
Extended Data Fig. 4. Sequencing coverage and identification of significantly mutated genes.
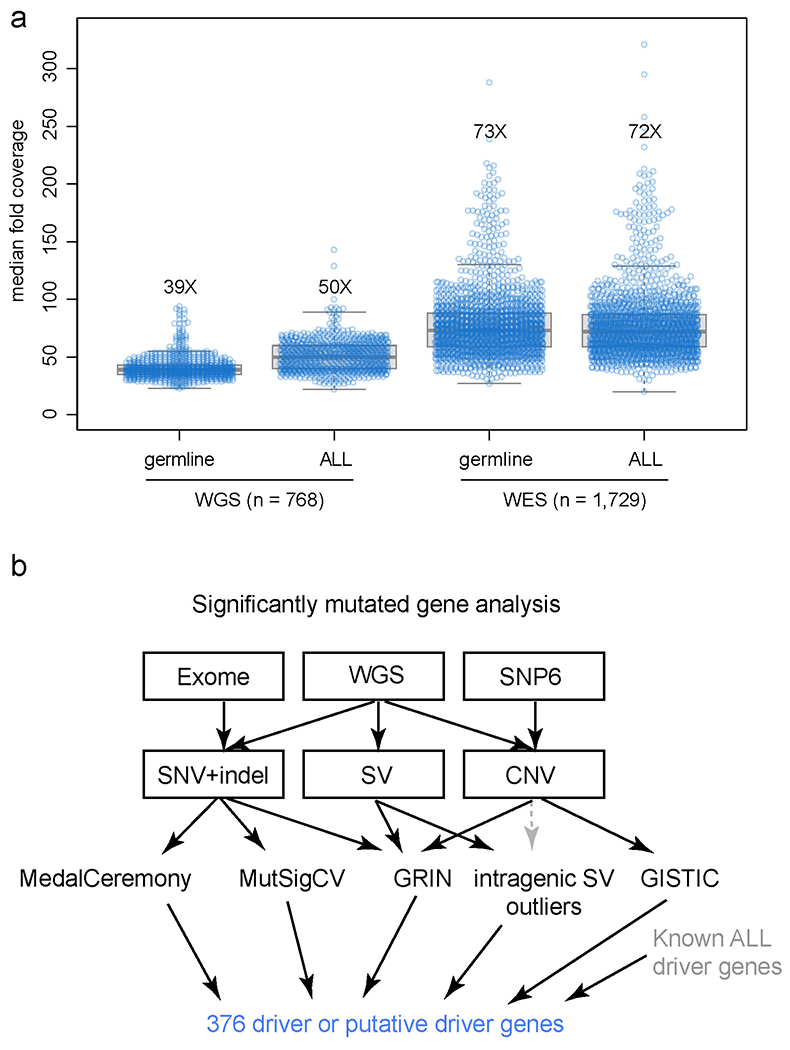
(a) Each sample’s median sequencing coverage based on WGS (n=768) or WES (n=1,729) is shown, including both germline and cancer (ALL) samples for each patient. Boxplot shows median (thick center line) and interquartile range (box). Whiskers are described in R boxplot documentation (a 1.5*interquartile range rule is used). The median coverage across all samples is indicated by text (e.g. “39X”). For WGS, the genome-wide coverage for each sample is indicated by each point. For WES, the median coverage in all protein-coding regions of exons (excluding 5’ and 3’ untranslated regions), as defined by the UCSC refGene.txt file, is shown. (b) Approach for identification of significantly mutated genes. The sequencing platform is shown on top, followed by the variant types detected by each platform below, and the third layer shows the tools used to identify significantly altered genes, with arrows indicating the variant types used as input to these tools. Intragenic SV outliers were identified initially by frequent SVs within the gene, and were corroborated manually with copy number analysis (dotted gray line) as the SVs were usually at the boundaries of focal deletions. All significantly mutated genes’ focal deletion and SNV/indel mutation site localization were manually inspected and those considered unlikely drivers were excluded. When combining the significantly mutated genes thus identified with the list of known drivers in ALL, a list of 376 driver or putative driver genes was identified.