
Figure 3. Current and Proposed Iron hypothesis of AD (simplified).
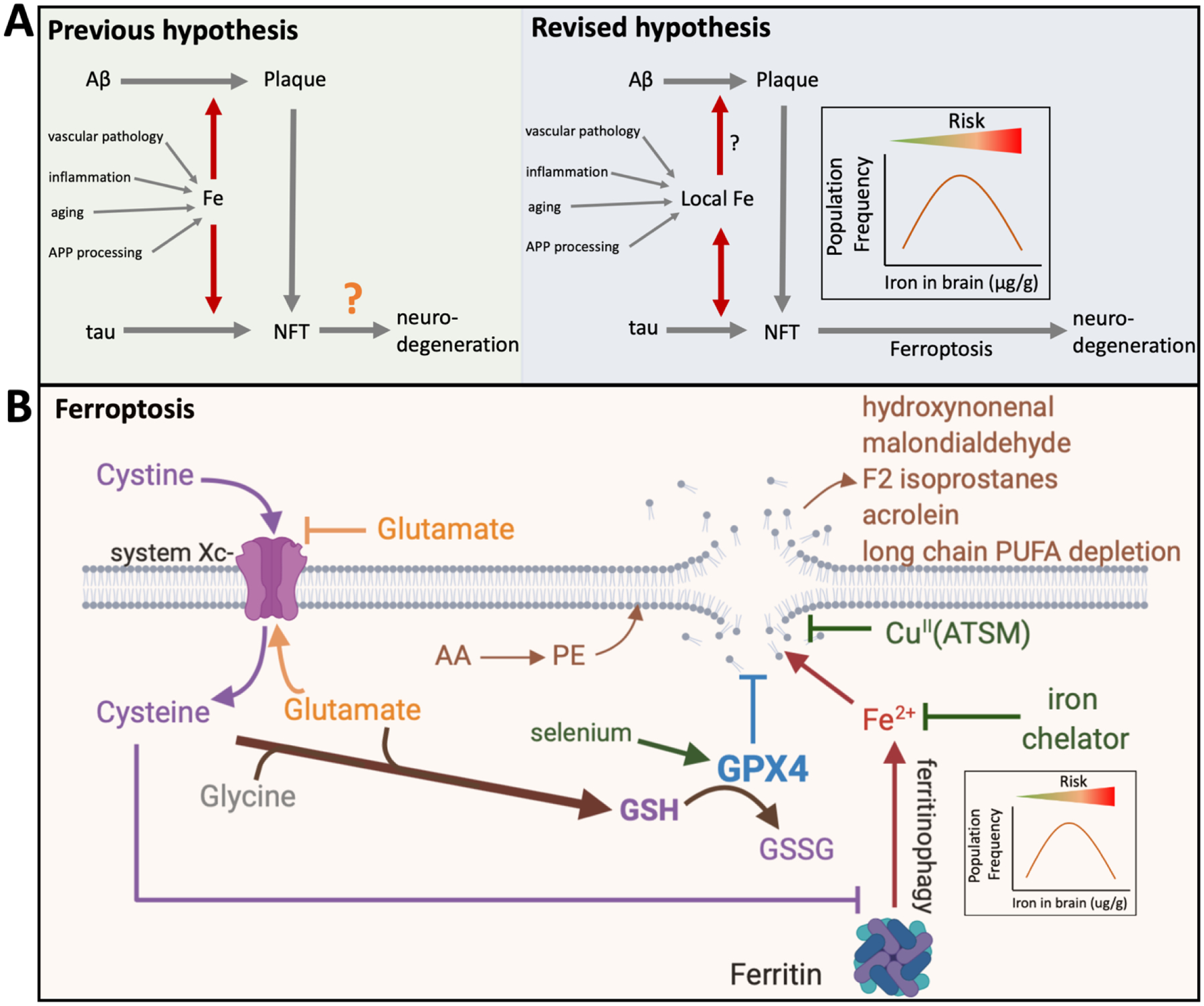
(A)The previous hypothesis was that iron promotes plaque and tangle formation by promoting APP and tau production as well as aggregation of Aβ and phospho-tau. Brain iron levels may be impacted by aging, changes in APP processing, inflammation and vascular damage. Iron trapped within these protein pathologies augmented toxicity through generating reactive oxygen species. Our current findings from a large longitudinal cohort confirmed that tissue iron burden may impact on the formation of NFTs, at least in the inferior temporal cortex. Our current investigation of bulk tissue levels of iron does not uphold a relationship between bulk tissue iron burden and amyloid formation. However, this study was unable to test the impact of iron in microvicinities, where previous findings indicate that iron is markedly enriched in plaques and tangles. Loss of tau function can, in its own right, cause neuronal iron accumulation. The new data lead us to further update the hypothesis, where iron has an additional role downstream of proteinopathy, by influencing the susceptibility of neurons to die (or cause synaptic damage) due to ferroptosis. Abnormal iron elevation does not itself cause ferroptosis, rather, the burden of iron in the tissue renders neurons more susceptible to ferroptotic stress. Therefore, people with relatively more iron, but still within the normal range, are likely to deteriorate faster during disease. (B) Ferroptosis pathways. Glutathione, which is depleted in AD brain tissue [51, 60, 68], prevents ferroptosis by providing substrate for the ferroptosis checkpoint selenoenzyme, GPX4. GPX4 detoxifies lipid hydroperoxides that are formed by cytoplasmic iron reacting with membrane PUFAs, otherwise cell rupture ensues. Cystine enters neurons via the system Xc− antiporter in exchange for glutamate that is exported. Cystine is reduced to cysteine within the cell, which is the rate limiting amino acid for glutathione synthesis. Low cysteine levels promote ferroptosis by both depleting glutathione, and by promoting ferritin degradation that releases cytoplasmic iron to fuel the peroxidation reaction [69]. The more iron in the cell, the greater the vulnerability toward ferroptosis. Lipid peroxides in ferroptosis lead to downstream products such as hydroxynonenal, malondialdehyde, F2-isoprostanes, acrolein, with subsequent depletion of long-chain PUFA depletion- all changes that are reported for AD brain tissue [70–76]. The sites of putative therapeutics are noted.