

Abstract

Abundant substituted catechols are emitted to, and created in, the atmosphere during wildfires and anthropogenic combustion and agro-industrial processes. While ozone (O3) and hydroxyl radicals (HO•) efficiently react in a 1 μs contact time with catechols at the air–water interface, the nighttime reactivity dominated by nitrate radicals (NO3) remains unexplored. Herein, online electrospray ionization mass spectrometry (OESI-MS) is used to explore the reaction of NO3(g) with a series of representative catechols (catechol, pyrogallol, 3-methylcatechol, 4-methylcatechol, and 3-methoxycatechol) on the surface of aqueous microdroplets. The work detects the ultrafast generation of nitrocatechol (aromatic) compounds, which are major constituents of atmospheric brown carbon. Two mechanisms are proposed to produce nitrocatechols, one (equivalent to H atom abstraction) following fast electron transfer from the catechols (QH2) to NO3, forming NO3– and QH2•+ that quickly deprotonates into a semiquinone radical (QH•). The second mechanism proceeds via cyclohexadienyl radical intermediates from NO3 attack to the ring. Experiments in the pH range from 4 to 8 showed that the production of nitrocatechols was favored under the most acidic conditions. Mechanistically, the results explain the interfacial production of chromophoric nitrocatechols that modify the absorption properties of tropospheric particles, making them more susceptible to photooxidation, and alter the Earth’s radiative forcing.
Keywords: phenols, dihydroxybenzene, nitrogen dioxide, ozone, nitrate radical, hydroxyl radical, secondary organic aerosol
Short abstract
Rapid oxidative browning of combustion and biomass burning substituted catechol pollutants at the air−water interface enabled by nitrate radicals.
1. Introduction
Industrial, biomass burning, and combustion processes release aromatic volatile organic compounds (VOCs), such as benzene, toluene, and anisole, to the atmosphere.1,2 We have previously explained that the gas-phase sequential oxidation of these VOCs by hydroxyl radicals (HO•) generates phenol, (ortho-, para- , and meta-) cresol and guaiacol first and water-soluble catechols (e.g., catechol, pyrogallol, 3- and 4-methylcatechol, and 3- and 4-methoxycatechol) second.3−5 The characterization of gas- and particle-phase samples collected over dissimilar locations during summer and winter indicated that catechols are precursors in the production of widespread nitroaromatic compounds6−11 found in atmospheric brown carbon.12−14 Brown carbon absorbing radiation efficiently in the near-UV (300–400 nm) and visible (400–700 nm) regions12 has attracted considerable attention as it contributes up to 48% to the overall radiative forcing from light-absorbing aerosols.15
Catechol oxidation by NO3 follows a similar order of reactivity in the gas and aqueous phases16−18 as that observed for their reactions with HO• at the air–water interface.3,4,19 In this context, polar and surface active catechols are considered to favorably partition onto the particle phase of aerosols, accommodating on the interface for heterogenous chemistry to proceed. Gas phase reaction rates between nitrate radical (NO3) and methoxyphenols or cresol are 106–107 higher than for O3.20,21 The surface reactions of catechols with HO• and O3 proceed with the participation of semiquinone and cyclohexadienyl radicals.3−5,19 In the case of exposure to gaseous NO3 at nighttime, the production of nitrocatechols at the air–water interface of particles can provide an efficient formation pathway of nitrocatechols and an unaccounted source of atmospheric nitroaromatic compounds.
Herein, the oxidation of catechols is studied at the air–water interface using surface-sensitive online electrospray ionization (OESI) mass spectrometry (MS) with ultra-fast contact (tc = 1 μs) and detection times (td = 1 ms).3−5,22−24 Studying these oxidations at the air–water interface is important to understand what occurs in atmospheric waters, where this unique interface may accelerate reactions and trigger unique chemistry compared to bulk water.25,26 The OESI-MS experiments below supported by cyclic voltammetry measurements of redox potentials reveal the propensity of catechols’ interfacial oxidation by NO3 to generate nitroaromatic compounds through reactions that should be operative in areas affected by pollution from biomass burning and combustion emissions.
2. Experimental Section
2.1. Sample Preparation
Working 100 μM solutions of known pH were made by dilution of stock solutions with 1.00 mM of catechol (Sigma-Aldrich, 99.9%), pyrogallol (Acros Organics, 99.7%), 3-methylcatechol (TCI America, 99.8%), 4-methylcatechol (Acros Organics, 97.6%), or 3-methoxycatechol (Alfa Aesar, 99.4%) prepared daily in degassed ultrapure water (ELGA PURELAB Flex, 18.2 MΩ cm–1). A dropwise addition of 1.00 or 10.0 mM HCl (EMD Millipore, 37.67%) and 1.00 or 10.0 mM NaOH (AMRESCO, ≥97%) was used for pH adjustment, which was registered with a calibrated pH meter (Thermo Scientific). The partitioning of 5–50 ppbv catechol detected over biomass burning plumes27 onto aqueous particles predicts that 22–223 μM solutions are formed based on Henry’s law constant Hocatechol = 4600 M atm–1.28 Thus, the predicted concentration range covers the atmospheric relevant concentration studied of 100 μM. Furthermore, the pH range from 4 to 8 studied here directly applies to large intervals of those existing for cloud water (pH 3–8)29−32 and aerosol particles (pH 1.5 to 7.7).33−37
2.2. OESI-MS Experiments
A 100 μM solution was pneumatically aerosolized into a calibrated OESI-MS at a flow rate of 50 μL/min (Scheme S1, Supporting Information).23,38 This instrument was customized from an MSQ Plus (Thermo Scientific) system to provide an ambient pressure flow-through reactor that enables the oxidation study at the air–water interface of a mist of generated microdroplets in tc = 1 μs and a td = 1 ms detection time, as explained previously.3−5,23,24,38
Ozone was generated using a spark discharge ozone generator (Ozone Solutions) fed with 500 mL min–1 O2 (Scott-Gross, UHP) and diluted using 100 to 2000 mL min–1 N2 (Scott-Gross, UHP). The concentration of O3 was monitored in the UV spectrum using a photodiode array (PDA) detector (Thermo Scientific), with a 10 cm-pathlength cuvette made of quartz, using a deuterium lamp. Nitrogen dioxide (NO2) flows of 60 to 1000 L min–1 were provided from gas cylinders of 100, 200, or 214 ppmv (Scott-Gross, UHP, nitrogen balance) to prepare mixtures with O3(g) in a 4 L amber flask with the reactive species of interest by reactions R1 and R2:
| R1 |
| R2 |
The input molar ratios of O3 and NO2 mixed in the 4 L amber flask are provided in Table S1 (Supportive Information), where the excess NO2 was aimed to decrease direct ozonolysis side reactions. The output O3 molar ratio after mixing (Table S1, Supportive Information) was determined down the gas transmission line spectrophotometrically and used to calculate the production rate of NO3 (PNO3):
| 1 |
with rate constant kNO3 = 3.2 × 10–17 cm3 molecule–1 s–1 (≡ 7.89 × 10–7 ppbv–1 s–1).39−41 The mixture was allowed to equilibrate for ≥30 min before directing a flow of 200 mL min–1 through a short tube to the OESI-MS reactor, where it was finally diluted 61 times by the N2(g) nebulizing gas (12.0 L min–1).
The effective concentration of NO3 to which the surface of microdroplets was exposed is obtained by integrating PNO3 over the residence time tr = 8.77 s; the mixture of O3(g) and NO2(g) is in the quartz cuvette before its transfer to the final PTFE line directing the flow to impinge the microdroplets. In addition, the wall loss of NO3,
| R3 |
taking place inside the quartz (kR3 ≃ 0.11 s–1)42,43 cuvette and the PTFE (kR3 = 7.1 × 10–3 s–1)44 transfer line,42,45 indicates that a cumulative transmission efficiency of 3.0% of the produced NO3 reaches the microdroplets’ surface. Only dry gases are used in the generation and dilution of oxidizer mixtures, which impinge the microdroplet generated at a fixed pH. The environmental constraints of the experiments under low molar ratios of NO3 are provided in the Supporting Information followed by the background information of how the effective NO3 concentration was estimated from PNO3 in eq 1.
The reactions of NO3 with the catechols on the surface of the microdroplets are monitored by the generated anions at reported mass-to-charge ratios (m/z–), without wall losses due to the flow-through nature of this surface-sensitive OESI-MS reactor.3,4,22−24 This setup was demonstrated to be surface sensitive to monitor reactions in real time by MS.3−5,22−24,38 OESI-MS only detects products from interfacial reactions of the selected molecules (catechols in this case) occurring at the most external layers of the aqueous microdroplets. The uptake of oxidizer molecules onto the liquid interface must take place for the catechols to react and later detect the ions of products.3−5,23 No ions for assigned products are detected in the absence of either water, oxidizer mix, or the catechols. Thus, any exclusive gas-phase reaction between NO3 and catechol would remain undetected.
The same experimental conditions from previous studies with catechols were used:3,4 nebulizer pressure, 70 psi; nebulizer voltage, −1.9 kV; cone voltage, −50 V. Reported normalized ion count values (Im/z), unless indicated otherwise, correspond to solvent background subtracted raw data acquired at fixed time intervals (e.g., time ≥30 s). All experiments are performed in duplicate. The large aqueous microdroplets produced in this setup and the ultrafast contact time (tc = 1 μs) discard any possible contribution from solvent evaporation in the surface reactions studied.3−5,22,23 Diffusion limitations have been discarded in this surface-sensitive setup for catechol reactions.3,4 The conditions selected not only minimize any cluster formation but also are extremely soft to prevent any artifacts that could conduct to ionize molecules in the gas phase.3−5,23 Furthermore, the controls performed confirm that the products are not the result from redox chemistry in the probe but from the interfacial reactions examined.3−5,23,24,38 In more detail, spontaneous reactions observed in other ESI probes with a fused silica capillary (e.g., to form HO•,46 H2O2,47 or facilitated electron transfer48 in water) are discarded by the controls performed and by previous studies with this OESI MS setup and complementary techniques.3−5,19,23,24,38,49 For the quantification of nitroaromatic products, the method of standard addition was used as detailed in the Supporting Information with standards and pseudo-standards for commercially unavailable species.
2.3. Redox Potential Measurement
The redox potentials were determined at pH 4, 6, 8, and 10 by cyclic voltammetry with an electrochemical workstation (CHI-660A, CH Instruments, Austin, Texas) in a three-electrode configuration cell. More details of redox potential measurements are provided in the Supporting Information.
3. Results and Discussions
3.1. OESI-MS Analysis of Oxidized Catechols
The top panel of Figure 1 shows the OESI-MS spectra of 100 μM catechol at pH 8.04 exposed to 200 mL min–1 of 1 atm N2(g), 7.6 pptv NO3, and 1.62 ppbv NO3. Catechol is detected as a monoanion (C6H5O2–) at m/z– 109 after loss of a proton, while co-formed nitrate ion, NO3–, at m/z– 62 is generated either from reaction of NO3 with water or by hydrolysis of N2O5.50−53 The peak at m/z– 163 present in the control and experiments should correspond to a cluster of catechol with a magic number of three molecules of water. When the aerosolized solution of catechol is exposed to 7.6 pptv NO3, new peaks at m/z– 123, m/z– 125, and m/z– 154 are observed. The peak at m/z– 154 indicates the reaction of catechol with NO3 forms 4-nitrocatechol, or less likely, due to steric hindrance, the isomer 3-nitrocatechol. The observed peak at m/z– 124 is experimentally confirmed with the use of a standard under variable cone voltages to be a fragment from collisional-induced dissociation (CID) of the parent 4-nitrocatechol anion at m/z– 154, in agreement with previous work.54 The species at m/z– 125 and 123 separated by 2 amu were previously described as 1,2,3- or 1,2,4-trihydroxybenzene and 3-hydroxy-o-quinone or 4-hydroxy-o-quinone, respectively, from the attack of in situ generated HO• to catechol.3
Figure 1.

OESI-MS spectra of 100 μM (top) catechol (m/z– 109) at pH 8.04 and (bottom) 3-methoxycatechol (m/z– 139) at pH 8.03 exposed to 200 mL min–1 of (A) 1 atm N2(g), (B) 7.6 pptv NO3, and (C) 1.62 ppbv NO3. Ion count values (Im/z) are normalized percentages relative to the parent ions I109 and I139 representing 100%. Molar ratios of O3(g) and NO2(g) are provided in Table S1.
In addition, the pair of peaks at m/z– 139 and 141 observed for a high NO3 molar ratio (top panel in Figure 1) indicate that a dihydroxy-o-quinone and a tetrahydroxybenzene, respectively, can be produced from the hydroxylation of the hydroxy-o-quinone (m/z– 123) and trihydroxybenzene (m/z– 125).3 The conversion of terephthalic acid to 2-hydroxyterephthalic acid during the oxidation of catechol was used previously to confirm the in situ production of HO• at the air–water interface in this setup, which conducted to the formation of the products at m/z– 123 and 125.4 However, the cis,cis-muconic acid formed during the ring cleavage of catechol is expected to dominate the signal at m/z– 141, as shown in Figure 1.3 The process in the presence of such an oxidizer mixture with NO3 could still yield similar products to those reported from heterogenous ozonolysis previously.3,4
Importantly, the second largest peak in the spectrum (Figure 1) at a high NO3 molar ratio occurs at m/z– 147, with a normalized ion count of 20% as shown in the top panel of Figure 1, which is assigned to dihydroxymaleic acid. Moreover, during the oxidation of cis,cis-muconic acid (m/z– 141), a nitro product (e.g., 2-nitromuconic acid) is registered at m/z– 186 (Figure S1, Supporting Information), and the formation of the species at m/z– 147 is confirmed as the dominant product. Therefore, the cyclohexadienyl (or allyl) radicals generated by NO3 addition to the catechols’ ring (or to the conjugated double bond of cis,cis-muconic acid) participate in the formation of heavy intermediates with covalent bonds at unsaturated positions in the presence of NO3 and O2.11 The nitrocatechols formed can undergo oxidative denitroxidation,55 demethylation,56 and demethoxylation57 by ipso addition of HO•, resulting in new and similar hydroxylated products independently of the starting substituents in the catechols. Next, oxidative reactions yield hydroxylated muconic acids, which may undergo acid-catalyzed hydration (e.g., see Scheme 3 in ref (19), and further fragmentation reactions to form dihydroxymaleic acid or its isomer, dihydroxyfumaric acid.
For the case of aerosolized 100 μM pyrogallol solutions at pH 8.09, the OESI-MS spectra in Figure S2 (Supporting Information) in the absence of an oxidizer shows a peak for its anion at m/z– 125. Upon oxidation of pyrogallol, two small peaks are detected at m/z– 141 for tetrahydroxybenzene and m/z– 170 for 4-nitropyrogallol (or an isomer). Moreover, the cleavage of the aromatic ring11 should result in the characteristic peak of dihydroxymaleic acid at m/z– 147, which becomes the dominant product for high concentrations of the oxidizer.
The bottom panel of Figure 1 displays the OESI-MS spectra of 3-methoxycatechol, whose anion is detected at m/z– 139 with a CID fragment at m/z– 124 from −CH3 loss typical of methoxyphenols, also described during a related analysis.54,58,59 The most characteristic products’ peaks from 3-methoxycatechol exposed to the oxidizer mix with NO3 appear at m/z– 147 again for dihydroxymaleic acid and m/z– 184 for 3-methoxy-5-nitrocatechol (or less likely 3-methoxy-4-nitrocatechol) that also yields a small CID fragment from −CH3 loss at m/z– 169, which is characteristic of methoxyphenols.
Figure 2 shows the OESI-MS spectra of 4-methylcatechol at pH 8.06 and 3-methylcatechol at pH 8.03 exposed to 200 mL min–1 of 1 atm N2(g), 7.6 pptv NO3, and 1.62 ppbv NO3. Both 4- and 3-methylcatechol are detected as anions at m/z– 123. For 4-methylcatechol (top panel in Figure 2), a new peak is observed at m/z– 168 upon exposure to 7.6 pptv NO3, which is assigned to 4-methyl-5-nitrocatechol (or an isomer).10,60−62 The peak at m/z– 138 corresponds to the CID fragment of the parent peak at m/z– 168, as observed with a standard in this work and also reported during chromatography studies.54 Peaks at m/z– 137 and m/z– 139 correspond to the formation of 4-methyl-5-hydroxy-o-quinone and 4-methyl-pyrogallol, or their isomers.4 The bottom panel of Figure 2 shows the OESI-MS spectra of 3-methylcatechol (m/z– 123) for the 1 atm N2(g) control as well as for low and high NO3 molar ratios. Exposure to NO3 generates the product peak for 3-methyl-4-nitrocatechol or 3-methyl-5-nitrocatechol at m/z– 168. The product ratio of the former and latter isomers was 1:10 in the aqueous phase when using nitrous acid (HNO2) for nonradical-driven nitration.60 Similarly, in our radical system, the production of 3-methyl-5-nitrocatechol should be dominant. Other peaks such as m/z– 137 and m/z– 139 correspond to 3-methyl-4-hydroxy-o-quinone and 3-methyl-1,2,4-trihydroxybenzene, respectively.4
Figure 2.

OESI-MS spectra of 100 μM (top) 4-methylcatechol (m/z– 123) at pH 8.06 and (bottom) 3-methylcatechol (m/z– 123) at pH 8.03 exposed and to 200 mL min–1 of (A) 1 atm N2(g), (B) 7.6 pptv NO3, and (C) 1.62 ppbv NO3. Ion count values (Im/z) are normalized percentages relative to the parent ion I123 representing 100%. Molar ratios of O3(g) and NO2(g) are provided in Table S1.
3.2. Effects of Oxidizer Concentration and pH
Figure 3 shows the ion count of the catechols relative to their initial value before addition of an oxidizer, Im/z/Im/z,0, for increasing NO3 levels and at variable pH. Panels A through E display the monotonic decay of Im/z/Im/z,0 for catechol (m/z– 109), 4-methylcatechol (m/z– 123), 3-methylcatechol (m/z– 123), 3-methoxycatechol (m/z– 139), and pyrogallol (m/z– 125) exposed to increasing molar ratios of NO3, 7.6, 235.2, 1621.1, and 1986.3 pptv, during experiments at pH 4.05 (black circle), 5.04 (red square), 6.08 (blue diamond), 7.07 (pink hexagon), and 8.04 (green triangle). The drop of Im/z/Im/z,0 for higher NO3 molar ratios in Figure 3 for all molecules seems roughly the same at pH ≥5.04, but it may be slightly pronounced at pH 4.05. Such a change may arise from the acid–base properties of an intermediate or reactive oxidizer, which is discussed later. It must be noted that control experiments adding only a flow of NO2(g) or O3(g) to impinge the aerosolized solutions of catechols show no contribution to the formation nitroaromatic compounds.
Figure 3.

Ion count of (A) catechol (m/z– 109), (B) 4-methylcatechol (m/z– 123), (C) 3-methylcatechol (m/z– 123), (D) 3-methoxycatechol (m/z– 139), and (E) pyrogallol (m/z– 125) relative to its initial value before NO3 addition, Im/z/Im/z,0, of 7.6, 235.2, 1621.1, and 1986.3 pptv NO3 during experiments at pH 4.05 (black circle), 5.04 (red square), 6.08 (blue diamond), 7.07 (pink hexagon), and 8.04 (green triangle). Molar ratios of O3(g) and NO2(g) are provided in Table S1. Dashed lines are provided as a guide to the eye only.
Figure 4 shows the growing concentration of the produced nitrocatechols with increasing molar ratio of the oxidizer and variable pH from the same experiments in Figure 3. The panels from top to bottom (A through E) in Figure 4 display the production of 4-nitrocatechol (4NC) from catechol, 4-methyl-5-nitrocatechol (4M5NC) from 4-methylcatechol, 3-methyl-5-nitrocatechol (3M5NC) from 3-methylcatechol, 3-methoxy-5-nitrocatechol (3MT5NC) from 3-methoxycatechol, and 4-nitropyrogallol (4NPG) from pyrogallol. The ion count for NO3– at m/z– 62 (I62) during the oxidations is also provided in Figure 4. The highest concentration of 4NC and 4M5NC in Figure 4 occurs under acidic conditions (at pH 4.05), which is better displayed in Figure S3 (Supporting Information), while the pH dependence of other species is less consistent. In addition to the favorable NO3– production that is maximum at pH 4.05, a different mechanism must slightly affect the process as the pH increases from 5.04 to 8.04 (Figure S3, Supporting Information). This additional mechanism can be explained by electron transfer to NO3 to also proceed from the small amount of catecholates appearing as the pH → 8 (in the range from ∼3 to 7%).
Figure 4.

Concentration of (A) 4-nitrocatechol (4NC), (B) 4-methyl-5-nitrocatechol (4M5NC), (C) 3-methyl-5-nitrocatechol (3M5NC), (D) 3-methoxy-5-nitrocatechol (3MT5NC), and (E) 4-nitropyrogallol (4NPG) from the oxidation of the corresponding catechols in panels A through E in Figure 3 with 7.6, 235.2, 1621.1, and 1986.3 pptv NO3 during experiments at pH 4.05 (black circle), 5.04 (red square), 6.08 (blue diamond), 7.07 (pink hexagon), and 8.04 (green triangle). (F) Ion count for nitrate at m/z– 62 (I62) vs the molar ratio of oxidizers at variable pH. Molar ratios of O3(g) and NO2(g) are provided in Table S1. Dashed lines are provided as a guide to the eye only.
While organic nitrates such as those derived from isoprene undergo rapid hydrolysis under typical acidic aerosol conditions,63−65 the produced nitroaromatic compounds in Figure 4 form a stable C–N bond and are not lost in this way under the short time scale studied here. The increased production of the oxidizer, as reflected by the molar ratios in the experiments shown in Figure 4, results in a quasi-linear rise in I62 in the full pH range, which is maximum for pH 4.05. The large I62 values in Figure 4 demonstrate the efficient loss of the oxidizer mixture once in contact with the outermost layers of the aqueous interface. This is a heterogeneous process that involves hydrolysis of NO3 and N2O5 on particle surfaces.66−70 For example, reaction R4 reflects the loss of adsorbed NO3 through the direct reaction with water to generate HNO3(aq):50,51
| R4 |
which must significantly decrease the surface pH given the low pKa, HNO3 = −1.38.71 Henry’s law constant of NO3 in water at 293 K has been estimated to be H0, NO3 = 0.2 (± 0.1) M atm–1.51,72,73 In the atmospheric chemistry context, the large production of NO3– resulting from the impinging oxidizer mix can recreate observations for this anion to be the most abundant water-soluble inorganic species available in PM2.5 influenced by large NOx emissions.74 While NO3 can undergo electron transfer with ions such as halides, HSO3–, SO3, HCOO–, CH3OO–, and HO– to also form NO3–,50,69 such a redox process with catechols has remained unnoticed and is discussed in the next section.
Reaction R5(52,75) describes the loss of adsorbed N2O5 through heterogeneous hydrolysis on the aqueous surface of the microdroplets:
| R5 |
A computational study of the adsorption of N2O5(g) to form HNO3 by hydrolysis on the air–water interface favored the role played by the high free energy of adsorption (3.4 kcal mol–1) as compared to the corresponding solvation free energy (1.3 kcal mol–1).66Reaction R5 favored on wet aerosol surfaces under acidic conditions constitutes an important atmospheric sink for N2O5.67,70,76−79 The direct uptake of NO2(g), with Henry’s law constant H0, NO2 = 7.0 × 10–3 M atm,80 also theoretically contributes to acidify the interface by reaction R6, which creates not only nitric acid but also nitrous acid (pKa, HNO2 = 3.35):28
| R6 |
However, control experiments impinging the aerosolized solutions only with NO2(g) indicate a negligible contribution of reaction R6 to the generation of ions at m/z– 62 in this reactor. For example, for catechol at pH 4.05, reaction R6 produced <0.09% of the detected I62 for the whole NO2 concentration range studied in the presence of O3. For the oxidizer mixture with O3(g) + NO2(g), the detected NO3– ions should be largely originated from hydrolysis reactions R4 + R5, reflecting a stoichiometric amount of nitrate radical equivalents partitioning to the surface of microdroplets.
3.3. Redox Potentials at Variable pH and Thermodynamic Analysis
Previous oxidation studies of catechols and phenolic aldehydes by O3 and HO• at the air–water interface3,4,38 indicate that the speciation of catechols plays a fundamental role in determining the preferred reactivity channels. The fraction of catechols (α) available in the fully protonated form (H2Q) or the partially dissociated form (HQ–) is presented in Table 1 based on the first and second dissociation constants reported or calculated in our previous work as pKa1 and pKa2:4,28,81−86
| R7 |
| R8 |
Table 1. Physical Constants, Fraction of Dissociation, Measured Redox Potential, and Fee Energy Change for Electron Transfer.
| name | MW (amu) | pKa1 | pKa2 | pH | αQH2 | αQH– |
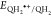 (V)d (V)d
|
ΔGO3°e,f(kJ mol–1) | ΔGNO3°e,g(kJ mol–1) |
|---|---|---|---|---|---|---|---|---|---|
| catechol | 110.11 | 9.34a | 12.60a | 4.02 | 1.000 | 0.000 | 0.564 | –44.96 | –183.51 |
| 6.13 | 0.999 | 0.001 | 0.562 | –45.15 | –183.71 | ||||
| 8.00 | 0.956 | 0.043 | 0.562 | –45.15 | –183.71 | ||||
| 9.97 | 0.190 | 0.809 | 0.565 | –44.87 | –183.42 | ||||
| 4-methylcatechol | 124.14 | 9.59b | 12.66c | 4.07 | 1.000 | 0.000 | 0.491 | –52.01 | –190.56 |
| 6.07 | 1.000 | 0.000 | 0.489 | –52.20 | –190.75 | ||||
| 8.08 | 0.967 | 0.033 | 0.491 | –52.01 | –190.56 | ||||
| 10.11 | 0.215 | 0.782 | 0.502 | –50.94 | –189.50 | ||||
| 3-methylcatechol | 124.14 | 9.59b | 12.69c | 4.08 | 1.000 | 0.000 | 0.506 | –50.56 | –189.11 |
| 6.07 | 1.000 | 0.000 | 0.507 | –50.46 | –189.01 | ||||
| 8.04 | 0.973 | 0.027 | 0.508 | –50.37 | –188.92 | ||||
| 10.05 | 0.257 | 0.741 | 0.539 | –47.37 | –185.93 | ||||
| 3-methoxycatechol | 140.14 | 9.59b | 12.69c | 4.07 | 1.000 | 0.000 | 0.506 | –50.56 | –189.11 |
| 6.08 | 1.000 | 0.000 | 0.498 | –51.33 | –189.88 | ||||
| 7.99 | 0.975 | 0.025 | 0.495 | –51.62 | –190.17 | ||||
| 10.08 | 0.242 | 0.756 | 0.500 | –51.14 | –189.69 | ||||
| pyrogallol | 126.11 | 9.12a | 11.19a | 4.06 | 1.000 | 0.000 | 0.511 | –50.08 | –188.63 |
| 6.08 | 0.999 | 0.001 | 0.518 | –49.40 | –187.95 | ||||
| 8.02 | 0.926 | 0.074 | 0.539 | –47.37 | –185.93 | ||||
| 10.18 | 0.074 | 0.844 | 0.521 | –49.11 | –187.66 |
Measured in this work with Ag/AgCl at the given pH values (Tables S2–S6, Supporting Information) and converted to standard hydrogen electrode (SHE) using the equation Ered (SHE) = Ered (Ag/AgCl sat. KCl) + 0.197.89
ΔG° = – nFΔEi with F = 96.485 kJ mol–1V–1 for electron transfer of n = 1 electron to aqueous species i, where
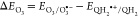 with EO3/O3•–= 1.03 V,88 and
with EO3/O3•–= 1.03 V,88 and
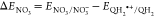 with ENO3/NO3–= 2.466 V.88
with ENO3/NO3–= 2.466 V.88
For the range from pH 4 to 8, >92%
of the catechols in Table 1 remain fully protonated
and are therefore the major species available to react with the adsorbed
oxidant molecules.38 Thus, we report in Table 1 the measured first
redox potential, EH2Q•+/H2Q, for the H2Q form of the catechols,
which are also the dominant species at the pH of atmospheric particles. Table 1 also provides the
free energy change obtained from the relationship ΔG° = −nFΔE, determined from the change in redox potential (ΔE) for the one-electron transfer (n = 1)
from the QH2 form of the catechols to produce the corresponding
radical cation, 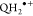 (pKa ≈
−1.62),87 that deprotonates in picoseconds,
in the presence of adsorbed O3 and NO3 that
are converted to O3•– and NO3–, respectively. The redox potentials of
the last two aqueous species are EO3/O3•–= 1.03 V and ENO3/NO3–=
2.466 V.88 The ΔG° values (Table 1) for reactions between catechols and NO3 are four times larger than with O3. Thus, in the actual
atmosphere, the more favorable thermodynamics for electron transfer
could partially compensate for the lower probability of encounters
with NO3 than with most abundant O3. An analogous
thermodynamic analysis can be applied to expand the information in Table 1 using the second
redox potentials, Ered,2, for HQ– of the catechols, which are reported in Tables S2–S6 (Supporting Information). Lastly, although electron
transfer from QH2 to NO2 should be thermodynamically
favorable, the fact that the reaction did not proceed in the control
with NO2 only suggests it is kinetically prevented.
(pKa ≈
−1.62),87 that deprotonates in picoseconds,
in the presence of adsorbed O3 and NO3 that
are converted to O3•– and NO3–, respectively. The redox potentials of
the last two aqueous species are EO3/O3•–= 1.03 V and ENO3/NO3–=
2.466 V.88 The ΔG° values (Table 1) for reactions between catechols and NO3 are four times larger than with O3. Thus, in the actual
atmosphere, the more favorable thermodynamics for electron transfer
could partially compensate for the lower probability of encounters
with NO3 than with most abundant O3. An analogous
thermodynamic analysis can be applied to expand the information in Table 1 using the second
redox potentials, Ered,2, for HQ– of the catechols, which are reported in Tables S2–S6 (Supporting Information). Lastly, although electron
transfer from QH2 to NO2 should be thermodynamically
favorable, the fact that the reaction did not proceed in the control
with NO2 only suggests it is kinetically prevented.
3.4. Environmental Implications of Proposed Reaction Pathways
It is clear from the discussion above and the products observed that the catechols react with the oxidizer mixture by at least two competitive mechanisms. In one thermodynamically favorable process, reaction R1 (Scheme 1) initiates the process by electron transfer between QH2 and NO3. The created NO3– immediately accepts the proton released in picoseconds from QH•+ by reaction R2 (Scheme 1), resulting in a semiquinone radical, which, given their approximate pKa, QH• = 5,57 should be present as QH• in typical atmospheric waters. Completely undistinguishable from the above explanation for electron and proton transfers, a hydrogen (H) atom abstraction from QH2 by NO3 can create QH• and HNO3, in a process that describes the sum of reactions R1 + R2. Finally, the semiquinone radical can recombine with NO2 carried in the oxidizer mix via reaction R3 (Scheme 1) to create the nitrocatechols after recovering aromaticity.
Scheme 1. Electron and Proton Transfer Mechanism of Nitrocatechol Formation at the Air–Water Interface.

Hydrogen atom abstraction with the participation of semiquinone radical intermediates was determined to proceed with a high molar yield on dry particles (<1% relative humidity), i.e., 91% for catechol.90 The exothermic H atom abstraction from the phenolic −OH group of catechol has no energy barrier to form a semiquinone radical in the gas phase, in opposition to the thermodynamic unfavorable (endothermic) abstraction of an aromatic H atom.91 The ipso-addition of NO3 to the −OH substituent of the ring was proposed in the gas phase to form a six-membered transition state, which eliminates HNO3 while generating the phenoxy radical (semiquinone radical in our case) originally depicted by Atkinson et al.92 However, based on our thermodynamic determinations in Table 1 and the fact that the reactions proceed on the surface of water in this work, the pathway with the participation of semiquinone radical intermediates formed by electron and proton transfers (Scheme 1) is preferred to explain present findings. The proposed mechanism has eluded consideration in the atmospheric chemistry literature and should clearly contribute to explain the production of 4NC (m/z– 154), 4M5NC (m/z– 168), 3M5NC (m/z– 168), 3MT5NC (m/z– 184), and 4NPG (m/z– 170) in this work, as well as to explain the abundance of nitrocatechols in biomass burning plumes.93 In favor of this mechanism, the production of 4NC, 4M5NC, 3M5NC, and 4NPG in Figure S3 was clearly enhanced at pH 4, when the fraction of QH• was maximized relative to pH 5 (pKa, QH• = 5).57
A second mechanism to produce the nitrocatechols observed proceeds by electrophilic addition of NO3 (reaction R1, Scheme 2) to the most activated (and with less steric hindrance) aromatic ring position. A cyclohexadienyl radical intermediate results from reaction R1 (Scheme 2). This first intermediate is equivalent to that resulting by HO• attack to the catechols at the air–water interface.3,4,19 The termination reaction is provided in Scheme 2 by reaction R2 for the encounter of the cyclohexadienyl radical with dissolved NO2, which forms a second intermediate. The new intermediate undergoes rearrangement in reaction R3 (Scheme 2), eliminating HNO3 to produce the nitrocatechols as similarly explained before for nitrophenols in the gas phase.94 In support of this radical mechanism in Scheme 2, the nitration of cis,cis-muconic acid (Figure S2, Supporting Information) cannot proceed by the first pathway in Scheme 1 but should go forward through a related allyl radical to the cyclohexadienyl radical intermediate.
Scheme 2. Nitrate Radical Electrophilic Addition Mechanism of Nitrocatechol Formation at the Air–Water Interface.

A third pathway that could be considered to enhance the nitration of catechols at pH 4 (the lowest pH studied) is that resulting from N2O5 directly. Because only 20% of the N2O5 reaching the surface produces nitric acid directly via reaction R5, the remaining molecules of N2O5 can dissociate into nitronium ions (NO2+) and NO3–,95,96
| R9 |
Despite the short half-lifetime of NO2+ ∼1 ns in water to form nitric acid,
| R10 |
the fast electrophilic nitration may proceed by addition of NO2+ to the aromatic ring97 forming a cationic intermediate in a rate-limiting step, followed by fast deprotonation en route to generate the nitrocatechols.98 Instead, any consideration of adsorbed N2O5 acting as a precursor to HNO299 can be ruled out to contribute to the observed nitrocatechols as explained below.
While the formation of HNO2 (MW 47.01) is possible at pH 4, for the highest molar ratio of oxidants studied, the detection of trace peaks at m/z– 46 indicates that this pathway is of minor importance. Thus, N2O5 is not a major source of HNO2 in the experiments at the air–water interface as assessed by the small average ratio of I46/I62 ≤ 0.13% detected for the five catechols. A further discussion of the nonradical oxidation by the HNO2 /NO2– pair is provided in the Supporting Information. Finally, the reaction HNO3 + HO• → NO3 + H2O has been modeled in a cluster of 21 water molecules,100 implying the participation of a HNO3···OH complex with water, from which the products are obtained through a coupled proton–electron transfer mechanism.100 This pathway can contribute an additional source of NO3 for the oxidation of catechols at the air–water interface, which becomes more important under strong acidic conditions.100
The oxidation of catechols, tracers from biomass burning and anthropogenic processes, by nitrate radical proceeds efficiently at the air–water interface to produce nitrocatechols in a few microseconds. This fast production of nitroaromatic compounds alters the absorption properties and reactivity of catechols. The reactions operative on the surface of water are confirmed to proceed through thermodynamically favorable electron and proton transfers, forming semiquinone radical intermediates, or by electrophilic substitution via cyclohexadienyl radical intermediates. Subsequent attack of adsorbed NO2 to the intermediates forms the nitrocatechols by the respective mechanisms shown in Schemes 1 and 2, while coproducing nitric acid. A primary concern from the incorporation of a nitro group into the aromatic ring is the increased hydrophobicity of the products, rendering molecules that are more toxic toward humans and animals.91
Nitrocatechols’ higher tendency to partition into the particle phase (e.g., up to 68% for 4NC)90 increases the solar absorption of organic matter in aerosol particles, contributing to the complexity of atmospheric brown carbon SOA and their role in radiative forcing.7,12,14,101−104 In contrast, the produced nitrocatechols can also be photobleached losing their absorption, followed by functionalization and ring-cleavage reactions.105 Nitrocatechols are prone to further photooxidation, forming oligomers,106 and as other aromatics can be expected to fragment into multifunctional carboxylic acids.3,4 Importantly, the larger production of 4NC we observed under acidic pH suggests an enhanced atmospheric presence of this chromophore is possible, relative to high pH conditions, that is accompanied by a bathochromic effect.107
While the production of NO3 from the oxidation of NO2(g) by O3(g) is well understood, future experimental efforts could aim to quantify it at the air–water interface. In the gas phase, NO3 can be quantified in real time, e.g., by differential optical absorption spectroscopy, whose measurements could be estimated by offline matrix isolated electron spin resonance.65,108 The later technique is complex65 but could be potentially adapted to advance the understanding of this surface reaction. Future interfacial studies should evaluate the uptake of oxidizer molecules in the presence of catechols by exploring heterogeneous reaction kinetics at variable relative humidity.
Acknowledgments
Funding from the USA National Science Foundation under award 1903744 to M.I.G. is greatly acknowledged.
Glossary
Abbreviations
- amu
atomic mass unit
- CID
collisional induced dissociation
- EH2Q•+/H2Q
redox potential for the reduction of catechols to radical cations
- F
Faraday constant
- H0,i
Henry’s law constant of species i
- HO•
hydroxyl radical
- HNO2
nitrous acid
- HNO3
nitric acid
- Im/z
ion count at value m/z
- m/z–
mass-to-charge ratio of anion
- MS
mass spectrometry
- MW
molecular weight
- n
number of electrons transferred
- NO3
nitrate radical
- NO3–
nitrate anion
- NO2
nitrogen dioxide
- N2O5
dinitrogen pentoxide
- O3
ozone
- O3•–
ozonide radical anion
- OESI
online electrospray ionization
- PDA
photodiode array
- PNO3
production rate of NO3 radical
- ppbv
parts per billion by volume
- pptv
parts per trillion by volume
- QH2
fully protonated form of the catechols
- QH–
mono-dissociated form of the catechols
- Q2–
fully dissociated form of the catechols
- QH•+
radical cation of the catechols
- QH•
semiquinone radical of the catechols
- SHE
standard hydrogen electrode
- SOA
secondary organic aerosol
- tc
contact time
- UHP
ultrahigh purity
- α
fraction of dissociation
- ΔE
change of redox potential
- ΔGi°
free energy change for electron transfer of catechols to species i
- 3M5NC
3-methyl-5-nitroctaechol
- 3MT5NC
3-methoxy-5-nitrocatechol
- 4NC
4-nitroctaechol
- 4M5NC
4-methyl-5-nitrocatechol
- 4NPG
4-nitropyrogallol
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.2c05640.
Online electrospray ionization mass spectrometry (OESI-MS) reactor diagram; OESI-MS spectra for control and oxidation; cyclic voltammograms; redox potentials (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Henze D.; Seinfeld J.; Ng N.; Kroll J.; Fu T.-M.; Jacob D. J.; Heald C. Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: high-vs. low-yield pathways. Atmos. Chem. Phys. 2008, 8, 2405–2420. 10.5194/acp-8-2405-2008. [DOI] [Google Scholar]
- Schauer J. J.; Kleeman M. J.; Cass G. R.; Simoneit B. R. T. Measurement of emissions from air pollution sources. 3. C1–C29 organic compounds from fireplace combustion of wood. Environ. Sci. Technol. 2001, 35, 1716–1728. 10.1021/es001331e. [DOI] [PubMed] [Google Scholar]
- Pillar E. A.; Camm R. C.; Guzman M. I. Catechol oxidation by ozone and hydroxyl radicals at the air–water interface. Environ. Sci. Technol. 2014, 48, 14352–14360. 10.1021/es504094x. [DOI] [PubMed] [Google Scholar]
- Pillar E. A.; Guzman M. I. Oxidation of substituted catechols at the air–water interface: Production of carboxylic acids, quinones, and polyphenols. Environ. Sci. Technol. 2017, 51, 4951–4959. 10.1021/acs.est.7b00232. [DOI] [PubMed] [Google Scholar]
- Pillar-Little E. A.; Guzman M. An overview of dynamic heterogeneous oxidations in the troposphere. Environments 2018, 5, 104. 10.3390/environments5090104. [DOI] [Google Scholar]
- Iinuma Y.; Böge O.; Gräfe R.; Herrmann H. Methyl-nitrocatechols: Atmospheric tracer compounds for biomass burning secondary organic aerosols. Environ. Sci. Technol. 2010, 44, 8453–8459. 10.1021/es102938a. [DOI] [PubMed] [Google Scholar]
- Li M.; Wang X.; Lu C.; Li R.; Zhang J.; Dong S.; Yang L.; Xue L.; Chen J.; Wang W. Nitrated phenols and the phenolic precursors in the atmosphere in urban Jinan, China. Sci. Total Environ. 2020, 714, 136760 10.1016/j.scitotenv.2020.136760. [DOI] [PubMed] [Google Scholar]
- Mohr C.; Lopez-Hilfiker F. D.; Zotter P.; Prévôt A. S. H.; Xu L.; Ng N. L.; Herndon S. C.; Williams L. R.; Franklin J. P.; Zahniser M. S.; Worsnop D. R.; Knighton W. B.; Aiken A. C.; Gorkowski K. J.; Dubey M. K.; Allan J. D.; Thornton J. A. Contribution of nitrated phenols to wood burning brown carbon light absorption in detling, United Kingdom during winter time. Environ. Sci. Technol. 2013, 47, 6316–6324. 10.1021/es400683v. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Shen Z.; Zhang L.; Zeng Y.; Ning Z.; Zhang T.; Lei Y.; Wang Q.; Li G.; Sun J.; Westerdahl D.; Xu H.; Cao J. Investigation of primary and secondary particulate brown carbon in two chinese cities of Xi’an and Hong Kong in wintertime. Environ. Sci. Technol. 2020, 54, 3803–3813. 10.1021/acs.est.9b05332. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Hu M.; Wang Y.; Zheng J.; Shang D.; Yang Y.; Liu Y.; Li X.; Tang R.; Zhu W.; Du Z.; Wu Y.; Guo S.; Wu Z.; Lou S.; Hallquist M.; Yu J. Z. The formation of nitro-aromatic compounds under high NOx and anthropogenic VOC conditions in urban Beijing, China. Atmos. Chem. Phys. 2019, 19, 7649–7665. 10.5194/acp-19-7649-2019. [DOI] [Google Scholar]
- Salvador C. M. G.; Tang R.; Priestley M.; Li L.; Tsiligiannis E.; Le Breton M.; Zhu W.; Zeng L.; Wang H.; Yu Y.; Hu M.; Guo S.; Hallquist M. Ambient nitro-aromatic compounds – biomass burning versus secondary formation in rural China. Atmos. Chem. Phys. 2021, 21, 1389–1406. 10.5194/acp-21-1389-2021. [DOI] [Google Scholar]
- Laskin A.; Laskin J.; Nizkorodov S. A. Chemistry of atmospheric brown carbon. Chem. Rev. 2015, 115, 4335–4382. 10.1021/cr5006167. [DOI] [PubMed] [Google Scholar]
- Hoffmann E. H.; Tilgner A.; Wolke R.; Böge O.; Walter A.; Herrmann H. Oxidation of substituted aromatic hydrocarbons in the tropospheric aqueous phase: kinetic mechanism development and modelling. Phys. Chem. Chem. Phys. 2018, 20, 10960–10977. 10.1039/C7CP08576A. [DOI] [PubMed] [Google Scholar]
- Li C.; He Q.; Hettiyadura A. P. S.; Käfer U.; Shmul G.; Meidan D.; Zimmermann R.; Brown S. S.; George C.; Laskin A.; Rudich Y. Formation of secondary brown carbon in biomass burning aerosol proxies through NO3 radical reactions. Environ. Sci. Technol. 2020, 54, 1395–1405. 10.1021/acs.est.9b05641. [DOI] [PubMed] [Google Scholar]
- Zeng L.; Zhang A.; Wang Y.; Wagner N. L.; Katich J. M.; Schwarz J. P.; Schill G. P.; Brock C.; Froyd K. D.; Murphy D. M.; Williamson C. J.; Kupc A.; Scheuer E.; Dibb J.; Weber R. J. Global measurements of brown carbon and estimated direct radiative effects. Geophys. Res. Lett. 2020, 47, e2020GL088747 10.1029/2020GL088747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olariu R. I.; Barnes I.; Becker K. H.; Klotz B. Rate coefficients for the gas-phase reaction of OH radicals with selected dihydroxybenzenes and benzoquinones. Int. J. Chem. Kinet. 2000, 32, 696–702. . [DOI] [Google Scholar]
- Olariu R. I.; Bejan I.; Barnes I.; Klotz B.; Becker K. H.; Wirtz K. Rate coefficients for the gas-phase reaction of NO3 radicals with selected dihydroxybenzenes. Int. J. Chem. Kinet. 2004, 36, 577–583. 10.1002/kin.20029. [DOI] [Google Scholar]
- Barzaghi P.; Herrmann H. Kinetics and mechanisms of reactions of the nitrate radical (NO3) with substituted phenols in aqueous solution. Phys. Chem. Chem. Phys. 2004, 6, 5379–5388. 10.1039/b412933d. [DOI] [Google Scholar]
- Pillar E. A.; Zhou R.; Guzman M. I. Heterogeneous oxidation of catechol. J. Phys. Chem. A 2015, 119, 10349–10359. 10.1021/acs.jpca.5b07914. [DOI] [PubMed] [Google Scholar]
- Yang B.; Zhang H.; Wang Y.; Zhang P.; Shu J.; Sun W.; Ma P. Experimental and theoretical studies on gas-phase reactions of NO3 radicals with three methoxyphenols: Guaiacol, creosol, and syringol. Atmos. Environ. 2016, 125, 243–251. 10.1016/j.atmosenv.2015.11.028. [DOI] [Google Scholar]
- Zein A. E.; Coeur C.; Obeid E.; Lauraguais A.; Fagniez T. Reaction kinetics of catechol (1,2-benzenediol) and guaiacol (2-methoxyphenol) with ozone. J. Phys. Chem. A 2015, 119, 6759–6765. 10.1021/acs.jpca.5b00174. [DOI] [PubMed] [Google Scholar]
- Guzman M. I.; Athalye R. R.; Rodriguez J. M. Concentration effects and ion properties controlling the fractionation of halides during aerosol formation. J. Phys. Chem. A 2012, 116, 5428–5435. 10.1021/jp3011316. [DOI] [PubMed] [Google Scholar]
- Pillar E. A.; Guzman M. I.; Rodriguez J. M. Conversion of iodide to hypoiodous acid and iodine in aqueous microdroplets exposed to ozone. Environ. Sci. Technol. 2013, 47, 10971–10979. 10.1021/es401700h. [DOI] [PubMed] [Google Scholar]
- Eugene A. J.; Pillar E. A.; Colussi A. J.; Guzman M. I. Enhanced acidity of acetic and pyruvic acids on the surface of water. Langmuir 2018, 34, 9307–9313. 10.1021/acs.langmuir.8b01606. [DOI] [PubMed] [Google Scholar]
- Ruiz-Lopez M. F.; Francisco J. S.; Martins-Costa M. T. C.; Anglada J. M. Molecular reactions at aqueous interfaces. Nat. Rev. Chem. 2020, 4, 459–475. 10.1038/s41570-020-0203-2. [DOI] [PubMed] [Google Scholar]
- Zhang X. Mass spectrometry at the air-water interface. Int. J. Mass Spectrom. 2021, 462, 116527 10.1016/j.ijms.2021.116527. [DOI] [Google Scholar]
- Veres P.; Roberts J. M.; Burling I. R.; Warneke C.; Gouw J. d.; Yokelson R. J.. Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry. J. Geophys. Res. 2010, 115 (), 10.1029/2010JD014033. [DOI] [Google Scholar]
- Haynes W. M.; Lide D. R.; Bruno T. J.. CRC Handbook of Chemistry and Physics; 93rd ed.; CRC Press/Taylor and Francis: Boca Raton, Fl., 2013, 2664. [Google Scholar]
- Sun M.; Wang Y.; Wang T.; Fan S.; Wang W.; Li P.; Guo J.; Li Y.. Cloud and the corresponding precipitation chemistry in south China: Water-soluble components and pollution transport. J. Geophys. Res.: Atmos. 2010, 115 (), 10.1029/2010JD014315. [DOI] [Google Scholar]
- Benedict K. B.; Lee T.; Collett J. L. Cloud water composition over the southeastern Pacific Ocean during the VOCALS regional experiment. Atmos. Environ. 2012, 46, 104–114. 10.1016/j.atmosenv.2011.10.029. [DOI] [Google Scholar]
- Budhavant K. B.; Rao P. S. P.; Safai P. D.; Granat L.; Rodhe H. Chemical composition of the inorganic fraction of cloud-water at a high altitude station in West India. Atmos. Environ. 2014, 88, 59–65. 10.1016/j.atmosenv.2014.01.039. [DOI] [Google Scholar]
- Pye H. O. T.; Nenes A.; Alexander B.; Ault A. P.; Barth M. C.; Clegg S. L.; Collett J. L. Jr.; Fahey K. M.; Hennigan C. J.; Herrmann H.; Kanakidou M.; Kelly J. T.; Ku I. T.; McNeill V. F.; Riemer N.; Schaefer T.; Shi G.; Tilgner A.; Walker J. T.; Wang T.; Weber R.; Xing J.; Zaveri R. A.; Zuend A. The acidity of atmospheric particles and clouds. Atmos. Chem. Phys. 2020, 20, 4809–4888. 10.5194/acp-20-4809-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y.; Murphy J. G. The sensitivity of PM2.5 acidity to meteorological parameters and chemical composition changes: 10-year records from six Canadian monitoring sites. Atmos. Chem. Phys. 2019, 19, 9309–9320. 10.5194/acp-19-9309-2019. [DOI] [Google Scholar]
- Jia S.; Chen W.; Zhang Q.; Krishnan P.; Mao J.; Zhong B.; Huang M.; Fan Q.; Zhang J.; Chang M.; Yang L.; Wang X. A quantitative analysis of the driving factors affecting seasonal variation of aerosol pH in Guangzhou, China. Sci. Total Environ. 2020, 725, 138228 10.1016/j.scitotenv.2020.138228. [DOI] [PubMed] [Google Scholar]
- Masiol M.; Squizzato S.; Formenton G.; Khan M. B.; Hopke P. K.; Nenes A.; Pandis S. N.; Tositti L.; Benetello F.; Visin F.; Pavoni B. Hybrid multiple-site mass closure and source apportionment of PM2.5 and aerosol acidity at major cities in the Po Valley. Sci. Total Environ. 2020, 704, 135287 10.1016/j.scitotenv.2019.135287. [DOI] [PubMed] [Google Scholar]
- Ding J.; Zhao P.; Su J.; Dong Q.; Du X.; Zhang Y. Aerosol pH and its driving factors in Beijing. Atmos. Chem. Phys. 2019, 19, 7939–7954. 10.5194/acp-19-7939-2019. [DOI] [Google Scholar]
- Shi G.; Xu J.; Shi X.; Liu B.; Bi X.; Xiao Z.; Chen K.; Wen J.; Dong S.; Tian Y.; Feng Y.; Yu H.; Song S.; Zhao Q.; Gao J.; Russell A. G. Aerosol pH Dynamics During Haze Periods in an Urban Environment in China: Use of Detailed, Hourly, Speciated Observations to Study the Role of Ammonia Availability and Secondary Aerosol Formation and Urban Environment. J.Geophys. Res.: Atmos. 2019, 124, 9730–9742. 10.1029/2018JD029976. [DOI] [Google Scholar]
- Rana M. S.; Guzman M. I. Oxidation of phenolic aldehydes by ozone and hydroxyl radicals at the air–water interface. J. Phys. Chem. A 2020, 124, 8822–8833. 10.1021/acs.jpca.0c05944. [DOI] [PubMed] [Google Scholar]
- Brown S. S.; Stutz J. Nighttime radical observations and chemistry. Chem. Soc. Rev. 2012, 41, 6405–6447. 10.1039/c2cs35181a. [DOI] [PubMed] [Google Scholar]
- Burkholder J. B.; Sander S. P.; Abbatt J. P. D.; Barker J. R.; Cappa C.; Crounse J. D.; Dibble T. S.; Huie R. E.; Kolb C. E.; Kurylo M. J.. Chemical kinetics and photochemical data for use in atmospheric studies; evaluation number 19; Jet Propulsion Laboratory, National Aeronautics and Space: Pasadena, CA, 2020. [Google Scholar]
- Sander S. P.; Friedl R. R.; Barker J. R.; Golden D. M.; Kurylo M. J.; Wine P. H.; Abbatt J.; Burkholder J. B.; Kolb C. E.; Moortgat G. K.; Huie R. E.; Orkin V. L.. Chemical kinetics and photochemical data for use in Atmospheric Studies: Evaluation Number 17; Jet Propulsion Laboratory, National Aeronautics and Space Administration: Pasadena, CA, 2009. [Google Scholar]
- Wood E. C.; Wooldridge P. J.; Freese J. H.; Albrecht T.; Cohen R. C. Prototype for In Situ Detection of Atmospheric NO3 and N2O5 via Laser-Induced Fluorescence. Environ. Sci. Technol. 2003, 37, 5732–5738. 10.1021/es034507w. [DOI] [PubMed] [Google Scholar]
- Dubé W. P.; Brown S. S.; Osthoff H. D.; Nunley M. R.; Ciciora S. J.; Paris M. W.; McLaughlin R. J.; Ravishankara A. R. Aircraft instrument for simultaneous, in situ measurement of NO3 and N2O5 via pulsed cavity ring-down spectroscopy. Rev. Sci. Instrum. 2006, 77, 034101 10.1063/1.2176058. [DOI] [Google Scholar]
- Ren Y.; Zhou L.; Mellouki A.; Daële V.; Idir M.; Brown S. S.; Ruscic B.; Paton R. S.; McGillen M. R.; Ravishankara A. R. Reactions of NO3 with aromatic aldehydes: gas-phase kinetics and insights into the mechanism of the reaction. Atmos. Chem. Phys. 2021, 21, 13537–13551. 10.5194/acp-21-13537-2021. [DOI] [Google Scholar]
- Lambe A. T.; Wood E. C.; Krechmer J. E.; Majluf F.; Williams L. R.; Croteau P. L.; Cirtog M.; Féron A.; Petit J. E.; Albinet A.; Jimenez J. L.; Peng Z. Nitrate radical generation via continuous generation of dinitrogen pentoxide in a laminar flow reactor coupled to an oxidation flow reactor. Atmos. Meas. Technol. 2020, 13, 2397–2411. 10.5194/amt-13-2397-2020. [DOI] [Google Scholar]
- Xing D.; Meng Y.; Yuan X.; Jin S.; Song X.; Zare R. N.; Zhang X. Capture of hydroxyl radicals by hydronium cations in water microdroplets. Angew. Chem., Int. Ed. 2022, 61, e202207587 10.1002/anie.202207587. [DOI] [PubMed] [Google Scholar]
- Lee J. K.; Walker K. L.; Han H. S.; Kang J.; Prinz F. B.; Waymouth R. M.; Nam H. G.; Zare R. N. Spontaneous generation of hydrogen peroxide from aqueous microdroplets. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 19294–19298. 10.1073/pnas.1911883116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Song X.; Gong C.; Zhang D.; Wang R.; Zare R. N.; Zhang X. Sprayed water microdroplets containing dissolved pyridine spontaneously generate pyridyl anions. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2200991119 10.1073/pnas.2200991119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana M. S.; Guzman M. I., Surface Oxidation of Phenolic Aldehydes: Fragmentation, Functionalization, and Coupling Reactions. Submitted 2022. [DOI] [PMC free article] [PubMed]
- Rudich Y.; Talukdar R. K.; Imamura T.; Fox R. W.; Ravishankara A. R. Uptake of NO3 on KI solutions: rate coefficient for the NO3 + I– reaction and gas-phase diffusion coefficients for NO3. Chem. Phys. Lett. 1996, 261, 467–473. 10.1016/0009-2614(96)00980-3. [DOI] [Google Scholar]
- Rudich Y.; Talukdar R. K.; Ravishankara A. R.; Fox R. W. Reactive uptake of NO3 on pure water and ionic solutions. J. Geophys. Res. 1996, 101, 21023–21031. 10.1029/96JD01844. [DOI] [Google Scholar]
- Mentel T. F.; Sohn M.; Wahner A. Nitrate effect in the heterogeneous hydrolysis of dinitrogen pentoxide on aqueous aerosols. Phys. Chem. Chem. Phys. 1999, 1, 5451–5457. 10.1039/a905338g. [DOI] [Google Scholar]
- Tang S.; Li F.; Tsona N. T.; Lu C.; Wang X.; Du L. Aqueous-phase photooxidation of vanillic acid: A potential source of humic-like substances (HULIS). ACS Earth Space Chem. 2020, 4, 862–872. 10.1021/acsearthspacechem.0c00070. [DOI] [Google Scholar]
- Kitanovski Z.; Grgić I.; Yasmeen F.; Claeys M.; Čusak A. Development of a liquid chromatographic method based on ultraviolet–visible and electrospray ionization mass spectrometric detection for the identification of nitrocatechols and related tracers in biomass burning atmospheric organic aerosol. Rapid Commun. Mass Spectrom. 2012, 26, 793–804. 10.1002/rcm.6170. [DOI] [PubMed] [Google Scholar]
- Greenstock C. L.; Dunlop I.; Neta P. Radiation chemical studies of the oxidation and reduction of nitrofurans. Oxidative denitration by hydroxyl radicals. J. Phys. Chem. 1973, 77, 1187–1190. 10.1021/j100628a021. [DOI] [Google Scholar]
- North G. R.; Pyle J. A.; Zhang F. (ed.), Encyclopedia of Atmospheric Sciences; Elsevier Science, 2014; p 1998. [Google Scholar]
- Steenken S.; O’Neill P. Oxidative demethoxylation of methoxylated phenols and hydroxybenzoic acids by the hydroxyl radical. An in situ electron spin resonance, conductometric pulse radiolysis and product analysis study. J. Phys. Chem. 1977, 81, 505–508. 10.1021/j100521a002. [DOI] [Google Scholar]
- Kitanovski Z.; Čusak A.; Grgić I.; Claeys M. Chemical characterization of the main products formed through aqueous-phase photonitration of guaiacol. Atmos. Meas. Technol. 2014, 7, 2457–2470. 10.5194/amt-7-2457-2014. [DOI] [Google Scholar]
- Doerge D. R.; Divi R. L.; Churchwell M. I. Identification of the colored guaiacol oxidation product produced by peroxidases. Anal. Biochem. 1997, 250, 10–17. 10.1006/abio.1997.2191. [DOI] [PubMed] [Google Scholar]
- Vidović K.; Lašič Jurković D.; Šala M.; Kroflič A.; Grgić I. Nighttime aqueous-phase formation of nitrocatechols in the atmospheric condensed phase. Environ. Sci. Technol. 2018, 52, 9722–9730. 10.1021/acs.est.8b01161. [DOI] [PubMed] [Google Scholar]
- Vidović K.; Kroflič A.; Jovanovič P.; Šala M.; Grgić I. Electrochemistry as a tool for studies of complex reaction mechanisms: The case of the atmospheric aqueous-phase aging of catechols. Environ. Sci. Technol. 2019, 53, 11195–11203. 10.1021/acs.est.9b02456. [DOI] [PubMed] [Google Scholar]
- Vidović K.; Kroflič A.; Šala M.; Grgić I. Aqueous-phase brown carbon formation from aromatic precursors under sunlight conditions. Atmosphere 2020, 11, 131. 10.3390/atmos11020131. [DOI] [Google Scholar]
- Hu K. S.; Darer A. I.; Elrod M. J. Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates. Atmos. Chem. Phys. 2011, 11, 8307–8320. 10.5194/acp-11-8307-2011. [DOI] [Google Scholar]
- Liu S.; Shilling J. E.; Song C.; Hiranuma N.; Zaveri R. A.; Russell L. M. Hydrolysis of organonitrate functional groups in aerosol particles. Aerosol Sci. Technol. 2012, 46, 1359–1369. 10.1080/02786826.2012.716175. [DOI] [Google Scholar]
- Ng N. L.; Brown S. S.; Archibald A. T.; Atlas E.; Cohen R. C.; Crowley J. N.; Day D. A.; Donahue N. M.; Fry J. L.; Fuchs H.; Griffin R. J.; Guzman M. I.; Herrmann H.; Hodzic A.; Iinuma Y.; Jimenez J. L.; Kiendler-Scharr A.; Lee B. H.; Luecken D. J.; Mao J.; McLaren R.; Mutzel A.; Osthoff H. D.; Ouyang B.; Picquet-Varrault B.; Platt U.; Pye H. O. T.; Rudich Y.; Schwantes R. H.; Shiraiwa M.; Stutz J.; Thornton J. A.; Tilgner A.; Williams B. J.; Zaveri R. A. Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol. Atmos. Chem. Phys. 2017, 17, 2103–2162. 10.5194/acp-17-2103-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galib M.; Limmer D. T. Reactive uptake of N2O5 by atmospheric aerosol is dominated by interfacial processes. Science 2021, 371, 921. 10.1126/science.abd7716. [DOI] [PubMed] [Google Scholar]
- Hallquist M.; Stewart D. J.; Stephenson S. K.; Anthony Cox R. Hydrolysis of N2O5 on sub-micron sulfate aerosols. Phys. Chem. Chem. Phys. 2003, 5, 3453–3463. 10.1039/b301827j. [DOI] [Google Scholar]
- Hirshberg B.; Rossich Molina E.; Götz A. W.; Hammerich A. D.; Nathanson G. M.; Bertram T. H.; Johnson M. A.; Gerber R. B. N2O5 at water surfaces: binding forces, charge separation, energy accommodation and atmospheric implications. Phys. Chem. Chem. Phys. 2018, 20, 17961–17976. 10.1039/C8CP03022G. [DOI] [PubMed] [Google Scholar]
- Imamura T.; Rudich Y.; Talukdar R. K.; Fox R. W.; Ravishankara A. R. Uptake of NO3 on water solutions: Rate coefficients for reactions of NO3 with cloud water constituents. J. Phys. Chem. A 1997, 101, 2316–2322. 10.1021/jp962787e. [DOI] [Google Scholar]
- Thornton J. A.; Braban C. F.; Abbatt J. P. D. N2O5 hydrolysis on sub-micron organic aerosols: the effect of relative humidity, particle phase, and particle size. Phys. Chem. Chem. Phys. 2003, 5, 4593–4603. 10.1039/b307498f. [DOI] [Google Scholar]
- Dean J. A. (ed.), Lange’s Handbook of Chemistry. 15 ed.; McGraw-Hill: New York, NY, 1999; p 1538. [Google Scholar]
- Schütze M.; Herrmann H. Uptake of the NO3 radical on aqueous surfaces. J. Atmos. Chem. 2005, 52, 1–18. 10.1007/s10874-005-6153-8. [DOI] [Google Scholar]
- Exner M.; Herrmann H.; Zellner R. Laser-based studies of reactions of the nitrate radical in aqueous solution. Ber. Phys. Chem. 1992, 96, 470–477. 10.1002/bbpc.19920960347. [DOI] [Google Scholar]
- Jin Z.; Qian L.; Shi Y.; Fu G.; Li G.; Li F. Quantifying major NOx sources of aerosol nitrate in Hangzhou, China, by using stable isotopes and a Bayesian isotope mixing model. Atmos. Environ. 2021, 244, 117979 10.1016/j.atmosenv.2020.117979. [DOI] [Google Scholar]
- Mentel T. F.; Bleilebens D.; Wahner A. A study of nighttime nitrogen oxide oxidation in a large reaction chamber—the fate of NO2, N2O5, HNO3, and O3 at different humidities. Atmos. Environ. 1996, 30, 4007–4020. 10.1016/1352-2310(96)00117-3. [DOI] [Google Scholar]
- Badger C. L.; Griffiths P. T.; George I.; Abbatt J. P. D.; Cox R. A. Reactive uptake of N2O5 by aerosol particles containing mixtures of humic acid and ammonium sulfate. J. Phys. Chem. A 2006, 110, 6986–6994. 10.1021/jp0562678. [DOI] [PubMed] [Google Scholar]
- Mogili P. K.; Kleiber P. D.; Young M. A.; Grassian V. H. N2O5 hydrolysis on the components of mineral dust and sea salt aerosol: Comparison study in an environmental aerosol reaction chamber. Atmos. Environ. 2006, 40, 7401–7408. 10.1016/j.atmosenv.2006.06.048. [DOI] [Google Scholar]
- Chang W. L.; Brown S. S.; Stutz J.; Middlebrook A. M.; Bahreini R.; Wagner N. L.; Dubé W. P.; Pollack I. B.; Ryerson T. B.; Riemer N. Evaluating N2O5 heterogeneous hydrolysis parameterizations for CalNex 2010. J. Geophys. Res.: Atmospheres 2016, 121, 5051–5070. 10.1002/2015JD024737. [DOI] [Google Scholar]
- Russell A. G.; Cass G. R.; Seinfeld J. H. On some aspects of nighttime atmospheric chemistry. Environ.Sci.Technol. 1986, 20, 1167–1172. 10.1021/es00153a013. [DOI] [Google Scholar]
- Lee Y. N.; Schwartz S. E. Reaction kinetics of nitrogen dioxide with liquid water at low partial pressure. J. Phys. Chem. 1981, 85, 840–848. 10.1021/j150607a022. [DOI] [Google Scholar]
- Perrin D. D.; Dempsey B.; Serjeant E. P. (ed.), pKa Prediction for organic acids and bases; 1st ed., Springer Netherlands: London, 1981; p 146. [Google Scholar]
- Wishart D. S.; Jewison T.; Guo A. C.; Wilson M.; Knox C.; Liu Y.; Djoumbou Y.; Mandal R.; Aziat F.; Dong E.; Bouatra S.; Sinelnikov I.; Arndt D.; Xia J.; Liu P.; Yallou F.; Bjorndahl T.; Perez-Pineiro R.; Eisner R.; Allen F.; Neveu V.; Greiner R.; Scalbert A. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Shrivastava S.; Hassanali M.; Stothard P.; Chang Z.; Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinn R. G.; Huang J.; Weiss R. F.; Cunnold D. M.; Fraser P. J.; Simmonds P. G.; McCulloch A.; Harth C.; Reimann S.; Salameh P.; O’Doherty S.; Wang R. H. J.; Porter L. W.; Miller B. R.; Krummel P. B. Evidence for variability of atmospheric hydroxyl radicals over the past quarter century. J. Geophys. Res. Lett. 2005, 32, 7. 10.1029/2004GL022228. [DOI] [Google Scholar]
- Zhu X.-Q.; Wang C.-H.; Liang H. Scales of oxidation potentials, pKa, and BDE of various hydroquinones and catechols in DMSO. J. Org. Chem. 2010, 75, 7240–7257. 10.1021/jo101455m. [DOI] [PubMed] [Google Scholar]
- Steenken S.; Neta P. Electron transfer rates and equilibriums between substituted phenoxide ions and phenoxyl radicals. J. Phys. Chem. 1979, 83, 1134–1137. 10.1021/j100472a005. [DOI] [Google Scholar]
- Steenken S.; Neta P., Properties of Phenoxyl Radicals. In The Chemistry of Phenols; Rappoport Z. Ed., John Wiley & Sons: New York, 2003; p 1000. [Google Scholar]
- Armstrong D. A.; Huie R. E.; Koppenol W. H.; Lymar S. V.; Merényi G.; Neta P.; Ruscic B.; Stanbury D. M.; Steenken S.; Wardman P. Standard electrode potentials involving radicals in aqueous solution: inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1139–1150. 10.1515/pac-2014-0502. [DOI] [Google Scholar]
- Bard A. J.; Faulkner L. R. (ed.), Electrochemical methods: Fundamentals and applications. 2nd ed., John Wiley & Sons, Inc.: Phoenix, 2001; p 864. [Google Scholar]
- Finewax Z.; de Gouw J. A.; Ziemann P. J. Identification and quantification of 4-nitrocatechol formed from OH and NO3 radical-initiated reactions of catechol in air in the presence of NOX: Implications for secondary organic aerosol formation from biomass burning. Environ. Sci. Technol. 2018, 52, 1981–1989. 10.1021/acs.est.7b05864. [DOI] [PubMed] [Google Scholar]
- Wei B.; Sun J.; Mei Q.; An Z.; Wang X.; He M. Theoretical study on gas-phase reactions of nitrate radicals with methoxyphenols: Mechanism, kinetic and toxicity assessment. Environ. Pollut. 2018, 243, 1772–1780. 10.1016/j.envpol.2018.08.104. [DOI] [PubMed] [Google Scholar]
- Atkinson R.; Aschmann S. M.; Arey J. Reactions of hydroxyl and nitrogen trioxide radicals with phenol, cresols, and 2-nitrophenol at 296±2 K. Environ. Sci. Technol. 1992, 26, 1397–1403. 10.1021/es00031a018. [DOI] [Google Scholar]
- Decker Z. C. J.; Zarzana K. J.; Coggon M.; Min K.-E.; Pollack I.; Ryerson T. B.; Peischl J.; Edwards P.; Dubé W. P.; Markovic M. Z.; Roberts J. M.; Veres P. R.; Graus M.; Warneke C.; de Gouw J.; Hatch L. E.; Barsanti K. C.; Brown S. S. Nighttime chemical transformation in biomass burning plumes: A box model analysis initialized with aircraft observations. Environ. Sci. Technol. 2019, 53, 2529–2538. 10.1021/acs.est.8b05359. [DOI] [PubMed] [Google Scholar]
- Harrison M. A. J.; Barra S.; Borghesi D.; Vione D.; Arsene C.; Iulian Olariu R. Nitrated phenols in the atmosphere: a review. Atmos. Environ. 2005, 39, 231–248. 10.1016/j.atmosenv.2004.09.044. [DOI] [Google Scholar]
- Behnke W.; George C.; Scheer V.; Zetzsch C. Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments. J. Geophys. Res. Atmos. 1997, 102, 3795–3804. 10.1029/96JD03057. [DOI] [Google Scholar]
- Schütze M.; Herrmann H. Determination of phase transfer parameters for the uptake of HNO3, N2O5 and O3 on single aqueous drops. Phys. Chem. Chem. Phys. 2002, 4, 60–67. 10.1039/b106078n. [DOI] [Google Scholar]
- Taylor R. (ed.), Electrophilic aromatic substitution; John Wiley & Sons: New York, 1990; p 530. [Google Scholar]
- Ridd J. H.; Pletcher D.; Qiao X.; Pascal R. A. Jr.; Bard A. J.; Francis G. W.; Szúnyog J.; Långström B. Some unconventional pathways in aromatic nitration. Acta Chem. Scand. 1998, 52, 11–22. 10.3891/acta.chem.scand.52-0011. [DOI] [Google Scholar]
- Bunton C. A.; Hughes E. D.; Ingold C. K.; Jacobs D. I. H.; Jones M. H.; Minkoff G. J.; Reed R. I. 512. Kinetics and mechanism of aromatic nitration. Part VI. The nitration of phenols and phenolic ethers: the concomitant dealkylation of phenolic ethers. The role of nitrous acid. J. Chem. Soc. 1950, 0, 2628–2656. [Google Scholar]
- Anglada J. M.; Martins-Costa M. T. C.; Francisco J. S.; Ruiz-López M. F. Reactivity of undissociated molecular nitric acid at the air–water interface. J. Am. Chem. Soc. 2021, 143, 453–462. 10.1021/jacs.0c11841. [DOI] [PubMed] [Google Scholar]
- Li X.; Yang Y.; Liu S.; Zhao Q.; Wang G.; Wang Y. Light absorption properties of brown carbon (BrC) in autumn and winter in Beijing: Composition, formation and contribution of nitrated aromatic compounds. Atmos. Environ. 2020, 223, 117289 10.1016/j.atmosenv.2020.117289. [DOI] [Google Scholar]
- Lin G.; Penner J. E.; Flanner M. G.; Sillman S.; Xu L.; Zhou C. Radiative forcing of organic aerosol in the atmosphere and on snow: Effects of SOA and brown carbon. J. Geophys. Res. Atmos. 2014, 119, 7453–7476. 10.1002/2013JD021186. [DOI] [Google Scholar]
- Liebmann J.; Karu E.; Sobanski N.; Schuladen J.; Ehn M.; Schallhart S.; Quéléver L.; Hellen H.; Hakola H.; Hoffmann T.; Williams J.; Fischer H.; Lelieveld J.; Crowley J. N. Direct measurement of NO3 radical reactivity in a boreal forest. Atmos. Chem. Phys. 2018, 18, 3799–3815. 10.5194/acp-18-3799-2018. [DOI] [Google Scholar]
- Lin P.; Bluvshtein N.; Rudich Y.; Nizkorodov S. A.; Laskin J.; Laskin A. Molecular chemistry of atmospheric brown carbon inferred from a nationwide biomass burning event. Environ. Sci. Technol. 2017, 51, 11561–11570. 10.1021/acs.est.7b02276. [DOI] [PubMed] [Google Scholar]
- Zhao R.; Lee A. K. Y.; Huang L.; Li X.; Yang F.; Abbatt J. P. D. Photochemical processing of aqueous atmospheric brown carbon. Atmos. Chem. Phys. 2015, 15, 6087–6100. 10.5194/acp-15-6087-2015. [DOI] [Google Scholar]
- Hems R. F.; Abbatt J. P. D. Aqueous phase photo-oxidation of brown carbon nitrophenols: Reaction kinetics, mechanism, and evolution of light absorption. ACS Earth Space Chem. 2018, 2, 225–234. 10.1021/acsearthspacechem.7b00123. [DOI] [Google Scholar]
- Chen J. Y.; Rodriguez E.; Jiang H.; Chen K.; Frie A.; Zhang H.; Bahreini R.; Lin Y.-H. Time-Dependent Density Functional Theory Investigation of the UV–Vis Spectra of Organonitrogen Chromophores in Brown Carbon. ACS Earth Space Chem. 2020, 4, 311–320. 10.1021/acsearthspacechem.9b00328. [DOI] [Google Scholar]
- Geyer A.; Alicke B.; Mihelcic D.; Stutz J.; Platt U. Comparison of tropospheric NO3 radical measurements by differential optical absorption spectroscopy and matrix isolation electron spin resonance. J. Geophys. Res. Atmos. 1999, 104, 26097–26105. 10.1029/1999JD900421. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.