

Abstract
Multi-resistant pathogenic strains of non-lactose fermenting Escherichia coli (NLF E. coli) are responsible for various intestinal and extraintestinal infections. Although several studies have characterised such strains using conventional methods, they have not been comprehensively studied at the genomic level. To address this gap, we used whole-genome sequencing (WGS) coupled with detailed microbiological and biochemical testing to investigate 17 NLF E. coli from a diagnostic centre (icddr,b) in Dhaka, Bangladesh. The prevalence of NLF E. coli was 10%, of which 47% (8/17) exhibited multi-drug resistant (MDR) phenotypes. All isolates (17/17) were confirmed as E. coli and could not ferment lactose sugar. WGS data analysis revealed international high-risk clonal lineages. The most prevalent sequence types (STs) were ST131 (23%), ST1193 (18%), ST12 (18%), ST501 (12%), ST167 (6%), ST73 (6%) and ST12 (6%). Phylogenetic analysis corroborated a striking clonal population amongst the studied NLF E. coli isolates. The predominant phylogroup detected was B2 (65%). The blaCTX-M-15 extended-spectrum beta-lactamase gene was present in 53% of isolates (9/17), whilst 64.7% (11/17) isolates were affiliated with pathogenic pathotypes. All extraintestinal pathogenic E. coli pathotypes demonstrated β-hemolysis. Our study underscores the presence of critical pathogens and MDR clones amongst non-lactose fermenting E. coli. We suggest that non-lactose fermenting E. coli be considered equally capable as lactose fermenting forms in causing intestinal and extraintestinal infections. Further, there is a need to undertake systematic, unbiased monitoring of predominant lineages amongst non-lactose fermenting E. coli that would help in better treatment and prevention strategies.
Keywords: genomic epidemiology, high risk clone, carbapenem resistance, ESBL – Escherichia coli, ST131 and non-ST131 lineages, non lactose fermenter
Introduction
Escherichia coli is a versatile gram-negative bacterium with huge genetic diversity (Nia Santos Braz et al., 2020). It is the most common organism responsible for opportunistic infections. Its primary habitat includes the lower intestinal tract of humans and animals (Nia Santos Braz et al., 2020). However, infections with variant strains of E. coli are responsible for various clinical manifestations ranging from diarrhoea, urinary tract infections, and life-threatening septicemia, resulting in over 2 million deaths every year globally (Hussain et al., 2012; Huang et al., 2021). Moreover, these infections are often associated with cephalosporin and carbapenem-resistant strains, impacting the mortality of patients and imparting huge healthcare costs (Logan and Weinstein, 2017).
Escherichia coli are non-pathogenic facultative anaerobic flora of the intestinal tract in humans. The gram-negative E. coli bacilli ferments lactose to produce hydrogen sulfide. However, up to 20% of E. coli isolates from patients are reported to be atypical, which are slow or non-lactose fermenters due to the deficiency in enzyme lactose permease encoded by the lacY gene. (Nicoletti et al., 1988; Hossain, 2012; Chang et al., 2014; Yaratha et al., 2017; Johnson et al., 2019). Non-lactose fermenting (NLF) E. coli can be identified as colourless, transparent colonies on MacConkey agar and by negative lactose (sugar) fermentation tests (Hossain, 2012; Siqueira et al., 2021). Various strains of E. coli are equipped to cause different forms of enteric and extraintestinal infections in human hosts with varying propensities (Kaper et al., 2004). The pathogenic strains of E. coli belong to both lactose fermenting and non-lactose fermenting E. coli types. However, little attention is given to these atypical strains of E. coli, particularly the non-lactose fermenting (NLF) E. coli in routine diagnostic testing laboratories.
Escherichia coli is a well-established etiological agent of diarrhoea (Colonna et al., 1992; Nataro and Kaper, 1998; Hossain, 2012). Diarrheagenic strains of E. coli fall into different categories possessing distinct pathogenic mechanisms, the three important E. coli strains include enteropathogenic E. coli (EPEC), enterohemorrhagic E. coli (EHEC), and enteroaggregative E. coli (EAEC) (Nataro and Kaper, 1998). These E. coli categories cause a range of clinical syndromes such as watery diarrhoea in children, persistent diarrhoea, traveller’s diarrhoea and hemolytic uremic syndrome (Nataro and Kaper, 1998). A few studies have reported that NLF E. coli strains were identified as typical pathogenic E. coli isolates, having a possible diarrheagenic role (Nicoletti et al., 1988; Colonna et al., 1992; Hossain, 2012).
Urinary tract infections (UTIs) represent one of the most common bacterial infections. It is a serious public health problem affecting around 150 million people worldwide (Flores-Mireles et al., 2015). E. coli is the most common pathogen responsible for (un)complicated urinary tract infections (Flores-Mireles et al., 2015). Such strains harbour multiple virulence factors, which play their role in the pathophysiology of UTIs. These virulence factors affect bacterial colonisation and evasion of host defences (Hussain et al., 2014; Mazumder et al., 2020a). Recent reports suggest that clonal groups of multi-resistant, multi-virulent E. coli are increasingly responsible for UTIs, presenting a challenge for their treatment (Riley, 2014). In recent years, a few studies have demonstrated that NLF E. coli have been increasingly isolated from urine specimens in microbiology laboratories and have reported their clinical significance concerning antibiotic resistance, virulence and emerging clones (Chang et al., 2014; Chakraborty et al., 2016; Wu et al., 2017).
Rapid identification of bacterial pathogens in microbiology laboratories is critical for initiating successful infection treatment. Screening of gram-negative bacteria from urine and stool samples is routinely performed on MacConkey agar, and the colourless transparent NLF E. coli variants are usually missed in the screening process as little attention is given to these atypical strains of E. coli. Although reports of such isolates are limited worldwide, a few studies have reported the occurrence and characterisation of NLF E. coli from clinical specimens. They have characterised the strains using only conventional methods. This study aims to decipher the biochemical profiles, population structure, and genomic characteristics of NLF E. coli from stool and urine samples isolated at a referral diagnostic centre (International Center for Diarrheal Disease Research, Bangladesh; icddr,b) in Dhaka, Bangladesh. Studies such as these will inform us about the evolutionary trajectories of atypical E. coli variants and shed light on their clinical and public health significance.
Materials and methods
Bacterial isolates and their biochemical and serological characterisation
A total of 175 E. coli isolates were sourced from urine (n = 98; 56%) and stool (n = 77; 44%) culture plates. The isolates were collected randomly from the Clinical Microbiology Laboratory of the icddr,b, based in Dhaka, Bangladesh. The isolates were sampled between 2019 and 2020 as part of a larger 1% AMR surveillance study. From this collection of 98 urine and 77 stool E. coli isolates, we identified 9 (9.2%) and 8 (10.4%) NLF E. coli isolates, respectively. These 17 NLF E. coli were subjected to complete biochemical profiling and whole-genome sequencing (WGS) using an Illumina platform (NextSeq 500). NLF E. coli were presumptively identified by their inability to ferment lactose on the MacConkey agar. The presence of Shigella spp. was ruled out by serotyping using slide agglutination test employing a commercial Shigella grouping antisera kit that includes group A (S. dysenteriae), B (S. flexineri), C (S. boydii) and group D (S. sonnei) antisera from Denka Seiken Co. Ltd. Tokyo, Japan. Further, confirmation was made by standard biochemical methods that include motility, indole and urease tests (using MIU agar), triple sugar iron (TSI) agar test to visualise glucose and lactose fermentation reactions, catalase test (using 3% H2O2), oxidase test (using 1% aqueous solution of tetra-methyl-p-phenylenediamine dihydrochloride), citrate utilisation test (using Simmons citrate agar) and acetate utilisation test (using Acetate agar) (Hawkey, 2006; Aditi et al., 2017). As mentioned in Table 1, fermentation of other sugars was confirmed using carbohydrate broth with bromothymol blue as a pH indicator, employing previously described methods (Bopp et al., 2003). Voges-Proskauer test was performed in MRVP broth and the reaction was visualised by adding 5% α naphthol and 40% potassium hydroxide (KOH) (Hawkey, 2006). Gelatine liquefaction and ONPG tests were performed as described elsewhere (Hawkey, 2006; Nusrat et al., 2017). Decarboxylation reaction of amino acids was performed on decarboxylase broth supplemented by 1% of the appropriate amino acids as described earlier (Brooks and Sodeman, 1974). The Analytical profile index was evaluated using the API 20E kit (bioMérieux) as per the manufacturer’s instructions (Table 1). Hemolytic properties of strains were evaluated using 5% sheep blood agar plates. Clearing zones around the colonies were suggestive of beta-haemolysis.
Table 1.
Microbiological and biochemical characteristics of 17 NLF E. coli isolates.
| Sl. no. | Biochemical tests | No. (%) | |
|---|---|---|---|
| Positive | Negative | ||
| 1 | Catalase test | 17 (100) | 0 |
| 2 | Oxidase test | 0 | 17 (100) |
| 3 | TSI agar | ||
| a. Acid production in slant | 17 (100) | 0 | |
| b. Acid production in butt | 17 (100) | 0 | |
| c. Hydrogen sulfide production(H2S) | 0 | 17 (100) | |
| d. Gas production | 17 (100) | 0 | |
| 4 | Motility Indole Urease Test (MIU) | ||
| a. Motility | 17 (100) | 0 | |
| b. Indole Production | 17 (100) | 0 | |
| c. Urea hydrolysis | 0 | 17 (100) | |
| 5 | Simmons Citrate reaction test | 0 | 17 (100) |
| 6 | Acetate | 17 (100) | 0 |
| 7 | Sugar fermentation | ||
| a. Glucose | 17 (100) | 0 | |
| b. Lactose | 0 | 17 (100) | |
| c. Sucrose | 11 (65) | 6(35) | |
| e. Mannose | 17 (100) | 0 | |
| f. Arabinose | 17 (100) | 0 | |
| g. Sorbitol | 17 (100) | 0 | |
| h. Mannitol | 17 (100) | 0 | |
| i. Inositol | 0 | 17 (100) | |
| 8 | Ortho-Nitrophenyl-β-galactoside (ONPG) | 17 (100) | 0 |
| 9 | Gelatine liquefaction | 0 | 17 (100) |
| 10 | Vogas-proskauer | 0 | 17 (100) |
| 11 | Lysine decarboxylase | 17 (100) | 0 |
| 12 | Ornithine decarboxylase | 14(82) | 3 (18) |
| 13 | Arginine Dihydrolase | 11 (65) | 6(35) |
| API 20E Results [API profile: isolates] | |||
| 1 | API profile: 7144532 | A3, A4, A5, A7, U1, U7, U8, U12, U13, U14, U15 | |
| 2 | API profile: 5144572 | A6, U4, U6 | |
| 3 | API profile: 5044552 | A1, A8, A9 | |
| Bacteriological Characteristics [17 (100%)] | |||
| 1 | Growth on MacConkey Agar | Colourless and transparent (NLF colony) | |
| 2 | Growth on SS agar | Colourless and transparent (NLF colony) | |
| 3 | Growth on CHROMagar™ Orientation | Pink colour colony | |
| 4 | Growth Temperature | (26–42°C) | |
| 5 | Gram’s staining | Gram-negative rods | |
Antimicrobial susceptibility testing
The Genome Centre at icddr,b reassessed 17 NLF E. coli isolates with an extensive panel of 18 antibiotics (Oxoid, US) by the Kirby-Bauer disc diffusion method. The results were interpreted using the 2019 Clinical and Laboratory Standards Institute (CLSI) guidelines. Upon antibiotic susceptibility testing, the NLF E. coli isolates identified as intermediate and resistant were considered as resistant. They were classified as multi-drug resistant (MDR) if they were non-susceptible to at least one agent belonging to three different antimicrobial classes (Magiorakos et al., 2012).
Whole-genome sequencing
Total bacterial DNA was isolated and purified from the overnight grown bacterial cultures using the QIAmp DNA Mini kit (Qiagen, Germany). DNA QC and quantification were performed employing a Nanopore spectrophotometer (Thermo Fisher Scientific, United States) and Qubit 4.0 fluorometer (Life Technologies; Baddam et al., 2020). DNA libraries for the short-read paired-end sequencing were prepared using the Nextera DNA Flex library prep kit (Illumina). Size selected and pooled libraries were sequenced at the icddr,b Genome Centre in Illumina NextSeq 500 system using a NextSeq v2.5 reagent kit (2 × 150 bp) (Mazumder et al., 2020b).
Sequence assembly and annotation
High-quality reads were obtained by filtering the paired-end data using fastp (Chen et al., 2018). De novo assemblies were generated using SPAdes (v.3.11.11) (Bankevich et al., 2012). NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016) was used for annotating the genome assemblies. The genome statistics from the resulting file were gleaned using the QUAST software (v5.2.0) (Gurevich et al., 2013).
In silico analysis
Species identification was confirmed using Kmer Finder software (v3.2) (Hasman et al., 2014; Larsen et al., 2014; Clausen et al., 2018). Phylogenetic groups of the genomes were inferred using the Clermon Typing tool (Beghain et al., 2018). The sequence types (STs) were determined by submitting the contigs to MLST (v2.0; scheme #1) (Larsen et al., 2012). SerotypeFinder 2.0 (Joensen et al., 2015) and CHTyper 1.0 (Roer et al., 2018) software tools were used for elucidating serotypes and clonotypes, respectively.
Resistance genes were screened by BLASTn analysis of the genome assemblies against the data downloaded from the ResFinder database (Bortolaia et al., 2020). The 70% query coverage and 90% identity indicated a positive hit in the genome. Plasmid incompatibility groups were determined by PlasmidFinder (v2.1) (Carattoli et al., 2014). Mobile genetic elements associated with acquired antimicrobial resistance (AMR) genes were determined using Mobile Element finder v1.0.3 (Johansson et al., 2021). Chromosomal point mutations conferring fluoroquinolone resistance were identified by PointFinder software (Zankari et al., 2017). Contigs with blaNDM-5 and blaCTX-M-15 were analysed by BLASTn analysis against the database in NCBI to identify the chromosomal or plasmid origin of these genes.
Virulence gene content of strains was determined by BLASTn analysis of 17 genomes against the E. coli database selected in the virulence factor database (VFDB; Chen et al., 2005). The genes with 90% identity and 70% query coverage were considered present. Isolates were classified as extraintestinal pathogenic E. coli based on Johnson’s criteria (Johnson and Stell, 2000). The in-silico analysis described above was performed on default parameters unless otherwise stated.
Phylogenomic and pangenomic analysis
The phylogenetic tree was inferred with the reference sequence alignment-based phylogeny builder (REALPHY) software (Bertels et al., 2014). E. coli K-12 MG1655 genome (accession no.U00096) was used as the single reference sequence and PhyML (Guindon et al., 2010) was used as the treebuilder. The phylogenetic tree was later visualised using the Interactive Tree of Life (iTOL; Letunic and Bork, 2016). Pan-genome analysis was conducted using the Anvi’o (v7.1) software package (Eren et al., 2015) following the microbial pan-genomics analysis workflow.
Results
Bacterial and biochemical characteristics
The colourless NLF colonies on the MacConkey agar plate appeared like Shigella spp. colonies. Colonies were colourless even in 72-h old MacConkey agar cultures, removing the possibility of late lactose fermenters. Serology of all colonies revealed negative reactions to the four serogroups of Shigella: S. flexneri, S. dysenteriae, S. boydii, and S. sonnei. Further, streaking of colonies on CHROMagar Orientation media revealed small, pink-red colonies typical of E. coli. The isolates were then tested by routine biochemical tests as described in Table 1 and identified as E. coli. Notably, all isolates were O-nitrophenyl-beta-D-galactopyranoside positive and did not ferment lactose sugar. Further confirmation was done by generating a biochemical profile for each isolate using 20 reactions of the API 20E strip. The API results gave three distinct profiles: 7144532 (n = 11), 5,044,552 (n = 3) and 5,144,572 (n = 3) and identified all isolates as E. coli with 99.8% probability (Table 1). Gram staining confirmed the presence of gram-negative bacilli. The optimum growth temperate of NLF E. coli isolates ranged from 26° to 42°C.
Molecular analysis and phylogenomics of NLF Escherichia coli isolates
WGS of the 17 NLF E. coli study isolates yielded an average genome size of 5,160,336 bp (range 4,890,228–5,480,336) and the average GC content was 50.6% (range 50.3–50.7%). KmerFinder software results confirmed all the isolates as E. coli. On average, the genome assemblies had a genome coverage of 86-fold (range: 62–124-fold; Table 2).
Table 2.
Genomic features of the 17 whole-genome sequenced NLF E. coli isolates.
| Sl no. | Isolate ID | Strain ID | Source | Isolation year | Genome coverage (X) | Contig no. (>500 bp) | Genome size (bp) | No. CDS | GC % | Accession no. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A1 | BDEc_NLF-A1 | Stool | 2019 | 91 | 106 | 5,305,815 | 5,077 | 50.5 | JAMKCS000000000 |
| 2 | A3 | BDEc_NLF-A3 | Stool | 2019 | 101 | 103 | 5,480,336 | 5,335 | 50.6 | JAMKCT000000000 |
| 3 | A4 | BDEc_NLF-A4 | Stool | 2019 | 96 | 96 | 5,019,286 | 4,772 | 50.7 | JAMKCU000000000 |
| 4 | A5 | BDEc_NLF-A5 | Stool | 2019 | 63 | 77 | 5,074,445 | 4,830 | 50.5 | JAMKCV000000000 |
| 5 | A6 | BDEc_NLF-A6 | Stool | 2019 | 121 | 84 | 4,890,228 | 4,656 | 50.6 | JAMKCW000000000 |
| 6 | A7 | BDEc_NLF-A7 | Stool | 2020 | 90 | 70 | 4,978,296 | 4,710 | 50.7 | JAMKCX000000000 |
| 7 | A8 | BDEc_NLF-A8 | Stool | 2020 | 124 | 151 | 5,033,902 | 4,934 | 50.6 | JAMKCY000000000 |
| 8 | A9 | BDEc_NLF-A9 | Stool | 2020 | 91 | 43 | 5,068,010 | 4,850 | 50.6 | JAMKCZ000000000 |
| 9 | U-1 | BDEc_NLF-U1 | Urine | 2019 | 71 | 44 | 5,073,930 | 4,893 | 50.6 | JAMKDA000000000 |
| 10 | U-4 | BDEc_NLF-U4 | Urine | 2019 | 62 | 53 | 5,251,806 | 4,988 | 50.4 | JAMKDF000000000 |
| 11 | U-6 | BDEc_NLF-U6 | Urine | 2019 | 79 | 44 | 5,054,491 | 4,847 | 50.6 | JAMKDG000000000 |
| 12 | U-7 | BDEc_NLF-U7 | Urine | 2019 | 88 | 109 | 5,302,191 | 5,061 | 50.5 | JAMKDH000000000 |
| 13 | U-8 | BDEc_NLF-U8 | Urine | 2020 | 71 | 56 | 5,186,272 | 5,002 | 50.6 | JAMKDI000000000 |
| 14 | U-12 | BDEc_NLF-U12 | Urine | 2020 | 83 | 57 | 5,021,789 | 4,793 | 50.7 | JAMKDB000000000 |
| 15 | U-13 | BDEc_NLF-U13 | Urine | 2020 | 84 | 69 | 5,262,816 | 5,008 | 50.5 | JAMKDC000000000 |
| 16 | U-14 | BDEc_NLF-U14 | Urine | 2020 | 70 | 145 | 5,304,507 | 5,153 | 50.7 | JAMKDD000000000 |
| 17 | U-15 | BDEc_NLF-U15 | Urine | 2020 | 75 | 79 | 5,417,595 | 5,190 | 50.36 | JAMKDE000000000 |
According to in silico MLST, isolates were classified into nine distinct sequence types (STs) and they represented some of the international high-risk lineages (Figure 1). ST131 was the predominant ST that accounted for 4 (23%) of the 17 NLF E. coli isolates. ST1193 and ST12 were present in equal proportions, 3 (18%) each, followed by two ST501 strains (12%). Other significant STs detected were ST167 (6%), ST73 (6%) and ST12 (6%). The predominant phylogroup detected was B2 (65%) which comprised all isolates affiliated with ST131, ST1193, ST12 and ST73. This was followed by group A (n = 2: 1ST167; 1 ST2089); other phylogroups were represented by single isolates (Figure 1). By serogroup, the 4 ST131 isolates were either O16: H4 (n = 2: fumC40: fimH41) or O25: H4 (n = 2: fumC40: fimH30; Figure 1). All ST1193 isolates were O75: H5 with fumC14: fimH64. All ST12 isolates were O4: H1and had fumC13. Other STs exhibited diverse serotypes and CH types (Figure 1).
Figure 1.

Phylogenetic relationships amongst sequenced NLF E. coli genomes. The maximum likelihood phylogenetic tree is based on the alignment of detected core genome SNPs of 17 NLF E. coli genomes with the MG1655 genome strain as a reference genome. The phylogroups, clinical source, blaCTX-M-15 status, beta-hemolysis, MDR status, STs, serogroups, CH types and blaNDM-5 status are shown to the right of the tree.
Based on the alignment of detected core genome SNPs, a phylogenetic tree was built for 17 NLF E. coli from our study with the MG1655 genome as the reference genome. There was relatively low diversity of the studied NLF E. coli as most strains clustered together, resulting in clades and subclades. The analysis clustered the 17 isolates into two major clades, one tight cluster corresponding to phylogroup B2 and the other loosely clustered clade included strains from A, B1, D and F phylogroup. Compared to NLF E. coli from stool, the urine E. coli isolates formed a tight clade with strains belonging to ST12, ST1193 and ST131. Three strains from stool origin clustered closely with urine strains reinforcing the gut as a possible reservoir of UTI-causing agents. The blaCTX-M-15, MDR phenotype and β-hemolysis did not correspond with the clusters indicating their widespread presence across E. coli lineages. However, the sequence types, serogroups, and CH-types corresponded closely with the phylogenetic clusters of E. coli genomes. The ST131 strains with O16: H5 (fumC40: fimH41) formed a subcluster with ST131 strains having O25: H4 (fumc40: fimH30), indicating there are sub-lineages within ST131.
We also analysed the pan-genomes of the 17 NLF E. coli genomes (Figure 2). The number of core and accessory genes identified was 3,400 and 30,563, respectively. Contrary to the core-genome-based phylogenetic tree, no two genomes shared the same gene content with respect to the pan-genome. A closer look into the pan-genome dendrogram reflects a clustering pattern that mimics the clades of the core genome-based phylogenetic tree (Figure 2). These observations indicate that the 17 NLF E. coli genomes do not differ drastically, even regarding their accessory gene content. They all have a few unique gene families (Figure 2).
Figure 2.

Pan-genome analysis of 17 NLF E. coli genomes from Anvi’o pan-genomics suite. The circles represent individual genomes arranged as per their pan-genomic phylogenetic relationship depicted by the outside dendrogram. In the circles, dark colour indicates the presence of a gene group and light colour its absence.
Virulence genes
Our analysis identified 73 virulence-associated genes out of 335 genes analysed. The distribution of 73 virulence genes amongst 17 NLF E. coli isolates is shown in Figure 3. Overall, 65% (11/17) of all NLF E. coli isolates were assigned to different pathotypes, as classified by the presence of specific virulence markers. Most urine NLF E. coli isolates were assigned to the extraintestinal pathogenic pathotype (67%, 6/9). Suspected diarrheagenic variants comprised 62% (5/8) of the stool NLF E. coli isolates. The most prevalent pathotype amongst diarrheagenic variants was enteroaggregative E. coli (38%), which harboured both aggR and aatA genes, followed by enteropathogenic E. coli (13%) that showed the presence of intimin gene (eae). One isolate from stool origin was also classified as extraintestinal pathogenic E. coli. The remaining six isolates (6/17) that did not belong to any pathotype included three strains from ST1193, two from ST131 and one from ST501. Out of four ST131 studied strains, two O25: H4 serogroup (fimH30) strains qualified as extraintestinal pathogenic and two strains with O16: H5 serogroup (fimH41) did not qualify as extraintestinal pathogenic. Amongst all the pathotypes, the seven (100%) ExpEC strains produced β-hemolysis in 5% sheep blood agar. This hemolytic phenotype was strongly associated with alpha-hemolysin genes (hlyA, hlyB and hlyD). Although the three ST1193 isolates did not qualify as extraintestinal pathogenic E. coli pathotypes, they all carried a similar set of virulence genes, including adhesins (fimH), toxins (sat, hlyE/clyA), siderophores (chuA, fyuA, sitABCD, iutA) and others. The NLF E. coli sourced from urine had a higher prevalence of virulence genes, with higher aggregate virulence scores (median 90; range 72–114) compared to those of stool NLF E. coli isolates (median 73; range 62–116).
Figure 3.

Heat map depicting the distribution of 73 virulence genes amongst 17 NLF E. coli genomes. Dark brown represents the presence, and light brown blocks represent a virulence gene’s absence.
Antimicrobial resistance
The phenotypic and genotypic trends of AMR of the studied NLF E. coli strains were investigated (Figure 4). Overall, the NLF E. coli strains from the stool and urine samples showed high antimicrobial drug resistance rates to nalidixic acid (100%), ceftazidime (94%), cefradine (94%), followed by ciprofloxacin (88%), ampicillin (82%), cefotaxime (65%) and cefepime (65%). The resistance rates to cefuroxime (59%), ceftriaxone (53%), co-trimoxazole (41%), imipenem (35%) were moderate and the resistance rates to gentamicin (18%), amikacin (12%), doxycycline (6%), meropenem (6%) were low. In contrast, there appeared to be no resistance to chloramphenicol, fosfomycin and tigecycline. Compared to the stool isolates, the urine NLF E. coli isolates had relatively low resistance to amikacin, cefepime, ceftriaxone and cefuroxime; consequently, there were 63% (5/8) MDR isolates in the stool category and 33% (3/9) MDR isolates in urine category.
Figure 4.
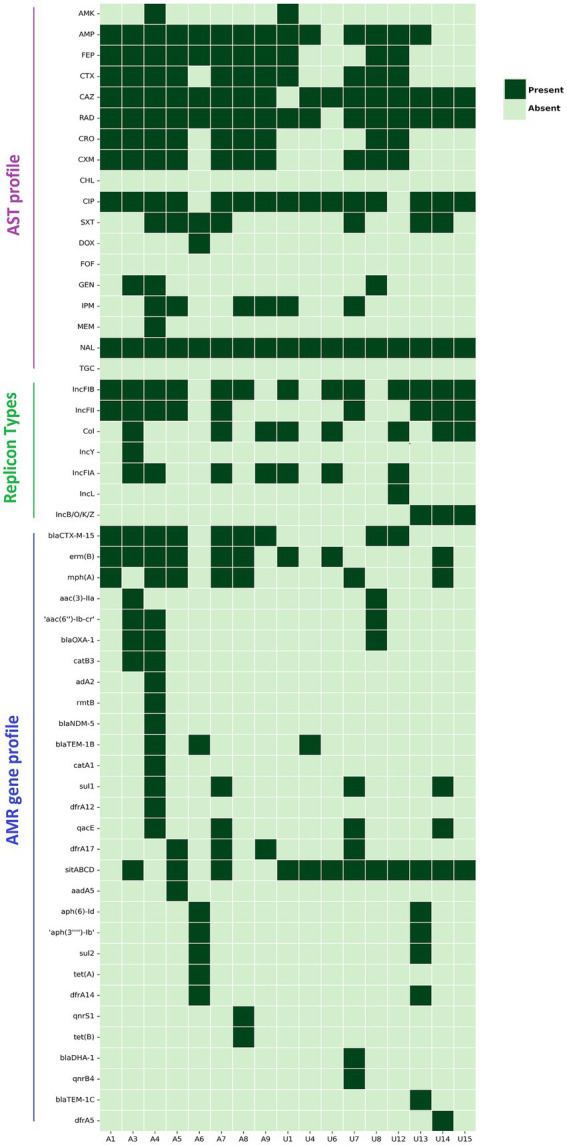
Heat map demonstrating the AST profile, plasmid replicon types and acquired AMR gene profile of 17 NLF E. coli isolates/genomes. Dark green represents resistance/presence, and light green blocks represent the sensitivity/absence of a particular trait. Definition of abbreviated antibiotics for AST: AMK, amikacin; Amp, ampicillin; FEP, cefepime; CTX, cefotaxime; CAZ, ceftazidime; RAD, cephradine; CRO, ceftriaxone; CXM, cefuroxime; CHL, chloramphenicol; CIP, ciprofloxacin; SXT, trimethoprim-sulfamethoxazole; DOX, doxycycline; FOF, fosfomycin; GEN, gentamicin; IPM, imipenem; MEM, meropenem; NAL, nalidixic acid; TGC, tigecycline.
We detected 31 unique AMR gene alleles belonging to different classes, with most genomes exhibiting at least 6 AMR genes (Figure 4). The AMR genes had a relatively similar distribution between the stool and urine NLF E. coli isolates, with strains from both the groups having a minimum of 6 AMR genes per genome, albeit with variation amongst isolates (E. coli range: stool 3–15; urine 2–13). No specific AMR profile was associated with isolates from a particular clinical source.
A variety of ESBL gene variants encoded cephalosporin resistance. Amongst the identified ESBL genes in NLF E. coli isolates, blaCTX-M-15, was predominant (9/17, 53%), but we also identified blaOXA-1, blaTEM1B, blaDHA-1 and blaTEM-1C genes in several isolates (Figure 4). We observed perfect concordance (100%) between the presence of blaCTX-M-15 gene and resistance phenotypes to the following cephalosporin antibiotics, cefradine, cefotaxime, ceftazidime, ceftriaxone, cefuroxime and cefepime. One blaCTX-M-15 positive stool NLF E. coli harboured a blaNDM-1 gene; this strain (A4) demonstrated difficult-to-treat resistance (DTR) as it was resistant to 14 of 18 antibiotics tested, including all first-line agents. It was sensitive to chloramphenicol, doxycycline, fosfomycin and tigecycline. The probable genome locus of the blaCTX-M-15 gene was detected to be a plasmid for 4 out of 9 NLF E. coli isolates (Table 3). The genetic environment of the blaCTX-M-15 gene in most isolates (56%, 5/9) consisted of the insertion element ISEcp1preceeding the locus (Table 3). The blaCTX-M-15 gene was mainly associated with the epidemiological significant clonal E. coli lineages such as ST131, ST167, ST1193 and ST6303. Interestingly, these strains were all NLF.
Table 3.
Characteristics of CTX-M-15 associated NLF E. coli isolates.
| Isolate ID | Source | ST | Phylogroup | Genome locus | MGEsa | Plasmid replicon |
|---|---|---|---|---|---|---|
| A1 | Stool | 6303 | D | Plasmid | ISEcp1 | IncFIB, IncFII |
| A3 | Stool | 131 | B2 | Plasmid | NDb | Col, IncFIA, IncFIB, IncFII, IncY |
| A4 | Stool | 167 | A | Chromosome | ISEcp1 | IncFIA, IncFIB, IncFII |
| A5 | Stool | 501 | D | Chromosome | ISEcp1 | IncFIB, IncFII |
| A7 | Stool | 131 | B2 | Chromosome | NDb | Col, IncFIA, IncFIB, IncFII |
| A8 | Stool | 2089 | A | Plasmid | ISKpn19 | IncFIB |
| A9 | Stool | 1193 | B2 | Chromosome | ISEcp1 | Col, IncFIA |
| U-8 | Urine | 131 | B2 | Plasmid | IS3 | NDb |
| U-12 | Urine | 131 | B2 | Chromosome | ISEcp1 | Col, IncFIA, IncFIB, IncL |
Mobile genetic elements.
Not detected.
We screened for mutations in the quinolone-resistance-determining regions (QRDRs) of gyrA, parC and parE. We identified amino acid substitutions at codon positions S83I (16/17 isolates), D87N (9/17 isolates) in the amino acid sequence of gyrA and S80I (9/17 isolates), S57T (one isolate), E84V (4/17 isolates), E84G (two isolates) and S80R (two isolates) in parC. We also identified mutations at codon positions S45A, I529L and L416F in the parE gene. The gyrA S83L mutation was strongly associated with resistance to ciprofloxacin. The ESBL gene blaCTX-M-15 and gyrA S83L, parC E84V were strongly linked in ST131 and ST167 lineage strains. Additionally, the isolates also harboured acquired AMR genes encoding fluoroquinolone resistance, including Qnrs1 (one isolate), Qnrs13 (one isolate) and aac(6′)-Ib-cr gene (3 isolates). All the three acquired fluroquinolone resistance genes were associated with ciprofloxacin resistance.
MDR was frequently observed in isolates with multiple AMR genes identified in eight (47%) isolates. Cephalosporin resistance genes and the gyrA S83L mutation were strongly associated with the MDR phenotype. PlasmidFinder identified seven unique plasmid replicon groups, with each isolate carrying an average of 2.5 plasmid groups (Figure 4). FIB was the most commonly encountered plasmid group (n = 13/17), followed by FII (n = 9/17), CoI (n = 8/12) and FIA (n = 7/17). FIA, FIB and CoI plasmids were strongly associated with ST131 E. coli. The New Delhi metallo-β-lactamase (NDM)-positive E. coli strain (A4) concomitantly hosted three IncF-type replicons, FIA, FIB, and FII groups. We could not associate AMR genes with known plasmid replicons in two strains (A6 and U8) as no replicons were detected in them. Surprisingly, these strains belong to well-known ESBL-producing lineages (ST501 and ST131, respectively).
Discussion
We found a high prevalence of epidemiologically significant clonal lineages amongst NLF E. coli isolates from patients with suspected UTIs and enteric infections in a referral diagnostic centre (icddr,b) in Dhaka, Bangladesh. None of the previous studies has focused on the genomic epidemiology of NLF E. coli. In the current study, we carried out a comprehensive characterisation of 17 NLF E. coli isolates to delineate their microbiological, biochemical, and molecular characterisation using WGS to identify their population composition and detect their molecular markers of AMR and virulence. This first report forms a baseline study on the genomic epidemiology of clinical NLF E. coli isolates. Knowledge of such strains’ virulence and resistance properties is important to help direct researchers, clinicians, and laboratory technologists screen, analyse and report atypical variants of E. coli recovered from clinical samples.
In line with previous reports (Hossain, 2012; Yaratha et al., 2017), our study revealed that the prevalence rate of NLF E. coli in patients in the community is around 10%. This makes it important to identify NLF E. coli in clinical specimens, which would help initiate appropriate antimicrobial therapy. Given the difficulty in differentiating between NLF E. coli and Shigella spp., multiple tests need to be performed as described in this study, including serology, biochemical identification (manual and API) and molecular methods like WGS. It is reported that routine MALDI-TOF MS analysis cannot reliably differentiate between E. coli and Shigella species (Ling et al., 2019). It can be noted here that the chromogenic media-CHROMagar Orientation approach cannot distinguish between lactose fermenting and NLF E. coli and can be used to isolate/identify both types of E. coli in a single step.
The predominant sequence types detected in this study were epidemiologically significant high-risk clones, including ST131, ST1193, ST12, ST73 and ST167. Our group previously reported these clones from Bangladesh amongst ESBL-producing lactose fermenting E. coli (Mazumder et al., 2021). None of the studies has reported them from NLF E. coli except for ST1193 (Wu et al., 2017). The dominance of clonal STs amongst this collection of NLF E. coli is an interesting finding. It warrants close attention because the inclusion of isolates in this study was not based on any AMR phenotypes and genotypes. These STs are predominantly reported to be associated with extraintestinal infections (Pitout, 2012; Riley, 2014; Manges et al., 2019). Also, a review article on global extraintestinal pathogenic E. coli STs enlisted these STs to be responsible for the enormous burden of extraintestinal human infections globally (Manges et al., 2019). These highly successful clonal groups are a major mode of spreading AMR by the mechanism of global expansion (Shaik et al., 2017; Tchesnokova et al., 2019).
ST131 E. coli is an established pandemic extraintestinal MDR pathogen (Rogers et al., 2011; Hussain et al., 2012, 2014; Nicolas-Chanoine et al., 2014). The four ST131 isolates identified in our collection were of two O and CH types, two ST131 E. coli exhibited O25:H4 with fumC40:fimH30 and the other two ST131 E. coli had O16:H5 serogroup and fumC40:fimH41 CH type. The two strains with O25:H4 serogroup belonged to the subclade C2, known as the H30Rx sub-lineage, which is the highly drug-resistant and successful sub-lineage of the ST131 clone. ST1193 is reported to be an emerging multi-resistant clonal group (Johnson et al., 2019). The three ST1193 isolates in this study carried the same set of three nonsynonymous chromosomal mutations in gyrA (S83L), gyrA (D87N) and parC (S80I) that confer fluoroquinolone resistance and had O75:H5 serogroup with fumC14: fimH64 CH type, they were all ciprofloxacin-resistant. ST1193 prevalence is increasing rapidly worldwide and is expected to replace the most successful clone, ST131, in the near future (Pitout et al., 2022). The three ST12 isolates in this collection were qualified as ExPEC pathogens and were ciprofloxacin-resistant. ST12 is reported to be one of the predominant STs that cause bloodstream infections (Kallonen et al., 2017; Manges et al., 2019). The ST73 isolate detected in this study harboured 104 of 335 virulence genes, including several markers of extraintestinal pathogenic and sepsis-associated E. coli. In contrast, it carried a smaller number of AMR genes. It demonstrated β-hemolysis on 5% sheep blood agar and had the CHtype-fumC24:fimH30. THE ST167 strain we identified showed a difficult-to-treat phenotype and carried blaNDM-5 gene. It qualified as an EAEC pathotype and this strain has been associated with a global spread of ESBL-E. coli in humans (Ewers et al., 2012). ST167 associated with blaNDM-5 gene was reported as an emerging carbapenem-resistant high-risk clone (Garcia-Fernandez et al., 2020).
Overall, the NLF E. coli isolates carried the AMR genes and were moderately resistant to antibiotics, but a higher number were assigned to different pathotypes. The isolates carried extensive virulence genes that belonged to different categories, including adhesion, invasion, colonisation, and iron uptake ability. Nevertheless, the combination of resistance and virulence in these NLF E. coli lineages could contribute to their increased fitness and potential to expand globally.
This study has multiple limitations. First, limited clinical and epidemiological data were available. Second, a small sample size constrained the power to generalise our findings and confirm statistical associations, particularly with the high-risk clones. The main gain of the study is that emphasis on inclusion was not laid on MDR or ESBL phenotypes; this has led to a clear understanding of the important extraintestinal pathogenic lineages within NLF E. coli. The WGS approach helped to provide a greater resolution to the study observations.
In conclusion, the prevalence of NLF E. coli variants isolated at a referral diagnostic centre (icddr,b) in Dhaka, Bangladesh, was moderate (10%). Our study revealed multi-resistant strains and high-risk clonal groups, including ST131, ST1193, ST12, ST73 and ST167, emerging within NLF E. coli isolates. To our knowledge, this is the first report on such a phenomenon in NLF E. coli; most studies focus on pathotypes encompassing single STs. These high-risk clones may further evolve by acquiring carbapenem and colistin resistance genes to cause difficult-to-treat infections. Therefore, strengthening microbiology laboratories to detect and report NLF E. coli isolates is important to accomplish successful patient treatment and to feed data to AMR surveillance programs. Further national and regional multicentre “one health” studies are required to ascertain the significance of NLF E. coli as significant pathogens impacting public health.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.
Author contributions
RM and AH designed the study and drafted the manuscript. RM performed genome sequencing. AM, AH, and RM carried out the bioinformatics analyses and interpretation of results and prepared tables and figures. RM and SH performed microbiological tests. JP, SC, MA, DA, TC, and DM contributed to the discussions and reviewed the manuscript. DM supervised the study. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
icddr,b is grateful to the governments of Bangladesh, Canada, Sweden, and the United Kingdom for providing core/unrestricted support for its operations and research. The work is funded through a Royal Society International Collaboration Award (ref. ICA\R1\191309).
References
- Aditi F. Y., Rahman S. S., Hossain M. M. (2017). A study on the microbiological status of mineral drinking water. Open Microbiol. J. 11, 31–44. doi: 10.2174/1874285801711010031, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddam R., Sarker N., Ahmed D., Mazumder R., Abdullah A., Morshed R., et al. (2020). Genome dynamics of vibrio cholerae isolates linked to seasonal outbreaks of cholera in Dhaka, Bangladesh. MBio 11:e03339-19. doi: 10.1128/mBio.03339-19, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beghain J., Bridier-Nahmias A., Le Nagard H., Denamur E., Clermont O. (2018). Clermon typing: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genomics 4:192. doi: 10.1099/mgen.0.000192, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertels F., Silander O. K., Pachkov M., Rainey P. B., Van Nimwegen E. (2014). Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 31, 1077–1088. doi: 10.1093/MOLBEV/MSU088, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bopp C. A., Brenner F. W., Fields P. I., Wells J. G., (2003). Manual of Clinical Microbiology. 8th Edn.. Washington, DC: ASM Press. [Google Scholar]
- Bortolaia V., Kaas R. S., Ruppe E., Roberts M. C., Schwarz S., Cattoir V., et al. (2020). Res finder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. doi: 10.1093/jac/dkaa345, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks K., Sodeman T. (1974). A rapid method for determining decarboxylase and dihydrolase activity. J. Clin. Pathol. 27, 148–152. doi: 10.1136/JCP.27.2.148, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carattoli A., Zankari E., García-Fernández A., Voldby Larsen M., Lund O., Villa L., et al. (2014). In Silico detection and typing of plasmids using plasmid finder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A., Adhikari P., Shenoy S., Saralaya V. (2016). Virulence factor profiles, phylogenetic background, and antimicrobial resistance pattern of lactose fermenting and nonlactose fermenting Escherichia coli from extraintestinal sources. Indian J. Pathol. Microbiol. 59, 180–184. doi: 10.4103/0377-4929.182032, PMID: [DOI] [PubMed] [Google Scholar]
- Chang J., Yu J., Lee H., Ryu H., Park K., Park Y. J. (2014). Prevalence and characteristics of lactose non-fermenting Escherichia coli in urinary isolates. J. Infect. Chemother. 20, 738–740. doi: 10.1016/J.JIAC.2014.07.005, PMID: [DOI] [PubMed] [Google Scholar]
- Chen L., Yang J., Yu J., Yao Z., Sun L., Shen Y., et al. (2005). VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhou Y., Chen Y., Gu J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen P. T. L. C., Aarestrup F. M., Lund O. (2018). Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19, 1–8. doi: 10.1186/S12859-018-2336-6/TABLES/2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna B., Ranucci L., Fradiani P. A., Casalino M., Calconi A., Nicoletti M. (1992). Organisation of aerobactin, hemolysin, and antibacterial resistance genes in lactose-negative Escherichia coli strains of serotype O4 isolated from children with diarrhea. Infect. Immun. 60, 5224–5231. doi: 10.1128/IAI.60.12.5224-5231.1992, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren A. M., Esen O. C., Quince C., Vineis J. H., Morrison H. G., Sogin M. L., et al. (2015). Anvi’o: an advanced analysis and visualisation platformfor’omics data. Peer J 2015:1319. doi: 10.7717/PEERJ.1319/SUPP-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers C., Bethe A., Semmler T., Guenther S., Wieler L. H. (2012). Extended-spectrum β-lactamase-producing and AmpC-producing Escherichia coli from livestock and companion animals, and their putative impact on public health: a global perspective. Clin. Microbiol. Infect. 18, 646–655. doi: 10.1111/J.1469-0691.2012.03850.X, PMID: [DOI] [PubMed] [Google Scholar]
- Flores-Mireles A. L., Walker J. N., Caparon M., Hultgren S. J. (2015). Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 13, 269–284. doi: 10.1038/nrmicro3432, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Fernandez A., Villa L., Bibbolino G., Bressan A., Trancassini M., Pietropaolo V., et al. (2020). Novel insights and features of the NDM-5-producing Escherichia coli sequence type 167 high-risk clone. mSphere 5:e00269-20. doi: 10.1128/MSPHERE.00269-20/ASSET/6E7B35EC-775E-40A6-B00C-FDACD968538C/ASSETS/GRAPHIC/MSPHERE.00269-20-F0003.JPEG, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/SYSBIO/SYQ010, PMID: [DOI] [PubMed] [Google Scholar]
- Gurevich A., Saveliev V., Vyahhi N., Tesler G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/BIOINFORMATICS/BTT086, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasman H., Saputra D., Sicheritz-Ponten T., Lund O., Svendsen C. A., Frimodt-Moller N., et al. (2014). Rapid whole-genome sequencing for detection and characterisation of microorganisms directly from clinical samples. J. Clin. Microbiol. 52, 139–146. doi: 10.1128/JCM.02452-13, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkey P. M. (2006). Identification of Enterobacteriaceae. 3rd Edn. Minneapolis: Burgess Publishing Co. [Google Scholar]
- Hossain A. (2012). Presence and pattern of virulence genes in non-lactose fermenting Escherichia coli strains isolated from stools of children <5 years in rural and urban Bangladesh. Int. J. Infect. Dis. 16:e395. doi: 10.1016/J.IJID.2012.05.525 [DOI] [Google Scholar]
- Huang J., Zhao Z., Zhang Q., Zhang S., Zhang S., Chen M., et al. (2021). Phylogenetic Analysis Reveals Distinct Evolutionary Trajectories of the Fluoroquinolones-Resistant Escherichia coli ST1193 From Fuzhou, China. Front. Microbiol. 12:3370. doi: 10.3389/FMICB.2021.746995/BIBTEX, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A., Ewers C., Nandanwar N., Guenther S., Jadhav S., Wieler L. H., et al. (2012). Multi-resistant uropathogenic Escherichia coli from a region in India where urinary tract infections are endemic: genotypic and phenotypic characteristics of sequence type 131 isolates of the CTX-M-15 extended-spectrum-β-lactamase-producing lineage. Antimicrob. Agents Chemother. 56, 6358–6365. doi: 10.1128/AAC.01099-12, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A., Ranjan A., Nandanwar N., Babbar A., Jadhav S., Ahmed N. (2014). Genotypic and phenotypic profiles of Escherichia coli isolates belonging to clinical sequence type 131 (ST131), clinical non-ST131, and fecal non-ST131 lineages from India. Antimicrob. Agents Chemother. 58, 7240–7249. doi: 10.1128/AAC.03320-14, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensen K. G., Tetzschner A. M. M., Iguchi A., Aarestrup F. M., Scheutz F. (2015). Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 53, 2410–2426. doi: 10.1128/JCM.00008-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M. H. K., Bortolaia V., Tansirichaiya S., Aarestrup F. M., Roberts A. P., Petersen T. N. (2021). Detection of mobile genetic elements associated with antibiotic resistance in salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 76, 101–109. doi: 10.1093/JAC/DKAA390, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. J., Elnekave E., Miller E. A., Munoz-Aguayo J., Figueroa C. F., Johnston B., et al. (2019). Phylogenomic analysis of Extraintestinal pathogenic Escherichia coli sequence type 1193, an emerging multidrug-resistant clonal group. Antimicrob. Agents Chemother. 63:e01913-18. doi: 10.1128/AAC.01913-18, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. R., Stell A. L. (2000). Extended virulence genotypes of Escherichia coli strains from patients with urosepsis in relation to phylogeny and host compromise. J. Infect. Dis. 181, 261–272. doi: 10.1086/315217, PMID: [DOI] [PubMed] [Google Scholar]
- Kallonen T., Brodrick H. J., Harris S. R., Corander J., Brown N. M., Martin V., et al. (2017). Systematic longitudinal survey of invasive Escherichia coli in England demonstrates a stable population structure only transiently disturbed by the emergence of ST131. Genome Res. 27, 1437–1449. doi: 10.1101/GR.216606.116/-/DC1, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper J. B., Nataro J. P., Mobley H. L. T. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140. doi: 10.1038/nrmicro818 [DOI] [PubMed] [Google Scholar]
- Larsen M. V., Cosentino S., Lukjancenko O., Saputra D., Rasmussen S., Hasman H., et al. (2014). Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 52, 1529–1539. doi: 10.1128/JCM.02981-13/SUPPL_FILE/ZJM999093347SO4.PDF, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. V., Cosentino S., Rasmussen S., Friis C., Hasman H., Marvig R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J., Wang H., Li G., Feng Z., Song Y., Wang P., et al. (2019). A novel short-term high-lactose culture approach combined with a matrix-assisted laser desorption ionisation-time of flight mass spectrometry assay for differentiating Escherichia coli and Shigella species using artificial neural networks. PLoS One 14:e0222636. doi: 10.1371/JOURNAL.PONE.0222636, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan L. K., Weinstein R. A. (2017). The epidemiology of carbapenem-resistant enterobacteriaceae: the impact and evolution of a global menace 215, S28–S36. doi: 10.1093/infdis/jiw282, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiorakos A. P., Srinivasan A., Carey R. B., Carmeli Y., Falagas M. E., Giske C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/J.1469-0691.2011.03570.X, PMID: [DOI] [PubMed] [Google Scholar]
- Manges A. R., Geum H. M., Guo A., Edens T. J., Fibke C. D., Pitout J. D. D. (2019). Global extraintestinal pathogenic Escherichia coli (Expec) lineages. Clin. Microbiol. Rev. 32:e00135-18. doi: 10.1128/CMR.00135-18, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder R., Abdullah A., Ahmed D., Hussain A. (2020a). High prevalence of blactx-m-15 gene among extended-spectrum β-lactamase-producing Escherichia coli isolates causing extraintestinal infections in Bangladesh. Antibiotics. 9:796. doi: 10.3390/antibiotics9110796, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder R., Abdullah A., Hussain A., Ahmed D., Mondal D. (2020b). Draft genome sequence of Chromobacterium violaceum RDN09, isolated from a patient with a wound infection in Bangladesh. Microbiol. Resour. Announc. 9:e00957-20. doi: 10.1128/mra.00957-20, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder R., Hussain A., Abdullah A., Islam M. N., Sadique M. T., Muniruzzaman S. M., et al. (2021). International high-risk clones among extended-Spectrum β-lactamase–producing Escherichia coli in Dhaka, Bangladesh. Front. Microbiol. 12:2843. doi: 10.3389/FMICB.2021.736464/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataro J. P., Kaper J. B. (1998). Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201. doi: 10.1128/CMR.11.1.142/FORMAT/EPUB, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nia Santos Braz V., Melchior K., Gallina Moreira C. (2020). Escherichia coli as a multifaceted pathogenic and versatile bacterium. Front. Cell. Infect. Microbiol. 10:548492. doi: 10.3389/fcimb.2020.548492, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas-Chanoine M.-H., Bertrand X., Madec J.-Y. (2014). Escherichia coli ST131, an intriguing clonal group. Clin. Microbiol. Rev. 27, 543–574. doi: 10.1128/CMR.00125-13, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti M., Superti F., Conti C., Calconi A., Zagaglia C. (1988). Virulence factors of lactose-negative Escherichia coli strains isolated from children with diarrhea in Somalia. J. Clin. Microbiol. 26, 524–529. doi: 10.1128/JCM.26.3.524-529.1988, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusrat N., Ridwan B. R., Anowara B., Humaira A. (2017). Isolation, identification and molecular characterisation of rhizobium species from Sesbania bispinosa cultivated in Bangladesh. Afr. J. Agric. Res. 12, 1874–1880. doi: 10.5897/AJAR2017.12321 [DOI] [Google Scholar]
- Pitout J. D. D. (2012). Extraintestinal pathogenic Escherichia coli: a combination of virulence with antibiotic resistance. Front. Microbiol. 3:9. doi: 10.3389/fmicb.2012.00009, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitout J. D. D., Peirano G., Chen L., DeVinney R., Matsumura Y. (2022). Escherichia coli ST1193: following in the footsteps of E. coli ST131. Antimicrob. Agents Chemother. 66:e0051122. doi: 10.1128/AAC.00511-22, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley L. W. (2014). Pandemic lineages of extraintestinal pathogenic Escherichia coli. Clin. Microbiol. Infect. 20, 380–390. doi: 10.1111/1469-0691.12646, PMID: [DOI] [PubMed] [Google Scholar]
- Roer L., Johannesen T. B., Hansen F., Stegger M., Tchesnokova V., Sokurenko E., et al. (2018). CHTyper, a web tool for subtyping of Extraintestinal pathogenic Escherichia coli based on the fumC and fimH alleles. J. Clin. Microbiol. 56, 63–81. doi: 10.1128/JCM.00063-18, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers B. A., Sidjabat H. E., Paterson D. L. (2011). Escherichia coli O25b-ST131: a pandemic, multi-resistant, community-associated strain. J. Antimicrob. Chemother. 66, 1–14. doi: 10.1093/JAC/DKQ415, PMID: [DOI] [PubMed] [Google Scholar]
- Shaik S., Ranjan A., Tiwari S. K., Hussain A., Nandanwar N., Kumar N., et al. (2017). Comparative genomic analysis of globally dominant ST131 clone with other epidemiologically successful Extraintestinal pathogenic Escherichia coli (ExPEC) lineages. MBio 8:e01596-17. doi: 10.1128/mBio.01596-17, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siqueira F. M., De Carli S., Lopes C. E., Machado L., Vieira T. R., Pöppl G., et al. (2021). Non-lactose-fermenting uropathogenic Escherichia coli from dogs: virulence profile characterization. Lett. Appl. Microbiol. 72, 596–603. doi: 10.1111/LAM.13454, PMID: [DOI] [PubMed] [Google Scholar]
- Tatusova T., Dicuccio M., Badretdin A., Chetvernin V., Nawrocki E. P., Zaslavsky L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/NAR/GKW569, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchesnokova V., Radey M., Chattopadhyay S., Larson L., Lee Weaver J., Kisiela D., et al. (2019). Pandemic fluoroquinolone resistant Escherichia coli clone ST1193 emerged via simultaneous homologous recombinations in 11 gene loci. Proc. Natl. Acad. Sci. U. S. A. 116, 14740–14748. doi: 10.1073/pnas.1903002116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Lan F., Lu Y., He Q., Li B. (2017). Molecular characteristics of ST1193 clone among phylogenetic group B2 non-ST131 fluoroquinolone-resistant Escherichia coli. Front. Microbiol. 8:2294. doi: 10.3389/FMICB.2017.02294/BIBTEX, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaratha G., Perloff S., Changala K. (2017). Lactose vs non-lactose fermenting E. coli: epidemiology, clinical outcomes, and resistance. Open Forum Infect. Dis. 4, S589–S590. doi: 10.1093/OFID/OFX163.1546 [DOI] [Google Scholar]
- Zankari E., Allesøe R., Joensen K. G., Cavaco L. M., Lund O., Aarestrup F. M. (2017). PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 72, 2764–2768. doi: 10.1093/jac/dkx217, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.