

Abstract
Lysine (K)-specific demethylase 6A (KDM6A) is a frequently mutated tumor suppressor gene in pancreatic ductal adenocarcinoma (PDAC). However, the impact of KDM6A loss on the PDAC tumor immune microenvironment is not known. This study used a genetically engineered, pancreas-specific Kdm6a-knockout PDAC mouse model and human PDAC tissue samples to demonstrate that KDM6A loss correlates with increased tumor-associated neutrophils (TAN) and neutrophil extracellular traps (NET) formation, which are known to contribute to PDAC progression. Genome-wide Bru-seq analysis to evaluate nascent RNA synthesis showed that the expression of many chemotactic cytokines, especially CXC motif chemokine ligand 1 (CXCL1), were upregulated in KDM6A-knockout PDAC cells. KDM6A-deficient PDAC cells secreted higher levels of CXCL1 protein, which in turn recruited neutrophils. Furthermore, in a syngeneic orthotopic mouse model, treatment with a CXCL1 neutralizing antibody blocked the chemotactic and NET-promoting properties of KDM6A-deficient PDAC cells and suppressed tumor growth, confirming CXCL1 as a key mediator of chemotaxis and PDAC growth driven by KDM6A loss. These findings shed light on how KDM6A regulates the tumor immune microenvironment and PDAC progression and suggests that the CXCL1-CXCR2 axis may be a candidate target in PDAC with KDM6A loss.
Statement of significance:
KDM6A loss in pancreatic cancer cells alters the immune microenvironment by increasing CXCL1 secretion and neutrophil recruitment, providing a rationale for targeting the CXCL1-CXCR2 signaling axis in tumors with low KDM6A.
Keywords: epigenetics, histone demethylase, pancreatic ductal adenocarcinoma, cytokine, tumor microenvironment
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal cancers with a 5-year survival of less than 11%, which leads to 432,242 yearly new deaths worldwide (1,2). Recent pancreatic cancer genome sequencing demonstrated an accumulation of genetic alterations in epigenetic regulating genes in addition to the common oncogenes and tumor suppressor genes (e.g., KRAS, TP53, CDNK2A, and SMAD4) (3,4). In particular, KDM6A (Lysine (K)-specific demethylase 6A), also known as UTX (ubiquitously transcribed X chromosome tetratricopeptide repeat protein), has emerged as an important epigenetic regulator in PDAC. KDM6A is located on the X chromosome, but it escapes X inactivation in mice and humans (5). Functionally, KDM6A catalyzes the demethylation of trimethylated histone H3 lysine 27 (H3K27me3). Together with MLL methyltransferase and CBP/P300 in the COMPASS (COMplex of Proteins Associated with Set1)-like complex, KDM6A regulates the transcription and expression of downstream genes, thereby regulating cell fate and cell functional characteristics, which is particularly important for normal pancreatic development (6,7). The deletion of KDM6A promotes proliferation, invasion, and metastasis of PDAC, and it is an independent prognostic factor of PDAC(8,9). Our previous study had shown that loss of KDM6A induces an aggressive undifferentiated subtype of PDAC by promoting epithelial-mesenchymal transition (EMT) via a p38-dependent non-canonical activin A signaling using a genetically engineered PDAC mouse model (10). Meanwhile, we also discovered that KDM6A deficiency promotes persistent acinar-to-ductal metaplasia (ADM) and inflammation in a well-established cerulein-induced chronic pancreatitis mouse model, which may explain its role in accelerating KRAS-induced pancreatic tumorigenesis (10). This finding implied that loss of KDM6A may lead to an abnormal immune response to tissue damages and interfere with normal tissue repair (11). Immune cells are crucial elements in the pancreatic tumor microenvironment (TME) (12). However, how KDM6A deficiency affects the immune microenvironment of PDAC remains unknown.
Recent studies showed that epigenetics impact the remodeling of the immune microenvironment by regulating the expression of cytokines or chemokines in tumor cells (13,14). Moreover, emerging studies suggest that KDM6A deficiency may promote the development of tumors by remodeling the tumor immune microenvironment. For example, in bladder cancer, the deficiency of KDM6A in cancer cells promoted the polarization of macrophages into M2 type by activating the cytokine and chemokine pathways and led to the development of bladder cancer (13). In medulloblastoma, deletion of KDM6A significantly reduced the recruitment of CD8+ T lymphocytes (14).
In this study, we used human PDAC samples, genetically engineered mouse PDAC models, and in vitro PDAC cell lines to explore the role of KDM6A deficiency in the tumor immune microenvironment and find potential therapeutic targets in PDACs with KDM6A loss.
Materials and Methods
Tissue microarrays
All Hematoxylin and Eosin (H&E) stained slides were reviewed, diagnosed, and confirmed by an experienced board-certified and fellowship-trained gastrointestinal pathologist (JS). Corresponding areas were carefully selected and marked. Duplicated 1 mm diameter tissue cores from a total of 213 patient tissue samples were selectively punched and transferred to recipient tissue array blocks. Five tissue microarrays (TMAs) were set up according to a standard protocol, as previously described (14,15). H&E staining was performed on each TMA block using a standard protocol, and unstained slides were prepared for immunohistochemical staining.
Animal study
To generate the genetically engineered pancreas specific Kdm6a knockout (KO) mouse model, we first crossed Ptf1aCre mice (kindly shared by Dr. Christopher Wright) with Kdm6afl/fl (female) or Kdm6afl/Y (male) mice (Jackson Laboratory, #024177) to get Ptf1aCre Kdm6afl/fl (female homozygous Kdm6a KO) or Ptf1aCre Kdm6afl/Y (male hemizygous Kdm6a KO). Then we crossed LSL-KrasG12D/wt;LSL-p53R172H/wt (kindly shared by Dr. Tyler Jacks) with Ptf1aCre Kdm6afl/fl or Ptf1aCre Kdm6afl/Y to generate five different genotypes of mice (KPC- Kdm6aY/+, KPC- Kdm6aY/fl, KPC- Kdm6a+/+, KPC- Kdm6a+/fl and KPC- Kdm6afl/fl). All the animal studies were performed under the reviewed and approved animal protocol (#PRO00007450) by the Unit of Laboratory Animal Medicine (ULAM) at the University of Michigan.
For orthotopic mouse models, 5×104 KPC 7940 cells (a gift from Dr. Gregory Beatty) in 40 μl Matrigel and cell culture media (1:1) were injected into the pancreata of 6- to 8-week-old C57BL/6J mice. IgG or anti-CXCL1 antibody (4 mg/kg, MAB453-500, R&D Systems) was intraperitoneally injected twice a week from the 8th day of tumor injection. Four weeks after the tumor cell injection, mice were euthanized, and tumor tissues were collected and fixed in formalin.
Cell lines
PANC-1 cells (ATCC) and mouse KPC 7940 cells were cultured in DMEM (GIBCO) supplemented with 10% FBS (GIBCO) and 1% penicillin/streptomycin (GIBCO). HPAF-Ⅱ cells (ATCC) were cultured in EMEM with 10% FBS (GIBCO) and 1% penicillin/streptomycin (GIBCO). HPDE cells (a gift from Dr. Craig Logsdon) were cultured in Keratinocyte SFM with EGF, bovine pituitary extract (Invitrogen), and 1% penicillin/streptomycin (GIBCO). All cells were cultured at 37°C with 5% CO2. PLB-985 cells (kindly provided by Dr. Alan Smrcka) were cultured in RPMI 1640+GlutaMAX® (GIBCO) supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin (GIBCO). For PLB-985 cell differentiation, 0.5×106 cells/mL was cultured in fully supplemented RPMI media with 1.3% DMSO. The media was changed every 2 days. After 5 days, differentiated PLB-985 cells were ready for the chemotaxis assay.
Cell lines were authenticated by DNA fingerprinting using the AmpFISTR Amplification or AmpFISTR Profiler PCR Amplification protocols (Life Technologies) and were negative for mycoplasma.
KDM6A-knockout PANC-1 cells were generated by using Edit-R Lentiviral Cas9 nuclease vectors with synthetic CRISPR RNAs (Dharmacon). Briefly, PANC-1 cells were transduced with lentiviral Cas9 nuclease expression particles and then cultured in the selection medium with blasticidin. The stable PANC-1 cell line expressing the Cas9 nuclease was transfected with Edit-R Human KDM6A (7403) crRNA (Dharmacon, #CM-014140-04-0002) and Edit-R CRISPR-Cas9 Synthetic tracrRNA (Dharmacon, #U-002005-05) by using DharmaFECT transfection reagents. The phenotype of KDM6A-knockout isolated clones was analyzed by western blots and their genotype was confirmed by Sanger DNA sequencing.
Knockdown of KDM6A in HPAF-Ⅱ and HPDE cells was achieved by direct transfection of KDM6A siRNAs or the scramble siRNA (Dharmacon) (50 nM) using Lipofectamine RNA iMAX reagent (Invitrogen) according to the manufacturer’s directions.
Stable knockdown of Kdm6a in KPC 7940 cells was established by transducing GIPZ lentivirus (Horizon Discovery, #RHS4346 and #V2LMM_196734) mouse Kdm6a shRNA into cells. Briefly, lentivirus was produced in 293FT cells by transfecting plasmid DNA and packaging plasmids (pMD2.G and psPAX2, Addgene). Lentivirus-containing supernatant was collected after 24 hours. KPC 7940 cells were incubated with lentivirus in DMEM with polybrene. Selection with puromycin was started 48–72 hours after infection. Targeted sequences of crRNAs, siRNAs, and shRNA are provided in Supplementary Table 1.
Bromouridine sequencing (Bru-seq) and Gene Set Enrichment Analysis (GSEA)
Bru-seq was performed as previously described (14,16). Briefly, PANC-1 cells were incubated in bromouridine (Bru) (2 mM, Sigma-Aldrich) contained media for 30 minutes at 37°C. Cells were then lysed in Trizol, and total RNA was isolated. Bru-labeled RNA was immunocaptured using anti-BrdU antibodies (BD Pharmingen, 555627). Then strand-specific cDNA libraries were prepared using the Illumina TruSeq kit (Illumina), followed by deep sequencing using the Illumina sequencing platform as previously described (14,17,18).
The difference in expression profile was pre-ranked based on rLogFC value with a cut-off of 300 bp to eliminate the signal from noise or background. The pre-ranked file was loaded into the GSEA software tool (Broad Institute, Inc., Massachusetts Institute of Technology, and Regents of the University of California) for analysis of the upregulated pathways that are enriched in a positive or negative manner. The list of the top 10 gene sets is represented by a bar graph based on their Normalized enrichment score (NES).
RNA extraction and quantitative real-time RT-PCR
Total RNAs were isolated using the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s directions. cDNA was prepared using the SuperScript ® III First-Strand Synthesis kit (Invitrogen). Quantitative RT-PCR of RNA was performed with SYBR Green reagents (Applied Biosystems) in MicroAmp Optical 96-well reaction plates (Applied Biosystems). Primer sequences are listed in Supplementary Table 2.
Chromatin immunoprecipitation (ChIP) and real-time PCR
ChIP was performed following the manufacturer’s instructions (Cell Signaling Technology, #9003S). Briefly, cells were fixed with formaldehyde and lysed. Chromatin was fragmented using Micrococcal nuclease. Antibodies against KDM6A (Cell Signaling Technology, #33510), H3K27me3 (Cell Signaling Technology, #9733), H3K27ac (Active Motif, #39060), H3K4me3 (Active Motif, #39060) and H3K4me (Abcam, #ab8580) were used for chromatin immunoprecipitation following the manufacturer’s recommendation. Protein-DNA cross-link was reversed, and DNA was isolated for real-time quantitative PCR.
Western Blot
Proteins were prepared using whole-cell lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 2 mM ethylenediaminetetraacetic acid (EDTA), 1 mM PMSF, 1X proteinase inhibitor, and 1.5% NP-40) followed by concentration measurement using the Bradford assay (Bio-Rad). Prepared protein samples were separated on sodium dodecyl sulfate (SDS)-PAGE and transferred to PVDF membranes (Millipore). After one hour of blocking, the membranes were incubated with the primary antibodies overnight at 4°C, followed by the incubation with peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) for 30 minutes. Then the proteins were visualized using an ECL detection kit (Thermo Scientific).
Enzyme-linked immunosorbent assay (ELISA)
ELISA of related chemokines was performed using DuoSet ELISA Development Systems (R&D Systems) according to the manufacturer’s recommended protocol modified to incorporate overnight sample incubation. The supernatant of human PDAC cell lines including PANC-1, HPAF-Ⅱ, and HPDE cells and murine KPC 7940 cells were collected for test after 1 day or 3 days of incubation.
Immunohistochemistry (IHC) and immunofluorescence (IF)
Formalin-fixed, paraffin-embedded tissue sections were used for IHC and IF analysis. For IHC, primary antibodies CD3 (#A0452, Dako Agilent), B220 (CD45R, #14-0452-82, Thermo Fisher Scientific), F4/80 (#70076S, Cell Signaling Technology), myeloperoxidase (MPO, #AF3667, R & D Systems), CXCL1 (#ab86436, Abcam), and CK19 (#ab52625, Abcam) were used. All tissue sections were deparaffinized and rehydrated, and antigen retrieval was performed in EDTA buffer (pH 9.0) or citrate buffer (pH 6.0). Sections were incubated with 3% hydrogen peroxide for 10 minutes to block endogenous peroxidase activity and blocked with 10% Bovine Serum albumin prepared in Tris Buffered Saline (TBS) or 20 minutes. Then, sections were incubated with the above primary antibodies overnight at 4°C, respectively. The next day, after washing 3 times with TBS, slides were incubated with secondary antibody (SignalStain® Boost IHC Detection Reagent, HRP, Rabbit #8114 and Mouse #8125, Cell Signaling Technology; Goat #ab97110, Abcam; Rat #ab6734, Abcam) for 30 minutes at room temperature, stained with DAB substrate kit (SignalStain DAB chromogen substrate, Cell Signaling Technologies), counterstained with hematoxylin, and mounted with mounting medium from Signal Stain mounting medium (Cell Signaling Technology #14177s). Positive cells of B220 and MPO in the peritumoral and intertumoral hot spots (3-5 hot spots per sample) were blindly quantified manually by two authors (JY and LJ). Positive cells of CD3 and F4/80 in the peritumoral and intertumoral hot spots (3-5 hot spots per sample) were quantified via ImageJ software (NIH; http://imagej.nih.gov/ij/) according to a standard procedure for the quantification of images. The percentage and intensity of CXCL1 positive cells were scored, as previously described (19). For IF staining, primary antibodies myeloperoxidase (MPO, #AF3667, R&D Systems), citrullinated histone H3 (Cit-histone H3, #ab5103, Abcam), and CXCR2 (#ab14935, Abcam) were used. Alexa Fluor 488 goat anti-rabbit IgG (H+L) Cross-Adsorbed secondary antibody (#A-11008, Invitrogen) and NorthenLights anti-goat IgG-NL557 (#NL001, R&D Systems) were used as the second antibodies. The nuclei were counterstained with ProLong™ Gold Antifade Mountant with DAPI (#P36031, Invitrogen). Confocal microscope ZENS 800 was used to scan z-stack images for every IF section (3 z-stack images/sample, magnification 63×). Colocalization analysis for MPO/Cit-histone H3 and CXCR2/MPO was performed via ZENS 2.3 software. The mean colocalization area per sample was calculated.
Mouse primary neutrophil isolation
Mouse primary neutrophils were isolated from mouse femur bone marrow of wild-type C57BL/6J mice. After washing with Hank’s buffered salt solution (HBSS) without Ca2+/Mg2+, bone marrow cells were resuspended in ammonium-chloride-potassium (ACK) buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM Na2-EDTA, pH 7.4) and washed again with HBSS without Ca2+/Mg2+. Neutrophils were separated from mononuclear cells by layering 1 mL of the cell suspension on 3 mL of Histopaque-1077 (density, 1.077 g/ml) and 3 mL of Histopaque-1119 (density, 1.119 g/ml), followed by centrifuging at 400 x g for 30 min at room temperature. The middle layer enriched for neutrophils was washed twice in HBSS without Ca2+/Mg2+.
Chemotaxis assay
The 24-well plate and 3 μm polyester membrane inserts were coated with 2% BSA solution for 1 hour at 37°C, followed by washing with DPBS (GIBCO). 0.4×106 differentiated PLB-985 cells or mouse primary neutrophils (in 100 μl HBSS) were seeded in the upper chamber. 600μl conditioned media was added into the lower chamber. For CXCL1 blockage, 1 μg/ml anti-CXCL1 antibody (MAB453-500, R&D Systems) was added into the lower chamber. After incubating at 37°C for 2 hours, the membrane was stained and cell numbers in the lower chamber were counted.
Flow cytometry
Single-cell suspensions were filtered through a 40 μM nylon mesh cell strainer and washed twice with a 2% FBS/PBS mix. Blocking was done with anti-Mouse CD16/CD32 antibodies (BD, 553142) and cells were stained with Live/Dead dye (Thermo Fisher, NC0584313) for 10 minutes at room temperature. Cells were then stained with the following antibody at 4°C for 30 minutes: anti-CD11b (Biolegend, 101256), anti-Ly6G (Biolegend, 127628). Data were analyzed with FlowJo v10 software.
Statistical analyses
All statistical analyses in our study were performed using GraphPad Prism 7. An unpaired t-test or Mann-Whitney test was used to make comparisons between two groups. Multiple group comparisons were performed using a one-way ANOVA test. The results are shown as mean ± standard deviation (SD) or mean ± standard error of the mean (SEM). Significant differences are noted by P-value ≤ 0.05. The number of independent experiments (N) is indicated in the figure legends.
Data Availability
The data generated in this study are available within the article and publicly available in Gene Expression Omnibus (GEO) at GSE202873.
Results
Loss of KDM6A is associated with tumor-associated neutrophils (TANs) accumulation in human and murine PDACs
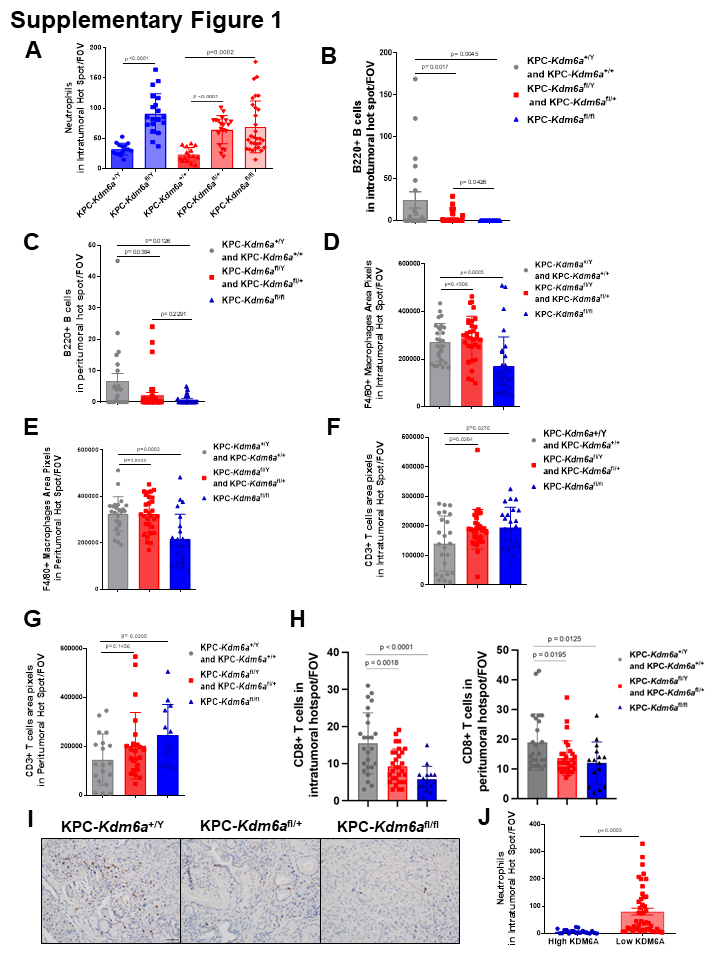
To determine whether KDM6A loss influence immune TME in PDAC, we first performed IHC staining of various immune cells, including T cells (CD3+), CD8+ T cells, B cells (B220+), macrophages (F4/80+), and neutrophils (MPO+) in a genetically engineered PDAC mouse model with pancreas-specific Kdm6a KO (KPC-Kdm6a) (Fig. 1A). We found tumor associated neutrophils (TANs) were significantly increased in Kdm6a knockout PDACs (KPC-Kdm6afl/Y, KPC-Kdm6afl/+, and KPC-Kdm6afl/fl) compared to Kdm6a wild-type (KPC-Kdm6a+/Y and KPC-Kdm6a+/+) (Fig. 1B-C, Supplementary Fig. 1A). No statistically significant change in neutrophil numbers was observed in peritumoral areas (Fig. 1D). B220-positive B cells were decreased in Kdm6a knockout groups compared with Kdm6a wild-type groups in both intratumoral and peritumoral areas (Supplementary Fig. 1B-C). F4/80-positive macrophages in both intra- and peri-tumoral areas were decreased in homozygous Kdm6a knockout mice (KPC-Kdm6afl/fl) but not in heterozygous Kdm6a knockout mice (KPC-Kdm6afl/Y, KPC-Kdm6afl/+) (Supplementary Fig. 1D-E). CD3-positive T cells were slightly increased in Kdm6a knockout mice in both intra- and peri-tumoral areas (Supplementary Fig. 1F-G). However, CD8+ T cells were decreased in Kdm6a knockout groups compared with Kdm6a wild-type groups in both intratumoral and peritumoral areas (Supplementary Fig. 1H-I). Because neutrophils were the only immune cells that were consistently and specifically increased in the intratumoral areas in Kdm6a knockout tumors, we decided to focus our studies on TANs.
Figure 1. Loss of KDM6A is associated with increased tumor-associated neutrophils (TANs) in human and murine PDACs.

(A) Genetic schematic of Ptf1aCre; LSL-KrasG12D/wt; LSL-p53R172H/wt; Kdm6afl/Y/ Kdm6afl/fl (KPC-Kdm6a) mouse model. (B) Representative images of H&E and IHC staining of KDM6A and MPO in KPC-Kdm6a mice pancreata, scale bar 50 μm. (C-D) Quantification of intratumoral (C) and peritumoral (D) MPO+ TANs (n=3 mice for KPC-Kdm6a+/+ and KPC-Kdm6a+/Y, n=4 mice for KPC-Kdm6afl/+ and KPC-Kdm6afl/Y, and n=6 mice for KPC-Kdm6afl/fl; mean±SD, unpaired t-test). Five hot spots per sample were quantified. FOV: field of view. (E) Representative images of H&E and IHC staining of KDM6A and MPO in human PDAC samples, scale bar 100 μm. (F) Quantification of MPO+ TANs (n = 4 samples for high KDM6A and n = 9 samples for low KDM6A; mean±SEM, unpaired t-test). Five intratumoral hot spots per sample were taken and quantified. FOV: field of view.
To investigate if the increased TANs in KPC-Kdm6a KO mice applied to human PDACs, we compared TANs in human PDACs with high or low KDM6A protein expression. Consistent with our data from the animal models, we observed increased TANs in KDM6A-low tumors compared to KDM6A-high tumors (Fig. 1E-F and Supplementary Fig. 1J). These results suggested that TAN accumulation in human and murine PDACs is associated with KDM6A loss.
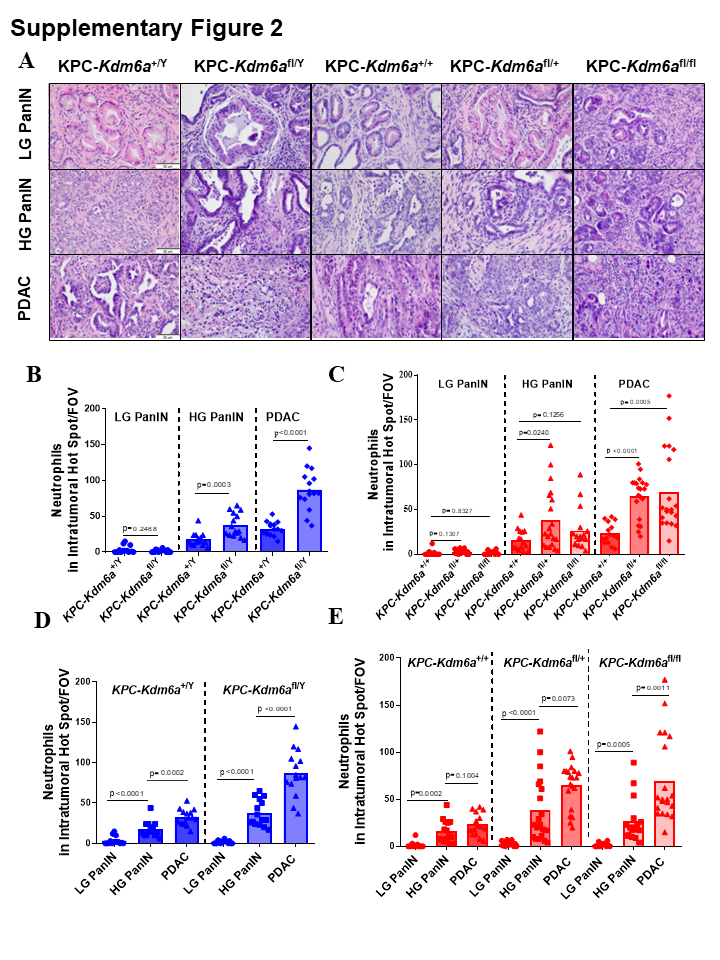
Pancreatic intraepithelial neoplasia (PanIN) is a precursor lesion of PDAC. To determine at what stage TANs were recruited during PDAC progression, we quantified TANs in both low-grade (LG) and high-grade (HG) PanIN lesions comparing to PDACs using our KPC-Kdm6a mouse model. In both female and male mice, TANs were significantly increased in HG PanINs and PDACs in Kdm6a knockout mice compared with wild-type group, while no significance was observed in LG PanIN between groups (Supplementary Fig. 2A-C). These results imply that KDM6A loss is associated with increased TANs infiltration starting at the late stages of PDAC initiation and prior to PDAC development. When we further compared TANs infiltration in LG PanIN, HG PanIN, and PDAC in the same genotype, we found that the number of TANs increased from LG PanIN to HG PanIN to PDACs in both Kdm6a knockout and Kdm6a wild-type groups (Supplementary Fig. 2A, D-E). However, the degree of TANs increase appeared to be higher in Kdm6a knockout groups (23 and 55-fold vs. 5 and 10-fold increase in HG PanIN and PDAC in male Kdm6a knockout and wild-type respectively, and 16 and 43-fold vs. 12 and 17-fold increase in HG PanIN and PDAC in female homozygous Kdm6a knockout and wild-type respectively) (Supplementary Fig. 2A, D-E). These results suggest that increased TANs are closely related with PDAC initiation and progression and loss of KDM6A may accelerate this process.
Loss of KDM6A is associated with neutrophil extracellular trap (NET) formation in human and murine PDAC
Many recent studies have highlighted that neutrophils play an important role in tumor immunology (20). Neutrophil can release NETs when it is activated by various stimuli, including chemokines and cytokines secreted by tumor cells (21). NETs are defined as web-like structures formed by decondensed chromatin (histones and DNA) and antimicrobial components, such as neutrophil elastase and MPO (22,23). Multiple studies showed that NETs could promote tumor metastasis, epithelial-mesenchymal transition (EMT), thrombosis, and awakening cancer dormancy (24,25). Given that TANs were significantly increased in both human and murine PDAC with low KDM6A, we speculated that the formation of NETs (NETosis) would increase as well. IF staining of NETosis markers, MPO and citrullinated histone H3 (CitH3) (26), was performed in human and mouse PDAC samples (Fig. 2A-D). Colocalization analysis confirmed that CitH3 was increased and colocalized with MPO in KDM6A-low human or Kdm6a knockout mouse PDAC samples. These results implied that loss of KDM6A in tumor cells is associated with increased TANs infiltration and NETs generation.
Figure 2. Loss of KDM6A induces neutrophil extracellular traps (NETs) formation in human and murine PDACs.

(A) Representative images of IF staining for MPO (red), CitH3 (green), and DAPI (blue) in KPC-Kdm6a mice PDACs. Scale bar = 10 μm. (B) Quantification of NETs in KPC-Kdm6a mice PDACs using ZENS software (n = 3 mice per group, mean±SEM, unpaired t-test). Three intratumoral hot spots per sample were analyzed in z stack images. FOV: field of view. (C) Representative images of IF staining for MPO (red), CitH3 (green), and DAPI (blue) in human PDAC samples. Scale bar = 10 μm. (D) Quantification of NETs in human PDAC samples using ZENS software (n=4 samples per group, mean±SEM, unpaired t-test). Three intratumoral hot spots per sample were analyzed in z stack images. FOV: field of view.
Loss of KDM6A in PDAC cells promotes neutrophil chemotaxis in vitro
To investigate whether KDM6A loss in PDAC cells is the cause of the TANs attraction, we performed the chemotaxis assay in vitro using differentiated PLB-985 cells and mouse primary neutrophils isolated from bone marrow. PLB-985 is a commonly used acute myeloid leukemia cell line (27) that can be differentiated into neutrophil-like cells with DMSO treatment (28-31). We first confirmed the differentiation of PLB-985 cells to mature neutrophil-like cells (PLB-985N) after 5 days of DMSO treatment (Fig. 3A). We then added the differentiated PLB-985N cells in the upper chamber and conditioned media from KDM6A knockout or knockdown PDAC cells into the lower chamber of transwell migration system (Fig. 3B). The conditioned media from KDM6A knockout or knockdown PDAC cells induced stronger migration in PLB-985N cells compared to the control cells (Fig. 3C-D). Next, we isolated neutrophils from mouse bone marrow and confirmed that 98.9% of the isolated cells were CD11b+/Ly6G+ by flow cytometry (Fig. 3E). Again, the migration of mouse neutrophils towards conditioned medium from murine KPC 7940 PDAC cells with Kdm6a-knockdown increased (Fig. 3F). Taken together, these results indicated that PDAC cells with KDM6A loss promote neutrophil recruitment in vitro.
Figure 3. Loss of KDM6A in PDAC cells promotes neutrophil chemotaxis in vitro.

(A) Histologic changes in PLB-985 cells after DMSO-induced differentiation. (B) Diagram showing the in vitro neutrophil chemotaxis assay using differentiated PLB-985N cells or primary mouse bone marrow neutrophils incubated with condition media from PDAC cells lack of KDM6A. (C) Representative images and quantifications of migrated PLB-985N cells in the conditional media from KDM6A knockout (KO) or control PANC-1 cells (mean±SEM, unpaired t-test). (D) Representative images and quantifications of migrated PLB-985N cells in the conditional media from KDM6A knockdown (siKDM6A) or control (siCtrl) HPAF-II PDAC cells (mean±SEM, unpaired t-test). (E) Flow cytometry result of mouse primary neutrophils isolated from bone marrow identified by CD11b+ and Ly6G+ cells. (F) Quantifications of migrated mouse primary neutrophils isolated from bone marrow in the conditional media from Kdm6a knockdown (shKdm6a) or control (shCtrl) KPC 7940 cells (mean±SEM, unpaired t-test). FOV: field of view.
KDM6A loss increases CXCL1 expression in PDAC cells
To explore the mechanism that KDM6A loss in tumor cells promotes neutrophils recruitment and NETs formation, we generated PANC-1 KDM6A knockout (PANC-1 KO) cells using CRISPR/Cas9 system (Fig. 4A). We then performed Bru-seq analysis (16,17) to determine the impact of KDM6A loss on nascent RNA transcription. GSEA showed that several inflammatory signaling pathways, including TNFα, inflammatory response, TGF beta, and interferon gamma response pathways, were enriched in PANC-1 KDM6A knockout cells (Supplementary Fig. 3A). More specifically, the expression of cytokines and chemokines contributing to neutrophil attraction, activation, and polarization, including CXCL1, CXCL2, CXCL8, CCL5, and VEGFA, was upregulated (Fig. 4B, Supplementary Table 3, and Supplementary Fig. 3B). Real-time RT-PCR analyses confirmed the increase in CXCL1 and CXCL2 transcriptions in both KDM6A KO cells (Fig. 4C). ELISA showed increased levels of CXCL1, CXCL2, CXCL8, and VEGF in the cell culture media of KDM6A KO PDAC cells compared to control cells (Fig. 4D).
Figure 4. Loss of KDM6A upregulates CXCL1 expression in human and mouse PDAC cell lines.

(A) Western blot of KDM6A in KDM6A knockout or control PANC-1 cells. β-actin was used as the loading control. (B) Heatmap showing the alterations of chemokine and cytokine gene expressions comparing KDM6A knockout with control PANC-1 cells based on Bru-Seq data. (C) Quantitative real-time RT-PCR of a panel of neutrophil-related chemokines and cytokines in KDM6A knockout and control PANC-1 cells. (D) ELISA result of neutrophil-related chemokine and cytokine proteins in KDM6A knockout and control PANC-1 cells. (E) Quantitative real-time RT-PCR of CXCL1 mRNA in KDM6A knockdown and control HPAF-II and HPDE cells. (F) ELISA of CXCL1 protein in KDM6A knockdown and control HPAF-II and HPDE cells. (G) Western blot of KDM6A in Kdm6a knockdown (shKdm6a) and control (shCtrl) murine KPC 7940 cells. GAPDH was used as the loading control. (H) Quantitative real-time RT-PCR of Cxcl1 in Kdm6a knockdown and control KPC 7940 cells. (I) ELISA of CXCL1 protein in Kdm6a knockdown and control KPC 7940 cells. (J) Quantification of migrated neutrophils in the conditional media from Kdm6a knockdown or control KPC 7940 cells treated with anti-CXCL1 neutralizing antibody or IgG isotype (mean±SEM, unpaired t-test) in a neutrophil chemotaxis assay.
Since CXCL1 is the most significantly and consistently upregulated chemokine and a well-known neutrophil-recruiting chemokine (32), we decided to focus on CXCL1. To explore the potential mechanism of transcriptional regulation by KDM6A and if there are any transcription factor involvement, we analyzed transcription factor binding targets using our Bru-seq data and compared them with transcription factor analyses based on publicly available database using PROMO (33) and GeneCards for CXCL1 gene. We found that the only transcription factor shared among all 3 analyses was CEBPB (Supplementary Fig. 3C). In addition, CEBPB is also the shared transcription factor for CXCL2, CXCL8, and VEGFA. Therefore, we postulate that the regulation of CXCL1 expression by KDM6A may be mediated by CEBPB. In addition, ChIP-PCR analysis using histone modification marks and KDM6A antibodies showed specific binding of KDM6A to CXCL1 promoter region and predominantly active CXCL1 enhancer/promoter in KO cells compared to WT cells (Supplementary Fig. 3D-E), confirming the increased expression of CXCL1 in KO cells is at the transcriptional level and is associated with decreased KDM6A binding and increased H3K27me3, H3K27ac, and H3K4me/me3 marks at the CXCL1 promoter region. We confirmed the upregulation of CXCL1 transcription and protein secretion in two additional human pancreatic cell lines (HPAF-II: PDAC cell line, HPDE: normal human pancreatic ductal epithelial cell line) and one murine PDAC cell line derived from KPC mouse with KDM6A knockdown (Fig. 4E-I and Supplementary Fig. 3F-G). To determine if the regulation of CXCL1 expression is dependent on the enzymatic activity of KDM6A, we treated cells with the KDM6A inhibitor, GSK-J4. CXCL1 expression was increased upon GSK-J4 treatment compared to vehicle control (Supplementary Fig. 3H), suggesting that the regulation of CXCL1 expression by KDM6A is at least in part enzyme-dependent. To investigate if CXCL1 is the main mediator of neutrophil recruitment, we used neutralizing CXCL1 antibody to block CXCL1 in the neutrophil chemotaxis assay. Indeed, the increased neutrophil recruitment by KDM6A loss was completely abolished by CXCL1 antibody (Fig. 4J). These results support that KDM6A deficiency up-regulates CXCL1 expression in PDAC cells and that CXCL1 is the major chemokine secreted by PDAC cells to recruit neutrophils.
CXCL1 expression is increased in PDACs with KDM6A deficiency in vivo
To determine whether CXCL1 expression is also increased in KDM6A deficient PDACs in vivo, we first performed CXCL1 and CK19 IHC staining with PDAC samples from KPC-Kdm6a mice. CXCL1 expression was increased significantly in Kdm6a knockout PDACs compared with wild-type PDACs (Fig. 5A-B). None of the tumors expressed squamous marker, p63 (Fig. 5A). Similar observation was made in human PDAC samples with either high or low KDM6A expression: KDM6A-low PDACs expressed much higher level of CXCL1 than KDM6A-high PDACs (Fig. 5C-D). These results supported our in vitro data and indicated that KDM6A loss is highly related to increased CXCL1 expression in PDAC cells.
Figure 5. The expression of CXCL1 protein is increased in human and murine PDACs/pancreata with KDM6A loss in vivo.

(A) Representative images of H&E and IHC staining of CXCL1,CK19, and p63 in KPC-Kdm6a mice. Scale bar=50 μm. (B) Quantification of CXCL1 IHC immunoreactivity (IR) score in KPC-Kdm6a mice pancreata (n = 3 for KPC-Kdm6a+/+ and KPC-Kdm6a+/Y, n = 4 for KPC-Kdm6afl/+and KPC-Kdm6afl/Y, and n = 6 for KPC-Kdm6afl/fl; mean±SEM, unpaired t-test). Five random areas per sample were evaluated. FOV: field of view. IR score=IHC staining intensity score × percentage of positive cells score. Intensity: 0=negative; 1=weak; 2=moderate; and 3=strong. Percentage of positive cells: 0, <5%; 1, 5-25%; 2, 26-50%; 3, 51-75%; 4, >75%. (C) Representative images of H&E and IHC staining of CXCL1 and CK19 in human PDAC samples with high or low KDM6A expression. Scale bar=50 μm. (D) Quantification of CXCL1 IR score in human PDAC samples (n = 4 for high KDM6A and n = 9 for low KDM6A; mean±SD, unpaired t-test).
Since CXCR2 is the high affinity receptor for CXCL1 (34), we then investigated whether TANs express CXCR2. We performed IF analyses of CXCR2 and MPO using both human and murine PDAC samples. Again, Kdm6a knockout murine PDACs and KDM6A-low human PDACs contained increased TANs which all expressed CXCL1 receptor CXCR2 (Fig. 6A-D).
Figure 6. CXCR2 expression in neutrophils in vivo.

(A) Representative images of IF staining for MPO (red), CXCR2 (green), and DAPI (blue) in KPC-Kdm6a mice (n = 3 for KPC-Kdm6a+/+, KPC-Kdm6afl/+, KPC-Kdm6a+/Y and KPC-Kdm6afl/Y, n = 4 for KPC-Kdm6afl/+, n=6 for KPC-Kdm6afl/fl). Scale bar = 10 μm. (B) Quantification of CXCR2+ TANs in KPC-Kdm6a mice using ZENS software. Three intratumoral hot spots per sample were analyzed in z stack images. FOV: field of view. Statistical difference was analyzed by unpaired t-test. (C) Representative images of IF staining for MPO (red), CXCR2 (green), and DAPI (blue) in human PDAC samples with low or high KDM6A expression (n=4 samples per group). Scale bar = 10 μm. (D) Quantification of CXCR2+ TANs in human PDAC samples using ZENS software. Three intratumoral hot spots per sample were analyzed in z stack images. Statistical difference was analyzed by unpaired t-test.
CXCL1 inhibition completely reverses NETosis and PDAC growth in vivo
To further confirm CXCL1 is the major mediator for KDM6A loss-induced TANs infiltration and tumor growth in vivo, we used neutralizing antibody to block CXCL1 in a syngeneic orthotopic PDAC mouse model. We orthotopically implanted murine KPC 7940-control (KPC shCtrl) or KPC-Kdm6a-deficient (KPC shKdm6a) cells to immunocompetent C57BL/6J mice. We then administrated CXCL1 neutralizing antibody or IgG control to the tumor bearing mice (Fig. 7A). Consistent with our previous study(10), Kdm6a-deficient tumors were larger compared to control tumors treated with IgG (Fig. 7B-C). Importantly, CXCL1 neutralization completely abolished Kdm6a-deficient tumor growth to the similar level of control tumor (Fig. 7B-C), indicating that CXCL1 is the main mediator of KDM6A loss-induced tumor growth in an immunocompetent animal model. Consistent with our in vitro data, Kdm6a-deficient tumors had more TAN infiltration and NETosis compared with control tumors treated with IgG (Fig. 7D-G). Anti-CXCL1 antibody treatment completely rescued these phenotypes (Fig. 7D-G). Together, our results highlighted CXCL1 as the central mediator of KDM6A-deficiency-induced TAN recruitment, NETosis, and tumor growth in PDAC, and a candidate therapeutic target in PDACs with KDM6A loss.
Figure 7. CXCL1 blockage suppresses TAN infiltration and PDAC growth in vivo.

(A) Diagram of experimental design. n=3-6 mice/group. (B) Images of tumors in each group, scale bar = 1 cm. x = no identifiable tumor. (C) Comparing tumor weights among groups (mean±SEM, Mann-Whitney test). (D) Representative images of MPO IHC staining of tumors from each group. Scale bar = 50 μm. (E) Quantification of TANs (n=3-6 mice/group; mean±SD, unpaired t-test). (F) Representative images of IF staining of MPO (red), CitH3 (green), and DAPI (blue) in tumors. Magnifications, 400×. (G) Quantification of MPO and CitH3 overlapped area (mean±SEM, Mann-Whitney test).
Discussion
It is well known that epigenetic deregulation and chromatin remodeling play important roles in PDAC tumorigenesis (35-37). Recent whole-exome sequencing discovered that in addition to the common gene mutations, such as KRAS, TP53, SMAD4, and CDKN2A, defects in epigenetic drivers that regulated chromatin accessibility were found in almost 40% of pancreatic cancer (3,4). Among the dysregulated epigenetic regulators, KDM6A is a tumor suppressor that plays a critical role in the tumorigenesis and development of PDAC (9,10). KDM6A dysfunction promotes the formation of squamous-like morphology that is closely associated with the poor prognosis of PDAC selectively in female KC mice (8,9). The observation that males who developed these tumors had a concomitant loss of UTY and KDM6A suggests overlapping and enzyme-independent tumor-suppressive roles (9). We previously reported that loss of Kdm6a in KPC mice promotes EMT, tumor growth, and metastasis (10). Different from the KC mouse model, there is no significant dosage effect of Kdm6a on PDAC progression or survival in our KPC mice. However, more female KPC-Kdm6a KO mice developed distant metastases and experienced slower recovery from pancreatitis compared to male mice. In this study, we did not observe significant gender differences in the primary tumor, consistent with our previous findings using the KPC mouse model.
PDAC is characterized by a TME with excessive deposition of extracellular matrix and immune cell infiltration, which are believed to support tumor progression and contribute to poor prognosis (38,39). Although several studies explored the mechanisms on how the loss of KDM6A promotes the initiation and progression of PDAC, whether and how KDM6A impacts the tumor immune microenvironment remains largely unknown. In this study, we focused on the impact of KDM6A deficiency on the PDAC immune microenvironment. Although there are changes in the numbers of other immune cell types (B cell, T cell, and macrophage), the neutrophil is the only immune cell type we investigated to have a consistent increase in the intratumoral areas but not in the peritumoral areas. Studies have shown that neutrophils promote metastasis by suppressing CD8+ T-cell and NK cell cytotoxicity at the metastatic site, indicating TANs interact with other effective immune cells (40,41). We found that CD8+ effector T cells are significantly decreased in KPC-Kdm6a KO mice despite the overall increased T cells, supporting an immunosuppressive and tumor-promoting microenvironment in these mice. Interestingly, B220+ B cells were also significantly decreased in the KPC-Kdm6a KO mice. Functional studies are needed to elucidate the role of B cells in KDM6A-deficient PDACs. However, the previous study has shown that B cells can support the adaptive antitumor immune response in the mouse PDAC model (42). Activated TANs can release extracellular DNA traps, called NETs, which contribute to tumor progression and metastasis (43,44). Our results also supported increased NETs formation in PDAC with low KDM6A expression, further suggesting TANs in PDAC with KDM6A loss likely play a pro-tumoral role.
PanIN is the main type of PDAC precancerous lesion (45). Most studies about TANs in PDAC focused on the relationship between TANs and tumor invasion and metastasis. This study compared TANs infiltration in PanINs and PDAC with or without Kdm6a loss. We found that the number of TANs increased at the late stages of PanIN (HG-PanIN) in both Kdm6a knockout and Kdm6a wild-type mice, and Kdm6a loss accelerated this process. These results suggested that increased TANs are closely related to PDAC initiation, and progression and loss of KDM6A may accelerate this process.
Neutrophils are recruited into tumors by cytokines and chemokines. CXCL1, CXCL2, CXCL3, CXCL5, CXCL7, and CXCL8 are responsible for recruiting neutrophils under normal physiological conditions (46). Our data indicated that loss of KDM6A significantly upregulates CXCL1 expression in tumoral cells, and CXCL1 is the key chemokine promoting TANs recruitment and, very likely, CD8+ T cell depletion in the tumor. Consistent with our data, the suppressive impact of tumor-cell-produced CXCL1 on CD8+ T cells was also shown in another study using congenic pancreatic cancer cell clones (47). In the meantime, we found KDM6A loss promotes TANs to activate and release extracellular DNA traps (NETs), which similarly contribute to tumor progression and metastasis (43,44), further suggesting TANs in KDM6A-deficient PDACs likely play a pro-tumoral role.
Growing evidence has implied that CXCLs–CXCR2 axis has a complex and important biological function in various tumors (48). It is not only closely associated with tumor angiogenesis, progression, and chemoresistance (49) but also involved in the regulation of the tumor immune microenvironment (48). Hence, over the last several decades, many therapeutic strategies targeting CXCLs–CXCR2 axis have been explored, and some have shown promising results (48,50,51). A study by Sano et al. showed that heterozygous knockout of Cxcr2 in a genetically engineered mouse model with pancreas epithelium-specific activation of KrasG12D and knockout of Tgfbr2 (Ptf1acre/+; LSL KrasG12D/+; Tgfbr2flox/flox) (PKF mice) prolonged survival and inhibited both tumor angiogenesis and PDAC microinvasion , suggesting blocking of the CXCLs–CXCR2 axis in tumor-stromal interactions could be a therapeutic approach against PDAC progression (51). Another study showed that genetic ablation or inhibition of CXCR2 abolished metastasis, but only inhibition attenuated tumorigenesis (50). CXCR2 inhibition improved T cell infiltration and augmented PD1 immunotherapy in mice with established PDAC. In our study, the anti-CXCL1 treatment effectively inhibited tumor growth by decreasing the infiltration of TANs and the formation of NETs in Kdm6a-deficient PDACs, suggesting blockade of the CXCL1–CXCR2 axis could be a candidate therapeutic approach against KDM6A-deficient PDACs.
In summary, we discovered novel signaling of KDM6A in preventing TANs infiltration and NETosis in PDAC development via CXCL1. Our findings provide novel insights into the biological function of KDM6A in immune TME and PDAC development and identified CXCL1-CXCR2 as candidate therapeutic targets to treat PDACs with KDM6A loss.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments:
We thank the University Michigan Biomedical Research Core facilities, including Flow Cytometry core, Vector core, and ELISA core, for providing service and resources needed for the study. We would like to acknowledge Dr. Yaqing Zhang for assistance with IHC, IF and flow. We also would like to acknowledge Drs. Phillip Palmbos and Yin Wang for assistance with confocal imaging and analysis. We thank Drs. Shuvasree SenGupta and Carole Parent for assistance with neutrophil isolation and chemotaxis assay. This study is supported by the National Cancer Institute of the National Institutes of Health under award number K08CA234222 and R37CA262209 (J. Shi).
Footnotes
Conflict of interest: The authors declare no potential conflict of interest.
Reference
- 1.Lippi G, Mattiuzzi C. The global burden of pancreatic cancer. Archives of medical science : AMS 2020;16:820–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: a cancer journal for clinicians 2021;71:7–33 [DOI] [PubMed] [Google Scholar]
- 3.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2016;30:355–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenfield A, Carrel L, Pennisi D, Philippe C, Quaderi N, Siggers P, et al. The UTX gene escapes X inactivation in mice and humans. Human molecular genetics 1998;7:737–42 [DOI] [PubMed] [Google Scholar]
- 6.Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007;318:447–50 [DOI] [PubMed] [Google Scholar]
- 7.Van der Meulen J, Speleman F, Van Vlierberghe P. The H3K27me3 demethylase UTX in normal development and disease. Epigenetics 2014;9:658–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe S, Shimada S, Akiyama Y, Ishikawa Y, Ogura T, Ogawa K, et al. Loss of KDM6A characterizes a poor prognostic subtype of human pancreatic cancer and potentiates HDAC inhibitor lethality. International journal of cancer 2019;145:192–205 [DOI] [PubMed] [Google Scholar]
- 9.Andricovich J, Perkail S, Kai Y, Casasanta N, Peng W, Tzatsos A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018;33:512–26.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi Z, Wei S, Jin L, Jeyarajan S, Yang J, Gu Y, et al. KDM6A Regulates Cell Plasticity and Pancreatic Cancer Progression by Noncanonical Activin Pathway. Cellular and molecular gastroenterology and hepatology 2022;13:643–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dougan SK. The Pancreatic Cancer Microenvironment. The Cancer Journal 2017;23:321–5 [DOI] [PubMed] [Google Scholar]
- 12.Parente P, Parcesepe P, Covelli C, Olivieri N, Remo A, Pancione M, et al. Crosstalk between the Tumor Microenvironment and Immune System in Pancreatic Ductal Adenocarcinoma: Potential Targets for New Therapeutic Approaches. Gastroenterol Res Pract 2018;2018:7530619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobatake K, Ikeda KI, Nakata Y, Yamasaki N, Ueda T, Kanai A, et al. Kdm6a Deficiency Activates Inflammatory Pathways, Promotes M2 Macrophage Polarization, and Causes Bladder Cancer in Cooperation with p53 Dysfunction. Clin Cancer Res 2020;26:2065–79 [DOI] [PubMed] [Google Scholar]
- 14.Yi J, Shi X, Xuan Z, Wu J. Histone demethylase UTX/KDM6A enhances tumor immune cell recruitment, promotes differentiation and suppresses medulloblastoma. Cancer Lett 2021;499:188–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen N, Bellile E, Thomas D, McHugh J, Rozek L, Virani S, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head & neck 2016;38:1074–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 2014;67:45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Tsan YC, et al. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc Natl Acad Sci U S A 2013;110:2240–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bedi K, Paulsen MT, Wilson TE, Ljungman M. Characterization of novel primary miRNA transcription units in human cells using Bru-seq nascent RNA sequencing. NAR genomics and bioinformatics 2020;2:lqz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuo C, Wu X, Li J, Hu D, Jian J, Chen C, et al. Chemokine (C-X-C motif) ligand 1 is associated with tumor progression and poor prognosis in patients with colorectal cancer. Bioscience reports 2018;38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masucci MT, Minopoli M, Carriero MV. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Frontiers in Oncology 2019;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erpenbeck L, Schön MP. Neutrophil extracellular traps: protagonists of cancer progression? Oncogene 2016;36:2483–90 [DOI] [PubMed] [Google Scholar]
- 22.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends in immunology 2009;30:513–21 [DOI] [PubMed] [Google Scholar]
- 23.Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front Immunol 2017;8:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cedervall J, Zhang Y, Olsson AK. Tumor-Induced NETosis as a Risk Factor for Metastasis and Organ Failure. Cancer research 2016;76:4311–5 [DOI] [PubMed] [Google Scholar]
- 25.Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018;361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun 2019;10:1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drexler HG, Dirks WG, Matsuo Y, MacLeod RA. False leukemia-lymphoma cell lines: an update on over 500 cell lines. Leukemia 2003;17:416–26 [DOI] [PubMed] [Google Scholar]
- 28.Servant G, Weiner OD, Neptune ER, Sedat JW, Bourne HR. Dynamics of a chemoattractant receptor in living neutrophils during chemotaxis. Molecular biology of the cell 1999;10:1163–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pivot-Pajot C, Chouinard FC, El Azreq MA, Harbour D, Bourgoin SG. Characterisation of degranulation and phagocytic capacity of a human neutrophilic cellular model, PLB-985 cells. Immunobiology 2010;215:38–52 [DOI] [PubMed] [Google Scholar]
- 30.Hauert AB, Martinelli S, Marone C, Niggli V. Differentiated HL-60 cells are a valid model system for the analysis of human neutrophil migration and chemotaxis. The international journal of biochemistry & cell biology 2002;34:838–54 [DOI] [PubMed] [Google Scholar]
- 31.Ear T, McDonald PP. Cytokine generation, promoter activation, and oxidant-independent NF-kappaB activation in a transfectable human neutrophilic cellular model. BMC Immunol 2008;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajarathnam K, Schnoor M, Richardson RM, Rajagopal S. How do chemokines navigate neutrophils to the target site: Dissecting the structural mechanisms and signaling pathways. Cell Signal 2019;54:69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farré D, Roset R, Huerta M, Adsuara JE, Roselló L, Albà MM, et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic acids research 2003;31:3651–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jablonska J, Wu CF, Andzinski L, Leschner S, Weiss S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. International journal of cancer 2014;134:1346–58 [DOI] [PubMed] [Google Scholar]
- 35.Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017;170:875–88.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 2017;49:367–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 2015;15:334–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science 1985;230:676–8 [DOI] [PubMed] [Google Scholar]
- 39.Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 2011;17:320–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015;522:345–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer discovery 2016;6:630–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spear S, Candido JB, McDermott JR, Ghirelli C, Maniati E, Beers SA, et al. Discrepancies in the Tumor Microenvironment of Spontaneous and Orthotopic Murine Models of Pancreatic Cancer Uncover a New Immunostimulatory Phenotype for B Cells. Front Immunol 2019;10:542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016;5:e1134073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takesue S, Ohuchida K, Shinkawa T, Otsubo Y, Matsumoto S, Sagara A, et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancerassociated fibroblasts. Int J Oncol 2020;56:596–605 [DOI] [PubMed] [Google Scholar]
- 45.Hruban Ralph H. AM, and Goggins Michael. Update on Pancreatic Intraepithelial Neoplasia.pdf. International journal of clinical and experimental pathology 2008;1:306–16 [PMC free article] [PubMed] [Google Scholar]
- 46.Fridlender Zvi G. JS, Mishalian Inbal, Singhal Sunil, Cheng Guanjun, Kapoor Veena, Horng Wenhwai GF, Bayuh Rachel, Worthen G. Scott, Albelda Steven M.. Transcriptomic Analysis Comparing Tumor-Associated Neutrophils with Granulocytic Myeloid-Derived Suppressor Cells and Normal Neutrophils. PloS one 2012;7:e31524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018;49:178–93.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng Y, Ma XL, Wei YQ, Wei XW. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochimica et biophysica acta Reviews on cancer 2019;1871:289–312 [DOI] [PubMed] [Google Scholar]
- 49.Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016;532:245–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steele C, Karim S, Leach J, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer cell 2016;29:832–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sano M, Ijichi H, Takahashi R, Miyabayashi K, Fujiwara H, Yamada T, et al. Blocking CXCLs–CXCR2 axis in tumor–stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment. Oncogenesis 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and publicly available in Gene Expression Omnibus (GEO) at GSE202873.