

Graphical abstract
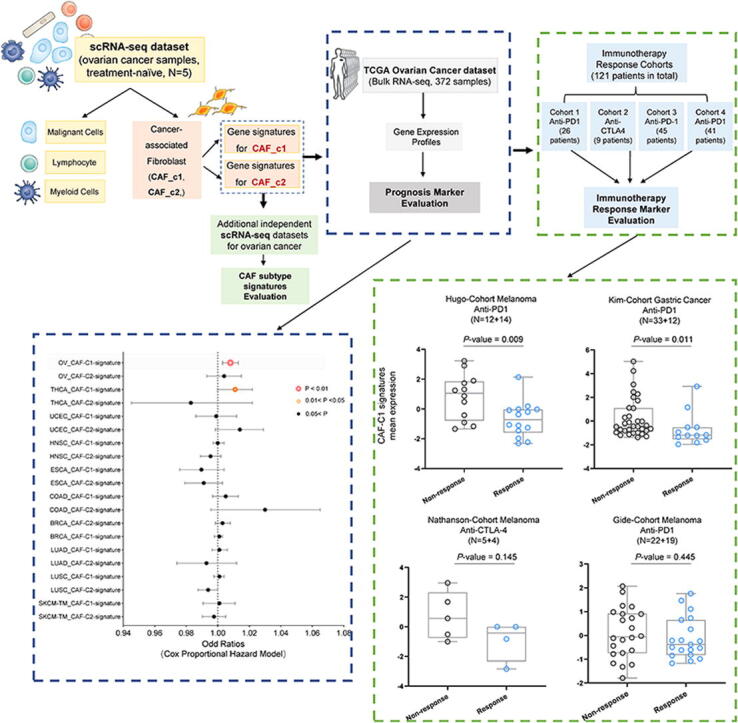
Abbreviations: scRNA-seq, Single-cell RNA-sequencing; CAFs, Cancer-associated fibroblasts; HGSOC, High-grade serous ovarian carcinoma; TCGA, The Cancer Genomics Atlas; LUAD, Lung adenocarcinoma; LUSC, Lung squamous cell carcinoma; SKCM, Skin cutaneous melanoma; HNSC, Head and neck squamous cell carcinoma; BRCA, Breast invasive carcinoma; ESCA, Esophageal carcinoma; COAD, Colon adenocarcinoma; UCEC, Uterine corpus endometrial carcinoma; OV, Ovarian serous cystadenocarcinoma; THCA, Thyroid cancer
Keywords: Single-cell sequencing, CAFs, Ovarian cancer, Gene signature, Bio-marker, Prognosis, Immunotherapy
Abstract
Accumulating evidence has recognized that cancer-associated fibroblasts (CAFs) are major players in the desmoplastic stroma of ovarian cancer, modulating tumor progression and therapeutic response. However, it is unclear regarding the signatures of CAFs could be utilized to predict the clinical outcomes of ovarian cancer patients. To fill in this gap, we explored the intratumoral compartment of ovarian cancer by analyzing the single-cell RNA-sequencing (scRNA-seq) datasets of ovarian carcinoma samples, and identified two distinct CAFs (tumor-promoting CAF_c1 subtype and myofibroblasts-like CAF_c2 subtype). The clinical significance of CAF subtypes was further validated in The Cancer Genomics Atlas (TCGA) database and other independent immunotherapy response datasets, and the results revealed that the patients with a higher expression of CAF_c1 signatures had a worse prognosis and showed a tendency of resistance to immunotherapy. This work uncovered the signatures of the CAF_c1 subtype that could serve as a novel prognostic indicator and predictive marker for immunotherapy.
1. Introduction
Ovarian cancer is the third most common gynecologic malignancy worldwide but accounts for the highest mortality rate among cancers [1]. Although aggressive frontline treatments with surgery and adjuvant chemotherapy, the 5-year survival rate is < 25 % for women diagnosed with stages III or IV disease [2], [3]. Although significant advancements have been achieved in the field of ovarian cancer research and treatment, clinical outcomes of ovarian cancer patients remain unsatisfactory [1], [3]. Previously, diverse cell type-specific expression patterns can be seen in different molecular subtypes of ovarian cancers, which suggested that the various cellular compositions of the tumor microenvironment (TME) have an important role in the heterogeneity of ovarian cancers [4]. Recently, accumulated pieces of evidence have revealed that ovarian cancer is characterized by an abundant stroma, a complex structure composed of different cell phenotypes including cancer cells, epithelial cells, endothelial cells, immune cells, and CAFs [5]. The crucial significance of TME in the initiation and progression of ovarian cancer as well as resistance to anti-tumor therapy is increasingly recognized.
The CAFs interact with both malignant cells and other stromal cells through a network of signaling pathways and mediators [6], [7]. These interactions contribute to tumor growth, invasion, metastasis, and resistance to therapy [8], [9], [7]. Much of the evidence came from previous studies that showed that CAFs are one of the most critical constituents of desmoplastic stroma in ovarian cancer, modulating tumorigenesis development and therapeutic response [10], [11]. Many works revealed that a lot of crucial protumorigenic processes such as chemoresistance, cancer stem cell renewal, tumor cell invasion, and immune cell polarization, are mediated by CAFs [10], [12], [13]. In the meanwhile, other studies suggested that CAFs can exert a tumor-suppressive role in some cancer types [12], [14]. These studies collectively raise the conclusion that CAFs are a highly heterogeneous population of cells and that, in order to assure accurate targeting of CAFs that promote tumors, a deeper understanding of CAFs heterogeneity through the discovery of particular markers is required [15]. The heterogeneity and dynamics among CAFs subtypes and their plasticity remain scarcely explored, and it is still an open question regarding whether the signatures of CAFs could be utilized to predict the clinical outcomes of ovarian cancer patients.
The TME has been a popular research area of cancer biology concerning novel biomarkers for diagnosis and prognosis, and therapeutic targets for drug discovery. And the high-throughput sequencing methods, especially the single-cell RNA-sequencing (scRNA-seq) technology, can be utilized to analyze the expression patterns of malignant tissues at single-cell levels and assess the details of cellular heterogeneity in the TME. In the present study, we performed an integrative analysis based on the scRNA-seq and bulk transcriptome data from ovarian carcinoma samples and highlighted the role of CAFs subsets in the prognosis and immunotherapy resistance for ovarian cancer patients. We investigated the relationship between molecular signatures of the CAFs subtype and the clinical outcome of ovarian cancer patients and identified a specific CAF subtype (CAF_c1) as a possible therapeutic target for ovarian treatment. In addition, these results promote the understanding of CAFs heterogeneity in the TME and provide a basis for biomarker development and precision treatment for ovarian carcinoma in the future.
2. Material and methods
2.1. TCGA pan-cancer patient cohort
The transcriptome data (RNA-seq) and clinical annotation (including overall survival time) of 4775 samples across 10 cancer types (LUAD: Lung adenocarcinoma, N = 493; LUSC: Lung squamous cell carcinoma, N = 494; SKCM-TM: Skin cutaneous melanoma—metastasis, N = 351; HNSC: Head and neck squamous cell carcinoma, N = 498; BRCA: Breast invasive carcinoma, N = 928; ESCA: Esophageal carcinoma, N = 161; COAD: Colon adenocarcinoma, N = 439; UCEC: Uterine corpus endometrial carcinoma, N = 537; OV: Ovarian serous cystadenocarcinoma, N = 372; THCA: Thyroid cancer, N = 502) from the largest publicly available cancer genomics database, namely TCGA with genomic, transcriptomic and clinical data. We accessed the TCGA data portal (https://cancergenome.nih.gov/, February 2021) and downloaded expression quantification profiles (HTSeq–FPKM). Clinical data files of cancer samples were downloaded from cBioPortal for Cancer Genomics [16] (https://www.cbioportal.org/, February 2021).
2.2. Immunotherapy patient cohort
The transcriptome data and clinical annotation of selected immunotherapy-treated patient cohorts [17], [18], [19], [20], which contained a total of 121 tumor samples were accessed from the Tumor Immune Dysfunction and Exclusion database [21] (https://tide.dfci.harvard.edu/, February 2021).
2.3. Data processing of scRNA-seq
We reanalyzed three independent public scRNA-seq datasets: the high-grade serous ovarian carcinoma data (included barcode files, gene annotation files, and raw count matrix files for each sample), the high-grade serous tubo-ovarian cancer data (included an integrated gene count matrix file of all samples and a metadata file), and the metastatic ovarian cancer data (included raw count matrix files for each sample). Then these raw data were imported into the R software (https://www.r-project.org/, version 3.5) using the Seurat (v.2.3.2) package for further data analysis. The first filtering was initially set up with the minimal cells of 3 and minimal genes of 200 per sample. Then, the cells with more than 5,000 genes or more than 25,000 unique molecular identifiers (UMIs) were removed, and the cells with less than 5% mitochondrial gene expression will be kept. Next, the raw counts of filtered cells were normalized by a factor of 10,000 and log-transformed to obtain log(T+1) values. Dimensionality reduction was performed using principal component analysis (PCA). The first 20 principal components were selected according to the PCA results for clustering, and the cell clusters were visualized by using the uniform manifold approximation and projection (UMAP) method. Differential gene expression analysis was performed by FindMarkers and FindAllMarkers in the Seurat package (Parameters: min.pct = 0.5, min.diff.pct = 0.1, logfc.threshold = 0.25, test.use = “Wilcox”). For the selection of signature genes of CAF_c1 and CAF_c2, firstly, we identified all the differentially expressed genes (DEGs) of CAF_c1 and CAF_c2 as marker genes (Table S1-2), and then we selected the representative genes as signatures if the following criteria were met: (1) with function associated with TME that has been reported according to previous studies, (2) both ranked top15 in the DEGs when compared with other CAF subset and all the other cell types, (3) with average expression value lower than 0.2 in tumor or endothelial cells. The gene set enrichment analysis (GSEA) based on the single-sample GSEA (ssGSEA) method implemented in GSVA (version 1.44.5) package of R software was used to calculate the GSEA score based on the curated signature genes of CAF and biological processes genesets in Gene Ontology using defaulted parameters.
2.4. SCENIC analysis
To investigate the transcription factor activity and gene regulatory network among different cell types in the TME of ovarian cancer based on the single-cell transcriptome data, the Single-Cell rEgulatory Network Inference and Clustering (SCENIC) [22] method was employed in this study. We selected the pySCENIC (version 0.10.2) with default parameters, a lightning-fast python implementation of the SCENIC pipeline, to perform the SCENIC analysis, and the gene-motif rankings (500 bp upstream or 100 bp downstream of the transcription start site [TSS]) were used to determine the search space around the TSS.
2.5. Cell–cell interaction network analysis
CellPhoneDB (version 2.1.2, https://www.cellphonedb.org) is a publicly available repository of curated receptors, ligands, and their interactions, that can be used to search for cell–cell interaction and receptor-ligand pairs among cell types [23]. Potential interactions between the two cell types were inferred by using the CellPhoneDB method based on expression quantification levels of receptor and ligand gene pairs through 1000 permutation tests. The resulting adjacency matrices for all cell–cell interactions were then created and displayed as heatmaps. Only gene pairs for receptor-ligand interactions in cell types of relevance were observed, and cell–cell interactions within identical biological lineages were omitted.
2.6. Quantification and statistical analysis
The specific tests used to analyze each set of experiments are indicated in the figure legends. The prognostic value of discrete variables was assessed using Kaplan-Meier survival curves for a patient's cohort-based study on the overall survival rate (5 years), and the log-rank test was used to determine the significance of various survival curves. GraphPad Prism software (GraphPad Software, San Diego, California), and R software (https://www.r-project.org/) were employed for the statistical calculations and figure visualization.
3. Results
3.1. Characterization of cellular heterogeneity in the TME of ovarian cancer
To investigate the detailed cellular heterogeneity in the TME of ovarian cancer samples, we accessed and reanalyzed a sophisticated scRNA-seq dataset of high-grade serous ovarian carcinoma (HGSOC, N = 5) with comprehensive infiltrated stromal cell profiling [24] (Fig. 1). After quality control and removal of low-quality cells, a total of 38,543 cells were retained for downstream analyses. Batch effects among the samples were observed and corrected by the canonical correlation analysis (CCA) method. Graph-based clustering of cells identified 10 major clusters with uniform manifold approximation and projection (UMAP) algorithm (Fig. 2A). The marker genes of each cluster based on the differential expression genes analysis were cross-referenced with canonical markers of cell phenotype from the published literature, and used to annotate the cell types (Fig. 2B). Interestingly, the CAFs in the ovarian tumor microenvironment had two distinct subclusters: CAF_c1 and CAF_c2 (Fig. 2A), which indicated that amongst these primary human HGSOC samples, at least two subtypes of CAFs existed. We also found that these two subtypes of CAFs existed in all 5 samples (Fig. S1A), although their proportion varied greatly (Fig. S1B). As shown in Fig. 2B, although all the CAFs could express the canonical markers of fibroblast (such as COL6A1 and COL6A2), two CAFs subtypes had different marker genes to distinguish (Table S1-2). We characterized the CAF_c1 as tumor-promoting fibroblasts by studying the discriminating marker genes of CAF_c1 (Fig. 2C, Fig. S2A): CCDC80 (promoting cell adhesion and matrix assembly [25]); SFRP2 (regulating cell growth and differentiation [26]); VCAN (promoting tumor progression and malignant transformation [27]) and COL8A1 (necessary for migration and proliferation of vascular smooth muscle cells [28]). Meanwhile, we defined CAF_c2 as myofibroblasts-like CAFs, because the gene markers of myofibroblasts and pericytes including RGS5, NOTCH3, and NDUFA4L2 were specifically expressed in the cells of the CAF_c2 (Fig. 2C, Fig. S2A) [29], [30], [31].
Fig. 1.

Illustration of study workflow. The flowchart of data collection and method implementation in this work.
Fig. 2.

Identification of CAFs subtypes in the TME of ovarian cancer. (A) Single-cell RNA-sequencing analysis of stromal cells in ovarian cancer (HGSOC). Uniformmanifold approximation and projection (UMAP) analysis showed 10 distinct clusters of cell phenotypes in the TME. (B) Dot plot of mean expression of canonical marker genes for each cell type. (C) Distribution plot of expression value of top marker genes for each CAFs subtype.
We also found that CAF_c1 is next to cancer cells in UMAP, which indicates that they have similar gene expression patterns. To further explore that, we derived CAF_c1 and all cancer cells and re-clustered them. The results showed that cancer cells could be divided into 2 subtypes: CancerCell_c1 and CancerCell_c2, in which CancerCell_c1 is close to CAF_c1 (Fig. S3A). CancerCell_c1 expressed some universe marker genes of cancer cells, like EPCAM, CD24, and KRT19, while CancerCell_c2 expressed some additional genes associated with cell cycle and cell proliferation, such as TOP2A and UBE2C (Fig. S3B). We then use the ssGSEA method to analyze the enriched GO BP of each cell type. The top 10 enriched processes showed CancerCell_c1 highly expressed genes related to immune cell activation and immune response, while CancerCell_c2 highly expressed cell cycle-associated genes (Fig. S3C).
Next, to determine the general significance of CAFs subtype identification in the TME of ovarian cancer, additional independent scRNA-seq datasets based on the high-grade serous tubo-ovarian cancer (HGSTOC) samples (N = 7) [32] and metastatic ovarian cancer samples (N = 6) [33] were collected and analyzed in the present study. As shown in Fig. 3, two identified CAFs subtypes including tumor-promoting CAFs (CAF_c1) and myofibroblasts-like CAFs (CAF_c2) were observed in the TME of 7 treatment-naive HGSTOC tumors (Fig. 3A) and 6 ovarian tumors resected from omental metastases (Fig. 3B). To further evaluate the consistency of CAF_c1 and CAF_c2 between these datasets, we calculated the correlation coefficients between each cell type in the two datasets. It showed a high correlation among these cell types in independent datasets (Fig. S4).
Fig. 3.

Validation of CAFs heterogeneity in the TME of ovarian cancer based on the additional scRNA-seq datasets. (A) UMAP plot (left) of profiled cell clusters signature distribution plot (right) of CAFs subtype signature genes based on the scRNA-seq dataset of 7 high-grade serous tubo ovarian cancer samples. (B) UMAP plot (left) of profiled cell clusters signature distribution plot (right) of CAFs subtype signature genes based on the scRNA-seq dataset of 6 metastatic ovarian cancer samples.
3.2. Identification of CAF subtype-specific gene regulatory transcription factors and cellular interaction networks
We next sought to identify transcription factors (TFs) to better understand how CAF subtypes are established and maintained. For this, we applied the algorithm SCENIC [22] to identify cell cluster-specific top10 TFs that are highly active in the CAF subtypes versus other cell populations of TME in ovarian cancer. We observed that CAF_c1 and CAF_c2 showed distinct patterns of highly expressed TFs (Fig. 4A). The homeobox genes including (HOXA10 and HOXD11) encode a highly conserved family of TFs that play an important role in morphogenesis [34] and were upregulated in the CAF_c2 subset. Furthermore, the PPARG, RARA, and TBX15, which have been implicated in the regulation of cell development and differentiation [35], [36], were also active in CAF_c2 (Fig. 4B). Remarkably, immunological TFs (STAT1, IRF1, and XBP1) [37] and NF-κB pathway-related TFs (JUN and RELB) [38] were enriched for the CAF_c1. In addition, ATF3 [39], KLF5 [40], and RELB, were induced by a variety of signals including many of those encountered by cancer cells, and are involved in the complex process of the cellular stress response, were all upregulated in CAF_c1 (Fig. 4B). These findings provide further insights into the gene regulatory networks underpinning CAF variability by identifying critical TFs responsible for regulating or sustaining the gene expression programs in the identified CAF subtypes.
Fig. 4.

Characterization of subtype-specific TFs and cellular interaction networks of CAFs. (A) Distribution plot of TFs using the SCENIC algorithm for the CAFs of ovarian cancer. (B) Heatmap of top10 activated subtype-specific TFs, the color indicates the relative expression level (Z-score) of TFs. (C) Heat map depicting the significant interactions among the identified major cell types, the color indicates the number of interactions between two specified TFs. (D) Overview of the selected ligand-receptor interactions. P-values (two-tailed permutation test) are indicated by circle size. The means of the average expression level of interacting molecule 1 in cluster 1 and interacting molecule 2 in cluster 2 are indicated by color.
Next, we used CellPhoneDB [23] to identify the expression of potential crosstalk signaling molecules between CAFs and other cell clusters based on ligand-receptor interactions. As shown in Fig. 4C, the endothelium cell populations, as well as the myeloid cell clusters showed the highest interaction numbers with CAFs, which suggests sufficient interactions between CAFs and immune and other stromal cells involving certain receptor-ligand gene pairs. For example, CAF cells express high levels of COL6A1, COL3A1, and FN1 as ligands, and the receptor aVb1 and a1b1 complex was expressed by endothelium cells as receptors (Fig. 4D). Interestingly, we found some specific gene pairs associated between the CAF_c1 subtype and other strongly interacting cell types. CAF_c1 cells can express high levels of CD55 to interact with many immune cell types (including lymphocytes and myeloid cells) by using the receptor of ADGRE5 (Fig. 4D). We also performed an extensive analysis of the ligand-receptor interactions on the CAF_c1 and more refined tumor cell sub-clusters (including CancerCell_c1 and CancerCell_c2). As shown in Fig. S3D-E, there was no significant difference between CancerCell_c1 and CancerCell_c2 in terms of their interaction with CAF_c1, and, interestingly, the expression of ligands FN1 in CAF_c1 had strong interaction potential with receptors integrin complex (a3b1, aVb1, and aVb5) expressed in both CancerCell_c1 and CancerCell_c2 sub-clusters.
Overall, our data revealed that the identified subtypes of CAFs were two distinct cell populations with different TF expression patterns and cellular interaction networks, reflecting the heterogeneity of molecular characteristics of CAFs in the TME of ovarian cancer.
3.3. Pan-cancer assessment of the biomarker utility of CAF signatures for clinical outcome
To determine if the abundance of CAF_c1 in the ovarian TME could serve as a predictive marker for clinical outcomes, we employed the Kaplan–Meier survival analysis by using the marker genes of CAF_c1, and the cox (proportional hazards) regression model by using the integrative CAF_c1 signature genes to explore their prognosis significance based on the TCGA patient cohort. The ovarian cancer patients with the higher expression of CAF_c1 marker genes all had a dramatically unfavorable 5-year overall survival, while no significant difference in survival curves was observed for CAF_c2 marker genes (Fig. 5A). Then, we used the integrative signature values of marker genes for each CAFs subtype to examine the prognosis value at the Pan-cancer level, and as shown in Fig. 5B, we found that the integrative CAF_c1 signatures were specifically significantly correlated with worse overall survival in ovarian cancer (P-value = 0.003), while no association was observed in any cancer types for CAF_c2 signatures.
Fig. 5.

The signature of the CAF_c1 subtype in ovarian cancer correlated with worse prognostic and immunotherapy responsiveness. (A) Kaplan–Meier survival curves comparing the 5-year overall survival for different marker genes of each CAFs subtype. (B) Hazard ratio estimates for overall survival in TCGA pan-cancer cohorts based on the integrative marker gene signatures of each CAFs subtype by using Cox regression. Plotting symbols give point estimates of HR and horizontal bars give 95% CIs. (C) The dot plot displays the distribution of CAF_c1 signature expression mean value among different immunotherapy response patient cohorts. The median values of each sample are indicated in horizontal bars.
Agents that alter the immunologic environment to incite an immune response against tumors have shown astonishing success in a number of solid malignancies, nevertheless, the responsiveness is not universal and overall response rates might be as low as 20 % [41]. While T cell-dependent mechanisms are crucial, other cellular subpopulations including some stromal cells in the TME could also play an important role in moderating the response to immunotherapy. Previous studies have suggested that CAFs are associated with this resistance [42], [43], [44], [45], therefore, to check if the signatures of CAF_c1 correlated with the immunotherapy response, we investigated the distribution of CAF_c1 signatures across four different published RNA-seq cohorts collected from multiple cancer patients prior to immunotherapy treatment [17], [18], [19], [20] to test whether it has a distinguishing power from clinical-benefit patients versus non-benefit patients of immunotherapy. Surprisingly, the expression of CAF_c1 signatures was enriched in the cancer patients that were resistant to immunotherapy, and that trend was observed in all the immune checkpoint inhibitor therapy cohorts of various cancer types (including anti-PD-(L)1 and anti-CTLA-4 immunotherapy, Fig. 5C). The expression value of CAF_c1 signatures showed the potential as a novel marker for both prognosis and immunotherapy responsiveness for clinical use.
4. Discussion
The plasticity of CAFs subtypes in the TME has been well demonstrated for various types of cancer including pancreatic [46] and breast cancer [47], and our investigation provides a new framework to develop omics data integration strategies to analyze the CAFs phenotype in the TME of ovarian cancer, to be combined with clinical outcome information. Our results painted the cellular heterogeneity of the CAFs population, and identified the signature genes and master regulatory network of each CAFs subcluster in the TME of ovarian cancer, which demonstrated the critical need to functionally characterize the pleiotropic effects of CAFs in terms of cancer progression, prognostic outcome, and response to immunotherapy.
In this study, by leveraging scRNA-seq data, TCGA database, and multiple immunotherapy patient cohorts, we mapped CAFs cell states that were associated with clinical outcomes, revealing a novel marker based on the identified CAFs subtype that has a prognostic value for clinical outcome and predictive value for immunotherapy response of melanoma. The expression value of signature derived from the CAF_c1 subtype that could predict immunotherapy responses was confirmed in several independent cohorts. Previous studies have found that CAFs could also be divided into 2 subsets, which were named inflammatory CAF (iCAF) and myo-CAF (mCAF), in some other cancer types, such as bladder urothelial carcinoma and pancreatic ductal adenocarcinoma [48], [49]. These results may suggest the consistent function of the CAFs in various cancer types. We believe that the specific type of CAFs have a selective function of anti-inflammatory and expected disadvantage during immune checkpoint inhibitor therapy. Additional prospective data with a sizable patient cohort will be required to confirm our findings and unambiguously link the CAFs signatures with the immunotherapy response of ovarian cancers.
There are a few limitations to our study that should be noted. First, because our study is in silicon analysis based on integrative omics data, it will be important and more convincing if we or others could characterize and validate the CAFs markers from our findings based on the newly collected cancer tissues of ovarian cancer by using experimental methods. Second, despite our best efforts to achieve the robustness of our CAFs clustering analysis, we suppose the heterogeneity results of CAFs in this paper could be further improved and refined with larger scRNA-seq and cancer genomics cohorts (more cells and more samples profiled). Third, since the new high-throughput technology such as spatial transcriptomics has been advancing rapidly in recent years, our findings could be effectively extended with other new-developed approaches.
To sum up, we have systematically characterized two distinct CAFs subtypes in the TME of ovarian cancer, and more importantly, we identify the abundance of the CAF_c1 subtype is related to unfavorable prognosis and immunotherapy resistance. We believe that when single-cell technologies become more widely used, more datasets containing CAFs from a variety of other cancer types will become accessible. Future studies might use the paradigm provided by our current study to characterize, compare, and investigate the functional and clinical aspects of CAFs heterogeneity across various cancer types.
CRediT authorship contribution statement
Yan Zhao: Data curation, Methodology, Writing – original draft, Writing – review & editing. Song Mei: Methodology, Writing – original draft, Writing – review & editing, Data curation. Yixuan Huang: Data curation, Visualization. Junru Chen: Data curation, Visualization. Xinlei Zhang: Visualization, Supervision. Peng Zhang: Conceptualization, Writing – original draft, Writing – review & editing, Funding acquisition, Supervision.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Yixuan Huang and Xinlei Zhang are employees of Beijing Cloudna Technology Co., Ltd., and the other authors declare no competing financial interests.
Acknowledgments
Acknowledgments
Not appliable.
Availability of data or materials
The materials of patient cohorts used for the present study were publicly available and can be assessed from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/, GSE154600, and GSE147082), VIB-KU Leuven Center for Cancer Biology website (https://lambrechtslab.sites.vib.be/en/high-grade-serous-tubo-ovarian-cancer-refined-single-cell-rna-sequencing-specific-cell-subtypes) and TCGA database (https://portal.gdc.cancer.gov/, https://www.cbioportal.org/). The analysis codes and processed data are accessible from the corresponding author upon reasonable request.
Author contributions
Y.Z. and S.M. analyzed the data and wrote the draft of the manuscript with the help of Y.H., J.C., and X.Z., P.Z. designed the studies, supervised the analysis, and revised the manuscript.
Funding
Beijing Nova Program (Z211100002121044), and Beijing Hospitals Authority Grant (QML20211205).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.11.025.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Kuroki L., Guntupalli S.R. Treatment of epithelial ovarian cancer. BMJ England. 2020;371:m3773. doi: 10.1136/bmj.m3773. [DOI] [PubMed] [Google Scholar]
- 2.Eisenhauer E.A. Real-world evidence in the treatment of ovarian cancer. Ann Oncol England. 2017;28:viii61–viii65. doi: 10.1093/annonc/mdx443. [DOI] [PubMed] [Google Scholar]
- 3.Odunsi K. Immunotherapy in ovarian cancer. Ann Oncol. 2017;28:viii1–viii17. doi: 10.1093/annonc/mdx444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Integrated genomic analyses of ovarian carcinoma Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thibault B., Castells M., Delord J.-P., Couderc B. Ovarian cancer microenvironment: implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev Netherlands. 2014;33:17–39. doi: 10.1007/s10555-013-9456-2. [DOI] [PubMed] [Google Scholar]
- 6.Bou-Tayeh B., Miller M.L. Ovarian tumors orchestrate distinct cellular compositions. Immunity United States. 2021;54:1107–1109. doi: 10.1016/j.immuni.2021.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Lheureux S., Oza A.M. Targeting the microenvironment in ovarian cancer. LancetOncol England. 2015;16:485–486. doi: 10.1016/S1470-2045(15)70157-9. [DOI] [PubMed] [Google Scholar]
- 8.Jiménez-Sánchez A., Memon D., Pourpe S., Veeraraghavan H., Li Y., Vargas H.A., et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. 2017;170:927–938.e20. doi: 10.1016/j.cell.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hornburg M., Desbois M., Lu S., Guan Y., Lo A.A., Kaufman S., et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell United States. 2021;39:928–944.e6. doi: 10.1016/j.ccell.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Gao Q., Yang Z., Xu S., Li X., Yang X., Jin P., et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J Exp Med. 2019;216:688–703. doi: 10.1084/jem.20180765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hussain A., Voisin V., Poon S., Karamboulas C., Bui N.H.B., Meens J., et al. Distinct fibroblast functional states drive clinical outcomes in ovarian cancer and are regulated by TCF21. J Exp Med. 2020;217 doi: 10.1084/jem.20191094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brechbuhl H.M., Finlay-Schultz J., Yamamoto T.M., Gillen A.E., Cittelly D.M., Tan A.-C., et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res. 2017;23:1710–1721. doi: 10.1158/1078-0432.CCR-15-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costa A., Kieffer Y., Scholer-Dahirel A., Pelon F., Bourachot B., Cardon M., et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell United States. 2018;33:463–479.e10. doi: 10.1016/j.ccell.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 14.Özdemir B.C., Pentcheva-Hoang T., Carstens J.L., Zheng X., Wu C.-C., Simpson T.R., et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galbo P.M.J., Zang X., Zheng D. Molecular features of cancer-associated fibroblast subtypes and their implication on cancer pathogenesis, prognosis, and immunotherapy resistance. Clin Cancer Res. 2021;27:2636–2647. doi: 10.1158/1078-0432.CCR-20-4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. CancerDiscov United States. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S.T., Cristescu R., Bass A.J., Kim K.-M., Odegaard J.I., Kim K., et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med United States. 2018;24:1449–1458. doi: 10.1038/s41591-018-0101-z. [DOI] [PubMed] [Google Scholar]
- 18.Gide T.N., Quek C., Menzies A.M., Tasker A.T., Shang P., Holst J., et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell United States. 2019;35:238–255.e6. doi: 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nathanson T., Ahuja A., Rubinsteyn A., Aksoy B.A., Hellmann M.D., Miao D., et al. Somatic mutations and neoepitope homology in melanomas treated with CTLA-4 blockade. Cancer Immunol Res. 2017;5:84–91. doi: 10.1158/2326-6066.CIR-16-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu J., Li K., Zhang W., Wan C., Zhang J., Jiang P., et al. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 2020;12:21. doi: 10.1186/s13073-020-0721-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. Cell PhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc England. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 24.Geistlinger L., Oh S., Ramos M., Schiffer L., LaRue R.S., Henzler C.M., et al. Multiomic analysis of subtype evolution and heterogeneity in high-grade serous ovarian carcinoma. Cancer Res. 2021;80:4335–4345. doi: 10.1158/0008-5472.CAN-20-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pei G., Lan Y., Lu W., Ji L., Hua Z. The function of FAK/CCDC80/E-cadherin pathway in the regulation of B16F10 cell migration. Oncol Lett. 2018 doi: 10.3892/ol.2018.9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Courtwright A., Siamakpour-Reihani S., Arbiser J.L., Banet N., Hilliard E., Fried L., et al. Secreted frizzle-related protein 2 stimulates angiogenesis via a calcineurin/NFAT signaling pathway. Cancer Res. 2009;69:4621–4628. doi: 10.1158/0008-5472.CAN-08-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitsui Y., Shiina H., Kato T., Maekawa S., Hashimoto Y., Shiina M., et al. Versican promotes tumor progression, metastasis and predicts poor prognosis in renal carcinoma. Mol Cancer Res. 2017;15:884–895. doi: 10.1158/1541-7786.MCR-16-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopes J., Adiguzel E., Gu S., Liu S.-L., Hou G., Heximer S., et al. Type VIII collagen mediates vessel wall remodeling after arterial injury and fibrous cap formation in atherosclerosis. Am J Pathol. 2013;182:2241–2253. doi: 10.1016/j.ajpath.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.James A.W., Hindle P., Murray I.R., West C.C., Tawonsawatruk T., Shen J., et al. Pericytes for the treatment of orthopedic conditions. Pharmacol Ther. 2017;171:93–103. doi: 10.1016/j.pharmthera.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi J., Xiao P., Liu X., Chen Y., Xu Y., Fan J., et al. Notch3 modulates cardiac fibroblast proliferation, apoptosis, and fibroblast to myofibroblast transition via negative regulation of the RhoA/ROCK/Hif1α axis. Front Physiol. 2020:11. doi: 10.3389/fphys.2020.00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mesa-Ciller C, Turiel G, Guajardo-Grence A, Lopez-Rodriguez AB, Egea J, de Bock K, et al. Unique expression of the atypical mitochondrial subunit NDUFA4L2 in cerebral pericytes fine tunes HIF activity in response to hypoxia. Journal of Cerebral Blood Flow & Metabolism. 2022;0271678X2211182. [DOI] [PMC free article] [PubMed]
- 32.Olbrecht S., Busschaert P., Qian J., Vanderstichele A., Loverix L., Van G.T., et al. High-grade serous tubo-ovarian cancer refined with single-cell RNA sequencing : specific cell subtypes influence survival and determine molecular subtype classification. Genome Med. 2021:1–30. doi: 10.1186/s13073-021-00922-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olalekan S., Xie B., Back R., Eckart H., Olalekan S., Xie B., et al. Article Characterizing the tumor microenvironment of metastatic ovarian cancer by single-cell transcriptomics ll ll Characterizing the tumor microenvironment of metastatic ovarian cancer by single-cell transcriptomics. Cell Reports ElsevierCompany. 2021;35 doi: 10.1016/j.celrep.2021.109165. [DOI] [PubMed] [Google Scholar]
- 34.Northcott J.M., Northey J.J., Barnes J.M., Weaver V.M. Fighting the force: Potential of homeobox genes for tumor microenvironment regulation. Biochim Biophys Acta. 2015;1855:248–253. doi: 10.1016/j.bbcan.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yasmeen R., Meyers J.M., Alvarez C.E., Thomas J.L., Bonnegarde-Bernard A., Alder H., et al. Aldehyde dehydrogenase-1a1 induces oncogene suppressor genes in B cell populations. Biochim Biophys Acta. 2013;1833:3218–3227. doi: 10.1016/j.bbamcr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee K.Y., Singh M.K., Ussar S., Wetzel P., Hirshman M.F., Goodyear L.J., et al. Tbx15 controls skeletal muscle fibre-type determination and muscle metabolism. Nat Commun. 2015;6:8054. doi: 10.1038/ncomms9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michalska A., Blaszczyk K., Wesoly J., Bluyssen H.A.R. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol. 2018;9:1135. doi: 10.3389/fimmu.2018.01135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tegowski M., Baldwin A. Noncanonical NF-κB in cancer. Biomedicines. 2018:6. doi: 10.3390/biomedicines6020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim D.E., Procopio M.-G., Ghosh S., Jo S.-H., Goruppi S., Magliozzi F., et al. Convergent roles of ATF3 and CSL in chromatin control of cancer-associated fibroblast activation. J Exp Med. 2017;214:2349–2368. doi: 10.1084/jem.20170724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei R, Zhou Y, Li C, Rychahou P, Zhang S, Titlow WB, et al. Ketogenesis attenuates KLF5-dependent production of CXCL12 to overcome the immunosuppressive tumor microenvironment in colorectal cancer. Cancer Res. United States; 2022. [DOI] [PMC free article] [PubMed]
- 41.O’Donnell J.S., Teng M.W.L., Smyth M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol England. 2019;16:151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 42.Dominguez C.X., Müller S., Keerthivasan S., Koeppen H., Hung J., Gierke S., et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15(+) myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov United States. 2020;10:232–253. doi: 10.1158/2159-8290.CD-19-0644. [DOI] [PubMed] [Google Scholar]
- 43.Kieffer Y., Hocine H.R., Gentric G., Pelon F., Bernard C., Bourachot B., et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov United States. 2020;10:1330–1351. doi: 10.1158/2159-8290.CD-19-1384. [DOI] [PubMed] [Google Scholar]
- 44.Mhaidly R., Mechta-Grigoriou F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev. 2021;302:259–272. doi: 10.1111/imr.12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mao X., Xu J., Wang W., Liang C., Hua J., Liu J., et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131. doi: 10.1186/s12943-021-01428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Öhlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A.S., Ponz-Sarvise M., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen X., Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov England. 2019;18:99–115. doi: 10.1038/s41573-018-0004-1. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z., Zhou L., Liu L., Hou Y., Xiong M., Yang Y., et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat Commun. 2020;11:5077. doi: 10.1038/s41467-020-18916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lavie D., Ben-Shmuel A., Erez N., Scherz-Shouval R. Cancer-associated fibroblasts in the single-cell era. Nat Cancer. 2022;3:793–807. doi: 10.1038/s43018-022-00411-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.