

Abstract
Sanfilippo syndrome type B (mucopolysaccharidosis type IIIB) is a recessive genetic disorder that severely affects the brain due to a deficiency in the enzyme α-N-acetylglucosaminidase (NAGLU), leading to intra-lysosomal accumulation of partially degraded heparan sulfate. There are no effective treatments for this disorder. In this project, we carried out an ex vivo correction of neural stem cells derived from Naglu−/− mice (iNSCs) induced pluripotent stem cells (iPSC) using a modified enzyme in which human NAGLU is fused to an insulin-like growth factor II receptor binding peptide in order to improve enzyme uptake. After brain transplantation of corrected iNSCs into Naglu−/− mice and long-term evaluation of their impact, we successfully detected NAGLU-IGFII activity in all transplanted animals. We found decreased lysosomal accumulation and reduced astrocytosis and microglial activation throughout transplanted brains. We also identified a novel neuropathological phenotype in untreated Naglu−/− brains with decreased levels of the neuronal marker Map2 and accumulation of synaptophysin-positive aggregates. Upon transplantation, we restored levels of Map2 expression and significantly reduced formation of synaptophysin-positive aggregates. Our findings suggest that genetically engineered iNSCs can be used to effectively deliver the missing enzyme to the brain and treat Sanfilippo type B-associated neuropathology.
Keywords: Sanfilippo type B, MPS, LSD, neural progenitor cells, cell therapy
Graphical abstract
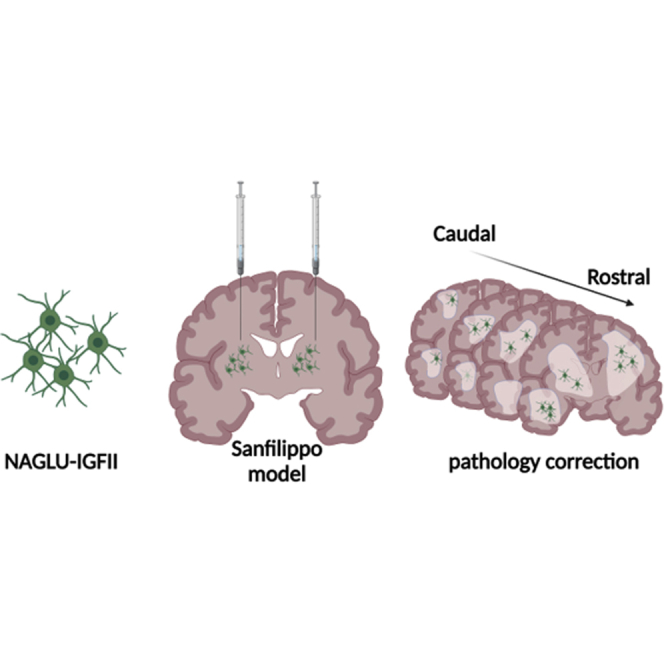
Neural progenitor cells overexpressing NAGLU-IGFII engraft and differentiate into neuron and astrocyte when implanted into Sanfilippo type B mouse model. Grafted cells secrete NAGLU-IGFII and correct pathology associated with Sanfilippo type B syndrome.
Introduction
Sanfilippo type B (mucopolysaccharidosis type IIIB, MPS IIIB), a sub-type of Sanfilippo syndrome, is a rare inherited lysosomal storage disease caused by the deficiency of the enzyme α–N-acetylglucosaminidase (EC 3.2.1.50 [NAGLU]), a lysosomal acid hydrolase involved in the breakdown of heparan sulfate (HS).1 HS glycosaminoglycans (GAGs) support multiple essential roles in cellular homeostasis such as ligand binding, cellular processes, and signaling pathway activation.2 Inherited mutations in the NAGLU gene result in a marked reduction of its enzymatic activity and the subsequent accumulation of HS-GAGs and monosialic gangliosides (GM2 and GM3) within lysosomes and other cellular compartments.3 This abnormal accumulation is thought to directly impair autophagy4 and endosomal trafficking,5 leading to a neuroimmune response affecting neuronal function and viability.6
Sanfilippo type B symptoms typically present in early childhood in the form of severe neurocognitive decline and progressive loss of speech and mobility, often leading to premature death before reaching adulthood.7 There are currently no effective treatments for Sanfilippo type B patients, beyond symptomatic or palliative approaches.8 To date, intra-cerebroventricular (ICV) enzyme replacement therapy (ERT),9 gene therapy,10 hematopoietic stem cell gene therapy,11 and substrate reduction12 have all been explored in preclinical studies as potential treatments for this disease. Although systemic ERT has shown promise in promoting functional recovery in MPS subtypes known to induce moderate neurological impairment (MPS I,13 II,14 IVA,15 and VII16), it poorly impacted both neurological deficit and cellular uptake in Sanfilippo type B17 because the recombinant human NAGLU (rhNAGLU) does not cross the blood-brain barrier.18 Currently, there is one ERT-based clinical trial (NCT03784287) testing the effect of the ICV implantation of an infusion port for direct delivery of the NAGLU enzyme to the brain. This trial was based on successful preclinical data9 and employs a fusion protein consisting of an IGFII receptor-binding peptide linked to the C-terminal of rhNAGLU in order to improve its cellular uptake via IGFII binding sites on the mannose-6-phosphate receptor (M6PR),9 in a glycosylation-independent lysosomal targeting fashion.19
Gene therapy represents a promising direct and long-term approach for the treatment of single-gene disorders such as Sanfilippo type B.20 To date, two distinct human clinical trials using adeno-associated viruses have been conducted using (1) a rAAV2/5-hNAGLU vector (NCT03300453), which showed amelioration of the disease,21 and (2) a rAAV2/9.CMV.hNAGLU vector (NCT03315182), which was terminated due to poor efficacy.
Hematopoietic stem cell22 transplantation is available as a treatment for MPS I patients with neurological symptoms. This approach was attempted for Sanfilippo type B23 but was not effective. A phase II clinical trial is currently ongoing in patients with Sanfilippo A syndrome (NCT04201405), in which hematopoietic stem cells are ex vivo corrected to overexpress the missing enzyme.
The direct implantation of neuronal stem cells (NSCs) into the CNS provides an alternative approach for sustained enzyme delivery to the brain and reduction of lysosomal storage, as shown in Sanfilippo type B mice,24 as well as in other lysosomal storage disorders.25,26,27 Recently this approach has also shown regenerative and therapeutic potential in neurodegenerative disorders such as Alzheimer disease.28 NSCs can be generated from patient induced pluripotent stem cells (iPSCs), allowing for autologous transplantation to improve compatibility and reduced risk for immune rejection.
Previously, we reprogrammed Naglu−/− mouse embryonic fibroblasts into iPSCs and corrected them ex vivo to overexpress the human NAGLU gene. We showed that corrected iNSCs (N-iNSCs) improved the brain neuropathology in Naglu−/− mice.24 Since hrNAGLU-IGFII is under clinical investigation for ERT, in this study, we have extended our previous work by using NAGLU-IGFII-secreting iNSCs (N-IGFII-iNSC) to determine whether NAGLU-IGFII can perform an effective cross-correction of Naglu−/− mouse neuropathology.
Results
iNSC cell engraftment and cell fate
To correct the Sanfilippo type B-associated genetic defects, we overexpressed full-length human NAGLU-IGFII cDNA in Naglu−/− iNSCs (N-IGFII-iNSC) (as previously described24), via lentiviral transduction. The viral construct carries a GFP reporter via internal ribosome entry site (IRES) sequence in order to facilitate the detection of engrafted cells within the brain. Following transduction, the cells were sorted using fluorescent-activated cell sorting to obtain pure un-silenced expression of NAGLU-IGFII (Figure S1A). We found that N-IGFII-iNSCs had a 4-fold increase in intracellular NAGLU-IGFII activity (p < 0.0001) and a 20-fold increase in secreted NAGLU-IGFII activity compared with unaffected cells (+/−) (p < 0.0001) (Figures S1B and S1C). Our construct was designed to address the limitation of poorly phosphorylated recombinant NAGLU-IGFII, which has previously demonstrated little to no intracellular uptake. To determine whether secreted NAGLU-IGFII from N-IGFII-iNSCs was able to enter Naglu−/− cells through mannose-6-phosphate (M6P) or IGFII-dependent signaling, we carried out cellular uptake assays using an excess amount of M6P and/or IGFII to inhibit NAGLU-IGFII uptake via M6P receptors (M6PR). We harvested the supernatant from N-IGFII-iNSCs iNSCs and applied it to Naglu−/− iNSCs in the presence of 5 mM M6P and 1 μM IGFII. After 4 h of treatment, we found a 3.5-fold increase in NAGLU-IGFII activity compared with Naglu−/− levels. Intracellular uptake was partially inhibited by M6P and completely inhibited by the presence of IGFII or a combination of both (Figure S1D). These data provide evidence that N-IGFII-iNSCs are capable of cross-correcting Naglu−/− cells through the secretion of sufficiently phosphorylated NAGLU-IGFII and M6PR-dependent signaling. To evaluate their therapeutic potential in vivo, we implanted corrected iNSCs into the brain of newborn mice and evaluated the long-term effect (9 months) of the transplantation on the disease phenotype. We have previously reported a higher level of iNSC engraftment at 9 months compared with 2 months post-transplantation, possibly due to continuous cell proliferation over the initial period after implantation.24 Therefore, we decided to focus this study on the longer-term effect at 9 months. First, we determined the post-transplantation fate of GFP-positive N-IGFII-iNSC in vivo using immunofluorescence staining of multiple phenotypic markers for NeuN and Map2 (neurons), GFAP (activated astrocytes), and O4 (oligodentrocytes) (Figure 1). We found that GFP-positive cells engrafted in the forebrain of Naglu−/− mice co-localized with NeuN, Map2, GFAP, and to a lower extent with O4, indicating that N-IGFII-iNSC cells retained their multipotency upon engraftment into the diseased brain and were capable of differentiating into neurons, astrocytes, and oligodentrocytes, while some GFP-positive iNSCs did not commit to either cell type.
Figure 1.

NAGLU-IGFII-corrected iNSC (N-IGFII-iNSCs) engraftment and cell fate 9 months after transplantation
Immunofluorescence staining of corrected N-IGFII-iNSCs in Naglu−/− brain slices (40-μm) positive for green fluorescent protein (GFP) (green). GFP-positive N-IGFII-iNSCs co-localized with Map2, NeuN, GFAP and oligodendrocyte marker O4. Nuclear staining with DAPI (blue) was overlaid. Scale bars represent 50 μm.
Pattern of iNSC cell engraftment and N-IGFII enzyme activity
Next, we evaluated the engraftment of N-IGFII-iNSCs by measuring the percentage area of GFP immunoreactivity along the rostrocaudal axis (Figure 2A). Transplanted GFP-positive cells were detected in all 12 mice, with a percentage area of GFP immunoreactivity measured for the whole hemisphere in each mouse. Expressed as a percentage of total area, the area of GFP immunoreactivity ranged from 0.0001% to 25.99%, with six mice displaying values of 0.25% and above (0.25%, 1.16%, 1.75%, 1.75%, 2.45%, 25.99%), while six mice showed less than 0.25% engraftment (Figure 2B). The engrafted iNSCs migrated from the site of injection to spread within four brain regions along the rostrocaudal axis. To evaluate the spread of transplanted cells, we chose three animals representing different patterns of N-IGFII-iNSC distribution regardless of their level of engraftment (Figure 2C). In animal #4763 (1.75% GFP area), the N-IGFII-iNSCs engrafted sporadically throughout the brain, but proportionally more cells engrafted within the caudal part of the midbrain in the substantia nigra. In contrast, animal #4871 (25.99% GFP area) showed a high number of cells located in the olfactory bulb with gradually decreasing number toward the midbrain, with a higher number of cells detected in the substantia nigra. In animal #4872 (1.16% GFP area), higher engraftment was observed within the cerebral cortex, through the rostral portion of the hippocampus, and in the substantia nigra (Figure 2C). Consistent with the distribution of engrafted iNSCs, we detected NAGLU-IGFII enzymatic activity in all 12 mice ranging from 0.007 to 0.492 units/mg protein (mean average enzymatic activity in the brain). Studies have shown that 1%–5% of wild-type enzyme activity is sufficient to correct storage accumulation in lysosomal storage disorders.29 In our study, all experimental animals reached 10% of carrier level (0.00775 units/mg protein red line) (Figure 2D). The overall enzyme activities correlated (Pearson R = 0.84) with the level of iNSC engraftment into the brain, as determined by assessing the area of GFP staining (Figure 2E).
Figure 2.

Engraftment and enzyme activity evaluation after 9 months post-transplantation
(A) Schematic illustration of a sagittal brain section (lateral 0.24 mm bregma) adapted from The Mouse Brain in Sterotaxic Coordinates (second edition) by Paxinos and Franklin.51 Black, spotted, striped, and plain white bars above schematic diagram correspond to specific regions of analysis along the rostrocaudal axis shown in (C). (B) Histogram showing total percentage area (%) (sum) GFP staining in each engrafted animal plotted along the x axis (12 animals in total). (C) Histogram showing GFP percentage area stained in three representative animals (#4763, #4871, #4872) for each individual section through the rostrocaudal axis, at the level of the olfactory bulb (black bars), cerebral cortex through to the beginning of the hippocampus (spots), thalamus (stripes), and midbrain (plain white). (D) Enzymatic activity of NAGLU-IGFII (N-IGFII) shown in units per milligram of protein, performed on brain lysates (mean average of the four sections depicted in F) following injections with N-IGFII-iNSCs in all 12 animals, using the NAGLU enzymatic activity in Naglu−/−mice as a background signal. Broken red line shows protein level at 100% of the carrier level; solid line shows 10% of the carrier level. (E) Correlation between level of engraftment and enzymatic activity of NAGLU-IGFII (R = 0.83) in transplanted animals. (F) Schematic diagram depicting how the brains were divided for each animal. Brains were dissected sagittally along the midline first, and then right hemisphere was further sectioned into four slices as shown. The black dots represent the approximate intraparenchymal injection sites. (G) Graph showing the percentage (Log10) of carrier-level enzymatic activity of NAGLU-IGFII in four segments (depicted in F) along the rostrocaudal axis in 12 engrafted brains. Brain segments 1 and 2 (1 + 2), and 5 and 6 (5 + 6) were pooled. Red dotted line represents carrier level (Log10), solid colored lines represent each engrafted brain. Values are shown as mean ± SEM of enzyme activity measured in each slice (n = 12).
Since our analysis of GFP-positive cells revealed that N-IGFII-iNSCs do not distribute along the rostrocaudal axis of the brain equally, we divided the right hemisphere into six segments along the rostrocaudal axis, including the cerebellum and the brain stem, and we measured the NAGLU-IGFII enzymatic activity in each of these segments (Figure 2F). Overall, the majority of injected mice displayed enzyme activity above 10% of carrier level (depicted by dotted red line) in all brain sections and ranged from 12%–3,285.7% of the carrier control (Figure 2G).
Effect of N-IGFII-iNSCs on glial activation and storage accumulation
Glial activation is a characteristic feature of the neuropathology observed in both Sanfilippo type B patients and Naglu−/− mice, and it is known to intensify with age.6,10 To determine the impact of engrafted N-IGFII-iNSCs upon the activation of microglia and astrocytes as part of the innate immune response, we stained sections of (1) unaffected Naglu+/− control mice (Unaffected +/−), (2) vehicle-injected Naglu−/− (Vehicle −/−), and (3) N-IGFII-iNSC-grafted Naglu−/− (T (N-IGFII)−/−) for the markers CD68 (microglia) and GFAP (astrocytes).
To determine how much impact N-IGFII-iNSC grafts had upon the neuropathology of Sanfilippo B, we carried out threshold image analysis of CD68, GFAP, and LAMP1 staining along the entire rostrocaudal axis. We found significantly increased CD68 and GFAP immunoreactivity in the forebrain of vehicle-injected Naglu−/− mice (Vehicle −/−), compared with control mice (Unaffected +/−) (Figures 3A and 3B), data that are consistent with our previous findings.24 We found a 72-fold increase of the CD68 staining in vehicle-injected mice compared with unaffected mice (p < 0.0001). However, the brain transplantation of N-IGFII-iNSC was associated with a 1.84-fold reduction of CD68 immunoreactivity in treated Naglu−/− mice compared with either vehicle-injected Naglu−/− mice (p = 0.0360) or with unaffected mice (p = 0.0193, Figure 3A). A similar trend was observed in GFAP immunoreactivity, with a 2.2-fold increase in vehicle-treated Naglu−/− mice compared with unaffected mice (p = 0.0007). Meanwhile, N-IGFII-iNSCs treatment of Naglu−/− mice was associated with reduced GFAP immunoreactivity compared with vehicle-treated Naglu−/− mice, resulting in GFAP immunoreactivity at comparable levels to those found in unaffected mice. These results indicate that N-IGFII-iNSCs have an overall suppressive effect on the glial reaction typically associated with Sanfilippo type B (Figure 3B).
Figure 3.

Correction of neuropathology and storage accumulation in Naglu−/− mice treated with NAGLU-IGFII-corrected iNSC (N-IGFII-iNSCs) at 9 months
Histograms (A–C) showing the percentage area of CD68 (B) and GFAP (C) immunoreactivity, and mean intensity of LAMP1 (D) immunoreactivity in every one-in-twelve sections, including all regions within the forebrain, for each group measured. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001 ∗∗∗∗p ≤ 0.0001; two-tailed, unpaired parametric t test. Values are shown as mean ± SEM (n = 3 mice per group). Representative images taken within the striatum (below). (D) Low magnification of representative bright-field images of coronal sections for representative vehicle-treated (Vehicle −/−) mice compared with three representatives N-IGFII treated (T (N-IGFII)−/−) mice to show the level of variation of pathology correction. Sections were taken at three levels along the rostrocaudal axis: rostral (R; at the level of the isocortex and olfactory areas), middle (M; at the level where the fimbria of the hippocampus appears), and caudal (C; at the level of the midbrain) of mice from engrafted Naglu−/− mice, and immunohistochemically stained for CD68 (first block), GFAP (second block), and LAMP1 (third block). 1–3 represent three different Naglu−/− mice injected with N-IGFII-iNSCs to demonstrate variation in CD68, GFAP, and LAMP1 immunoreactivity within the same treatment group. Red broken lines outline the areas where CD68 and GFAP immunoreactivity is reduced. (E) Representative adjacent sections from an engrafted Naglu−/− mouse immunohistochemically stained for GFP (top, color image) and CD68 (bottom, color image) taken at the rostral, medial, and caudal part of the brain. Red squares highlight the part that contains GFP staining and reduced CD68 staining, respectively. (F) Areas where CD68 staining was absent (no pixels) were modified to appear transparent and positioned on top of the corresponding GFP-stained section (black) as an overlay. Post-acquisition processing was applied to all images and included adjustments to brightness and contrast and RGB curves using Adobe Photoshop CS6 to improve visibility and consistency in color tone. Scale bars represent 200 μm (A-C), 1,000 μm (D).
LAMP1 is a lysosomal membrane protein that can be used as a surrogate marker for lysosomal storage accumulation.9 As expected, LAMP1 immunoreactivity was significantly greater (1.7-fold) in vehicle-injected Naglu−/− mice compared with unaffected controls (p = 0.0085). Strikingly, treatment with N-IGFII-iNSCs reduced LAMP1 immunoreactivity to levels comparable with those found in unaffected mice (p = 0.0080, Figure 3C). In addition, correction of the neuroimmune response and storage accumulation was most pronounced closer to the sites of engraftment (the olfactory bulb, white matter tracts, the ventral forebrain, and the substantia nigra), while showing varying degrees of positive impact beyond the site of engraftment (Figure 3D).
To determine the relationship between engraftment and correction of neuropathological changes, we compared the distribution of immunoreactivity of GFP-transplanted cells and of CD68 immunoreactivity in adjacent section. (Figure 3E). Next, GFP-stained images (imaged colored in black for contrast) were overlayed on CD68-stained section. We found a clear overlap between sites of engraftment that contained more GFP immunoreactivity and less CD68 immunoreactivity (Figure 3F).
Effect of N-IGFII-iNSCs on cytoskeletal pathology and synaptophysin aggregation in Naglu−/− mice
We have demonstrated that grafted N-IGFII-iNSCs positively impact the glial activation typically associated with Naglu deficiency. However, the human Sanfilippo type B syndrome is a neurodegenerative disorder that ultimately results in pronounced neuronal loss and brain atrophy.30 This extent of neuronal loss is not recapitulated in the Naglu−/− mouse model.6 Therefore, to provide a measure of neuronal integrity, we stained for the neuron-specific cytoskeletal protein microtubule-associated protein 2 (Map2), known to play an important role in stabilizing microtubules in dendritic processes.31 Downregulation of Map2 has been consistently reported in the studies using other neurodegenerative mouse models.32 To assess whether Naglu−/− mice display a similar phenotype, we quantified Map2 levels in whole brain by western analysis, but we did not detect any differences between unaffected, diseased, and N-IGFII-iNSC-treated animals (Figure 4A). Because regional effects on neuron integrity would be masked in whole-brain extracts, we explored the levels of Map2 via immunostaining sections (Figure 4B). We found a clear downregulation of Map2 signal in vehicle-treated Naglu−/− mice compared with unaffected mice in the hippocampus, which displays intense Map2 staining in unaffected animals (higher magnification images in red box). Treatment with N-IGFII-iNSC cells restored Map2 signals to levels similar to those found in unaffected controls (Figure 4C). Quantifying these effects revealed that Map2 immunoreactivity was significantly decreased in the hippocampus of vehicle-treated Naglu−/− mice compared with unaffected mice (p = 0.0017). Upon N-IGFII-iNSC treatment, Map2 signal was restored to levels comparable and not significantly different from those in unaffected mice. Our data provide novel evidence that Map2 is downregulated in the hippocampus of Naglu−/− mice. This may be associated with cytoskeletal dysfunction or disruption but is largely prevented by N-IGFII-iNSC treatment.
Figure 4.

Correction of Map2 downregulation in Naglu−/− mice at 9 months
(A) Map2 protein levels in whole-brain (n = 6 for each group) measured by western blot and quantification in heterozygous control mice (Unaffected) and Naglu−/− mice injected with vehicle (Vehicle −/−) or N-IGFII-iNSCs (T (N-IGFII)−/−). Red square shows the expected Map2 band size. (B) Representative bright-field images of 40-μm-thick half-brain coronal sections from immunohistochemically stained for Map2 in animal groups as in (A). Panels depicted by R, M, C show sections taken at three levels along the rostrocaudal axis: rostral (R; at the level of the isocortex and olfactory areas), middle (M; at the level where the fimbria of the hippocampus appears), and caudal (C; at the level of the midbrain). Panel highlighted in red box below shows high magnification images of hippocampus depicted for each group. Post-acquisition processing was applied equally to all images and included adjustments to brightness and contrast and RGB curves using Adobe Photoshop CS6 to improve visibility and consistency in color tone. Scale bars represent 1,000 μm (top) and 200 μm (bottom). (C) Histograms show the optical density (DAB staining intensity optical density) of Map2 immunohistochemical staining in the hippocampus of animal groups in (A). ∗∗p ≤ 0.01, two-tailed, unpaired parametric t test. Values are shown as mean ± SEM (n = 3 mice per group).
Synaptophysin is an abundant integral membrane glycoprotein that is found in presynaptic vesicles of neurons. It has been previously reported that the accumulation of synaptophysin in the corpus callosum is a reliable marker of axonal damage in inflammatory conditions.33 We found many intensely stained clusters of synaptophysin immunoreactivity in vehicle-treated Naglu−/− mice compared with both unaffected control mice and N-IGFII-iNSC-treated mice. These synaptophysin aggregates were most noticeable in the hippocampus, cortex, striatum, and entorhinal cortex of untreated mice (Figure 5A). To quantify these aggregates, we performed thresholding image of synaptophysin immunoreactivity based on these darkly stained spheroids and calculated the percentage area (sum) of synaptophysin-positive aggregates. Synaptophysin immunoreactivity was significantly greater (3-fold) in vehicle-injected Naglu−/− mice compared with unaffected controls (p = 0.0037). Interestingly, treating Naglu−/− mice with N-IGFII-iNSCs reduced this level of synaptophysin immunoreactivity to similar levels observed in unaffected carrier mice, leading to significantly different values compared with vehicle-treated Naglu−/− mice (p = 0.0146, Figure 5B). Next, we evaluated whether these synaptophysin aggregates accumulate within the lysosome of Naglu−/− mice. We found that large synaptophysin aggregates co-localized with LAMP1 staining (Figure 5C, white arrows) in untreated Naglu−/− mice, suggesting a lysosomal-mediated synaptophysin accumulation and mechanism of synaptic failure.34 Alternatively these aggregates may end up in the lysosome and may fail to be broken down.
Figure 5.

Correction of synaptophysin aggregation in Naglu−/− mice at 9 months
(A) Representative bright-field images of 40-μm-thick half-brain coronal sections from Naglu+/− mice (Unaffected) and Naglu−/− mice injected with saline (Vehicle −/−) or N-IGFII-iNSCs (T (N-IGFII) −/−) immunohistochemically stained for synaptophysin. High magnification images, panel below, show synaptophysin-positive aggregates (blue arrows) in the cortex, hippocampus, striatum, and entorhinal cortex (EC) of all treatment groups. Post-acquisition processing was applied equally to all images and included adjustments to brightness and contrast and RGB curves using Adobe Photoshop CS6 to improve visibility and consistency in color tone. Scale bars represent 1,000 μm low magnification, 200 μm higher magnification. (B) Histograms showing the percentage area of synaptophysin immunoreactivity in every one-in-twelve sections including all regions within the forebrain for each group measured. ∗p ≤ 0.0146, ∗∗p ≤ 0.0037; two-tailed, unpaired parametric t test. Values are shown as mean ± SEM (n = 3 mice per group). (C) Immunofluorescence staining showing co-localization (yellow) of LAMP1 (green) and synaptophysin (red) in 40-μm-thick half-brain coronal sections from Naglu−/− mice injected with vehicle (Vehicle −/−). Scale bars represent 50 μm.
Together, our data show an overall reduction of synaptophysin aggregates in N-IGFII-iNSC engrafted mice compared with vehicle-injected Naglu−/− mice. These results reveal a novel neuronal phenotype in Naglu−/− brains in which synaptophysin-positive aggregates alongside LAMP1 immunoreactivity represent markers of neuropathological outcome that can be used to evaluate the therapeutic efficacy of engrafted corrected iNSCs.
Discussion
Sanfilippo type B is caused by a deficiency in the enzyme NAGLU, known to be involved in the breakdown of HS, resulting in the accumulation of partially degraded GAGs in the lysosome. Pathological abnormalities in Naglu−/− brains provide a way to study the disease mechanisms that operate in Sanfilippo patients as Naglu−/− mice display abnormal lysosomal pathology, a pronounced neuroimmune response, and severe neuronal dysfunction.35,36 Previously, we showed that NAGLU produced in vitro by our N-iNSCs can be endocytosed by Naglu-deficient cells via the M6PR and cross-correct Sanfilippo-associated microglial activation, astrocytosis, and lysosomal storage accumulation in vivo.24 The current study shows that cross-correction can also be achieved using a modified NAGLU-IGFII enzyme. However, despite the use of NAGLU-IGFII, this modified enzyme did not completely correct the microglial activation. This result implies that limited cross-correction (due to insufficient enzyme distribution) might require more widespread engraftment of iNSCs or pharmacological means to dampen the neuroimmune response. Therefore, widespread dissemination of iNSCs or a combinatorial approach using an anti-inflammatory drug may prove more effective.
Previous studies on the post-transplantation fate of iNSCs in the adult brain have focused on direct migration toward focal areas of injury, e.g., ischemia or brain tumors.37,38 However, little is known about the way iNSCs migrate within the CNS following intraparenchymal transplantation into brains that have genetic deficiency rather than a physical lesion. Although transplanted N-IGFII-iNSCs were detected throughout the rostrocaudal axis in the majority of transplanted brains 9 months after treatment, their pattern of engraftment was highly variable, which can be attributed to the intrinsic characteristics of the donor cells. Overall, N-IGFII-iNSCs were found mostly within the ventral forebrain and substantia nigra (Figure 2). Future studies will be needed in order to evaluate the normal migratory behavior of transplanted N-IGFII-iNSCs in the brain of wild-type neonatal mice. In addition, additional safety studies will be required to assess the proliferation rate and cell death rate of donor cells in transplanted mice.
Map2 is a cytoskeletal protein found in the neuronal dendritic compartment where it plays an important role in the morphological stabilization of dendritic processes. It is a marker of structural integrity, and its expression has been linked to dendritic outgrowth, branching, and post-lesion remodeling and plasticity,39 known to play an important role in memory in the hippocampus.40 Downregulation of Map2 expression has been linked with impaired microtubule assembly and has been found in the hippocampus of the aging rat brain and Alzheimer disease.32,41 In our study, a reduced Map2 expression was significantly found within the hippocampus of Naglu−/− mice. Severe dementia is one of the primary characteristics of Sanfilippo type B disease in humans, and Naglu−/− mice perform poorly in water maze and radial-arm maze behavioral tests, indicating an impairment in spatial learning.42 Therefore, Map2 downregulation in the hippocampus of Naglu−/− mice may partially explain the dementia-like symptoms reported in Sanfilippo type B disease.7
Aggregation of abnormal cellular proteins into insoluble complexes is involved in the pathogenesis of most human neurodegenerative diseases.43 Partially degraded HS is the primary storage product in Naglu-deficient lysosomes. In addition, Naglu-deficient neurons were found to accumulate GM3 ganglioside,3 ubiquitin, and subunit c of mitochondrial ATP synthase.44 However, the mechanism by which abnormal protein aggregation leads to the degradation of different neuronal populations is not fully understood. In this study, we identified large aggregates of synaptophysin in the cells of Naglu−/− mice, a phenotype that has not previously been reported. Synaptophysin aggregation has been described as a reliable marker for axonal damage in conditions such as multiple sclerosis, CNS trauma, and multiple neuroinflammatory conditions.33 Decreased synaptophysin expression has also been reported in model of neurodegenerative disease.45 A previous study has demonstrated that synaptophysin expression is decreased in Sanfilippo type B as a result of storage material accumulation, and it can be reversed following correction by gene transfer. Several studies have reported that synaptophysin levels were generally reduced in the rostral cortex of Sanfilippo type B mice compared with controls as early as 10 days and that this reduction could not be accounted for by neuron loss.46 In addition, published literature has showed that synaptophysin does not accumulate in the lysosome and that dysregulation of synaptophysin levels is a result of enhanced proteasomal degradation induced by GAGs.6,46 In our study, we unexpectedly found that large synaptophysin-positive aggregates were present in multiple brain regions, which could be explained by the impaired degradation of this protein, despite a downregulation of its gene expression. The thresholds used in our image analysis were not set to detect the generalized downregulation of synaptophysin that occurs in Sanfilippo type B brains, instead to quantify the presence of the large intensely stained synaptophysin deposits that appear to accumulate within lysosomes. Studies suggest that the formation of such cytoplasmic inclusion bodies requires active, retrograde transport of aggregates or misfolded protein on microtubules. A microtubule-based apparatus, the aggresome, forms and isolates protein aggregates within the cytoplasm.47 Our Map2 and synaptophysin data suggest that the downregulation of Map2 observed in our study may be indicative of microtubule dysfunction that results in the aggregation of synaptophysin.
We previously transplanted mice with NAGLU-expressing iNSC.24 In this previous study, we assessed engraftment and distribution at 2 and 9 months post-transplantation and detected more engrafted cells and a broader dispersion throughout the brain at the 9-month time point, most likely due to higher proliferation of transplants over the course of weeks. In addition, we found that at this 9-month time point, almost complete correction of neuropathology was observed in some brain regions of low engraftment, an observation from which we concluded that the corrective capability of engrafted cells relies on the total level of engraftment throughout the brain rather than the precise distribution of the cells. One strong argument is found in the correlation between total cell engraftment and amount of secreted enzyme that is then available to cross-correct neighboring cells. Our approach using a modified NAGLU-IGFII enzyme reveals that this fusion protein can also be secreted and taken up, but the efficiency of cross-correction is potentially limited by the engraftment and dissemination of iNSCs within the postnatal brain. While 1%–5% of enzymatic activity may be sufficient to prevent storage accumulation (Figure 3C), it appears that higher levels of NAGLU-IGFII enzyme activity will be needed to counteract the Sanfilippo-associated neuroimmune response throughout the whole brain. As such, despite the use of NAGLU-IGFII, the amount of cross-correction provided was not sufficient to improve overall efficacy. It may be necessary to improve overexpression of NAGLU using a more powerful promoter, together with the use of the modified NAGLU-IGFII enzyme.
Despite N-IGFII-iNSC engrafted mice having a low level of chimerism, grafted cells distributed along the entire rostrocaudal axis and were capable of providing comparable levels of NAGLU-IGFII activity to those found in unaffected brains. This finding suggests that NAGLU-IGFII can be secreted at sufficient levels and distributed widely in order to potentially cross-correct neuropathological changes. This cross-correction was also reflected in positive impact upon multiple features of disease including glial activation, storage material accumulation, microtubular integrity system, and the aberrant accumulation of synaptic proteins. In conclusion, we have shown that the successful engraftment of iNSCs expressing a modified NAGLU protein can be achieved and is capable of partly preventing the accumulation of storage material and glial activation in 9-month post-treated Naglu−/− mice. We have also identified novel features of neuronal pathology in the form of Map2 immunoreactivity and synaptophysin accumulation that can be used to more rigorously assess the efficacy of potential treatments.
Material and methods
Lentivirus production
The human NAGLU gene ( Accession Number NM_000263.4) was engineered from a previously published NAGLU-IGFII48 sequence and further cloned into a lentiviral backbone plasmid, pSAM-GFP,24 at the XhoI and NotI sites. The resulting expression vector was named pSAM-NAGLU-IGFII-IRES-GFP. The lentiviral supernatant was produced with 293T cells (American Type Culture Collection, Manassas, Virginia) as described previously.24
Directed differentiation of iPSCs to iNSCs
iPSCs (previously obtained in Clarke et al.24) were grown feeder-free at a density of 8.0 × 104 cells/well in embryonic stem cell medium (knockout DMEM; Gibco, Carlsberg, CA) with 10% fetal bovine serum (FBS) (R&D systems, Minneapolis, MN) and 500 U/mL leukemia inhibitory factor (EMD Millipore, Burlington, MA). The following day, the medium was changed to NSC medium (DMEM/F12 supplemented with B27 without vitamin A and N2; Gibco, Carlsbad, CA) and grown for 12 days. On day 12, NSC medium, 20 ng/mL of epidermal growth factor, and 20 ng/mL basic fibroblast growth factor (bFGF) were added (PeproTech, Rocky Hill, NJ). Cells were expanded in NSC medium as monolayers.24
Enzyme activity assays
The catalytic activity of NAGLU-IGFII was determined by hydrolysis of the fluorogenic substrate 4-methylumbelliferyl-N-acetyl-α-glucosaminide (EMD Millipore, Burlington, MA) with a final concentration of 0.1 mM substrate in the incubation mixture as described previously.48 A unit of activity is defined as the release of 1 nmol of 4-methylumbelliferone (4MU) per hour at 37°C. Naglu activity signal in Naglu−/− cell and in Naglu−/− brain was used as background and subtracted from all other measurements. Protein concentration was estimated by the Bradford method, using BSA (Bio-Rad, Hercules, CA) as the standard. Fluorescence was measured on a microplate reader (Spectramax Paradigm, Molecular Devices, San Jose, CA) with λex 365 nm and λem 445 nm. Intracellular NAGLU-IGFII activity is presented as units per milligram of protein.
Experimental animals
Animal experiments were approved by the Institutional Animal Care and Use Committee at the Lundquist Institute at Harbor-UCLA Medical Center, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. The Naglu−/− knockout mouse was a gift from Dr. Neufeld from the University of California, Los Angeles (UCLA) and was back-bred onto C57BL6/J49 and maintained in Lundquist Institute. Genotyping was performed with the following primers: NAG5′, 5′-TGGACCTGTTTGCTGAAAGC-3′; NAG3′, 5′-CAGGCCATCAAATCTGGTAC-3′; Neo5′, 5′-TGGGATCGGCCATTGAACAA-3′; and Neo3′, 5′-CCTTGAGCCTGGCGAACAGT-3′. Naglu+/− females were crossed with Naglu−/− males to obtain homozygous affected mice and heterozygous (unaffected) controls. Experiments were performed on age-matched mice (usually littermates) of either gender.
Injection of iNSCs
2.5 × 105 NAGLU-IGFII-overexpressing iNSCs (N-IGFII-iNSC) in 3 μL of PBS or PBS alone were injected bilaterally into the striatum of postnatal day 0 (P0) or P1 Naglu−/− neonatal mice under cryo-anesthesia by Hamilton syringe with 32G needles. The injection site was approximately 3.0 mm rostral from bregma, 4.0 mm laterally from the midline, and at a depth of 3.0 mm as previously described.24 Injections were performed manually by the same experimenter using bregma and lamda landmarks. A plastic block was attached to the end of the syringe. The length of the needle entering the brain was limited by contact of the block with the top of the skull, thus ensuring the depth of injection was fixed and reproducible between injections.
Tissue harvesting, processing, and histological staining
After 9 months post-transplantation, brains were dissected sagittally along the midline, and left hemispheres were post-fixed overnight at 4°C in 4% paraformaldehyde (PFA) before dehydration and cryoprotection at 4°C in a solution of 30% sucrose in Tris-buffered saline (TBS). The right hemispheres were further sectioned into 2-mm-thick coronal slices using an adult mouse brain slicer matrix (Zivic Instruments, Pittsburgh, PA) and rapidly frozen and stored at −80°C until performing NAGLU-IGFII activity assay. For the fixed hemisphere, 40-μm frozen coronal sections were cut through the rostrocaudal extent of the cortical mantle (Microm HM 430 freezing microtome, Thermo Fisher Scientific, Waltham, MA). Sections were collected in a cryoprotectant solution (30% ethylene glycol/15% sucrose/0.05% sodium azide in TBS) and stored at 4°C before histological processing.24,50
Western blotting
Protein samples and dual plus molecular weight ladders were separated by SDS-PAGE and loaded on Precast stain-free gels with a 4%–15% gradient (Bio-Rad Hercules, CA). Proteins were transferred to PVDF membranes (Bio-Rad Hercules, CA) using the Bio-Rad Trans-Blot Turbo Transfer System (Bio-Rad Hercules, CA). Total proteins on membranes were detected using the ChemiDoc MP system (Bio-Rad Hercules, CA) gel-free staining. Membranes were blocked with 5% non-fat milk in TBS-T and incubated with primary antibody rabbit Anti-Map2 (1:1,000 dilution, Abcam, Cambridge, MA, catalog no. ab183830). Anti-Rabbit-HRP (Invitrogen G-21234) was used as a secondary antibody. Membranes were analyzed using ChemiDoc MP (Bio-Rad Hercules, CA).
Immunohistochemistry
To determine the identity of engrafted stem cells, single free-floating frozen sections were immunohistochemically stained using the following antibodies (all from Invitrogen Carlsbad, CA except where specified): Rabbit anti-GFP (1:10,000 dilution, catalog no. A10259); Rat anti-GFAP (1:400 dilution, catalog no. 13-0300). Secondary detection was performed with Streptavidin DyLight 488 nm (1:200 dilution, Vector Laboratories, Burlingame, CA, catalog no. SA-5488), Goat anti-Rat Alexa 546 nm (catalog no. A-11081), and Goat anti-Chicken Alexa 633 nm (catalog no. A-11040). All secondary antibodies were used at 1:500 dilution, with the exception of Streptavidin DyLight 488 nm. To examine the level of stem cell engraftment, the extent of glial activation, storage material accumulation, and neuronal markers, adjacent one-in-six (GFP) and one-in-twelve (GFAP, CD68, LAMP1, Map2, NeuN, synaptophysin) series of free-floating frozen sections were immunohistochemically stained using a standard immunoperoxidase staining protocol, described previously.24,50 GFP (as above), polyclonal rabbit anti-GFAP (1:8,000 dilution, Agilent Technologies, Santa Clara, CA; catalog no. Z0334), rat anti-CD68 (1:2,000, Bio-Rad, Hercules, CA; catalog no. MCA1957), rat anti-LAMP1 (1:4,000, DSHB, Iowa City, IA; catalog no. 1D4B), rabbit Anti-Map2 (1:40,000 dilution, Abcam, Cambridge, MA, catalog no. ab183830), Mouse anti-NeuN (1:100 dilution, EMD Millipore, Burlington, MA), and Rabbit anti-synaptophysin (1:2,000 dilution, Abcam, Cambridge, UK, catalog no. ab14692) were used, followed by the appropriate secondary antisera (all from Vector Laboratories, Burlingame, CA, except specified): biotinylated goat anti-rabbit IgG (#BA-1000), biotinylated swine anti-rabbit IgG (GFAP, Agilent Technologies, Santa Clara, CA; #E0353), biotinylated rabbit anti-rat IgG (CD68, catalog no. BA-4001), and biotinylated goat anti-rat IgG (LAMP1, #BA-9400), all 1:1,000 dilution. Sections were incubated in an avidin-biotin-peroxidase complex kit (Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, CA, #PK-6100). Immunoreactivity was visualized by incubation in 0.05% 3′, 3′-diaminobenzidine (DAB; Sigma-Aldrich, St. Louis, MO) and 0.01% H2O2 in TBS.
Quantitative analysis
To analyze the percentage area of engraftment (GFP/CD68/GFAP/LAMP1), all stained sections were scanned as follows: slides were imaged field by field at a magnification of ×10 (GFP, CD68, and GFAP) and ×20 (LAMP1), using a Zeiss Axio Imager M2 microscope and an automated stage (MBF Bioscience), then stitched using StereoInvestigator software (MBF Bioscience, Williston, VT). Thresholding analysis was performed with Image Pro-Premier software (Media Cybernetics, Rockville, MD) to report percentage area staining (sum) as described previously.50 The optical density (Intensity) was reported for Map2 and was defined by two intensity values where darker staining is read as opaque and no staining as transparent. The standard optical density formula, which assumes that light decays exponentially as it goes through light-transmitting material (OD(x,y) = –log ((I(x,y) – BL)/(IL – BL)), where BL and IL are the black level and the incident level respectively, and I(x,y) is the intensity at location x,y) can be used to create an intensity curve where the darkest staining (black pixels) equals 0 and the lightest staining (white pixels) equals 255. The regions of interest were defined according to neuroanatomical landmarks described by Paxinos and Franklin.51 For each sample, the appropriate threshold was applied to capture the different staining intensities for each of the areas stained (optical density).
Each section was outlined as a region of interest and analyzed together as one whole. For each sample, the appropriate threshold was applied to capture the different staining intensities for each of the areas stained according to color, background, and morphology.
For the overlay of GFP-stained sections and CD68 section, areas where CD68 staining was absent (no pixels) were modified to appear transparent and positioned on top of the corresponding GFP-stained section (black color) as an overlay using Adobe Photoshop CS6.
Statistical analysis
All statistical analyses were performed using Prism software (GraphPad, La Jolla, CA). A two-tailed, unpaired, parametric t test was used when two groups were compared. Results were considered statistically significant when p < 0.05. Unless otherwise stated, statistical comparisons are between untreated Naglu−/− mice and unaffected heterozygous controls and the treatment groups and untreated Naglu−/− mice. One-way ANOVA was used to compare groups greater than two.
Data and material availability
All data and material are available upon request.
Acknowledgments
We thank members of Dr Cooper’s Pediatric Storage Disorders Laboratory for helpful discussion and their technical support. This work was supported by grants from the NINDS (1R41NS092221-0181, 1R01 NS088766, 4R33NS096044-03), a traineeship from 5T32 GM8243-28 (to S.-h.K.) and by the Cure Sanfilippo Foundation.
Author contributions
Conceptualization: M.I. and P.I.D.; methodology: M.I., P.I.D., and J.D.C.; validation: Y.P., D.C., and S.-h.K; formal analysis: Y.P., D.C., S.-h.K., A.L., and L.R.N.; investigation: Y.P., D.C., S.-h.K., S.Q.L., V.S., I.P., and A.L.; resources: M.I., P.I.D., and J.D.C.; data curation: Y.P., D.C., S.-h.K.; writing – original draft, Y.P., D.C., and M.I.; writing – review & editing, Y.P., M.I., P.I.D., J.D.C., L.N., and S.-h.K.; visualization, Y.P.; supervision, M.I., P.I.D., and J.D.C.; project administration, M.I. and P.I.D.; funding acquisition: M.I. and P.I.D.
Declaration of interests
The authors declare no competing interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.10.013.
Supplemental information
References
- 1.Valstar M.J., Ruijter G.J.G., van Diggelen O.P., Poorthuis B.J., Wijburg F.A. Sanfilippo syndrome: a mini-review. J. Inherit. Metab. Dis. 2008;31:240–252. doi: 10.1007/s10545-008-0838-5. [DOI] [PubMed] [Google Scholar]
- 2.Iozzo R.V., Schaefer L. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55. doi: 10.1016/j.matbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGlynn R., Dobrenis K., Walkley S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004;480:415–426. doi: 10.1002/cne.20355. [DOI] [PubMed] [Google Scholar]
- 4.Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D.C., Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 5.Fraldi A., Annunziata F., Lombardi A., Kaiser H.J., Medina D.L., Spampanato C., Fedele A.O., Polishchuk R., Sorrentino N.C., Simons K., et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010;29:3607–3620. doi: 10.1038/emboj.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Wilkinson F.L., Holley R.J., Langford-Smith K.J., Badrinath S., Liao A., Langford-Smith A., Cooper J.D., Jones S.A., Wraith J.E., Wynn R.F., et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7:e35787. doi: 10.1371/journal.pone.0035787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fedele A.O. Sanfilippo syndrome: causes, consequences, and treatments. Appl. Clin. Genet. 2015;8:269–281. doi: 10.2147/tacg.s57672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrade F., Aldámiz-Echevarría L., Llarena M., Couce M.L. Sanfilippo syndrome: overall review. Pediatr. Int. 2015;57:331–338. doi: 10.1111/ped.12636. [DOI] [PubMed] [Google Scholar]
- 9.Kan S.H., Aoyagi-Scharber M., Le S.Q., Vincelette J., Ohmi K., Bullens S., Wendt D.J., Christianson T.M., Tiger P.M.N., Brown J.R., et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. USA. 2014;111:14870–14875. doi: 10.1073/pnas.1416660111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu H., Kang L., Jennings J.S., Moy S.S., Perez A., Dirosario J., McCarty D.M., Muenzer J. Significantly increased lifespan and improved behavioral performances by rAAV gene delivery in adult mucopolysaccharidosis IIIB mice. Gene Ther. 2007;14:1065–1077. doi: 10.1038/sj.gt.3302961. [DOI] [PubMed] [Google Scholar]
- 11.Holley R.J., Ellison S.M., Fil D., O'Leary C., McDermott J., Senthivel N., Langford-Smith A.W.W., Wilkinson F.L., D'Souza Z., Parker H., et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain. 2018;141:99–116. doi: 10.1093/brain/awx311. [DOI] [PubMed] [Google Scholar]
- 12.de Ruijter J., Valstar M.J., Narajczyk M., Wegrzyn G., Kulik W., Ijlst L., Wagemans T., van der Wal W.M., Wijburg F.A. Genistein in Sanfilippo disease: a randomized controlled crossover trial. Ann. Neurol. 2012;71:110–120. doi: 10.1002/ana.22643. [DOI] [PubMed] [Google Scholar]
- 13.Kakkis E.D., Muenzer J., Tiller G.E., Waber L., Belmont J., Passage M., Izykowski B., Phillips J., Doroshow R., Walot I., et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N. Engl. J. Med. 2001;344:182–188. doi: 10.1056/NEJM200101183440304. [DOI] [PubMed] [Google Scholar]
- 14.Muenzer J., Gucsavas-Calikoglu M., McCandless S.E., Schuetz T.J., Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome) Mol. Genet. Metab. 2007;90:329–337. doi: 10.1016/j.ymgme.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Hendriksz C.J., Burton B., Fleming T.R., Harmatz P., Hughes D., Jones S.A., Lin S.P., Mengel E., Scarpa M., Valayannopoulos V., et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 2014;37:979–990. doi: 10.1007/s10545-014-9715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LeBowitz J.H., Grubb J.H., Maga J.A., Schmiel D.H., Vogler C., Sly W.S. Glycosylation-independent targeting enhances enzyme delivery to lysosomes and decreases storage in mucopolysaccharidosis type VII mice. Proc. Natl. Acad. Sci. USA. 2004;101:3083–3088. doi: 10.1073/pnas.0308728100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitley C.B., Vijay S., Yao B., Pineda M., Parker G.J.M., Rojas-Caro S., Zhang X., Dai Y., Cinar A., Bubb G., et al. Final results of the phase 1/2, open-label clinical study of intravenous recombinant human N-acetyl-alpha-d-glucosaminidase (SBC-103) in children with mucopolysaccharidosis IIIB. Mol. Genet. Metab. 2019;126:131–138. doi: 10.1016/j.ymgme.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Weber B., Hopwood J.J., Yogalingam G. Expression and characterization of human recombinant and alpha-N-acetylglucosaminidase. Protein Expr. Purif. 2001;21:251–259. doi: 10.1006/prep.2000.1361. [DOI] [PubMed] [Google Scholar]
- 19.Morgan D.O., Edman J.C., Standring D.N., Fried V.A., Smith M.C., Roth R.A., Rutter W.J. Insulin-like growth factor II receptor as a multifunctional binding protein. Nature. 1987;329:301–307. doi: 10.1038/329301a0. [DOI] [PubMed] [Google Scholar]
- 20.Heldermon C.D., Qin E.Y., Ohlemiller K.K., Herzog E.D., Brown J.R., Vogler C., Hou W., Orrock J.L., Crawford B.E., Sands M.S. Disease correction by combined neonatal intracranial AAV and systemic lentiviral gene therapy in Sanfilippo Syndrome type B mice. Gene Ther. 2013;20:913–921. doi: 10.1038/gt.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tardieu M., Zérah M., Gougeon M.L., Ausseil J., de Bournonville S., Husson B., Zafeiriou D., Parenti G., Bourget P., Poirier B., et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017;16:712–720. doi: 10.1016/s1474-4422(17)30169-2. [DOI] [PubMed] [Google Scholar]
- 22.Kroshinsky D., Alloo A., Rothschild B., Cummins J., Tan J., Montecino R., Hoang M.P., Duncan L., Mihm M., Sepehr A. Necrotizing Sweet syndrome: a new variant of neutrophilic dermatosis mimicking necrotizing fasciitis. J. Am. Acad. Dermatol. 2012;67:945–954. doi: 10.1016/j.jaad.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 23.Vellodi A., Young E., New M., Pot-Mees C., Hugh-Jones K. Bone marrow transplantation for Sanfilippo disease type B. J. Inherit. Metab. Dis. 1992;15:911–918. doi: 10.1007/BF01800232. [DOI] [PubMed] [Google Scholar]
- 24.Clarke D., Pearse Y., Kan S.H., Le S.Q., Sanghez V., Cooper J.D., Dickson P.I., Iacovino M. Genetically corrected iPSC-derived neural stem cell grafts deliver enzyme replacement to affect CNS disease in Sanfilippo B mice. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2018;10:113–127. doi: 10.1016/j.omtm.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeyakumar M., Lee J.P., Sibson N.R., Lowe J.P., Stuckey D.J., Tester K., Fu G., Newlin R., Smith D.A., Snyder E.Y., et al. Neural stem cell transplantation benefits a monogenic neurometabolic disorder during the symptomatic phase of disease. Stem Cell. 2009;27:2362–2370. doi: 10.1002/stem.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meneghini V., Frati G., Sala D., De Cicco S., Luciani M., Cavazzin C., Paulis M., Mentzen W., Morena F., Giannelli S., et al. Generation of human induced pluripotent stem cell-derived bona fide neural stem cells for ex vivo gene therapy of metachromatic leukodystrophy. Stem Cells Transl. Med. 2017;6:352–368. doi: 10.5966/sctm.2015-0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor R.M., Lee J.P., Palacino J.J., Bower K.A., Li J., Vanier M.T., Wenger D.A., Sidman R.L., Snyder E.Y. Intrinsic resistance of neural stem cells to toxic metabolites may make them well suited for cell non-autonomous disorders: evidence from a mouse model of Krabbe leukodystrophy. J. Neurochem. 2006;97:1585–1599. doi: 10.1111/j.1471-4159.2006.03986.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhang T., Ke W., Zhou X., Qian Y., Feng S., Wang R., Cui G., Tao R., Guo W., Duan Y., et al. Human neural stem cells reinforce hippocampal synaptic network and rescue cognitive deficits in a mouse model of Alzheimer's disease. Stem Cell Rep. 2019;13:1022–1037. doi: 10.1016/j.stemcr.2019.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donsante A., Levy B., Vogler C., Sands M.S. Clinical response to persistent, low-level beta-glucuronidase expression in the murine model of mucopolysaccharidosis type VII. J. Inherit. Metab. Dis. 2007;30:227–238. doi: 10.1007/s10545-007-0483-4. [DOI] [PubMed] [Google Scholar]
- 30.Zafeiriou D.I., Savvopoulou-Augoustidou P.A., Sewell A., Papadopoulou F., Badouraki M., Vargiami E., Gombakis N.P., Katzos G.S. Serial magnetic resonance imaging findings in mucopolysaccharidosis IIIB (Sanfilippo's syndrome B) Brain Dev. 2001;23:385–389. doi: 10.1016/s0387-7604(01)00242-x. [DOI] [PubMed] [Google Scholar]
- 31.Garner C.C., Tucker R.P., Matus A. Selective localization of messenger RNA for cytoskeletal protein MAP2 in dendrites. Nature. 1988;336:674–677. doi: 10.1038/336674a0. [DOI] [PubMed] [Google Scholar]
- 32.Masliah E., Mallory M., Ge N., Alford M., Veinbergs I., Roses A.D. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp. Neurol. 1995;136:107–122. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- 33.Gudi V., Gai L., Herder V., Tejedor L.S., Kipp M., Amor S., Sühs K.W., Hansmann F., Beineke A., Baumgärtner W., et al. Synaptophysin is a reliable marker for axonal damage. J. Neuropathol. Exp. Neurol. 2017;76:109–125. doi: 10.1093/jnen/nlw114. [DOI] [PubMed] [Google Scholar]
- 34.Rebiai R., Givogri M.I., Gowrishankar S., Cologna S.M., Alford S.T., Bongarzone E.R. Synaptic function and dysfunction in lysosomal storage diseases. Front. Cell. Neurosci. 2021;15:619777. doi: 10.3389/fncel.2021.619777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiRosario J., Divers E., Wang C., Etter J., Charrier A., Jukkola P., Auer H., Best V., Newsom D.L., McCarty D.M., et al. Innate and adaptive immune activation in the brain of MPS IIIB mouse model. J. Neurosci. Res. 2009;87:978–990. doi: 10.1002/jnr.21912. [DOI] [PubMed] [Google Scholar]
- 36.Ohmi K., Greenberg D.S., Rajavel K.S., Ryazantsev S., Li H.H., Neufeld E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park K.I., Himes B.T., Stieg P.E., Tessler A., Fischer I., Snyder E.Y. Neural stem cells may be uniquely suited for combined gene therapy and cell replacement: evidence from engraftment of Neurotrophin-3-expressing stem cells in hypoxic-ischemic brain injury. Exp. Neurol. 2006;199:179–190. doi: 10.1016/j.expneurol.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Watson D.J., Walton R.M., Magnitsky S.G., Bulte J.W.M., Poptani H., Wolfe J.H. Structure-specific patterns of neural stem cell engraftment after transplantation in the adult mouse brain. Hum. Gene Ther. 2006;17:693–704. doi: 10.1089/hum.2006.17.693. [DOI] [PubMed] [Google Scholar]
- 39.Di Stefano G., Casoli T., Fattoretti P., Gracciotti N., Solazzi M., Bertoni-Freddari C. Distribution of map2 in hippocampus and cerebellum of young and old rats by quantitative immunohistochemistry. J. Histochem. Cytochem. 2001;49:1065–1066. doi: 10.1177/002215540104900818. [DOI] [PubMed] [Google Scholar]
- 40.Leuner B., Gould E. Structural plasticity and hippocampal function. Annu. Rev. Psychol. 2010;61:111–140.C1-3. doi: 10.1146/annurev.psych.093008.100359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matus A., Green G.D. Age-related increase in a cathepsin D like protease that degrades brain microtubule-associated proteins. Biochemistry. 1987;26:8083–8086. doi: 10.1021/bi00399a010. [DOI] [PubMed] [Google Scholar]
- 42.Kan S.H., Le S.Q., Bui Q.D., Benedict B., Cushman J., Sands M.S., Dickson P.I. Behavioral deficits and cholinergic pathway abnormalities in male Sanfilippo B mice. Behav. Brain Res. 2016;312:265–271. doi: 10.1016/j.bbr.2016.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sherman M.Y., Goldberg A.L. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 44.Ryazantsev S., Yu W.H., Zhao H.Z., Neufeld E.F., Ohmi K. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Mol. Genet. Metab. 2007;90:393–401. doi: 10.1016/j.ymgme.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong H., Martin M.V., Chambers S., Csernansky J.G. Spatial relationship between synapse loss and beta-amyloid deposition in Tg2576 mice. J. Comp. Neurol. 2007;500:311–321. doi: 10.1002/cne.21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vitry S., Ausseil J., Hocquemiller M., Bigou S., Dos Santos Coura R., Heard J.M. Enhanced degradation of synaptophysin by the proteasome in mucopolysaccharidosis type IIIB. Mol. Cell. Neurosci. 2009;41:8–18. doi: 10.1016/j.mcn.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 47.Johnston J.A., Ward C.L., Kopito R.R. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kan S.H., Troitskaya L.A., Sinow C.S., Haitz K., Todd A.K., Di Stefano A., Le S.Q., Dickson P.I., Tippin B.L. Insulin-like growth factor II peptide fusion enables uptake and lysosomal delivery of alpha-N-acetylglucosaminidase to mucopolysaccharidosis type IIIB fibroblasts. Biochem. J. 2014;458:281–289. doi: 10.1042/BJ20130845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H.H., Yu W.H., Rozengurt N., Zhao H.Z., Lyons K.M., Anagnostaras S., Fanselow M.S., Suzuki K., Vanier M.T., Neufeld E.F. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc. Natl. Acad. Sci. USA. 1999;96:14505–14510. doi: 10.1073/pnas.96.25.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bible E., Gupta P., Hofmann S.L., Cooper J.D. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol. Dis. 2004;16:346–359. doi: 10.1016/j.nbd.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 51.Paxinos G.F., Franklin K.B.J. Second ed. Accademic Press; 2001. The Mouse Brain in Stereotaxic Coordinates. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.