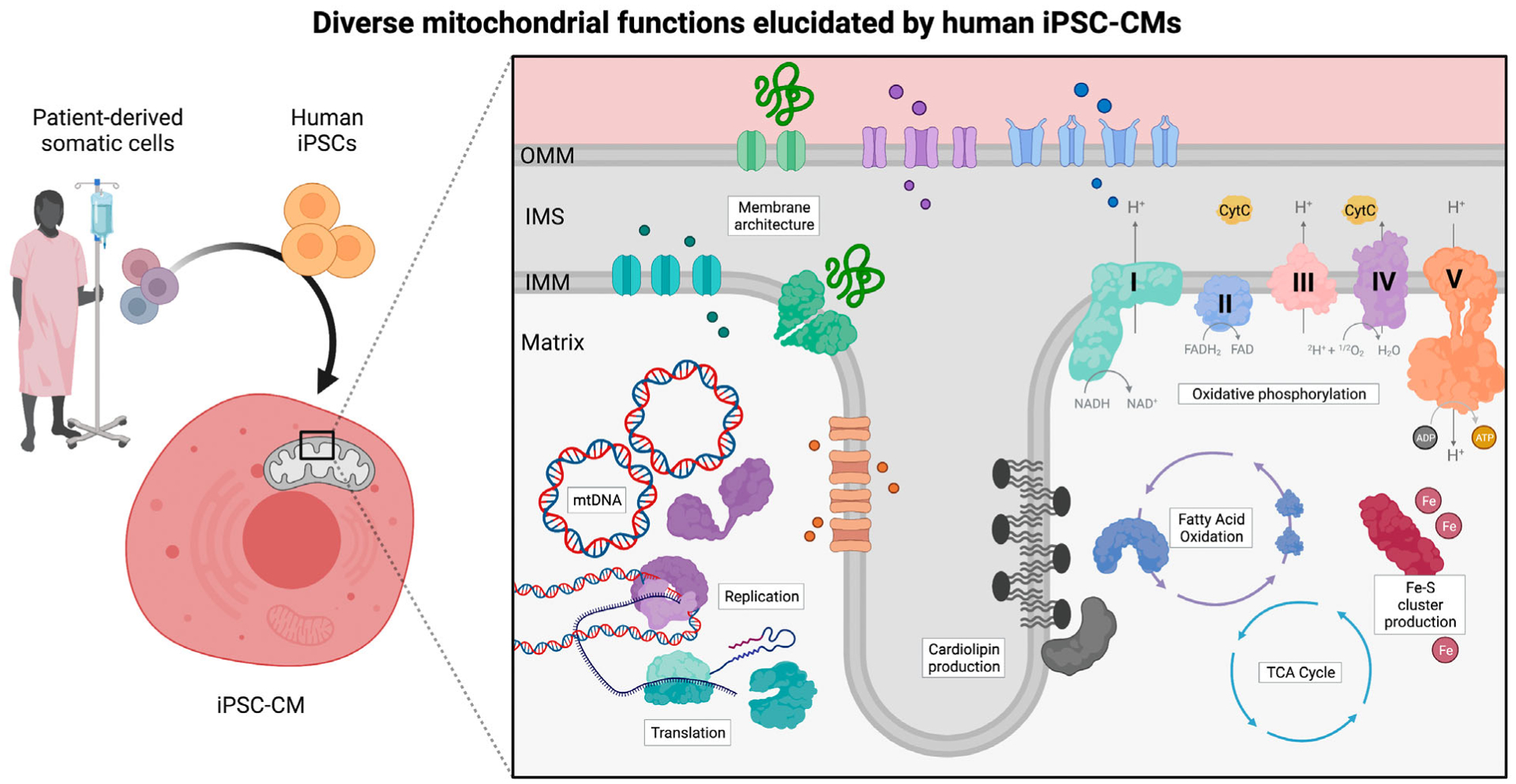
Summary of diverse mitochondrial functions elucidated using iPSC-CMs. Human iPSCs with mitochondrial mutations can be generated through patient-derived reprogramming of somatic cells or gene-editing strategies. To study the role of mitochondrial dysfunction in cardiac diseases, iPSCs can be robustly differentiated into cardiomyocytes using standard protocols. As summarized in this review, iPSC-CMs have been utilized as a platform to study a wide range of mitochondrial functions, from cardiolipin machinery to OXPHOS assembly factors. Created with BioRender.com.
Homeostasis of mitochondrial genome
The mitochondrion is an ancient organelle derived from the endosymbiotic relationship between the proteobacterium and its host [39]. During evolution, mitochondria forfeited most of their genes to the nuclear genome of eukaryotic cells [2,4], which drastically reduced the chromosomal mtDNA, whereas they acquired a myriad of new functions [2]. Contemporary human mtDNA is a circular molecule of 16 kilobases containing 37 genes encoding 13 subunits of Complexes I, III, IV, and V of the electron transport chain (ETC) as well as 22 tRNAs and 2 rRNAs that are necessary for mitochondrial translation machinery function [4,40] (Fig. 1).
Hypertrophic cardiomyopathies
Mutations in mtDNA or in its replication machinery play a role in hypertrophic cardiomyopathies (HCM). As reported by Li et al., m.2336 T > C led to decreased stability of 16S rRNA and the steady-state levels of its binding partners, impaired mitochondrial ribosomal assembly and an overall reduction in mitochondrial proteins in iPSC-CMs derived from HCM patients [41]. Similarly, mutations in DNA polymerase gamma (POLG) are indispensable for mtDNA replication and repair, resulting in diverse and disastrous mitochondrial phenotypes conserved during cellular reprogramming and differentiation of iPSCs [41,42].
Pathogenicity of mitochondrial heteroplasmy
Unlike nuclear chromosomes, the number of mtDNA copies varies between 500 and 6000 per cell in a tissue-specific manner [43]. Therefore, the coexistence of mtDNA mutations can affect either all mtDNA molecules (known as homoplasmy) or a proportion of the mtDNA molecules (heteroplasmy) [44]. The latter presents a unique bottleneck to understanding primary pathogenic mtDNA mutations. Combined with limited mitochondrial-targeted genetic strategies, it has been historically challenging to generate cellular and animal models with specific mutations in mtDNA using plasmid or gene-editing technology [41,45]. Genetic reprogramming of somatic donor cells into iPSCs circumvents these technical hurdles because patient-specific mtDNA defects are retained during this process. Thus, iPSCs provide a valuable tool for understanding pathogenic thresholds and their functional outcomes [41]. For example, two isogenic iPSC lines from the same patient harbouring low and high levels of m.3243 A > G in mt-TL1 had distinct cellular phenotypes. High proportions of m.3243 A > G, which exceeded the pathogenic threshold, strongly inhibited maturation and survival of iPSC-CMs via induction of mitochondrial respiratory dysfunction [46]. This study underscores intra-person variability in differentiation potential and gene expression of patient-derived iPSC lines induced by heteroplasmy [47,48]. Furthermore, segregation of mtDNA mutations proved to be a dynamic process, with some iPSC clones shifting towards wild-type mtDNA at a rate of ~ 12% per month when the initial mutation load was greater than 80%. In contrast, other iPSC clones showed no changes in heteroplasmy levels with prolonged (>1 month) culture. However, age-related accumulation of mtDNA mutation frequency is observed in iPSCs derived from elderly donors (60–72 years) and should be a clinical consideration for iPSC model generation [48,49].
Kearns–Sayre syndrome
High heteroplasmy levels of pathogenic mtDNA mutations also occur in some tumours, neurodegenerative diseases, diabetes and age-associated heart failure [43,47]. Large deletions in mtDNA may lead to multi-system disorders such as Kearns–Sayre syndrome (KSS). KSS is a slowly progressing disease with clinical manifestations predominantly in the cardiac, renal or central nervous systems, with few treatment strategies available. Deletions in mtDNA of KSS patients are primarily found in muscle tissues and are predominantly absent in the blood cells.
Nuclear reprogramming of peripheral blood mononuclear cells (PBMCs) purified from the blood of KSS patients into cardiomyocytes, neurons and fibroblasts generated ‘mutation-free’ isogenic sources of cell replacement therapies [40]. Thus, promoting mtDNA segregation ‘drift’ towards wild-type mtDNA or harnessing mitochondrial transplantation strategies [50] will be critical avenues for further therapeutic developments for KSS.
Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes
It is well-accepted that even healthy somatic mitochondria are heteroplasmic and possess extremely low levels of mtDNA variants of either inherited or single base-pair mutations. Indeed, next-generation sequencing of mtDNA (mtDNA NGS) from mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) patients demonstrated that heteroplasmy is conserved during nuclear reprogramming of iPSCs and did not affect cardiac differentiation potential into iPSC-CMs. Differentiated iPSC-CM progeny with conserved mtDNA mutations, such as m.9547G > A, has been shown to suppress mitochondrial respiratory function, further underscoring the need for mtDNA NGS as a selection tool during iPSC clonal selection [47] (Fig. 2).
Mitochondrial membrane architecture and dynamics
Mitochondria retain a sophisticated architecture comprised of an outer membrane (OMM) and inner membrane (IMM), which together divide the aqueous intermembrane space (IMS) from the inner matrix (Fig. 1). The individual lipid composition of each membrane is asymmetric to balance OMM fluidity and IMM structural integrity with mitochondrial dynamics of fission and fusion [51].
Barth syndrome
Cardiolipin is the hallmark lipid of the mitochondrial IMM, essential for dynamics, mitophagy, apoptosis, organization of supramolecular structure and function [2,52]. Mutations in the gene encoding taffazin (TAZ), an acyltransferase responsible for cardiolipin processing, cause Barth syndrome (BTHS), a subset of cardiac and mitochondrial muscle myopathies. To define the mechanistic link between abnormal cardiolipin biogenesis and cardiomyopathy, Wang et al. utilized both patient-derived and Cas9-mediated iPSC lines for BTHS. BTHS iPSC-CMs recapitulated patient phenotypes of diminished mature cardiolipin content by LC–MS and impaired mitochondrial respiration by Seahorse Analyzer (Agilent Technologies Inc., Santa Clara, CA, USA) (Fig. 2). When combined with heart-on-chip technology, BTHS iPSC-CM-engineered myocardium replicated the contractile pathophysiology clinically observed in patients. Furthermore, BTHS iPSC-CMs and engineered tissues can effectively model disease correction by reintroducing TAZ modRNA or treatment with linolenic acid, an essential unsaturated fatty acid precursor of mature cardiolipin [53].
Dilated cardiomyopathy with ataxia syndrome
The dual-membrane architecture of mitochondria retains an intricate translocase network to import mitochondrial proteins, of which 99% are nuclear-encoded [54]. Mitochondrial targeting signals direct precursor proteins to their sub-organellar destination. Proteins are imported through the translocase of the outer membrane (TOM) complex followed by the translocase of the inner membrane (TIM23) complex for transport into the IMM or matrix. Patients with dilated cardiomyopathy with ataxia syndrome (DCMA) were found to have mutations in the DNAJC19 gene and presented with Barth syndrome-like mitochondrial dysfunction. DNAJC19 is a mitochondrial chaperone homologous to yeast PAM18/TIM14 subunit, a critical component of TIM23. Splice mutations in DCMA patients were initially hypothesized to cause defects in mitochondrial import [55]. Patient-derived iPSC-CMs harbouring mutations in DNAJC19 had disorganized cristae and an imbalance of pro-fusion L-OPA1 to pro-fission S-OPA1 ratio. Imbalanced OPA1 processing led to short, less connected mitochondria and overall network fragmentation in DCMA iPSC-CMs, consistent with common clinical phenotypes [56].
Administration of elamipretide (SS-31), a mitochondrial-targeted synthetic tetrapeptide, to iPSC-CMs, rescued mitochondrial fragmentation and highlighted a potential therapy for DCMA. Furthermore, Rohani et al. demonstrated that iPSC-CMs are a viable platform for studying the regulation of human mitochondrial fission and fusion in vitro.
Mitochondrial energy metabolism during cardiac development, maturation and stress
The adult heart is metabolically flexible, toggling between various available carbon substrates as fuel sources to maintain the energy output necessary for constant contraction. Cardiac mitochondria play a central role in fluctuating fuel preferences throughout heart development, maturation and stress.
The fetal heart experiences a hypoxic environment rich in glucose and lactate that promotes rapid growth [57]. Shortly after birth, cardiomyocytes become post-mitotic and undergo hypertrophy, an enlargement of cell size and mass that promotes contraction force. In parallel, a burst in mitochondrial biogenesis greatly increases the oxidative capacity of the neonatal heart [58]. Maternal breastmilk supplies increased levels of circulating free fatty acids and triacylglycerides [59–61], and fatty acid oxidation (FAO) becomes the most prominent energy source in the post-natal heart [62]. Contributions from lactate, ketones and amino acids are considered minor due to their low circulating levels [57]. Under physiological stressors such as fasting or exercise, the healthy adult heart relies on metabolic plasticity to switch carbon sources to maintain ATP supply and cellular homeostasis [63–65], but the entire process remains incompletely understood. For example, Venkatesh et al. used proteomics and iPSC-CMs derived from dermal fibroblasts to elucidate the role of proteostasis—via LONP1 protease—as a negative regulator of FAO [66].
During pathological hypertrophy, the heart loses metabolic flexibility and makes an energetically unfavourable shift towards increased glucose utilization [57,67]. The precise mechanisms underlying how metabolic flexibility is maintained in health or impaired in disease remains an active area of investigation [68].
Sudden infant death syndrome
Recently, iPSC-CMs were used to study how FAO defects promote cardiac arrhythmias, such as sudden infant death syndrome (SIDS). SIDS manifests shortly after birth, once children begin nursing on lipid-rich breastmilk [69]. SIDS results from defects in the mitochondrial trifunctional protein (TFP), the central enzyme in the FAO pathway. Knockout of HADHA, the alpha subunit of TFP, leads to an accumulation of long-chain fatty acids, immature cardiolipin, mitochondrial dysfunction, aberrant calcium handling and beat rate abnormalities. SS-31 was found to rescue proton leak induced by fatty acid challenge in HADHA KO iPSC-CMs, highlighting a potential treatment for SIDS patients [69]. SS-31 has been reported to interact with a range of mitochondrial protein targets [70] and also was found to be beneficial in iPSC-CM DCMA [56]. Screening for positive effects of SS-31 across primary mitochondrial and cardiac diseases may be worthwhile.
Mitochondrial OXPHOS
Mature cardiomyocytes are the most mitochondria-rich cells in the body [11,13]. The adult human heart consumes nearly 6 kg of ATP per day, almost 15 times its weight [57,63]. Mitochondria are referred to as ‘powerhouses’ given their inherent ability to supply ATP, the energetic currency of the cell, primarily by OXPHOS. OXPHOS and glycolysis generate approximately 95% and 5% of the ATP in the adult heart, respectively [63]. The process of OXPHOS occurs when electrons are transferred from carriers to the ETC. The transfer of electrons is coupled to the pumping of protons from the matrix into the IMS, which generates an electrochemical gradient. The flow of electrons down this gradient is harnessed to regenerate ATP from ADP [3,12] (Fig. 1).
Defects in the ETC
The ETC is composed of large, multi-subunit protein assemblies known as Complex I (NADH/ubiquinone oxidoreductase, CI), Complex II (succinate dehydrogenase), Complex III (cytochrome c reductase, CIII) and Complex IV (cytochrome c oxidase, CIV). Of these, CI is the largest and most elaborate, containing 45 subunits amounting to a total mass of ~ 1 MDa. Mutations to the 13 ‘core’ protein subunits or the 31 accessory subunits account for nearly one-third of ETC deficiencies such as Leber’s hereditary optic neuropathy, Leigh syndrome and mitochondrial encephalomyopathy [71].
Friedrich’s ataxia
In addition, CI contains 8 iron–sulfur (Fe–S) clusters necessary for electron transfer across the IMM, spanning nearly 150 Å [72]. Therefore, biogenesis of Fe-S is especially pertinent to proper CI function and other mitochondrial enzymatic processes [73]. Friedrich’s ataxia (FRDA), arising from mutations in the frataxin gene, is a neurodegenerative disorder commonly associated with HCM. Deficiency of the frataxin protein, a mitochondrial activator of Fe–S biogenesis, in patients leads to mitochondrial dysfunction and increased sensitivity to oxidative stress. Electron microscopy (Fig. 2) showed that the mitochondrial ultrastructure of FRDA iPSC-CMs was aberrant with swollen cristae, suggestive of functional respiratory defects. However, standard features of patient autopsies, such as iron accumulation and sarcomere disorganization, were not observed in FRDA iPSC-CMs, suggesting that in vitro models recapitulate only the early stages of FRDA cardiomyopathy [74] but still provide useful insights into a complex and debilitating disorder.
Hypertrophic cardiomyopathy
CIV is the terminal end of the ETC, which accepts two electrons from cytochrome c (CYC) to reduce oxygen into water, forming the process known as ‘mitochondrial respiration’. Mammalian CIV consists of 14 subunits and three copper II ions [75]. Derangements in CIV are associated with deleterious effects in organs with high-energy demands, such as the heart. The human SCO2 gene encodes a metallochaperone that participates in copper delivery to CIV, and thus, mutations in SCO2 are among the most common sources of CIV dysfunction. CIV disorders can lead to early on-set HCM that causes death in infancy or childhood. Cardiac pathology of SCO2 deficiency was investigated in patient-derived iPSC-CM embryoid bodies [76]. Studies demonstrated age-dependent (15-, 30- and 45-day-old cultures) abnormalities in mitochondrial morphology, including enlargement of mitochondria, disorganized cristae, increased vacuoles, increased glycogen and lipid droplets. The iPSC-CM platform also revealed that SCO mutant iPSCs might have secondary sarcoplasmic reticulum calcium handling defects, highlighting a new potential mechanism for HCM [76].
Limitations of iPSC-CM technology
Patient-derived iPSCs provide a viable strategy to enhance our understanding of human diseases and enable personalized interrogation of mitochondrial diseases [77]. Still, several barriers limit the widespread use of this model, such as the lack of consistency, variable purity and immaturity of differentiated cells.
Maturation of iPSC-CMs
The level of cell differentiation and functional maturation greatly influences mitochondrial morphology and function. As such, differentiation and maturation protocols and media supplements are therefore important factors to be kept into consideration for mitochondrial studies relying on iPSC-CMs [78]. Current iPSC-CM differentiation protocols yield cell monolayers that often structurally and functionally resemble fetal cardiomyocytes, including a metabolic reliance on glucose utilization [79]. The immature phenotype of iPSC-CMs is a major limitation for metabolic studies, as they may lead to an incomplete understanding of in vivo bioenergetics and carbon fuel preference. The urgent need for methods that provide rapid and phenotypic maturation of iPSC-CMs [80] has been met on multiple fronts. Today, maturation protocols for iPSC-CMs are widespread and include long-term culture [29], genetic manipulations [69,81], fatty acid supplementation [82–84], three-dimensional aggregation [85], electromechanical training [86,87], mechanical stimulation [88,89] and combinatorial approaches [90] (Fig. 2).
Differences between iPSC-CMs and the human heart
Differences in morphology, electrophysiology, calcium signalling and adrenergic signalling phenotypes exist between adult primary cardiomyocytes and iPSC-CMs [91]. For example, mitochondrial density and volume are comparatively low in iPSC-CMs than in adult cardiomyocytes. Mitochondrial distribution is also perinuclear and irregular throughout iPSC-CMs and lacks high levels of fission and fusion protein machinery (DRP1, OPA, etc.). This is very different from the rigid longitudinal organization of mitochondrial rows between bundles of myofilaments observed in adult cardiomyocytes [79,92]. These broad differences should be considered when designing mitochondrial experiments using iPSC-CMs. They may suggest that the iPSC-CM model is currently more applicable to disorders that occur early during development.
Future applications of iPSC-CMs in mitochondrial biology
In recent years, mitochondrial assessment during iPSC-CM maturation has become of primary importance. Coincident with this goal, technology has advanced to include compatible, high-sensitivity assays such as single-cell sequencing, whole-cell mitochondrial respiration, metabolite profiling and next-generation sequencing of mtDNA for iPSC-CM assessment (Fig. 2). Today, common metrics for mitochondrial assessment include abundance, cristae morphology, mtDNA copy number, gene expression, energy source-specific respiration and protein expression of the OXPHOS machinery [66,83,84]. Evaluation of mitochondrial function provides new ground for improvements in diagnostic, prognostic and treatment potential in confounding primary mitochondrial defects and metabolic disorders. Furthermore, iPSC-CMs can be utilized for drug repurposing efforts and novel drug discovery for mitochondrial diseases using patient-derived cells and gene-edited isogenic cell lines, providing a new avenue for precision medicine. Considerations for de novo development of mitochondrial therapies must harness endogenous mitochondrial import mechanisms for high-specificity targeting and minimal off-target effects.
Conclusions
New technological advancements have enabled iPSC-CMs to emerge as a valuable platform to study primary mitochondrial disorders and their effects on cardiac health and disease. In the future, iPSC-CMs will further enable high-throughout, mitochondrial-targeted drug discovery and patient-specific studies.
Funding
This work was supported by the National Institutes of Health Grants R01 HL113006, R01 HL130020, R01 HL141371, R01 HL146690 (to JCW), and American Heart Association Postdoctoral Fellowship 908936 (to AC). Due to space limitations, we apologize in advance for the inability to include all references on this topic.
Abbreviations
- ACE
angiotensin-converting enzyme
- BTHS
Barth syndrome
- CYC
cytochrome c
- DCMA
dilated cardiomyopathy with ataxia syndrome
- ETC
electron transport chain
- FAO
fatty acid oxidation
- FRDA
Friedrich’s ataxia
- HCM
hypertrophic cardiomyopathy
- IMM
inner mitochondrial membrane
- IMS
intermembrane space
- iPSC
induced pluripotent stem cell
- iPSC-CM
iPSC-derived cardiomyocyte
- KSS
Kearns–Sayre syndrome
- MELAS
mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes
- OMM
outer mitochondrial membrane
- OXPHOS
oxidative phosphorylation
- PBMC
peripheral blood mononuclear cells
- POLG
polymerase gamma
- SIDS
sudden infant death syndrome
- TAZ
taffazin
- TFP
trifunctional protein
- TIM
translocase of the inner membrane
- TOM
translocase of the outer membrane
References
- 1.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–90. [DOI] [PubMed] [Google Scholar]
- 2.Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20:745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–83. [DOI] [PubMed] [Google Scholar]
- 5.Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol. 2018;19:7792. [DOI] [PubMed] [Google Scholar]
- 6.Sulaiman SA, Mohd Rani Z, Mohd Radin FZ, Abdul Murad NA. Advancement in the diagnosis of mitochondrial diseases. J Transl Genet Genom. 2020;4:159–87. [Google Scholar]
- 7.Kargaran PK, Mosqueira D, Kozicz T. Mitochondrial medicine: genetic underpinnings and disease modeling using induced pluripotent stem cell technology. Front Cardiovasc Med. 2021;7:604581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell OM, Gorman GS, Lightowlers RN, Turnbull DM. Mitochondrial diseases: hope for the future. Cell. 2020;181:168–88. [DOI] [PubMed] [Google Scholar]
- 9.Almannai M, El-Hattab AW, Ali M, Soler-Alfonso C, Scaglia F. Clinical trials in mitochondrial disorders, an update. Mol Genet Metab. 2020;131:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian R, Colucci WS, Arany Z, Bachschmid MM, Ballinger SW, Boudina S, et al. Unlocking the secrets of mitochondria in the cardiovascular system. Circulation. 2019;140:1205–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barth E, St€ammler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24:669–81. [DOI] [PubMed] [Google Scholar]
- 12.Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128:3716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14:238–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation. 2019;139:1435–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrmann G, Decherd M. The chemical nature of heart failure. Ann Intern Med. 1939;12:1233–44. [Google Scholar]
- 16.Ingwall JS, Weiss RG. Is the failing heart energy starved? Circ Res. 2004;95:135–45. [DOI] [PubMed] [Google Scholar]
- 17.Taegtmeyer H. Metabolism—the lost child of cardiology. J Am Coll Cardiol. 2000;36: 1386–8. [DOI] [PubMed] [Google Scholar]
- 18.Lopaschuk GD, Rebeyka IM, Allard MF. Metabolic modulation. Circulation. 2002;105:140–2. [PubMed] [Google Scholar]
- 19.Pfeffer MA, Braunwald E, Moy e LA, Basta L, Brown EJ Jr, Cuddy TE, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1992;327:669–77. [DOI] [PubMed] [Google Scholar]
- 20.Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N Engl J Med. 1996;334:1349–55. [DOI] [PubMed] [Google Scholar]
- 21.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709–17. [DOI] [PubMed] [Google Scholar]
- 22.Tariq S, Aronow WS. Use of inotropic agents in treatment of systolic heart failure. Int J Mol Sci. 2015;16:29060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Hardenberg A, Maack C. Mitochondrial therapies in heart failure. In: Bauersachs J, Butler J, Sandner P, editors. Heart failure. Cham: Springer International Publishing; 2017. p. 491–514. [DOI] [PubMed] [Google Scholar]
- 24.Russell LK, Finck BN, Kelly DP. Mouse models of mitochondrial dysfunction and heart failure. J Mol Cell Cardiol. 2005;38:81–91. [DOI] [PubMed] [Google Scholar]
- 25.Paik DT, Chandy M, Wu JC. Patient and disease–specific induced pluripotent stem cells for discovery of personalized cardiovascular drugs and therapeutics. Pharmacol Rev. 2020;72:320–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao B-Y, Zhang J Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci U S A. 2008;105:6987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riehle C, Bauersachs J. Small animal models of heart failure. Cardiovasc Res. 2019;115:1838–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pond AL, Scheve BK, Benedict AT, Petrecca K, Van Wagoner DR, Shrier A, et al. Expression of distinct ERG proteins in rat, mouse, and human heart: relation to functional I Kr channels. J Biol Chem. 2000;275:5997–6006. [DOI] [PubMed] [Google Scholar]
- 29.Ebert A, Joshi AU, Andorf S, Dai Y, Sampathkumar S, Chen H, et al. Proteasome-dependent regulation of distinct metabolic states during long-term culture of human iPSC-derived cardiomyocytes. Circ Res. 2019;125:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, et al. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. 2016;22:547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, et al. Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2018;11: e000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–9. [DOI] [PubMed] [Google Scholar]
- 35.Garg P, Oikonomopoulos A, Chen H, Li Y, Lam CK, Sallam K, et al. Genome editing of induced pluripotent stem cells to decipher cardiac channelopathy variant. J Am Coll Cardiol. 2018;72:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Lee J, Vincent LG, Wang Q, Gu M, Lan F, et al. Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised β-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell. 2015;17:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roger AJ, Muñoz-Gómez SA, Kamikawa R. The origin and diversification of mitochondria. Curr Biol. 2017;27:R1177–92. [DOI] [PubMed] [Google Scholar]
- 40.Lester Sequiera G, Srivastava A, Alagarsamy KN, Rockman-Greenberg C, Dhingra S. Generation and evaluation of isogenic iPSC as a source of cell replacement therapies in patients with Kearns Sayre syndrome. Cells. 2021;10:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li S, Pan H, Tan C, Sun Y, Song Y, Zhang X, et al. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem Cell Rep. 2018;10:808–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang KX, Kristiansen CK, Mostafavi S, Vatne GH, Zantingh GA, Kianian A, et al. Disease-specific phenotypes in iPSC-derived neural stem cells with POLG mutations. EMBO Mol Med. 2020;12:e12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elorza AA, Soffia JP. mtDNA heteroplasmy at the core of aging associated heart failure. An integrative view of OXPHOS and mitochondrial life cycle in cardiac mitochondrial physiology. Front Cell Dev Biol. 2021;9:625020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–42. [DOI] [PubMed] [Google Scholar]
- 45.Hussain S-RA, Yalvac ME, Khoo B, Eckardt S, McLaughlin KJ. Adapting CRISPR/Cas9 system for targeting mitochondrial genome. Front Genet. 2021;12:627050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokota M, Hatakeyama H, Ono Y, Kanazawa M, Goto YI. Mitochondrial respiratory dysfunction disturbs neuronal and cardiac lineage commitment of human iPSCs. Cell Death Dis. 2017;8:e2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perales-Clemente E, Cook AN, Evans JM, Roellinger S, Secreto F, Emmanuele V, et al. Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J. 2016;35:1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, et al. Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell. 2016;18:625–36. [DOI] [PubMed] [Google Scholar]
- 49.Hämäläinen RH. Mitochondrial DNA mutations in iPS cells: mtDNA integrity as standard iPSC selection criteria? EMBO J. 2016;35:1960–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lightowlers RN, Chrzanowska-Lightowlers ZM, Russell OM. Mitochondrial transplantation—a possible therapeutic for mitochondrial dysfunction? EMBO Rep. 2020;21:e50964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horvath SE, Daum G. Lipids of mitochondria. Prog Lipid Res. 2013;52:590–614. [DOI] [PubMed] [Google Scholar]
- 52.Richter-Dennerlein R, Korwitz A, Haag M, Tatsuta T, Dargazanli S, Baker M, et al. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with Prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014;20:158–71. [DOI] [PubMed] [Google Scholar]
- 53.Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–67. [DOI] [PubMed] [Google Scholar]
- 55.Davey KM, Parboosingh JS, DR ML, Chan A, Casey R, Ferreira P, et al. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet. 2006;43:385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rohani L, Machiraju P, Sabouny R, Meng G, Liu S, Zhao T, et al. Reversible mitochondrial fragmentation in iPSC-derived cardiomyocytes from children with DCMA, a mitochondrial cardiomyopathy. Can J Cardiol. 2020;36:554–63. [DOI] [PubMed] [Google Scholar]
- 57.Ritterhoff J, Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res. 2017;113:411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dorn GW, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015;29:1981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamosh M, Simon MR, Canter H, Hamosh P. Lipoprotein lipase activity and blood triglyceride levels in fetal and newborn rats. Pediatr Res. 1978;12:1132–6. [DOI] [PubMed] [Google Scholar]
- 60.Portman O, Behrman R, Soltys P. Transfer of free fatty acids across the primate placenta. Am J Physiol. 1969;216:143–7. [DOI] [PubMed] [Google Scholar]
- 61.Girard J, Ferre P, Pegorier J, Duee P. Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol Rev. 1992;72:507–62. [DOI] [PubMed] [Google Scholar]
- 62.Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56:130–40. [DOI] [PubMed] [Google Scholar]
- 63.Ingwall JS. ATP and the heart. New York: Springer Science & Business Media; 2002. [Google Scholar]
- 64.Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Venkatesh S, Baljinnyam E, Tong M, Kashihara T, Yan L, Liu T, et al. Proteomic analysis of mitochondrial biogenesis in cardiomyocytes differentiated from human induced pluripotent stem cells. Am J Phys Regul Integr Comp Phys. 2021;320:R547–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, et al. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res. 2020;126:182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metab. 2017;25:1027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miklas JW, Clark E, Levy S, Detraux D, Leonard A, Beussman K, et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat Commun. 2019;10: 4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chavez JD, Tang X, Campbell MD, Reyes G, Kramer PA, Stuppard R, et al. Mitochondrial protein interaction landscape of SS-31. Proc Natl Acad Sci U S A. 2020;117:15363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Padavannil A, Ayala-Hernandez MG, Castellanos-Silva EA, Letts JA. The mysterious multitude: structural perspective on the accessory subunits of respiratory complex I. Front Mol Biosci. 2022;8:798353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kampjut D, Sazanov LA. The coupling mechanism of mammalian respiratory complex I. Science. 2020;370: eabc4209. [DOI] [PubMed] [Google Scholar]
- 73.Rouault TA, Tong W-H. Iron–sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol. 2005;6:345–51. [DOI] [PubMed] [Google Scholar]
- 74.Hick A, Wattenhofer-Donzé M, Chintawar S, Tropel P, Simard JP, Vaucamps N, et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis Model Mech. 2013;6:608–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zong S, Wu M, Gu J, Liu T, Guo R, Yang M. Structure of the intact 14subunit human cytochrome c oxidase. Cell Res. 2018;28:1026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hallas T, Eisen B, Shemer Y, Ben Jehuda R, Mekies LN, Naor S, et al. Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell–derived cardiomyocytes. J Cell Mol Med. 2018;22:913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chanana AM, Rhee J-W, Wu JC. Human-induced pluripotent stem cell approaches to model inborn and acquired metabolic heart diseases. Curr Opin Cardiol. 2016;31:266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McKnight CL, Low YC, Elliott DA, Thorburn DR, Frazier AE. Modelling mitochondrial disease in human pluripotent stem cells: what have we learned? Int J Mol Sci. 2021;22:7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu JC, Garg P, Yoshida Y, Yamanaka S, Gepstein L, Hulot J-S, et al. Towards precision medicine with human iPSCs for cardiac channelopathies. Circ Res. 2019;125:653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tu C, Chao BS, Wu JC. Strategies for improving the maturity of human induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2018;123:512–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuppusamy KT, Jones DC, Sperber H, Madan A, Fischer KA, Rodriguez ML, et al. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc Natl Acad Sci U S A. 2015;112:E2785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, et al. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Rep. 2019;13:657–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horikoshi Y, Yan Y, Terashvili M, Wells C, Horikoshi H, Fujita S, et al. Fatty acid-treated induced pluripotent stem cell-derived human cardiomyocytes exhibit adult cardiomyocyte-like energy metabolism phenotypes. Cell. 2019;8:1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rana P, Anson B, Engle S, Will Y. Characterization of human-induced pluripotent stem cell–derived cardiomyocytes: bioenergetics and utilization in safety screening. Toxicol Sci. 2012;130:117–31. [DOI] [PubMed] [Google Scholar]
- 85.Shadrin IY, Allen BW, Qian Y, Jackman CP, Carlson AL, Juhas ME, et al. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat Commun. 2017;8:1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature. 2018;556:239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eng G, Lee BW, Protas L, Gagliardi M, Brown K, Kass RS, et al. Autonomous beating rate adaptation in human stem cell-derived cardiomyocytes. Nat Commun. 2016;7:10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Radisic M, Park H, Shing H, Consi T, Schoen FJ, Langer R, et al. Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proc Natl Acad Sci U S A. 2004;101:18129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abilez OJ, Tzatzalos E, Yang H, Zhao MT, Jung G, Zöllner AM, et al. Passive stretch induces structural and functional maturation of engineered heart muscle as predicted by computational modeling. Stem Cells. 2017;36:265–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, et al. Biowire: a platform for maturation of human pluripotent stem cell–derived cardiomyocytes. Nat Methods. 2013;10:781–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sayed N, Liu C, Wu JC. Translation of human-induced pluripotent stem cells: from clinical trial in a dish to precision medicine. J Am Coll Cardiol. 2016;67:2161–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hoppel CL, Tandler B, Fujioka H, Riva A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol. 2009;41:1949–56. [DOI] [PMC free article] [PubMed] [Google Scholar]