
Figure 7. H3K27M and EZHIP converge to restrict H3K27me3 to PRC2 nucleation sites.
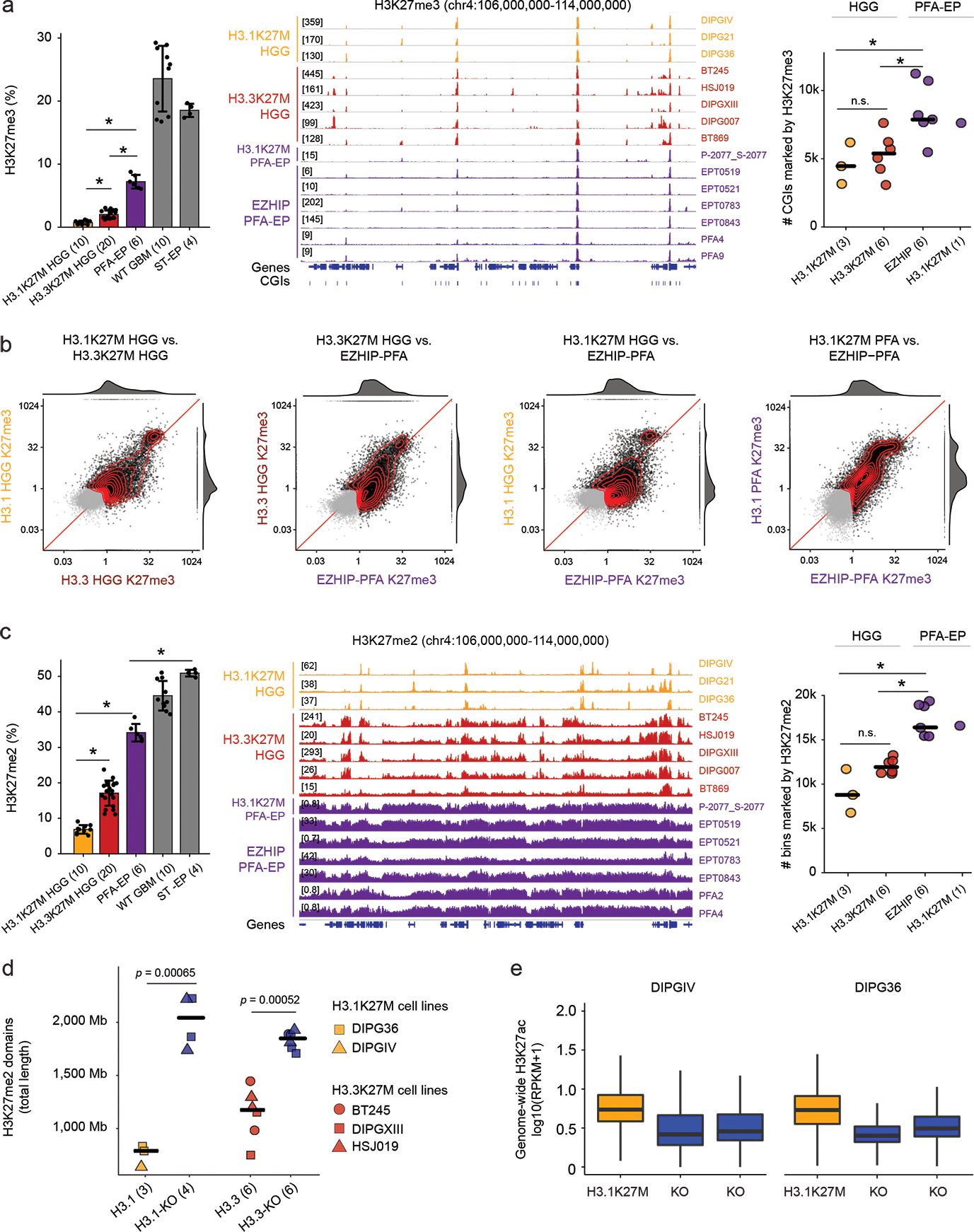
a. Profiling of H3K27me3. Left: Mass spectrometry data of H3K27me3 in cell lines. Number of biologically independent samples per group is indicated in parentheses. WT GBM: H3 wild-type glioblastoma. ST-EP: EZHIP wild-type supratentorial ependymoma. Error bars: mean +/− SD. P-values (Welch two-sample t-test): H3.1K27M vs H3.3K27M, p = 5.3×10−8; H3.1K27M vs PFA-EP, p = 1.6×10−5; H3.3K27M vs PFA-EP, p = 2.6×10−5. Middle: H3K27me3 ChIP-seq enrichment tracks over representative genomic region. Right: Number of H3K27me3-marked CGIs genome-wide. Crossbar indicates the median. P-values (Welch two-sample t-test): H3.1K27M vs. EZHIP PFA, p = 0.022; H3.1K27M vs. H3.3K27M, p = 0.55; H3.3K27M vs EZHIP PFA, p = 0.010.
b. Scatterplots of H3K27me3 signal over CGIs genome-wide in pairwise group comparisons. X- and Y- axes represent log2 mean RPKM value per group, normalized by input. Marked CGIs (mean RPKM > 1 in at least one groups in each comparison) are shown in black, while unmarked CGIs are shown in gray. Joint density and marginal distributions are calculated over marked CGIs only. Red line indicates the diagonal.
c. Profiling of H3K27me2. Left and middle panels: as in (a). P-values (Welch two-sample t-test): H3.1K27M HGG vs H3.3K27M HGG: p = 8.4×10−12; H3.1K27M vs PFA-EP: p = 1.0×10−7; PFA-EP vs ST-EP: p = 1.6×10-6. Right panel: Number of H3K27me2-marked 100kb-bins genome-wide. Crossbar indicates the median. P-values (Welch two-sample t-test without correction): H3.1K27M vs. EZHIP PFA, p = 0.014; H3.1K27M vs. H3.3K27M, p = 0.17; H3.3K27M vs EZHIP PFA, p = 1.8×10-5.
d. Total length of genome covered by H3K27me2 domains in K27M-mutant cell lines and isogenic K27M-KO lines. Domains were identified using a segmentation algorithm (see Methods). Crossbar indicates the median. P-values (Welch two-sample t-test without correction): H3.1K27M vs KO, p = 0.00065; H3.3K27M vs KO; p = 0.00052.
e. Distribution of H3K27ac in 1Mb bins genome-wide in isogenic H3.1K27M HGG cell lines DIPGIV and DIPG36.