

Abstract
The insulin–protamine interaction is at the core of the mode of action in many insulin formulations (Zn + insulin + protamine) and to treat diabetes, in which protamine is added to the stable form of hexameric insulin (Zn–insulin). However, due to the unavailability of quantitative data and a high-resolution structure, the binding mechanism of the insulin–protamine complex remains unknown. In this study, it was observed that Zn–insulin experiences destabilization as observed by the loss of secondary structure in circular dichroism (CD), and reduction in thermal stability in melting study, upon protamine binding. In isothermal titration calorimetry (ITC), it was found that the interactions were mostly enthalpically driven. This is in line with the positive ΔCm value (+880 cal mol–1), indicating the role of hydrophilic interactions in the complex formation, with the exposure of hydrophobic residues to the solvent, which was firmly supported by the 8-anilino-1-naphthalene sulfonate (ANS) binding study. The stoichiometry (N) value in ITC suggests the multiple insulin molecules binding to the protamine chain, which is consistent with the picture of the condensation of insulin in the presence of protamine. Atomic force microscopy (AFM) suggested the formation of a heterogeneous Zn–insulin–protamine complex. In fluorescence, Zn–insulin experiences strong Tyr quenching, suggesting that the location of the protamine-binding site is near Tyr, which is also supported by the molecular docking study. Since Tyr is critical in the stabilization of insulin self-assembly, its interaction with protamine may impair insulin’s self-association ability and thermodynamic stability while at the same time promoting its flexible conformation desired for better biological activity.
Introduction
Biomacromolecular interactions including protein–ligand interactions are routinely pursued to gain molecular insights into the structural–functional features of proteins. Insulin is a polypeptide hormone (Figure 1) that modulates glucose levels in the blood. Insulin in complex with Zn is stored in the pancreas as an R6 hexamer, which when released into the bloodstream readily dissociates into monomers.1−5 This is an extremely essential step as only a bioactive monomer can bind to the receptor to regulate blood glucose levels.1,5 Since the deficiency in insulin secretion causes diabetes,6,7 insulin is also used as a medical compound that is widely prescribed for the treatment of diabetes.8 However, the clinical application of insulin is often challenged by its precipitation, chemical degradation, and fibrillation.
Figure 1.
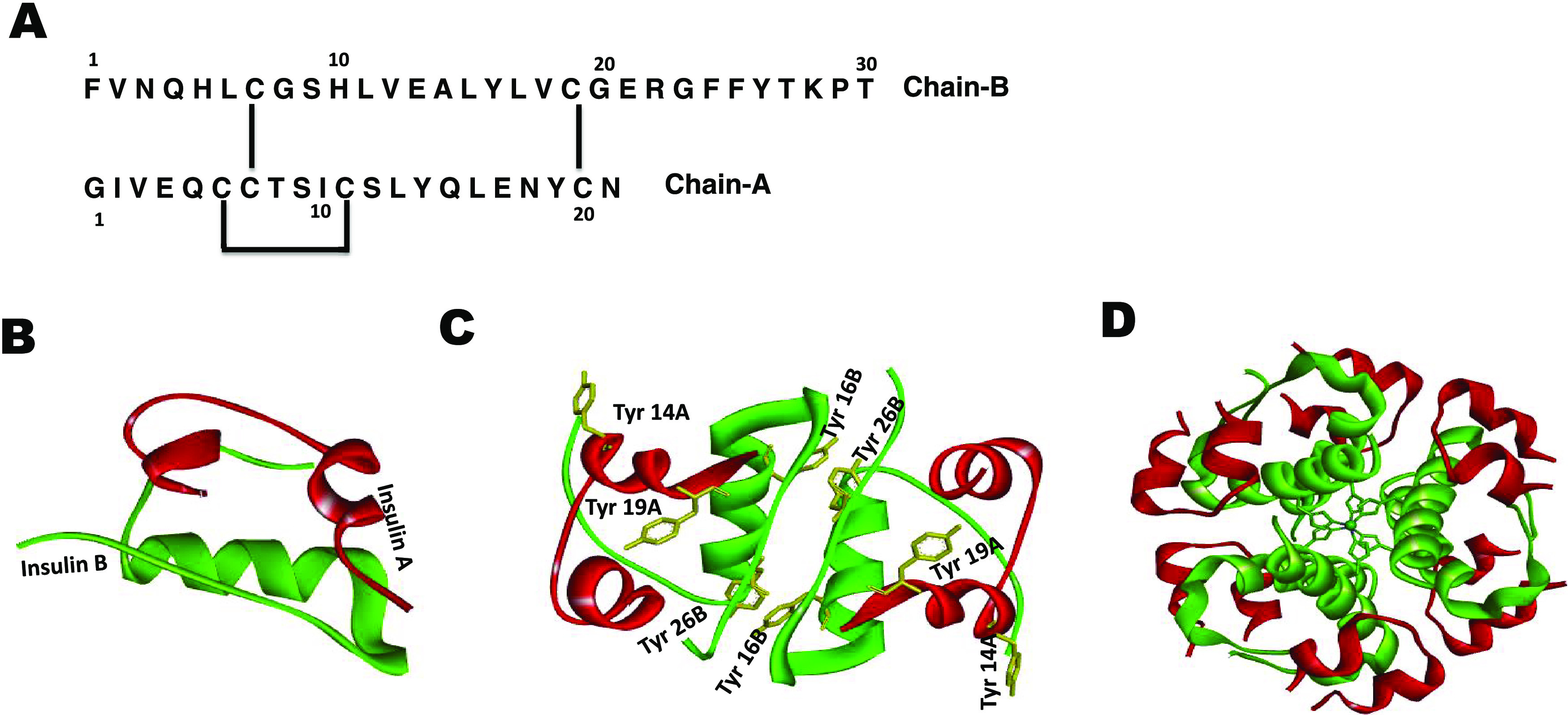
(A) Amino acid sequence of insulin-containing B-chain (upper) and A-chain (lower) with disulfide bridges as indicated, (B) insulin monomer (chain A in red and chain B in green; PDB 2JV1), and (C) insulin dimer, showing tyrosine residues as yellow sticks (PDB 2OMI). (D) Insulin hexamer assembly with the Zn ion at the center coordinated to the B10His residue from each monomer unit.
The development of an insulin–protamine formulation was an important advancement in circumventing some of these problems.9 The use of strongly basic proteins such as protamine, extracted from the nucleus of fish sperm, combined with Zn–insulin (hexamer) was shown to prolong the effect of insulin. Hagedorn and co-workers introduced insulin formulation for the first time containing insulin and protamine in “isophane” amounts (no excess of insulin or protamine) at a neutral pH, also having small amounts of zinc and phenol10 to support stability. There are several commercially available insulin-based formulations, which are heavily used even today. However, they are still far from mimicking the original physiological role of insulin. In view of this, studying the interaction of Zn–insulin with protamine, which is at the core of the mode of action in the formulation, is highly essential. Such a piece of information may contribute to important physical insights into the existing formulation. Also, it can help to unwind different proteins or peptides, which can interact with insulin in a similar manner.
Although Zn–insulin is well characterized,11 its interactions with protamine are unresolved. Here, we have attempted to cognize their interaction behavior using a variety of biophysical techniques. Previous studies indicate that, in solution, at low concentrations and physiological pH, insulin exists as a monomer or dimer.5 Numerous biophysical studies of insulin under variable conditions helped to understand its structure and function.3,4,7,12−19 The active monomeric form consists of two polypeptide chains, chain A, 21 residues, and chain B, 30 residues (Figure 1). These chains are held together by two interchain disulfide bonds (A7–B7 and A20–B19) and one intrachain disulfide bond in chain A (A6–A11). Chain B plays an important role in the association of insulin monomers into dimers and hexamers.4 The hexamer (Zn–insulin), which is the most stable form of insulin,7,14 is well characterized around neutral pH values.4,12 The hexamer is capable of transitioning among distinct conformational states, which have been given the nomenclature T6, T3R3, and R6,20 depending on the conformations of the monomer subunits. The binding of anions and phenols to allosteric sites of insulin is critical to bring conformational changes.
There are several studies reported on the interaction of insulin with a variety of ligands that have provided valuable insights into the possible ligand-binding mechanisms aimed at insulin.3,21,22 However, there are only a few reports on insulin–protamine complexes, including a crystallography study, which could not yield necessary atomic details due to poor resolution.9,23 Here, we focus on a comprehensive biophysical analysis of the forces that drive the molecular recognition process in the insulin–protamine complex. Based on experimental results, it was concluded that protamine indulges in the destabilization of Zn–insulin (from here on, Zn–insulin implies that it also contains phenol). Isothermal titration calorimetry (ITC) data helped to dissect the energetic basis for the interaction of protamine with free insulin and Zn–insulin. The fluorescence result revealed how Zn–insulin experiences Tyr quenching in the presence of protamine with a significant conformational change, supported by circular dichroism (CD) results. Atomic force microscopy (AFM) study helped to characterize the topology of insulin with a three-dimensional (3D) image using the cross-sectional height of a species visually. Molecular docking studies demonstrated the probable binding site of protamine at insulin. Finally, it was contemplated that protamine–insulin assembly may provide complementary structural adaptations to elude the formation of any toxic insulin state, such as fibrillation.
Material and Methods
Insulin human recombinant, expressed in yeast (≥98% purity, MW 5.8 kDa), protamine sulfate salt from salmon (MPRRRRSSSRPVRRRRRPRVSRRRRRRGGRRRR) (MW 5.1 kDa), 8-anilino-1-naphthalenesulfonic acid ammonium salt (ANS), and zinc sulfate monohydrate (ZnSO4) were purchased from Sigma-Aldrich. Na2HPO4, NaH2PO4, NaCl, and phenol were purchased from Merck Chemicals Ltd. All experiments were performed in phosphate buffer at pH 8.0 containing 10 mM Na2HPO4, 10 mM NaH2PO4, and NaCl. The concentration of insulin was measured using an absorbance of 276 nm on the Cary 100 UV–vis spectrophotometer. The molar extinction coefficient used for insulin was 6200 M–1cm–1.13
Native Poly(acrylamide) Gel Electrophoresis (PAGE)
Native PAGE was performed on the “Sure cast system, by Thermo Fisher Scientific”. The discontinuous Tris–glycine gels (8 × 8 cm2, 1 mm thickness, 10 wells) were used with a separating gel of 20% acrylamide and a stacking gel of 5% acrylamide. All of the buffers, resolving buffer (1.5 M Tris–HCl, pH = 8.8), stacking buffer (1 M Tris–HCl, pH = 6.8), sample loading buffer (50% glycerol, 0.5 M Tris–HCl, and 0.03% bromophenol blue), and running buffer (1× Tris–glycine, pH = 8.3), were freshly prepared using Milli-Q water. The samples (∼20 μL of loading volume per well) were loaded into the wells with the help of a microsyringe. The gel was then subjected to electrophoresis under a constant voltage of 100–120 V for ∼1 h. For the protein detection, the gel was stained with 0.1% Coomassie brilliant blue in 10% acetic acid and 45% methanol for 2–3 h and then destained in 10% methanol and 10% acetic acid for 7–8 h. The ultra-low-molecular-weight protein marker, M3546 was purchased from Sigma.
Isothermal Titration Calorimetry (ITC)
ITC measurements were performed to obtain thermodynamic parameters associated with the binding of protamine with free insulin and Zn–insulin (with phenol) in phosphate buffer with different salt (NaCl) concentrations of 50, 100, and 150 mM at 25 °C on a MicroCal iTC200 (Malvern Instruments Ltd., U.K.).24 Also, the temperature-dependent measurements were performed at 15, 25, and 35 °C in 10 mM phosphate-buffered saline (PBS) and 100 mM NaCl buffer. In the binding study, protamine (300 μM or 1.53 mg mL–1) was titrated into insulin (60 μM or 0.35 mg mL–1). In another experiment, the protamine was titrated into the insulin hexamer (Zn–insulin). Here, the concentrations of insulin, phenol, ZnSO4, and protamine were 40 μM (0.23 mg mL–1), 120 μM (0.011 mg mL–1), 20 μM (0.0035 mg mL–1), and 300 μM (1.53 mg mL–1), respectively. A total of 40 μL volume was titrated from the injection syringe to a sample cell containing 280 μL of sample. There was a total of 20 injections, with each injection of 2 μL of sample titrated into the cell containing buffer, and each injection was separated by 150 s intervals to allow the signal to return to the baseline. In the control experiment, the sample from the syringe was titrated into an ITC cell containing a working buffer (Figures S1 and S2). All of the experiments were done in at least triplets. The nonlinear data were fitted to the single or two sets of binding site model using MicroCal ORIGIN 7 software supplied by the manufacturer, yielding binding constant (KA), enthalpy change (ΔH), and entropy change (ΔS).24 ΔG was calculated using the relationship ΔG = ΔH – TΔS. Additionally, ΔCp was obtained from the slope of the temperature-dependent enthalpy plot.
The overall entropy change in the binding reaction has contributions from solvent entropy (ΔSsolv) and conformational entropy (ΔSconf). ΔSsolv can be estimated from the heat capacity change, ΔCp, using the relationship
| 1 |
where Ts* is the reference temperature (385 K) at which the hydrophobic contribution to S is zero.25,26
Circular Dichroism (CD)
The CD measurements were recorded with a CD spectrophotometer (Chirascan, Applied Photophysics). Far-UV (200–260 nm) CD was used to investigate the secondary structural changes in insulin with a cuvette cell of path length 0.1 cm. Spectra were recorded for free insulin and Zn–insulin in the presence and absence of protamine. Phenol was added to Zn–insulin in a 3:1 molar ratio to maintain a hexameric state. The concentrations of insulin and ZnSO4 were 20 μM (0.116 mg mL–1) and 10 μM (0.0017 mg mL–1), respectively. All of the experiments were performed in doublets. Changes observed in the helical content were calculated using online software BeStSel (β Structure Selection), as shown in Figure 2C.
Figure 2.
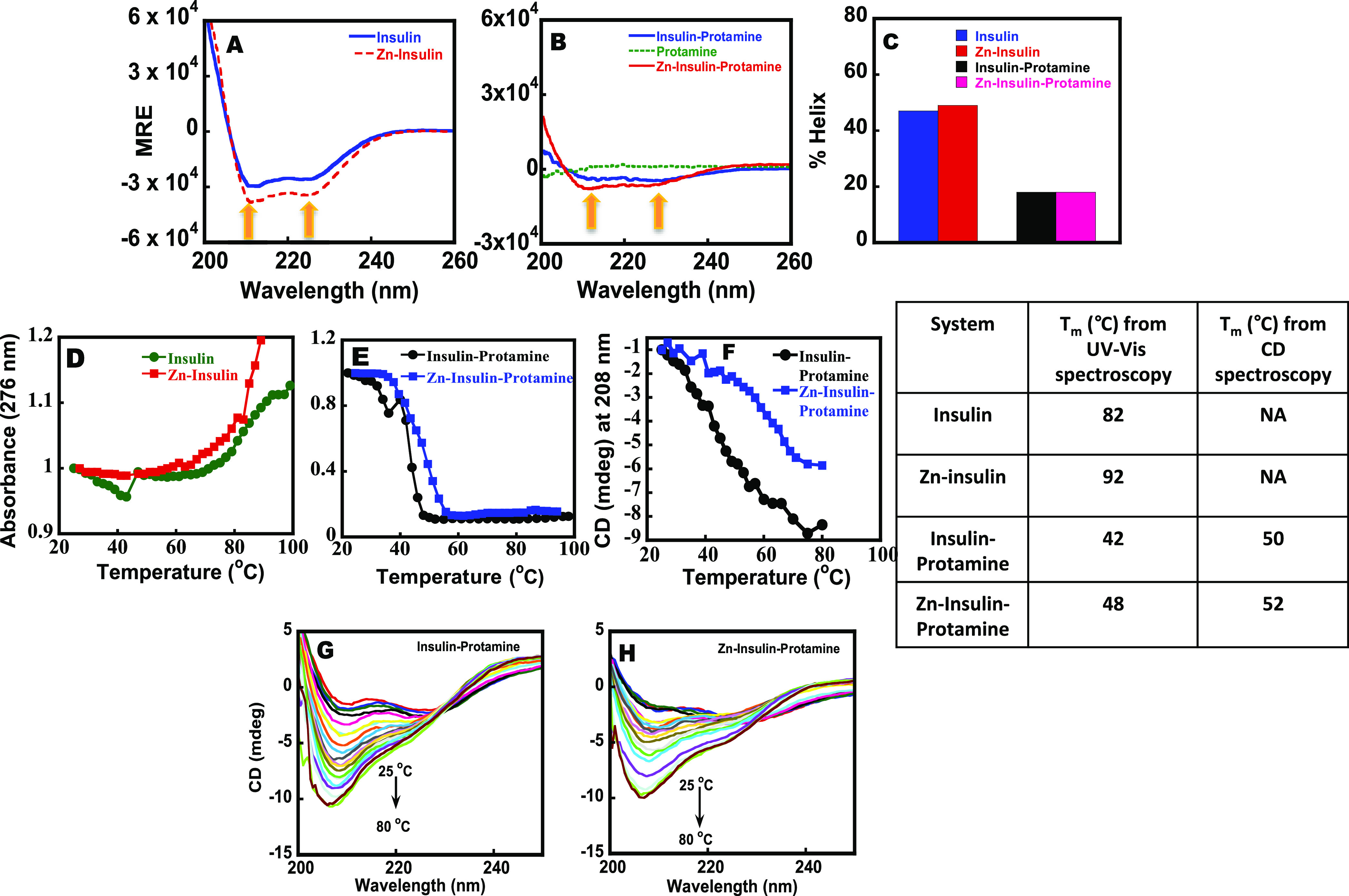
Far-UV CD of (A) free insulin and Zn–insulin and that of (B) insulin–protamine, Zn–insulin–protamine, and protamine. The concentrations of protamine, insulin, and ZnSO4 were 20, 20, and 10 μM, respectively. (C) % Helical content of insulin and Zn–insulin with and without protamine. Thermal stability curve of (D) free insulin and Zn–insulin and (E) insulin–protamine and Zn–insulin–protamine using ultraviolet–visible (UV–vis) spectrometer. (F) Plot of CD ellipticity at 208 nm vs temperature curve of insulin–protamine and Zn–insulin–protamine. Table showing a comparison of Tm values. Far-UV CD melting spectra of (G) insulin–protamine and (H) Zn–insulin–protamine. Phosphate buffer (10 mM) containing 100 mM NaCl at pH 8.0 was used. The insulin/Zn molar ratio was 1:0.5, and the insulin/phenol molar ratio was 1:3.
Intrinsic Fluorescence Spectroscopy
The fluorescence studies were performed using a fluorescence spectrophotometer from Cary Eclipse Varian. Here, steady-state experiments were performed using Tyr as an intrinsic probe. Protamine was titrated into free insulin and Zn–insulin. Concentrations of insulin, phenol, and ZnSO4 were 10 μM (0.058 mg mL–1), 30 μM (0.0028 mg mL–1), and 5 μM (0.0009 mg mL–1), respectively. All of the samples were excited at 276 nm, and emission spectra were recorded between 280 and 400 nm using a slit width of 5 nm. In the case of the binding of protamine to Zn–insulin, the Stern–Volmer equation was found to be nonlinear (upward curvature in the Stern–Volmer plot), and hence a modified Stern–Volmer27 equation was used.
| 2 |
where f0 and f are the relative fluorescence intensities of protein in the absence and presence of a quencher (Q), respectively; [Q] is the quencher concentration, KQ is the effective quenching constant for the accessible fluorophores, and fa is the fraction of accessible fluorophore. The plot of f0/(f0 – f) vs 1/[Q] yields 1/fa as the intercept and 1/(faKQ) as the slope. Protamine binding to Zn-phenol-insulin at different NaCl concentrations was plotted using both Stern–Volmer and modified Stern–Volmer equations.
For the dilution experiment, the samples were excited at 276 nm with a slit width of 10 nm. The initial concentrations of insulin, ZnSO4, and protamine for dilution were 20 μM (0.116 mg mL–1), 10 μM (0.0017 mg mL–1), and 20 μM (0.1 mg mL–1), respectively. To calculate the quenching constant in the case of binding of protamine to insulin and Zn–insulin, the Stern–Volmer equation was used.
| 3 |
where fo represents the fluorescence intensity of insulin in the absence of protamine, f represents the fluorescence intensity in the presence of protamine, KSV represents the Stern–Volmer quenching constant, and Q represents the concentration of protamine. When data are plotted, fo/f, i.e., emission intensity, vs Q, i.e., concentration, then upon linear fitting (y = mx + c), the value of slope will give the quenching constant KSV.
ANS Binding Study
The role of hydrophobic surfaces in the insulin–protamine complexes was determined by the ANS binding studies. Emission spectra were recorded in the range of 400–600 nm by exciting all of the samples at 388 nm.28,29 The excitation and emission slits were set at 5 nm, and the scan speed was 600 nm min–1. Here, the concentrations of insulin, protamine, phenol, and ZnSO4 were 10 μM (0.058 mg mL–1), 10 μM (0.05 mg mL–1), 30 μM (0.0028 mg mL–1), and 5 μM (0.0009 mg mL–1), respectively.
Melting Study
Melting studies were done using a Cary 100 UV–vis spectrophotometer. Here, the absorbance at 276 nm was recorded over a temperature range of 25–95 °C after 3 h incubation of samples at 4 °C. The concentrations of insulin and ZnSO4 were 30 μM (0.17 mg mL–1) and 15 μM (0.0026 mg mL–1), respectively. The experiments were performed with phenol using an insulin/phenol ratio of 1:3.
Far-UV CD melting experiments were performed on a JASCO-815 CD spectrophotometer equipped with a Peltier device. The temperature was increased from 25 to 80 °C, and the spectra were recorded in the wavelength range of 200–250 nm in a cuvette cell of 0.1 cm. Phenol was added to Zn–insulin in a 3:1 molar ratio to maintain the hexameric state. The concentrations of insulin and ZnSO4 were 20 μM (0.116 mg mL–1) and 10 μM (0.0017 mg mL–1), respectively.
Atomic Force Microscopy (AFM)
AFM images were recorded on a WITec alpha300 RA system. AFM is used for high-resolution nanoscale surface characterization by recording the interactive forces between the surface and a sharp cantilever tip. Through AFM, we have monitored the visual or structural changes in insulin and zinc-bound insulin and its 1:1 complexes with protamine in the presence of phenol in 10 mM phosphate buffer and 100 mM NaCl buffer. The concentrations of insulin and ZnSO4 were approximately 80 and 40 μM, respectively, and phenol was taken in the molar ratio of 1:3 with respect to insulin. The samples were freshly prepared and drop-cast on the freshly cleaved mica surface, and a gentle wash was given to remove the extra salt of buffer for obtaining a clear image. All images were taken two times to confirm the reproducibility of the results.
Molecular Docking
To explain the binding interaction between insulin and protamine, molecular docking was performed by utilizing HADDOCK2.2 (High Ambiguity Driven protein–protein DOCKing) program.30 The structure of the protamine sequence was converted to PDB format using I-TASSER online software from Zang lab.31 The crystal structure of the insulin monomer (PDB 2JV1) was downloaded from the protein data bank. All other docking parameters were set as default. After obtaining the docked structure, the analysis was done by Accelrys Discovery Studio 4.5. After the careful analysis of the parameters obtained for all four docked structures, the structure with the most accurate possibilities and minimum energy was taken for further analysis.
Results
Characterization of Insulin Using Gel
Native PAGE was used to analyze insulin and insulin hexamers (Figure S1). In lane 1, we used a low-molecular-weight marker for comparison. The free insulin in lane 2 shows a single intense band near 6 kDa, confirming the presence of majorly monomer insulin. In lane 3, the Zn–insulin sample shows the smears along its path, suggesting the formation of insulin hexamers (Figure 1D). The observed smearing of a band in lane 3 could be due to the following reason. The hexamer insulin is known to be stabilized by the formation of noncovalent interactions between the insulin dimer assemblies, and the electric field applied may disrupt these noncovalent interactions, thereby showing smearing with the intense band near ∼6 kDa representing the insulin monomer unit.
Insulin–Protamine Binding: UV–Vis and CD Study
Far-UV CD was employed to monitor the changes in the secondary structure of insulin in the presence of protamine. The CD spectrum of free insulin (Figure 2A) shows two minima, one at 208 nm and the other at 222 nm, which signifies the presence of α-helical content in agreement with the literature reports.7 The free insulin and Zn–insulin show identical helical contents (Figure 2A,B). Since free insulin forms a dimer and Zn–insulin forms a hexamer, the presence of identical helical contents suggests that the structure of the independent dimer is nearly identical to that of the dimer of the hexamer insulin. Similarly, protamine binding to Zn–insulin also resulted in a loss in the intensity of peak at 208 and 222 nm, indicating the alteration of the helical content in Zn–insulin.39
Thermal melting is useful to elucidate the stability of a protein in the presence of a ligand. Here, the samples were incubated for 3 h to equilibrate before performing the melting experiments. In Figure 2D, high Tm (temperature at which the protein is half denatured) values for free insulin (82 °C) and Zn–insulin (∼92 °C) helped to establish their respective dimer and higher-order status.4,13 In the case of Zn–insulin, the absorbance values continued increasing even beyond 90 °C (Figure 2D), indicating its strong stability. Free protamine did not give any melting transition as protamine is a randomly coiled structure (as shown in Figure 2B). The higher stability for Zn–insulin compared to free insulin is in agreement with the literature studies.35 On the contrary, the protamine binding to free insulin and Zn–insulin resulted in a downward curve with a reduced Tm of 42 and 50 °C, respectively (Figure 2E). As per the prior reports, based on experimental and simulated data, ligand stabilizers shift the protein melting temperature upward, whereas ligand destabilizers shift the Tm downward.36 Thus, it is obvious that the downward curve has resulted due to the destabilization of insulin and Zn–insulin from protamine binding.
We have also examined the melting behavior of the insulin–protamine and the Zn–insulin–protamine complexes by monitoring the secondary structure of insulin at 208 nm using CD (Figure 2E,F). In far-UV CD, the same Tm values reached around 48 and 52 °C for protamine binding with insulin and Zn–insulin, respectively (Table in Figure 2). The small differences in respective Tm values in UV and CD imply the presence of additional unfolding processes in CD not detected by UV–vis studies. As shown in Figure 2F, in the presence of protamine, the CD spectrum of insulin resulted in a change in the ratio of the intensity of peaks at 208 and 222 nm. In the classical paper,32 it has been suggested that a change in the ratio (208:222) can be observed when the percentage of α-helices decreases. Some studies suggested that the increase in the ratio of 208:222 nm indicates the formation a less-stable 310 helix.33,34 Such conformational changes would disturb the local environment, especially due to the four Tyr residues present in the insulin. Additionally, reversibility was checked by allowing the samples to cool down before reheating. It was observed that Zn–insulin–protamine has higher reversibility compared to the insulin–protamine complex (Figure S6).
Binding Thermodynamics Using ITC
The binding thermodynamics of protamine with insulin and Zn–insulin was explored by ITC (Figures 3 and 4). At 100 mM salt (Figure 3A), the protamine exhibited moderate binding affinity with insulin (KA = 1.5 × 105 M–1). The data were fitted using a model of a single set of binding sites. In Figure 3A, the solid line shows the best fit, and the model reproduces experimental data fairly well. Table 1 lists the thermodynamic parameters. Here, the binding was enthalpically favorable (ΔH = −10.6 kcal mol–1) and entropically unfavorable (TΔS = −3.6 kcal mol–1). Thus, the resultant Gibbs free energy change (ΔG) of −7.0 kcal mol–1 (Table 2) has a dominant contribution from enthalpy. The overall negative contribution of ΔS implies that entropic gain from desolvation entropy must be overcompensated by the entropy loss from conformational entropy.38 Next, as the salt concentration was increased to 150 mM (Figure 3B), the binding thermogram displayed a slight deviation from the standard shape. It appears from the ITC profile that at least two different types of binding interactions are present. After fitting the isotherm to different binding models, we found that single-site model was unable to account for the observed isotherm; instead, a sequential two-site model was best fitted that let us determine thermodynamic parameters. These two types of interactions were both exothermically driven with enthalpy changes of −2.3 kcal mol–1 (ΔH1) and −1.1 kcal mol–1 (ΔH2). The related changes ΔG of −6.4 kcal mol–1 (ΔG1) and −6.7 kcal mol–1 (ΔG2) (Table 1) indicated that the binding to either site was thermodynamically favorable. The binding affinity of the second site (KA2 = 2.9 × 105 M–1) was approximately fivefold stronger than that of the first (KA1 of 5.5 × 104 M–1). It was observed that as the salt concentration increased from 100 to 150 mM, the binding became enthalpically less favorable and entropically more favorable, suggesting that the complex formation is strongly driven by the release of counterions (solvation entropy).39
Figure 3.

ITC thermograms as a result of titration of protamine into insulin at (A) 100 mM and (B) 150 mM salt concentration, and also at different temperatures of (C) 15 °C, (D) 25 °C, and (E) 35 °C. Concentrations were as follows: protamine, 300 μM (1.53 mg mL–1), and insulin, 60 μM (0.35 mg mL–1). All of the experiments were performed in 10 mM phosphate buffer at pH 8.0 on a MicroCal iTC200.24
Figure 4.

ITC thermograms as a result of titration of protamine into Zn–insulin at (A) 100 mM and (B) 150 mM salt concentration and also at different temperatures of (C) 15 °C, (D) 25 °C, and (E) 35 °C. Concentrations were as follows: protamine, 300 μM or 1.53 mg mL–1; insulin, 40 μM or 0.23 mg mL–1; ZnSO4, 20 μM or 0.0035 mg mL–1; and phenol, 120 μM or 0.011 mg mL–1. All of the experiments were performed in 10 mM phosphate buffer at pH 8.0.
Table 1. Thermodynamic Parameters of Protamine Binding to Insulin.
| protamine into insulin | N | K1 (M–1) | ΔH1(kcal mol–1) | TΔS1(kcal mol–1) | ΔG1(kcal mol–1) | K2 (M–1) | Kavg (M–1) | ΔH2(kcal mol–1) | TΔS2(kcal mol–1) | ΔG2(kcal mol–1) | ΔGavg(kcal mol–1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Salt Dependent | |||||||||||
| NaCl 100 (mM) | 0.13 | (1.5 ± 0.7) × 105 | –10.6 ± 0.8 | –3.6 ± 1.1 | –7.0 ± 0.3 | ||||||
| NaCla 150 (mM) | (5.5 ± 1.0) × 104 | –2.3 ± 0.05 | 4.1 ± 0.2 | –6.4 ± 0.2 | (2.9 ± 1.5) × 105 | (1.7 ± 1.0) × 105 | –1.1 ± 0.2 | 5.6 ± 1.2 | –6.7 ± 3.1 | –6.5 ± 1.5 | |
| Temperature Dependent | |||||||||||
| 15 °C | 0.53 | (0.7 ± 0.2) × 104 | –6.2 ± 0.6 | –1.2 ± 0.4 | –5.1 ± 0.2 | ||||||
| 25 °C | 0.13 | (1.5 ± 0.7) × 105 | –10.6 ± 0.8 | –3.6 ± 1.1 | –7.0 ± 0.3 | ||||||
| 35 °C | 0.07 | (3.9 ± 0.05) × 104 | –15.3 ± 2.5 | –8.5 ± 2.3 | –6.7 ± 0.3 | ||||||
Constraint was put on N during the data fitting.
Table 2. Thermodynamic Parameters of Protamine Binding to Zn–Insulin.
| protamine into Zn–insulin | N | K1 (M–1) | ΔH1(kcal mol–1) | TΔS1(kcal mol–1) | ΔG1(kcal mol–1) | K2 (M–1) | Kavg (M–1) | ΔH2(kcal mol–1) | TΔS2(kcal mol–1) | ΔG2(kcal mol–1) | ΔGavg(kcal mol–1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Salt Dependent | |||||||||||
| NaCl 100 mM | 0.14 | (1.8 ± 0.35) × 105 | –12.8 ± 3.0 | –5.6 ± 3.1 | –7.2 ± 0.1 | ||||||
| NaCl 150 mM | (1.7 ±.4) × 105 | –8.1 ± 0.4 | –1.0 ± 0.6 | –7.1 ± 0.2 | (1.8 ± 0.2) × 105 | (1.7 ± 0.6) × 105 | 0.5 ± 0.2 | 7.6 ± 0.1 | –7.1 ± 0.1 | –7.1 ± 0.15 | |
| Temperature Dependent | |||||||||||
| 15 °C | 0.02 | (1.0 ± 0.12) × 105 | –19.2 ± 4.1 | –12.5 ± 4.1 | –6.7 ± 0.05 | ||||||
| 25 °C | 0.14 | (1.8 ± 0.4) × 105 | –12.8 ± 3.0 | –5.6 ± 3.1 | –7.2 ± 0.1 | ||||||
| 35 °C | 0.15 | (1.1 ± 0.16) × 106 | –1.6 ± 0.06 | 7.0 ± 0.1 | –8.5 ± 0.06 | ||||||
Figure 3C–E shows the temperature-dependent binding of protamine with insulin. At all of the studied temperatures, the binding was enthalpically favorable (<ΔH) but entropically unfavorable (<ΔS), as shown in Table 1. The reaction shows temperature dependence for both ΔH and TΔS. As shown in Figure 5, a linear dependence between ΔH and temperature was observed, giving a slope as the change in heat capacity (ΔCP; −450 cal mol–1 K–1). Negative ΔCP could be a result of hydrophobic residues being transferred to a more nonpolar environment,35 indicating the role of hydrophobic interactions in the complex formation. In further analysis, based on eq 1, we estimated a positive contribution (34.3 kcal mol–1) of TΔSsolv to the binding.40 Using this value, we estimated TΔSconf (−25.8 kcal mol–1), which suggests that the binding results in energetic costs from conformational entropy penalty. The negative contribution of TΔSconf suggests that the resultant complex is more rigid.
Figure 5.

Plot of ΔH vs temperature for insulin and Zn–insulin showing ΔCp (slope).
In Figure 4, the binding of protamine with Zn–insulin at 100 mM salt has resulted in decent binding affinity (Ka = 1.8 × 105 M–1). Table 2 lists the thermodynamic parameters. Here, the binding was enthalpically favorable (−12.8 kcal mol–1) and entropically unfavorable (−5.6 kcal mol–1), resulting in ΔG that has a dominating contribution from enthalpy. As the salt concentration was increased to 150 mM, the ITC thermogram showed a slight biphasic nature. The data show that the binding affinities for the first (KA1 = 1.7 × 105 M–1) and second sites (KA2 of 1.8 × 105 M–1) are approximately similar. These two binding sites show enthalpy changes of −8.1 kcal mol–1 (ΔH1) and +0.5 kcal mol–1 (ΔH2) and entropy changes of −1.0 kcal mol–1 (TΔS1) and 7.6 kcal mol–1 (TΔS2). The ΔG values (ΔG1 = −7.1 kcal mol–1 and ΔG2 =–7.1 kcal mol–1) indicated that the binding to either site was thermodynamically favorable. We presume that the first phase of the binding event was enthalpically driven that correlates with the specific interaction between protamine and Zn–insulin, and the second was entropically driven that correlates with the solvation effect. The relatively smaller effect of salt concentrations (100 and 150 mM) on binding affinity suggests the limited role of electrostatic interactions between the Zn–insulin and protamine. Instead, the large favorable contribution of enthalpy changes suggests that the hydrophilic interactions between protamine and Zn–insulin play an important role.
The temperature dependence of protamine complexation with Zn–insulin (Figure 4C–E) yielded a positive ΔCp value (+880 cal mol–1) (Figure 5). Such positive ΔCp for intermolecular interactions are reported in the interaction of MMP3c with the NT1 T98L mutant.41 It, prima facie, implicates the role of hydrophilic interactions in binding. The positive ΔCP leads to an estimate of −67.2 kcal mol–1 for TΔSsolv (eq 1) and +93.6 kcal mol–1 for TΔSconf. The positive TΔSconf suggests the formation of a conformationally flexible complex.
Also, the observed increase in binding affinity with an increase in the temperature may indicate that protamine favors binding to an open and partially unfolded insulin hexamer (Zn–insulin). This anomalous behavior can be explained on the basis of Sturtevant’s proposal, according to which several factors including conformational entropy contributes to the value of ΔCp.37,38 Thus, the binding of protamine with Zn–insulin is hydrophilic in nature and is also accompanied by conformational changes.
The binding stoichiometry resulting from the fitting was particularly interesting: each protamine molecule bound to multiple insulin molecules. In the case of insulin, 2, 8, 15 molecules were linked to protamine at 15, 25, and 35 °C, respectively, suggesting that at higher temperatures, protamine remains in the extended form and can link more insulin molecules. Similarly, in the case of Zn–insulin, multiple molecules were estimated to be linked to protamine. It is possible that the stoichiometry numbers have larger errors because of some uncertainty in the pretransition data in ITC. However, the molar ratio on the x-axis clearly supports the reported values obtained from fitting. Such a result with a single ligand bound to multiple protein molecules is unusual but possible because protamine is a sizable linear polypeptide (total 32 residues) rather than a compact ligand. Additionally, we checked the binding of Zn with insulin (Figure S5) using ITC. Zn binds to insulin under physiological conditions with a weak binding affinity (KA = 2.54 × 104 M–1; ΔH = 0.86 kcal mol–1). All of the control experiments in comparison with raw experiments of both the titrations of protamine into insulin and Zn–insulin at different salt concentrations and different temperatures are shown in Figures S2 and S3. Also, the control experiment for the binding between protamine and Zn, as shown in Figure S10, shows negligible binding.
Further, the plots of ΔH vs −TΔS of protamine binding to insulin and Zn–insulin are shown in Figure S4. The interaction has a good linear relationship between the ΔH and TΔS in both complexes (regression coefficient = 0.999; slope = 0.98 in insulin–protamine) and (regression coefficient = 0.98; slope = 1.03 in Zn–insulin–protamine). Thus, no significant deviation from the slope = 1.0 indicates the presence of enthalpy–entropy compensation phenomenon in both complexes.42
Probing the Role of Hydrophobic Interactions Using ANS Binding
To further confirm the exposure of hydrophobic surfaces in Zn–insulin–protamine complexation, ANS binding study was performed. The extrinsic fluorescence dye 8-anilino-1-naphthalene sulfonate (ANS) has a tendency to fluoresce upon binding to solvent-exposed hydrophobic surfaces on proteins and therefore is widely used for probing exposure of hydrophobic regions in proteins.43,44 As seen in Figure 6A,B, ANS binds very weakly to free insulin as well as to Zinc–insulin, suggesting that insulin under these conditions is intact with its hydrophobic regions mostly buried. Also, since there is no blue shift in the wavelength maxima of ANS (dotted line), this is another indicator of no exposure of hydrophobic residues in the Zn–insulin and free insulin states.
Figure 6.

Titration of increasing concentrations of ANS into (A) free insulin, (B) Zn–insulin, (C) Zn–insulin–protamine. The dotted line indicates free ANS excited at 388 nm. All of the experiments were performed in 10 mM phosphate buffer containing 100 mM NaCl at pH 8.0 at 25 °C. The insulin/Zn molar ratio was 1:0.5, and the insulin/phenol molar ratio was 1:3.
On the other hand, the binding of ANS to the Zn–insulin–protamine complex is very strong (Figure 6C), implying that protamine binding may have resulted in the dissociation and even partial unfolding of insulin, exposing the core hydrophobic pockets to the solvent.45 The positive heat capacity change observed in ITC, which confirmed the presence of hydrophilic interactions between protamine and Zn–insulin, and also far-UV CD melting, where protamine was clearly shown to destabilize insulin, support these results. A similar compact state for chymopapain has been reported earlier using ANS as a probe.46 The addition of ANS to free protamine in the control experiment did not yield any increase in the intensity. This is because protamine lacks proper structure and hence hydrophobic pockets to bind to ANS.
Fluorescence Study of the Protamine–Insulin Interaction
The fluorescence of aromatic amino acids in proteins acts as a probe for investigating the conformational changes in proteins upon ligand binding.47 Monomer insulin consists of four tyrosine residues, out of which two are involved in the helix of chain A (A14 and A19) and the other two are involved in the monomer–monomer interaction (B16 and B26).48 This suggests that tyrosine fluorescence could be exploited for probing the binding effect of protamine on structural rearrangements in free insulin and Zn–insulin. As shown in Figure 7, the gradual addition of protamine to free insulin or Zn–insulin resulted decreased fluorescence intensity. The quenching portrays the change in the microenvironment near Tyr residues, which can be attributed to the local conformational changes.27 Since no shift in the maximum emission wavelength was observed, the polarity of the tyrosine environment cannot be commented on. Generally, protein fluorescence is quenched when Tyr residues (Tyr16 and Tyr26) are buried in the hydrophobic core. However, here, insulin undergoes not only quenching but also destabilization (from Tm and CD results) upon protamine binding. Thus, the quenching could be attributed to the interaction of the Tyr with the polar amino acids of protamine such as Arg, whereas destabilization could be a consequence of these interactions disrupting the dimer interface. Figure 7 shows the modified Stern–Volmer plot for the binding of protamine with insulin and Zn–insulin. The Stern–Volmer constant (KQ) value represents the binding or association between a quencher and fluorophore (Table S1). The KQ for Zn–insulin–protamine (4.39 × 104 M–1) was approximately 5 times lower than that for insulin–protamine (22.38 × 104 M–1), indicating the relatively high affinity between the quencher (protamine) and fluorophore of free insulin. The strong quenching effect of protamine on free insulin indicates easy accessibility of Tyr, resulting in a binding that is strongly dominated near the site close to Tyr residue in the protamine–insulin complex.49
Figure 7.

Fluorescence titration of increasing concentration of protamine into (A) insulin and (B) Zn–insulin; (C) plot of [(fo/f – 1)] vs 1/[Q] using the modified Stern–Volmer equation to determine quenching constants. The intensity was recorded at 304 nm. All of the experiments were performed in 10 mM phosphate buffer at pH 8.0 and 25 °C. The insulin/Zn molar ratio was 1:0.5, and the insulin/phenol molar ratio was 1:3.
Additionally, a comparison of the fluorescence spectra of insulin under three different conditions is shown in Figure S8. The strength of the fluorescence intensity is in the following order: free insulin > Zn–insulin > Zn–insulin–protamine. The lowest intensity for the Zn–insulin–protamine complex further suggests that the protamine has a binding preference near Tyr residues.
To rule out fibril formation of insulin in the presence of protamine, the Zn–insulin–protamine complex was subjected to thioflavin T (ThT) treatment. ThT binding is widely used to probe protein fibrillation in various neurological disorders. ThT, when excited at 440 nm, has a characteristic emission maximum at 485 nm, which significantly increases upon binding with protein aggregates.50 In the ThT assay, as shown in Figure S9, the sample (ThT + Zn–insulin–protamine) was excited at 440 nm, and emission spectra were recorded for 18 h in the range 460–600 nm. The results clearly show that under given conditions, there is no significant increase in ThT fluorescence, indicating that the Zn–insulin–protamine complex is quite stable and does not undergo fibrillation.
Characterization of the Insulin–Protamine Complex Using AFM
AFM study was performed to acquire morphology maps to further characterize insulin, Zn–insulin, and Zn–insulin–protamine species (Figure 8). The experiments were repeated twice, and the data were quite reproducible. To the best of our knowledge, no AFM study has been reported on the insulin–protamine complexes previously. In the case of only insulin, a single-molecule statistical analysis revealed that the average population had a cross-sectional height of 10–12 nm (Figure 8A) with a broader distribution. We expect that the broadness of the peak is due to the presence of a mixture of monomeric and dimeric molecules of the insulin (without Zn) in the sample.4 Meanwhile, as shown in Figure 8B, in the Zn–insulin sample, the presence of the only distribution curve with a height of 20–22 nm indicates the presence of a single type of species, i.e., hexamer. On the other hand, in the presence of protamine (Figure 8C), the presence of two distribution curves (at 20 and 30 nm height) for Zn–insulin may indicate the formation of a heterogeneous assembly involving a somewhat unstructured form of insulin, resulting from the binding and crosslinking of multiple insulin molecules with protamine.
Figure 8.
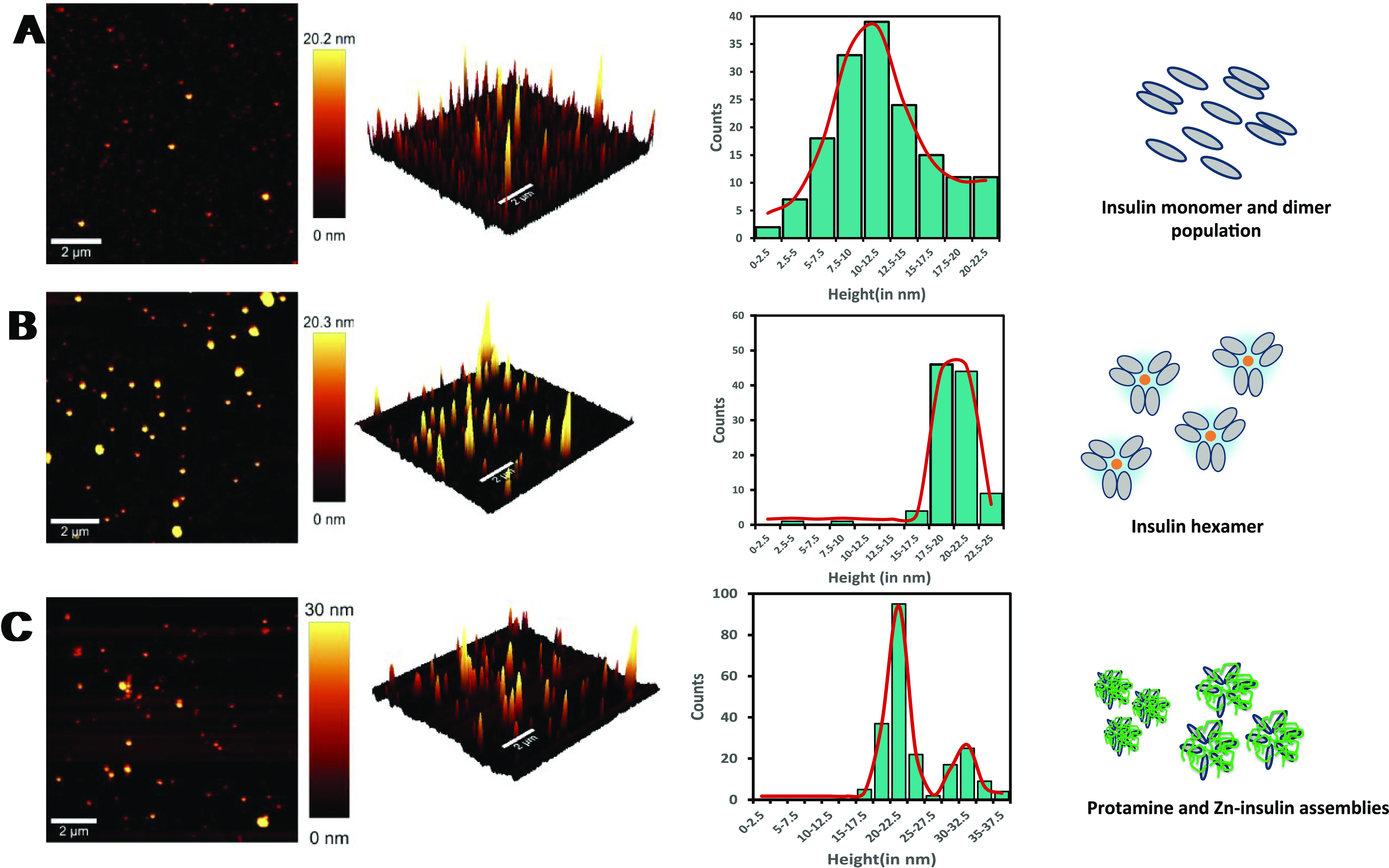
((A–C) From top to bottom) Topography of (A) insulin, (B) Zn–insulin, and (C) protamine–Zn–insulin assembly, followed by (in the respective row) 3D image of the topography, single-molecule statistical analysis with histogram distributions of cross-sectional height of a respective species. In the last column, it is shown how molecules get arranged at different stages in a pictorial form. Insulin units are represented in gray color (monomer, dimer, or hexamer) and protamine in green.
Probing Insulin–Protamine Interaction Using Molecular Docking Study
In the absence of high-resolution structural data, there is a lack of clarity on the binding interface involved in the protamine–insulin complex.9,23 Therefore, to support our quantitative experimental parameters and to find the most preferred amino acids responsible for the interaction between protamine and insulin, we attempted molecular docking studies. Figure 9A shows the amino acid residues at the monomer–monomer interface in insulin (PDB: 4INS). Figure 9B–D shows the docking results of the insulin–protamine complex, in which chain B of insulin is exclusively involved in protamine binding (Figure 9B). As shown in Figure 9B,C, protamine binding to insulin involved hydrogen bonding, electrostatic, and hydrophobic interactions (Table 3). The relevant details are further elaborated on in the Discussion section. Also, the biological significance of these interactions in the context of the insulin–receptor complex (Figure 9E,F) is discussed.
Figure 9.

(A) Insulin dimer interface showing residues (in pink) of chain B (from each monomer) responsible for monomer–monomer interactions (PDB: 4INS). (B) Docking result of insulin (PDB: 2JV1) with protamine (yellow surface). (C) Demonstration of the interactions at the binding interface of insulin and protamine. (D) Hydrophobic interactions between residues of insulin and protamine. (E) Insulin showing exposed hydrophobic residues (sticks) that are not interacting with protamine (yellow surface) and (F) the complex between insulin and receptor (αCT; 704–719 amino acids), showing the crucial residues of insulin involved in receptor binding; gray-shaded area represents the αCT surface1 (PDB: 4OGA). It can be seen that the hydrophobic residues, which are not involved in the binding in (E) are involved in receptor binding in (F).
Table 3. Docking Results of Insulin Monomer with Protamine, Displaying the Type of Interactions and Bond Distances between the Residues of Insulin and Protamine.
| residues of the insulin monomer | residue of protamine | type of bonding | bond distance |
|---|---|---|---|
| Glu B21 (COO–) | Arg 10 (NH2) | hydrogen bond | 1.92 |
| Glu B13 (COO–) | Arg 30 (NH2) | electrostatic | 4.23 |
| Phe B24 (CO) | Arg 14 (NH2) | hydrogen bond | 2.13 |
| Tyr B26 (OH) | Arg 15 (NH) | hydrogen bond | 2.55 |
| Leu B17 (CO) | Arg 23 (NH2) | hydrogen bond | 2.43 |
| Ser B9 (CO) | Arg 30 (NH2) | hydrogen bond | 2.09 |
| Val B12 (CH3–CH–CH3) | Pro 18 (ring) | hydrophobic | 4.97 |
| Tyr B16 (aromatic ring) | Val 12 (CH3–CH–CH3) | hydrophobic | 5.11 |
| Tyr B26 (aromatic ring) | Arg 14 (CH2–CH2) | hydrophobic | 4.54 |
| Phe B24 (aromatic ring) | Val 12 (CH3–CH–CH3) | hydrophobic | 4.33 |
Discussion
We have performed the first comprehensive biophysical analyses of the interaction between insulin and protamine in the presence and absence of Zn, which are important components of several insulin formulations. In a solution, free insulin exists as a dimer in the pH range of 5–8, whereas in the presence of Zn2+, it forms the hexamer assembly.4,51 In this study, we followed the appropriate experimental conditions to achieve the desired conformation of insulin.9,18,19,51 To analyze interactions under equilibrium conditions, samples containing insulin and protamine were prepared at equimolar ratios (as used in formulation conditions) and allowed to equilibrate after the addition of Zn. Insulin formulation is stabilized with respect to fibrillation by the addition of zinc and phenolic preservatives, which drive the assembly of the R6 hexamer.4 Here, all of the samples were soluble under experimental conditions, as we did not observe any precipitation. Further, insulin fibrillation was ruled out based on the ThT study.
Based on the results obtained from several complementary techniques, we found that Zn–insulin undergoes conformational alteration in the presence of protamine. Zn–insulin is mainly a storage form, which is required to undergo dissociation into active monomers during its endogenous delivery.5,52 The first question is: does protamine have any role in protecting insulin and facilitating its active state when necessary? In ITC, protamine binds to Zn–insulin with decent binding affinity. Here, the N value suggested multiple insulin molecules binding to protamine. Previously, protamine was shown to have a tendency to condense/aggregate DNA by sequestering it in the form of ordered aggregates.24,53 ITC suggesting the possibility of multiple insulin molecules binding to the protamine chain is consistent with this picture of how such condensation of insulin can occur in the presence of protamine. We assume that this is an ordered condensation and not aggregation or precipitation, as the data (AFM and ThT fluorescence) clearly do not support the latter. Further, in the case of aggregation, very large heat changes are generally expected in ITC,54 which we did not observe here. Here, protamine can be seen as a long polymer of closely spaced binding sites for insulin. In addition to multiple insulin molecules binding to a single protamine chain, multiple protamine chains can be cross-linked via the secondary sites on the insulin, generating a web of protamine chains binding insulin (Figure 8C). Since monomer insulin is prone to undergo fibrillation, here, the combination of protamine–insulin assembly may provide complementary structural adaptations to avoid the formation of such a toxic protein state as fibrillation. This is how protamine would shield insulin in the formulations before it can be released into the blood in an active form.
The second question is: what kind of interactions protamine may form with Zn–insulin? It is well established that Zn2+ coordination brings three insulin dimers together to form a hexamer. To dissociate the hexamer into monomers, Zn coordination needs to be destabilized. As we have seen, protamine and Zn2+ do not interact with each other, possibly due to their similar cationic nature. Therefore, we believe that the ability of protamine to recognize and disturb the dimer interface in insulin can lead to the weakening of Zn coordination leading to hexamer destabilization.
The presence of several basic Arg residues in protamine, which have the potential to promote multiple interactions with insulin,55 can play a crucial role in the binding mechanism. Arginine has aqueous pKa (∼13.8), indicating a strong tendency to remain charged at a physiological pH. Further, the guanidinium group on Arg has a unique ability to form multiple hydrogen bonds (H-bond), driving hydrophilic interactions propelled by favorable negative enthalpy (Table 2). A positive ΔCp value (+880 cal mol–1) (Figure 5) in the case of Zn–insulin and protamine binding also supports the role of hydrophilic interactions.56 Moreover, the positive TΔSconf indicates that the resultant complex is conformationally flexible, a very crucial property, which can help in the successful release of the final insulin into the blood. Previously, a crystallography study57 has shown that free Arg residues can bind specifically to insulin through Glu B13, but the interaction is not strong enough to destabilize insulin hexamer. On the contrary, here, as shown by our results (Figure 2D–G), protamine not only binds strongly to insulin but also has the ability to induce conformational changes in insulin. Here, the presence of multiple Arg residues along the polypeptide chain of protamine acts as key binding spots in successful contacts with insulin.
To confirm interactions at the molecular level between insulin and protamine, we have performed docking studies of protamine with biologically relevant monomer insulin. These results (Figure 9B,C) helped to dissect residue-level details as to how protamine might be interacting with insulin. As shown in Figure 9C, TyrB26 of insulin is directly involved in H-bonding with the side chain of Arg15 as well as hydrophobic interactions with the Arg14 of protamine. Also, PheB24 with Arg14 and GluB13 with Arg30 are involved through H-bond interactions (Table 3). This may directly hamper the H-bonding interaction between PheB24–TyrB26 of one monomer and TyrB26–PheB24 of the second monomer, which is responsible for the stability of insulin dimer.58 The fluorescence quenching observed in Figure 7 during protamine binding may also have its origin in the polar interaction between TyrB26 of insulin and Arg15 of protamine. Considering the vital role of TyrB26 in the self-assembly of insulin, its direct interaction with protamine should contribute to the dissociation of insulin assembly.59 The binding of protamine to Zn–insulin has resulted in the conformation of insulin with its hydrophobic residues exposed to the solvent (Figure 8E). As per the reports, hydrophobic amino acids (GlyA1, IleA2, ValA3, TyrA19, GlyB8, LeuB11 GlyB23, and PheB25) on the surface of insulin are highly essential for its interaction with insulin receptor (IR).1,17
Finally, what is the biological significance of these interactions? We know that Zn ions have a tendency to drive the insulin to an associative state, whereas protamine is shown to indulge in dissociation. Therefore, due to the opposing effects of Zn and protamine, insulin may adopt a conformation, which when delivered into blood undergoes controlled dissociation (into biologically active monomers).60,61 This may explain the long-lasting effect of insulin–protamine formulation in controlling blood glucose levels. Thus, the role of protamine seems to impair insulin’s self-association ability and thermodynamic stability while at the same time promoting flexible conformation with better receptor binding and therapeutic capability. Such an inverse correlation between stability and activity has been suggested in the modified analogues of insulin.62 Such co-optimization highlights the multidimensional role of protamine in insulin formulation. We will include a wide range of insulin analogues with different stabilities in future studies to test this hypothesis.
Acknowledgments
This research work was supported by grants from DST-SERB (ECR/2016/000942), IRHPA-SERB (IPA/2020/000007), and DST-DPRP (VI-D&P/569/2016-1/TDT), DST-Purse, and UPE II-JNU. The authors also acknowledge DST FIST funding for ITC facility. N.T. acknowledges research grant from the Council of Scientific and Industrial Research, Government of India. The authors also thank AIRF, JNU, for the instrument facility.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04419.
Insulin characterization using Native PAGE. Lane 1: ultra-low-molecular-weight marker, Lane 2: insulin, Lane 3: Zn–insulin (Figure S1); ITC thermogram showing control and raw titration of protamine into insulin at different salt concentrations, and at different temperatures (Figure S2); ITC thermogram showing control and raw titration of protamine into Zn–insulin at different salt concentrations, and at different temperatures (Figure S3); ΔH vs −TΔS plot of protamine titration into insulin and insulin hexamer (Figure S4); ITC thermogram showing titration of Zn2+ into insulin (Figure S5); UV absorbance spectra of insulin–protamine and Zn–Insulin–protamine at different temperatures from 25 to 80 °C (Figure S6); salt-based fluorescence titration of increasing concentration of protamine into Zn–insulin (Figure S7); fluorescence spectra for insulin, Zn–insulin, and Zn–insulin–protamine and free protamine (Figure S8); ThT fluorescence of ThT–Zn–insulin–protamine (Figure S9); ITC thermogram showing the titration of protamine into Zn experiment was performed in 10 mM PBS buffer at pH 8.0. Protamine was 300 μM and Zn was 20 μM in concentration (Figure S10); absorbance spectra of free protamine (Figure S11); parameters obtained from the modified Stern–Volmer plot (Table S1) (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Menting J. G.; Whittaker J.; Margetts M. B.; et al. How insulin engages its primary binding site on the insulin receptor. Nature 2013, 493, 241–245. 10.1038/nature11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen M. C.; Shulman G. I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter M. C.; Wilcox D. E. Thermodynamics of formation of the insulin hexamer: metal-stabilized proton-coupled assembly of quaternary structure. Biochemistry 2014, 53, 1296–1301. 10.1021/bi4016567. [DOI] [PubMed] [Google Scholar]
- Dunn M. F. Zinc-ligand interactions modulate assembly and stability of the insulin hexamer -- a review. BioMetals 2005, 18, 295–303. 10.1007/s10534-005-3685-y. [DOI] [PubMed] [Google Scholar]
- Brange J.; Owens D. R.; Kang S.; Vølund A. Monomeric insulins and their experimental and clinical implications. Diabetes Care 1990, 13, 923–954. 10.2337/diacare.13.9.923. [DOI] [PubMed] [Google Scholar]
- Archontogeorgis K.; Papanas N.; Nena E.; et al. Insulin Sensitivity and Insulin Resistance in Non-Diabetic Middle-Aged Patients with Obstructive Sleep Apnoea Syndrome. Open Cardiovasc. Med. J. 2017, 11, 159–168. 10.2174/1874192401711010159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi G. P.; Png C. Y.; Wilcox D. E. Thermodynamic contributions to the stability of the insulin hexamer. Biochemistry 2014, 53, 3576–3584. 10.1021/bi401678n. [DOI] [PubMed] [Google Scholar]
- Zaykov A. N.; Mayer J. P.; DiMarchi R. D. Pursuit of a perfect insulin. Nat. Rev. Drug Discovery 2016, 15, 425–439. 10.1038/nrd.2015.36. [DOI] [PubMed] [Google Scholar]; From NLM.
- Norrman M.; Hubálek F.; Schluckebier G. Structural characterization of insulin NPH formulations. Eur. J. Pharm. Sci. 2007, 30, 414–423. 10.1016/j.ejps.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Krayenbuhl C.; Rosenberg T. Crystalline protamine insulin. Rep. Steno. Mem. Hosp. Nord. Insulinlab. 1946, 1, 60–73. [Google Scholar]
- Smith G. D.; Swenson D. C.; Dodson E. J.; Dodson G. G.; Reynolds C. D. Structural stability in the 4-zinc human insulin hexamer. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 7093–7097. 10.1073/pnas.81.22.7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklepari M.; Rodger A.; Reason A.; Jamshidi S.; Prokes I.; Blindauer C. A. Biophysical characterization of a protein for structure comparison: methods for identifying insulin structural changes. Anal. Methods 2016, 8, 7460–7471. 10.1039/c6ay01573e. [DOI] [Google Scholar]
- Huus K.; Havelund S.; Olsen H. B.; van de Weert M.; Frokjaer S. Thermal dissociation and unfolding of insulin. Biochemistry 2005, 44, 11171–11177. 10.1021/bi0507940. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Mondal S.; Deshmukh A. A.; Gopal B.; Bagchi B. What Gives an Insulin Hexamer Its Unique Shape and Stability? Role of Ten Confined Water Molecules. J. Phys. Chem. B 2018, 122, 1631–1637. 10.1021/acs.jpcb.8b00453. [DOI] [PubMed] [Google Scholar]
- Brems D. N.; Alter L. A.; Beckage M. J.; et al. Altering the association properties of insulin by amino acid replacement. Protein Eng., Des. Sel. 1992, 5, 527–533. 10.1093/protein/5.6.527. [DOI] [PubMed] [Google Scholar]
- Ahmad A.; Millett I. S.; Doniach S.; Uversky V. N.; Fink A. L. Stimulation of insulin fibrillation by urea-induced intermediates. J. Biol. Chem. 2004, 279, 14999–15013. 10.1074/jbc.M313134200. [DOI] [PubMed] [Google Scholar]
- Mayer J. P.; Zhang F.; DiMarchi R. D. Insulin structure and function. Biopolymers 2007, 88, 687–713. 10.1002/bip.20734. [DOI] [PubMed] [Google Scholar]
- Attri A. K.; Fernández C.; Minton A. P. pH-dependent self-association of zinc-free insulin characterized by concentration-gradient static light scattering. Biophys. Chem. 2010, 148, 28–33. 10.1016/j.bpc.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia M.; Neves-Petersen M. T.; Jeppesen P. B.; Gregersen S.; Petersen S. B. UV-light exposure of insulin: pharmaceutical implications upon covalent insulin dityrosine dimerization and disulphide bond photolysis. PLoS One 2012, 7, e50733 10.1371/journal.pone.0050733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold H.; Hilgenfeld R. Binding of phenol to R6 insulin hexamers. Pept. Sci. 1999, 51, 165–172. 10.1002/(SICI)1097-0282(1999)51:2%3C165::AID-BIP6%3E3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Smith G. D.; Ciszak E.; Magrum L. A.; Pangborn W. A.; Blessing R. H. R6 hexameric insulin complexed with m-cresol or resorcinol. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2000, 56, 1541–1548. 10.1107/s0907444900012749. [DOI] [PubMed] [Google Scholar]
- Mohanty J.; Shinde M. N.; Barooah N.; Bhasikuttan A. C. Reversible Insulin Hexamer Assembly Promoted by Ethyl Violet: pH-Controlled Uptake and Release. J. Phys. Chem. Lett. 2016, 7, 3978–3983. 10.1021/acs.jpclett.6b01745. [DOI] [PubMed] [Google Scholar]
- Yip C. M.; Brader M. L.; Frank B. H.; DeFelippis M. R.; Ward M. D. Structural studies of a crystalline insulin analog complex with protamine by atomic force microscopy. Biophys. J. 2000, 78, 466–473. 10.1016/S0006-3495(00)76609-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Tiwari N.; Munde M. A Comprehensive Biophysical Analysis of the Effect of DNA Binding Drugs on Protamine-induced DNA Condensation. Sci. Rep. 2019, 9, 5891 10.1038/s41598-019-41975-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin R. L. Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 8069–8072. 10.1073/pnas.83.21.8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker B. M.; Murphy K. P. Prediction of binding energetics from structure using empirical parameterization. Methods Enzymol. 1998, 295, 294–315. 10.1016/s0076-6879(98)95045-5. [DOI] [PubMed] [Google Scholar]
- Lehrer S. Solute perturbation of protein fluorescence. Quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 1971, 10, 3254–3263. 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- Tanwar N.; Munde M. Thermodynamic and conformational analysis of the interaction between antibody binding proteins and IgG. Int. J. Biol. Macromol. 2018, 112, 1084–1092. 10.1016/j.ijbiomac.2018.01.208. [DOI] [PubMed] [Google Scholar]
- Asthana S.; Bhutia S. K.; Sahoo H.; Jha S. Chaotropes trigger conformational rearrangements differently in Concanavalin A. J. Chem. Sci. 2017, 129, 1267–1276. 10.1007/s12039-017-1333-z. [DOI] [Google Scholar]
- van Zundert G. C. P.; Rodrigues J. P. G. L. M.; Trellet M.; et al. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Roy A.; Kucukural A.; Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield N. J.; Fasman G. D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969, 8, 4108–4116. 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
- Bottorf L.; Rafferty S.; Sahu I. D.; McCarrick R. M.; Lorigan G. A. Utilizing Electron Spin Echo Envelope Modulation To Distinguish between the Local Secondary Structures of an α-Helix and an Amphipathic 3(10)-Helical Peptide. J. Phys. Chem. B 2017, 121, 2961–2967. 10.1021/acs.jpcb.7b00626. [DOI] [PubMed] [Google Scholar]
- Toniolo C.; Polese A.; Formaggio F.; Crisma M.; Kamphuis J. Circular Dichroism Spectrum of a Peptide 310-Helix. J. Am. Chem. Soc. 1996, 118, 2744–2745. 10.1021/ja9537383. [DOI] [Google Scholar]
- Manoharan C.; Singh J. Addition of Zinc Improves the Physical Stability of Insulin in the Primary Emulsification Step of the Poly(lactide-co-glycolide) Microsphere Preparation Process. Polymers 2015, 7, 836–850. 10.3390/polym7050836. [DOI] [Google Scholar]
- Cimmperman P.; Baranauskiene L.; Jachimoviciu̅te S.; et al. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J. 2008, 95, 3222–3231. 10.1529/biophysj.108.134973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S. P.; Blundell T. L.; Wollmer A.; Lazarus N. R.; Neville R. W. The relation of conformation and association of insulin to receptor binding; x-ray and circular-dichroism studies on bovine and hystricomorph insulins. Eur. J. Biochem. 1975, 55, 531–542. 10.1111/j.1432-1033.1975.tb02190.x. [DOI] [PubMed] [Google Scholar]
- Singh R. K.; Suzuki T.; Mandal T.; et al. Thermodynamics of Binding of Structurally Similar Ligands to Histone Deacetylase 8 Sheds Light on Challenges in the Rational Design of Potent and Isozyme-Selective Inhibitors of the Enzyme. Biochemistry 2014, 53, 7445–7458. 10.1021/bi500711x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.-Y.; Huang H.-M.; Lin C.-C.; Lin F.-Y.; Chan Y.-C. Effect of Temperature on Hydrophobic Interaction between Proteins and Hydrophobic Adsorbents: Studies by Isothermal Titration Calorimetry and the van’t Hoff Equation. Langmuir 2003, 19, 9395–9403. 10.1021/la034783o. [DOI] [Google Scholar]
- Hariharan P.; Balasubramaniam D.; Peterkofsky A.; Kaback H. R.; Guan L. Thermodynamic mechanism for inhibition of lactose permease by the phosphotransferase protein IIAGlc. Proc. Natl. Acad. Sci. 2015, 112, 2407–2412. 10.1073/pnas.1500891112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H.; Wu Y.; Brew K. Thermodynamic Basis of Selectivity in the Interactions of Tissue Inhibitors of Metalloproteinases N-domains with Matrix Metalloproteinases-1, -3, and -14. J. Biol. Chem. 2016, 291, 11348–11358. 10.1074/jbc.M116.720250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbazhagan V.; Sankhala R. S.; Singh B. P.; Swamy M. J. Isothermal titration calorimetric studies on the interaction of the major bovine seminal plasma protein, PDC-109 with phospholipid membranes. PLoS One 2011, 6, e25993 10.1371/journal.pone.0025993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A.; Uversky V. N.; Hong D.; Fink A. L. Early events in the fibrillation of monomeric insulin. J. Biol. Chem. 2005, 280, 42669–42675. 10.1074/jbc.M504298200. [DOI] [PubMed] [Google Scholar]
- Schönbrunn E.; Eschenburg S.; Luger K.; Kabsch W.; Amrhein N. Structural basis for the interaction of the fluorescence probe 8-anilino-1-naphthalene sulfonate (ANS) with the antibiotic target MurA. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 6345–6349. 10.1073/pnas.120120397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaLuz P. J.; Ackley B. L. Fluorescence investigation of hydrophobic pocket exposure in protein with 1-anilinonaphthalene-8-sulfonic acid. FASEB J. 2009, 23, 538.6 10.1096/fasebj.23.1_supplement.538.6. [DOI] [Google Scholar]
- Qadeer A.; Rabbani G.; Zaidi N.; Ahmad E.; Khan J. M.; Khan R. H. 1-Anilino-8-naphthalene sulfonate (ANS) is not a desirable probe for determining the molten globule state of chymopapain. PLoS One 2012, 7, e50633 10.1371/journal.pone.0050633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain F.; Birch D. J.; Pickup J. C. Glucose sensing based on the intrinsic fluorescence of sol-gel immobilized yeast hexokinase. Anal. Biochem. 2005, 339, 137–143. 10.1016/j.ab.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Antolíková E.; Žáková L.; Turkenburg J. P.; et al. Non-equivalent role of inter- and intramolecular hydrogen bonds in the insulin dimer interface. J. Biol. Chem. 2011, 286, 36968–36977. 10.1074/jbc.M111.265249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faisal Z.; Vörös V.; Fliszár-Nyúl E.; et al. Probing the Interactions of Ochratoxin B, Ochratoxin C, Patulin, Deoxynivalenol, and T-2 Toxin with Human Serum Albumin. Toxins 2020, 12, 392 10.3390/toxins12060392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins K. J.; Liu G.; Selmani V.; Lazo N. D. Conformational analysis of thioflavin T bound to the surface of amyloid fibrils. Langmuir 2012, 28, 16490–16495. 10.1021/la303677t. [DOI] [PubMed] [Google Scholar]
- Blundell T.; Dodson G.; Hodgkin D.; Mercola D.. Insulin: The Structure in the Crystal and its Reflection in Chemistry and Biology by. In Advances in Protein Chemistry; Anfinsen C. B.; Edsall J. T.; Richards F. M., Eds.; Academic Press, 1972; Vol. 26, pp 279–402. [Google Scholar]
- DeFelippis M. R.; Chance R. E.; Frank B. H. Insulin self-association and the relationship to pharmacokinetics and pharmacodynamics. Crit. Rev. Ther. Drug Carrier Syst. 2001, 18, 201–264. 10.1615/CritRevTherDrugCarrierSyst.v18.i2.30. [DOI] [PubMed] [Google Scholar]
- Mukherjee A.; de Izarra A.; Degrouard J.; et al. Protamine-Controlled Reversible DNA Packaging: A Molecular Glue. ACS Nano 2021, 15, 13094–13104. 10.1021/acsnano.1c02337. [DOI] [PubMed] [Google Scholar]
- Kandari D.; Joshi H.; Tanwar N.; Munde M.; Bhatnagar R. Delineation of the Residues of Bacillus anthracis Zinc Uptake Regulator Protein Directly Involved in Its Interaction with Cognate DNA. Biol. Trace Elem. Res. 2021, 199, 3147–3158. 10.1007/s12011-020-02427-x. [DOI] [PubMed] [Google Scholar]
- Harms M. J.; Schlessman J. L.; Sue G. R.; García-Moreno E. B. Arginine residues at internal positions in a protein are always charged. Proc. Natl. Acad. Sci. 2011, 108, 18954–18959. 10.1073/pnas.1104808108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. Heat capacity of hydrogen-bonded networks: an alternative view of protein folding thermodynamics. Biophys. Chem. 2000, 85, 25–39. 10.1016/s0301-4622(00)00136-8. [DOI] [PubMed] [Google Scholar]
- Březina K.; Duboué-Dijon E.; Palivec V.; et al. Can Arginine Inhibit Insulin Aggregation? A Combined Protein Crystallography, Capillary Electrophoresis, and Molecular Simulation Study. J. Phys. Chem. B 2018, 122, 10069–10076. 10.1021/acs.jpcb.8b06557. [DOI] [PubMed] [Google Scholar]
- Jørgensen A. M. M.; Olsen H. B.; Balschmidt P.; Led J. J. Solution structure of the superactive monomeric des-[Phe(B25)] human insulin mutant: elucidation of the structural basis for the monomerization of des-[Phe(B25)] insulin and the dimerization of native insulin. J. Mol. Biol. 1996, 257, 684–699. 10.1006/jmbi.1996.0194. [DOI] [PubMed] [Google Scholar]
- Pandyarajan V.; Phillips N. B.; Rege N.; Lawrence M. C.; Whittaker J.; Weiss M. A. Contribution of TyrB26 to the Function and Stability of Insulin: Structure-activity relationships at a conserved hormone-receptor interface. J. Biol. Chem. 2016, 291, 12978–12990. 10.1074/jbc.M115.708347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm L. H.; Ibsen H.; Dahlöf B.; et al. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002, 359, 1004–1010. 10.1016/S0140-6736(02)08090-X. [DOI] [PubMed] [Google Scholar]
- Heller S.; Kozlovski P.; Kurtzhals P. Insulin’s 85th anniversary--An enduring medical miracle. Diabetes Res. Clin. Pract. 2007, 78, 149–158. 10.1016/j.diabres.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Hua Q. X.; Nakagawa S.; Hu S. Q.; Jia W.; Wang S.; Weiss M. A. Toward the active conformation of insulin: stereospecific modulation of a structural switch in the B chain. J. Biol. Chem. 2006, 281, 24900–24909. 10.1074/jbc.M602691200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.