

Abstract
Pleuropulmonary blastomas (PPB) is the most common primary malignant neoplasm of the lung in children. Like the other solid dysontogenic neoplasms, this tumor typically presents before the age of seven years. The earliest manifestation is the presence of a lung cyst(s) which is usually recognized in the first year of life and is difficult on the basis of imaging studies to differentiate it from non-neoplastic cysts of early childhood. From a multilocular cyst, PPB has the potential to progress to a high grade multipatterned primitive sarcoma. Over 65% of all affected children have a heterozygous germline mutation in DICER1. The DICER1-PPB familial tumor predisposition syndrome is initially recognized in most cases on the basis of PPB alone, but also several other unique and characteristic extrapulmonary tumors including pediatric cystic nephroma, nasal chondromesenchymal hamartoma, nodular lesions of the thyroid, embryonal rhabdomyosarcoma of the cervix and ciliary body medulloepithelioma.
Keywords: Pleuropulmonary blastoma, lung, rhabdomyosarcoma, DICER1, cystic nephroma
Pleuropulmonary blastoma (PPB) is a neoplasm of the lung which presents in the first seven years of life and is generally familiar to most pediatric pathologists and surgeons. Because of the natural history of PPB, the initial clinical manifestation in the first year of life and even prenatally is a pulmonary cyst with the differential diagnosis including cystic lesions of the lung in the infancy period. Children beyond infancy are more likely to present with respiratory symptoms resulting from a mass which has filled the chest. These clinical extremes are correlated with the age at the time of diagnosis and with survival, since PPB progresses from a multicystic lesion to a solid high grade sarcoma.
The purpose of this paper is to review our experience with PPB which began almost 40 years ago when the first example of this neoplasm was seen by one of the authors (LPD). Over the arc of these years, the breadth and depth of our understanding of this neoplasm have evolved well beyond even our expectations at that time. Each stage of the process seemed to open another pathway in the odyssey of PPB.
Setting the stage.
It can be argued that the stage for the eventual recognition was set over 60 years when Barnard described a primary neoplasm of the lung in a 40-year-old female which was thought to resemble embryo-fetal lung and was designated an “embryoma” (1). Approximately 10 years later, Spencer reported three cases of a lung neoplasm in individuals between the ages of 19–59 years whose histopathologic features were reminiscent of Wilms tumor; he designated this tumor as “pulmonary blastoma” (PB) and suggested that it was the pathogenetic equivalent of Wilms tumor and the other dysontogenetic neoplasms presenting in childhood (2).
The hole in the story was the fact that unlike the other major dysontogenetic neoplasms of childhood, PB occurred almost exclusively in adults. Like others with bronchogenic carcinoma, there was a smoking history. The World Health Organization classification of lung neoplasms includes PB as a subtype of sarcomatoid carcinoma whereas PPB is classified with the mesenchymal tumors (3). In contrast to PB, most PPBs are diagnosed at or before seven years of age or detected on prenatal ultrasonography as a cystic lung lesion whose diagnosis of type I PPB is established after surgical resection (4,5).
It took almost 10 years to collect the first 11 cases of PPB; these tumors were all large masses occurring in children between the ages of 21/2 - 12 years with mean and median ages 5.5 and 3 years, respectively (Figure 1A,B) (6). Most tumors were unresectable at the time of diagnosis which accounts in part for the fact that almost 70% of patients had tumor-related deaths within two years of diagnosis.
Figure 1.
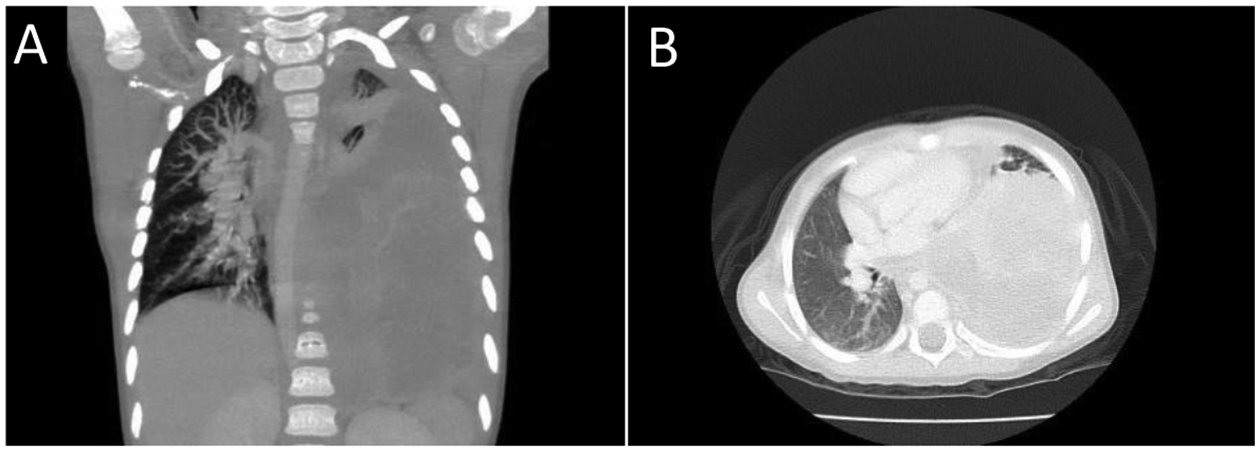
Pleuropulmonary blastoma presented in a 4-year-old girl with respiratory symptoms. A,B. The entire left thoracic cavity is occupied by a 15 × 9.5 × 9.7 cm. heterogeneous enhancing mass which has crossed the midline. Mass effect on the descending intrathoracic aorta, heart and left hemidiaphragm has occurred. This appearance is typical of type II or type III PPB. Both rhabdomyoblastic and chondroid elements were seen in the biopsy.
What more was to be learned about PPB?
Following the publication of our study of 11 cases of PPB, the major question to be answered was the direction of any future investigation. As with the study of any neoplasm in children, we knew almost by definition that PPB was rare since only 10–11,000 newly diagnosed malignancies are seen in children each year in the United States (7). In 1988, our collaborator, John Priest, M.D., suggested that a registry should be established and so the International Pleuropulmonary Blastoma Registry (IPPBR) (www.ppbregistry.org) was established. In 2003, with the advancement of the internet, the IPPBR began to proactively curate all peer-reviewed medical journal articles to identify likely examples of PPB. When a possible PPB case is identified, the IPPBR contacts the authors and requests the opportunity to review the pathology and obtain recent follow-up. Over time the IPPBR began to see more cases from pathologists and pediatric hematologists-oncologists of putative PPBs. It was established early in the development of the IPPBR that all suspected cases of PPB must have the diagnosis confirmed pathologically from the actual materials before entry into the IPPBR.
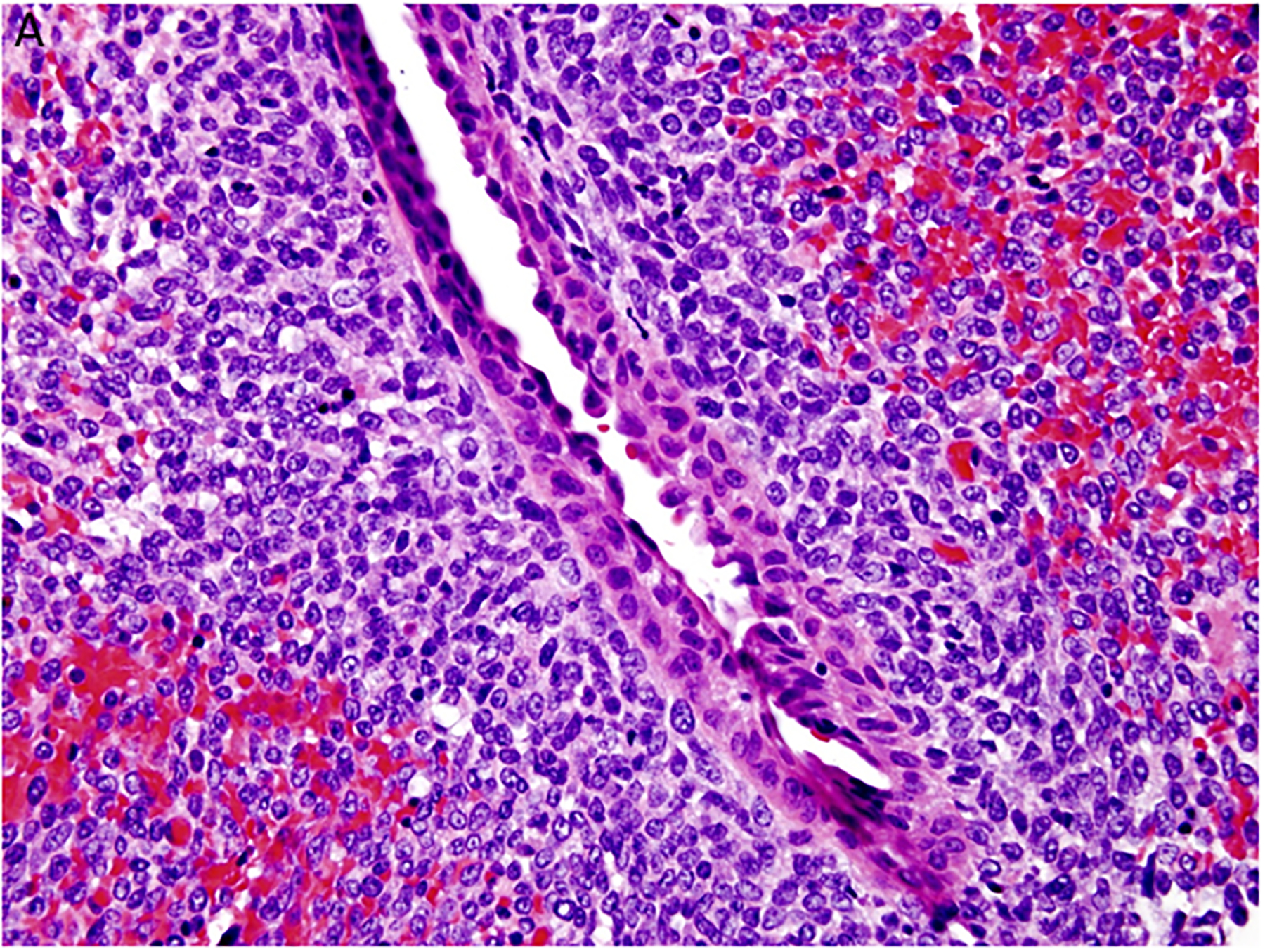
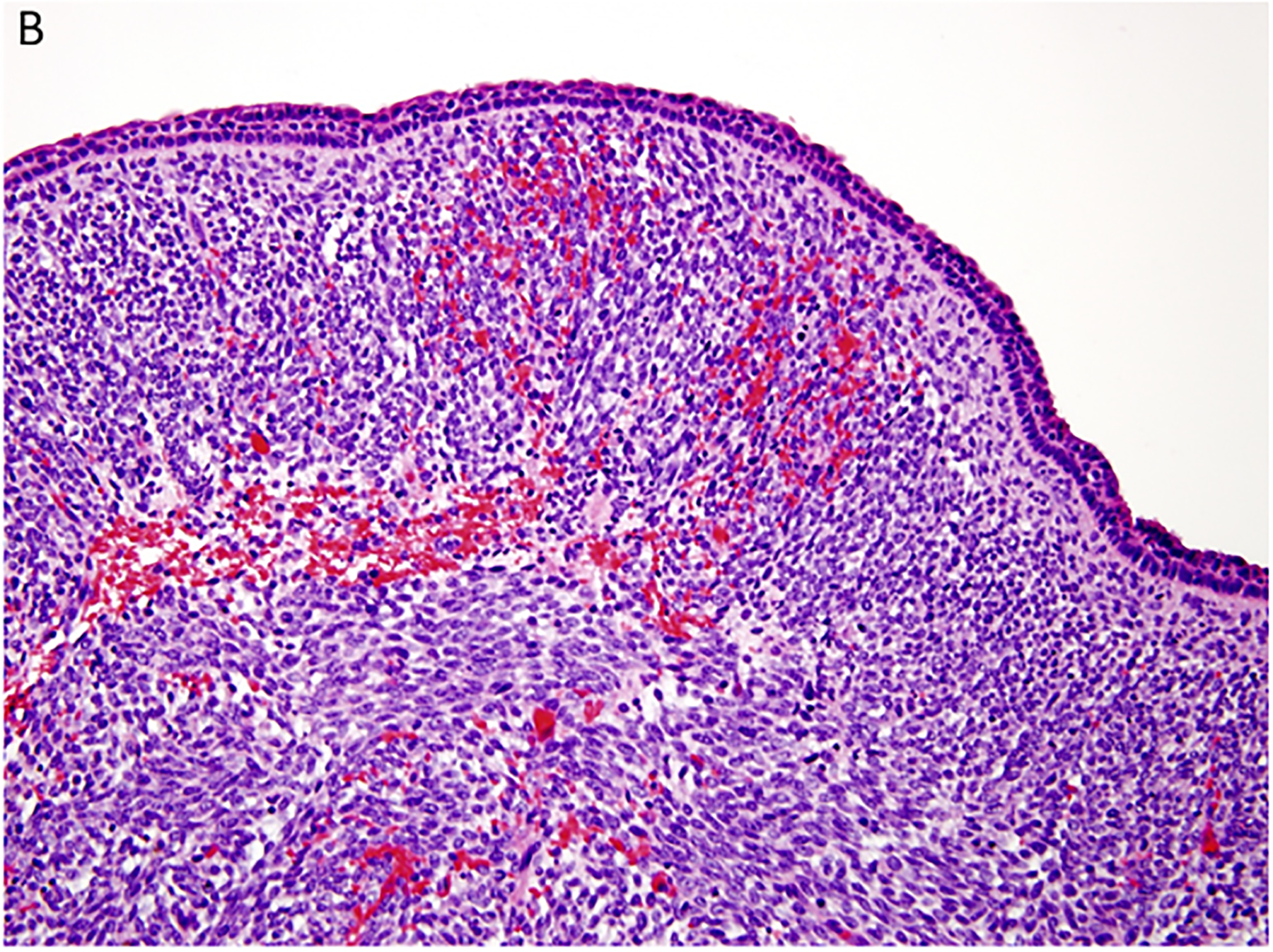
Two important findings emerged from these initial cases which were referred to the IPPBR and laid the foundation for future studies. The first of these related to the pathology of PPB. Our perspective from the first 11 cases was that PPB like the other solid malignancies presented as a fully developed pathologic process. However, it appeared that this mass stage was preceded by a multicystic lesion of the lung which was followed by a cystic and solid neoplasm whose histopathologic features in part resembled the purely cystic lesion whereas the solid areas had the complex collage of a mixed pattern primitive sarcoma (Figure 2A–D). These three lesions were regarded as progressive morphologic stages in the formation of PPB and were designated type I or purely multicystic PPB, type II or solid and cystic stage and the ultimate type III PPB or solid only stage. In the report of 50 cases of PPB, there was a remarkable correlation between the median age at diagnosis and the morphologic type of PPB: type I (10 mo), type II (34 months) and type III (44 months) (8). The age at diagnosis and the type of PPB in turn correlated with the prognosis.
Figure 2.
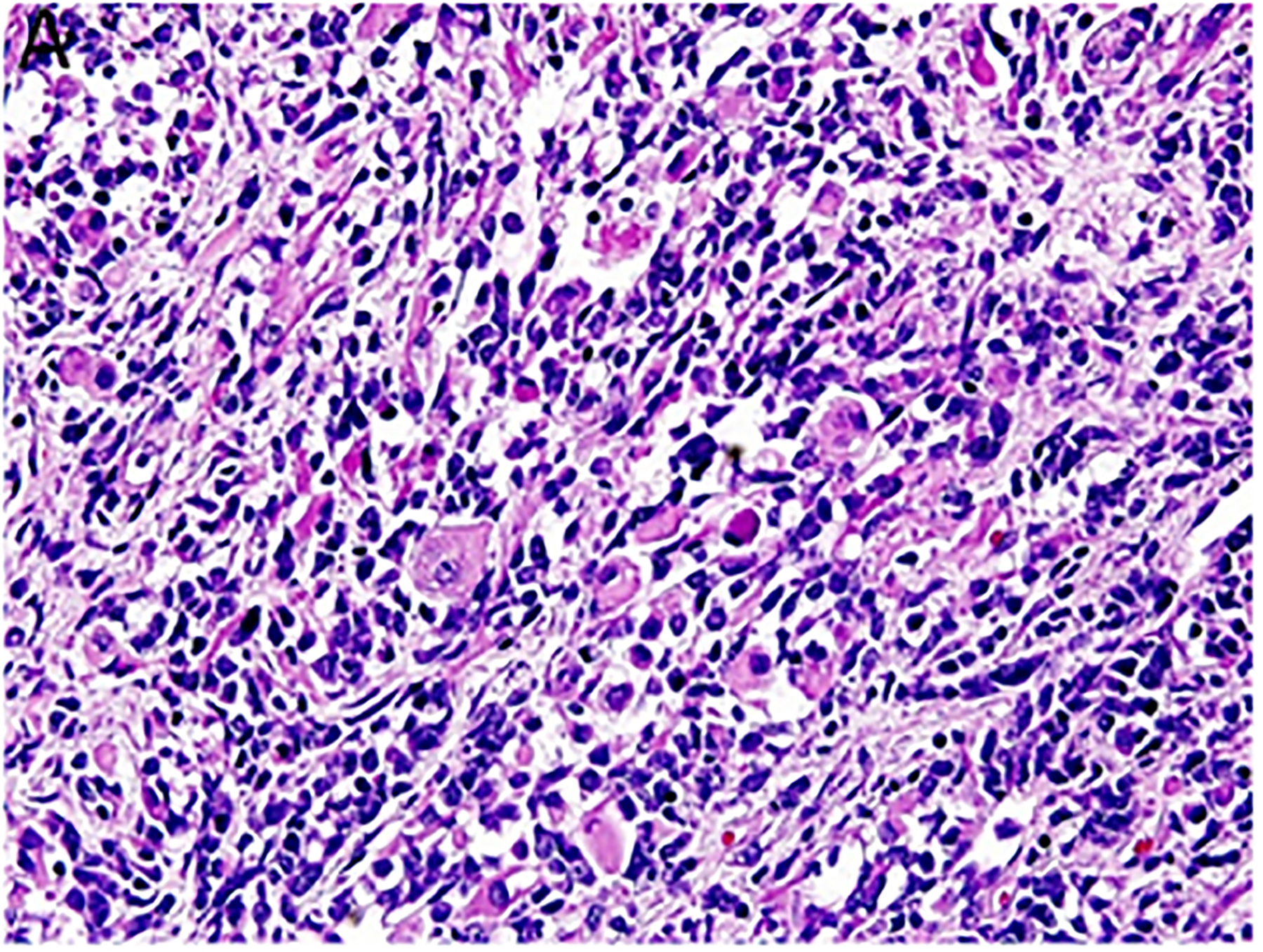
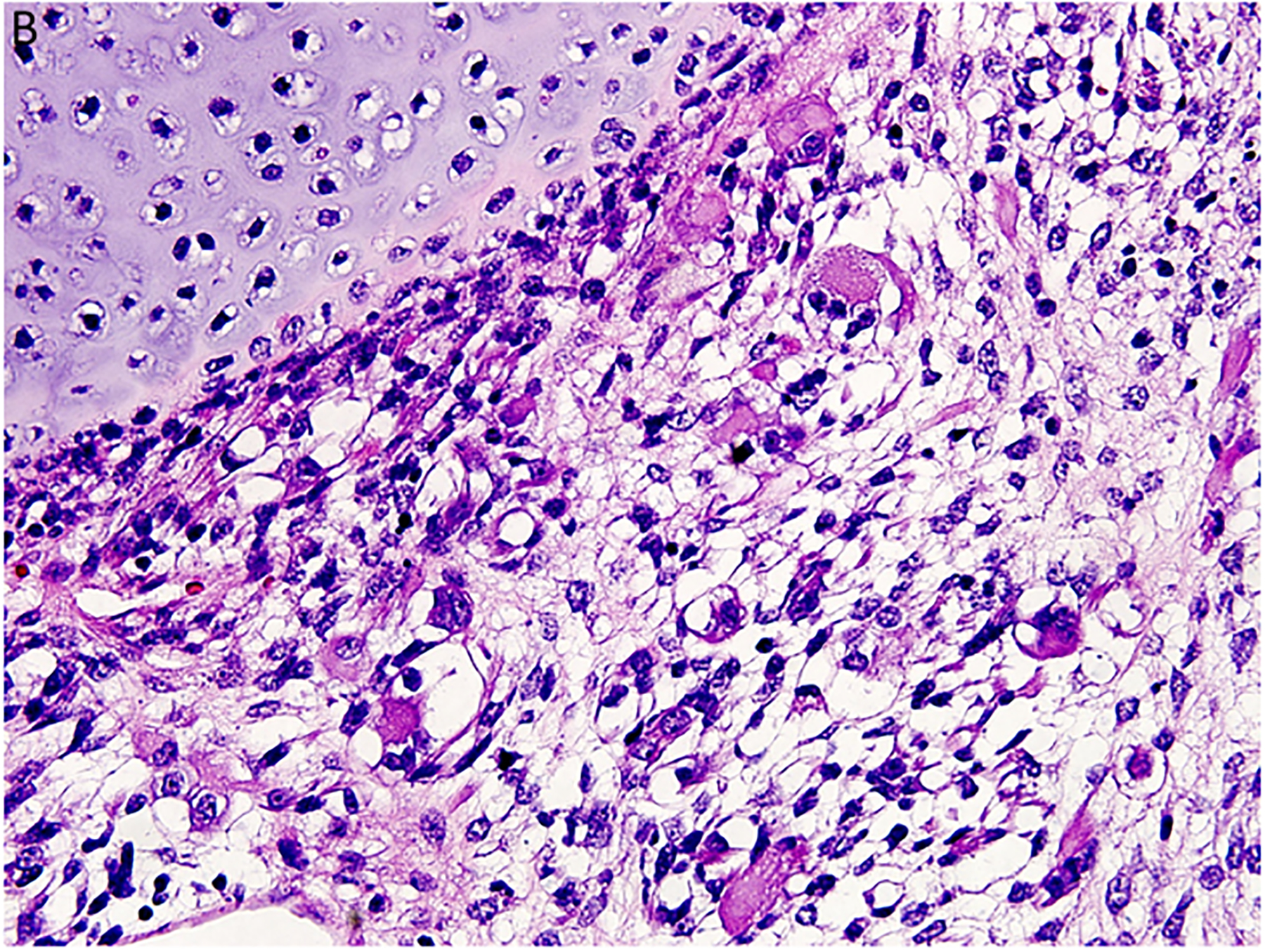
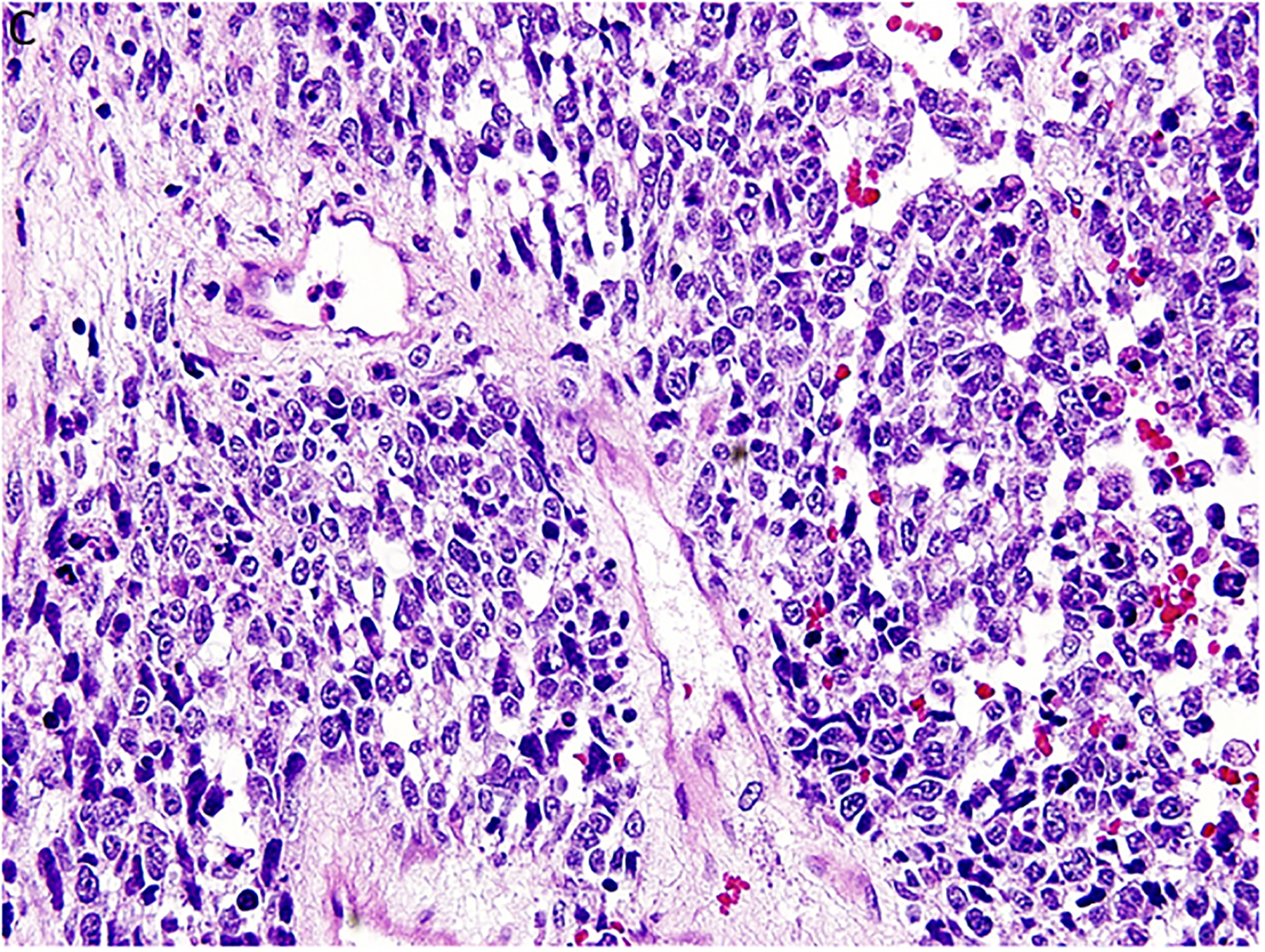
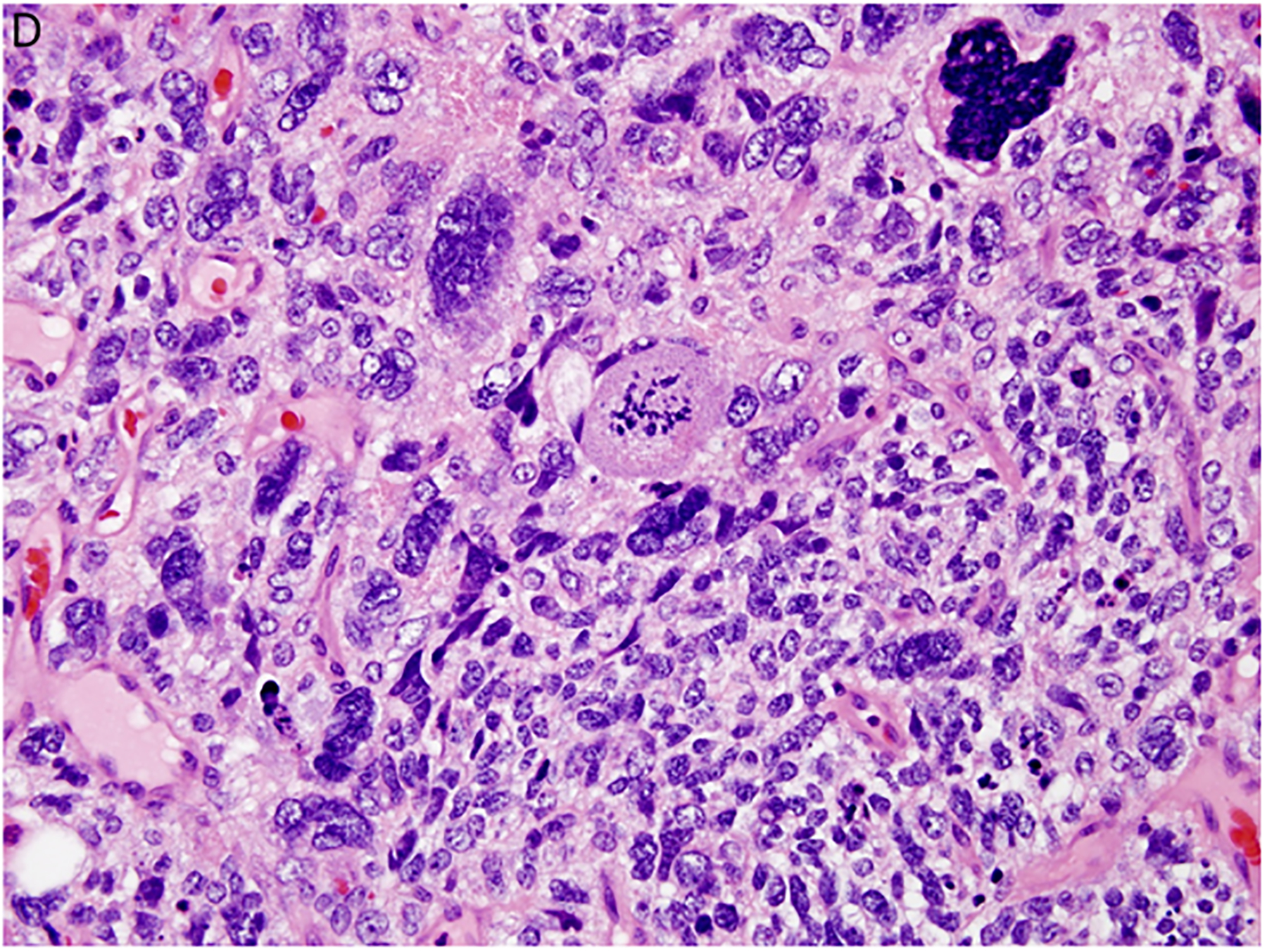
Pleuropulmonary blastoma has a heterogeneous appearance within the solid areas of either type II or type III PPB. A. Rhabdomyosarcomatous pattern includes large rhabdomyoblasts with intense eosinophilic cytoplasm surrounded by smaller, more primitive rhabdomyoblasts (x400). B. The periphery of a nodule of fetal appearing cartilage is adjacent to a focus of rhabdomyosarcoma (x400). C. Nests of primitive malignant small cells within poorly formed nests representing the blastemal pattern (x400). D. A focus of primitive sarcoma with enlarged, hyperchromatic tumor cells and bizarre mitotic figures representing anaplasia (x400).
The second notable observation related to the fact that approximately 25% of the initial 45 children had a history of other tumors or had young relatives who also had PPB, cystic nephroma (CN) and rhabdomyosarcoma as several examples (9). This finding was the initial clue to a possible familial tumor predisposition syndrome.
In order to pursue the heritable aspects of PPB, it was necessary to not only identify these families, but obtain a thorough pedigree analysis and to eventually acquire blood samples. In the first year of this genetic study, 45 families with multiple affected family members consented to participate in a study that would provide the opportunity to identify the underlying genetic defect that might not only benefit their family, but other families in the future. The strong support shown by the PPB families was critical in obtaining pilot funding from Hope Street Kids Foundation and the Children’s Discovery Institute at St. Louis Children’s Hospital. This funding supported history and sample collections, expertise of a genetic counselor and financial resources for SNP array testing on germline DNA for linkage analysis. A single narrow linkage peak was identified in the first four families tested containing only 72 genes (10).
A clue as to a candidate gene was provided in the study by Harris, et al (11); these authors reported that Dicer inactivation in murine lung epithelium resulted in branching arrest and the formation of a few “large epithelial pouches” with a resemblance to the earliest recognizable lesion of PPB, the type I or multicystic morphology. It was proposed that Dicer has a specific role in the branching morphogenesis of lung.
With the murine-DICER1 model in mind, the decision was made to sequence DICER1 in 11 probands with PPB. That decision proved to be a good one when all 11 probands showed deleterious heterozygous germline DICER1 mutations (10). The detection of a somatic mutation in the second DICER1 allele in PPB and other DICER1-related tumors conforms to the Knudson two-hit hypothesis (12,13). Later studies have expanded on Harris’ original observations in the Dicer deficient murine model. Conditional embryonic DICER1 loss leads to Fgf9 over-expression in the lung epithelium which drives cystic dilation and mesenchyme proliferation and produces a type I PPB phenotype (14). Another important finding in the pursuit of an experimental model of PPB has demonstrated that precise temporal and cell type specific loss of DICER1 is necessary and sufficient to recapitulate the pathology of type I PPB (15).
Natural history.
The most important stage at which to recognize PPB is the multicystic or type I lesion since it is the most “curable” phase (16). One complication in the pathologic diagnosis of type I PPB is the co-existence of congenital cystic adenomatoid malformation/congenital cystic pulmonary airway malformation type 4 (CCAM/CPAM 4) in the same morphologic niche as PPB (17). Some of the potential problems in diagnosis emerged in the study of malignant transformation of CCAM/CPAM and in particular CCAM/CPAM 4. One of the four children who were two years of age or less diagnosed with CCAM/CPAM developed a “grade 3 PPB” one year after the resection of a CCAM/CPAM 4 and “focal stromal hypercellularity” was noted in three of four cases in this young age group (18) (Table 1). The authors of that study concluded that “a distinction between grade 1 (type I PPB) pleuropulmonary blastomas and type 4 CPAMs was not possible using the conventional criteria for malignancy” (18). In an exchange of letters to the editor, MacSweeney et al. stated that “our views had advanced and it is likely that today, we would diagnose those cases showing stromal cellularity as type I pleuropulmonary blastoma” (19). It is critically important to the patient that a type I PPB should never be interpreted as CCAM/CPAM 4 since the opportunity for appropriate post-operative care and follow-up will be lost.
Table 1.
Malignant Transformation of “CCAM/CPAM 4”
| CCAM/CPAM | Ages At Presentation | Follow-up |
|---|---|---|
| TYPE 4 * | 4 cases (13, 18, 26, 28y) | PPB type Ir (2NR, 2LTFU) |
| 4 cases (1, 1, 2, 2y) | Focal stromal HC (3) | |
| (INR, 2 LTFU) 1 – “grade 3” PPB at 2y |
“TYPE 4 CCAMs show histologic overlap with grade 1 PPBs, and distinction between these entities may not be possible on histology alone.” Adapted from MacSweeney, et al. Am J Surg Pathol 2003; 21:1139–1146.
Abbreviations: y = year; NR = no recurrence; LTFU = lost to follow-up; HC = hypercellularity
Type I PPB is a multilocular cyst which is found at the periphery of the lung and is usually a collapsed structure when it is received for pathologic examination (20). The low magnification architecture is a multicystic or locular structure with a pleura at the periphery and lung parenchyma in most cases transitioned into a multilocular cyst (Figure 3A,B). There is no entrapped lung parenchyma within the cyst itself in contrast to its presence in most CCAM/CPAM types 1 and 2 (17). Once recognizing the architecture, the entire specimen should be submitted for microscopic evaluation.
Figure 3.
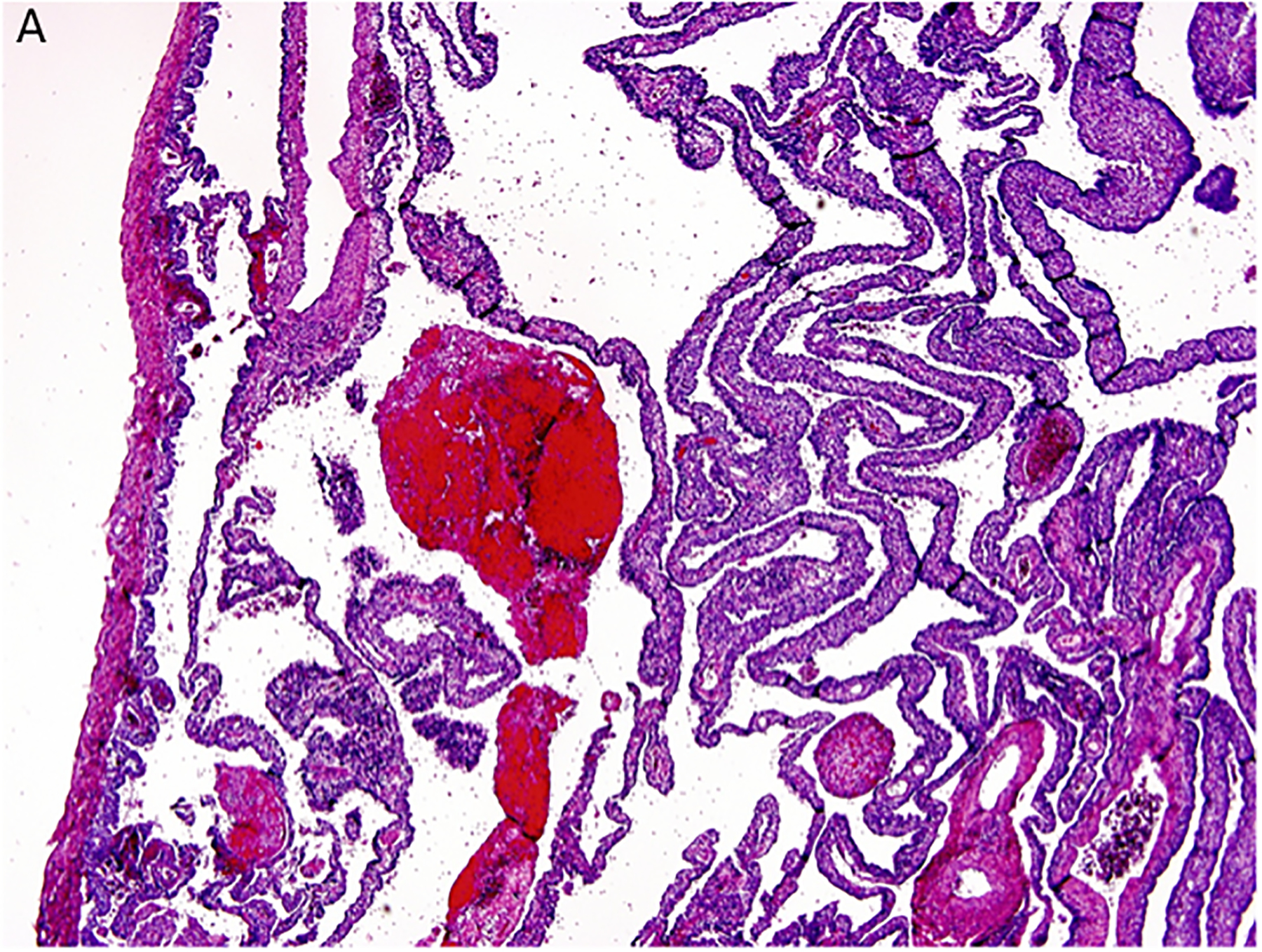
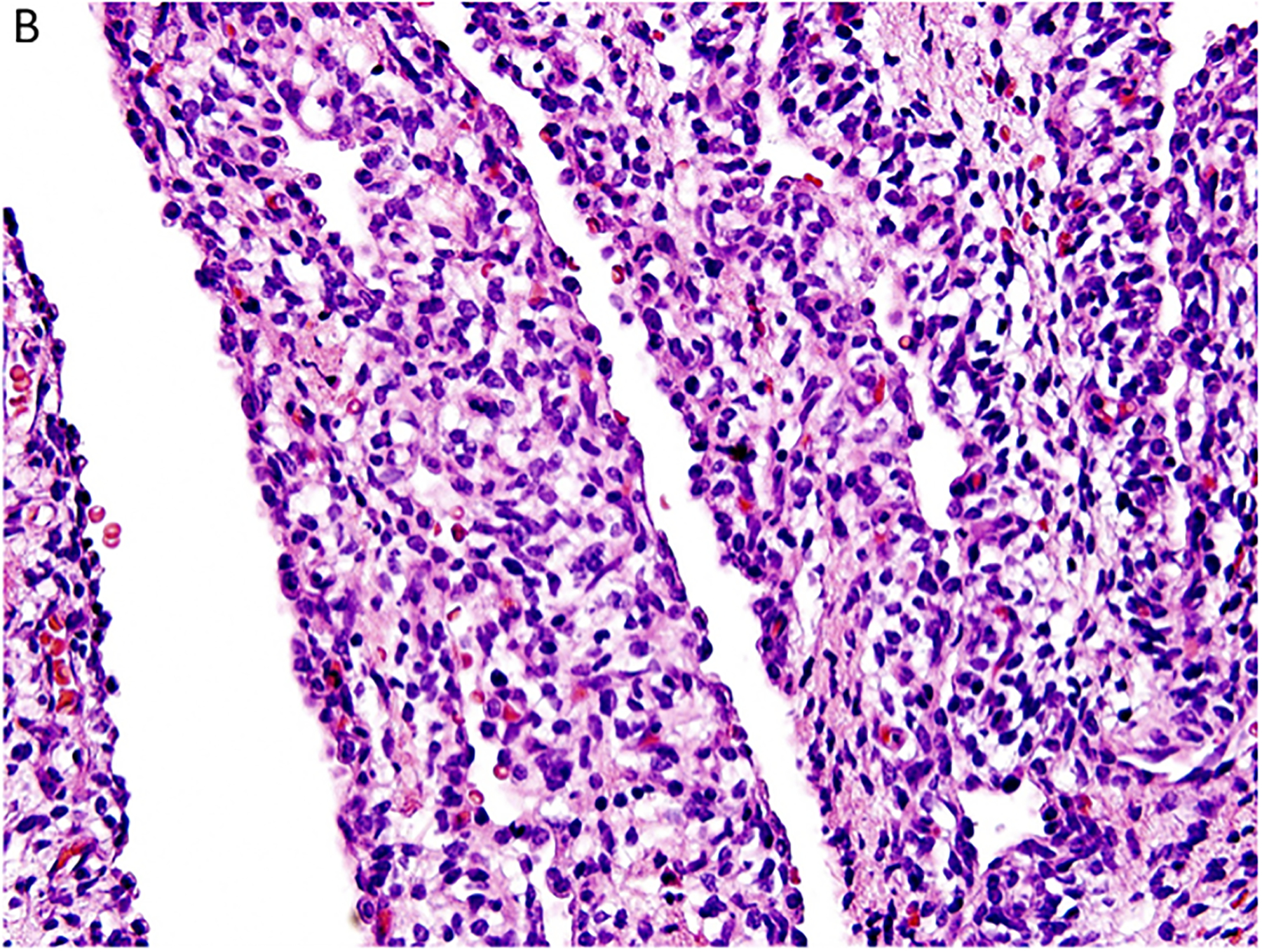
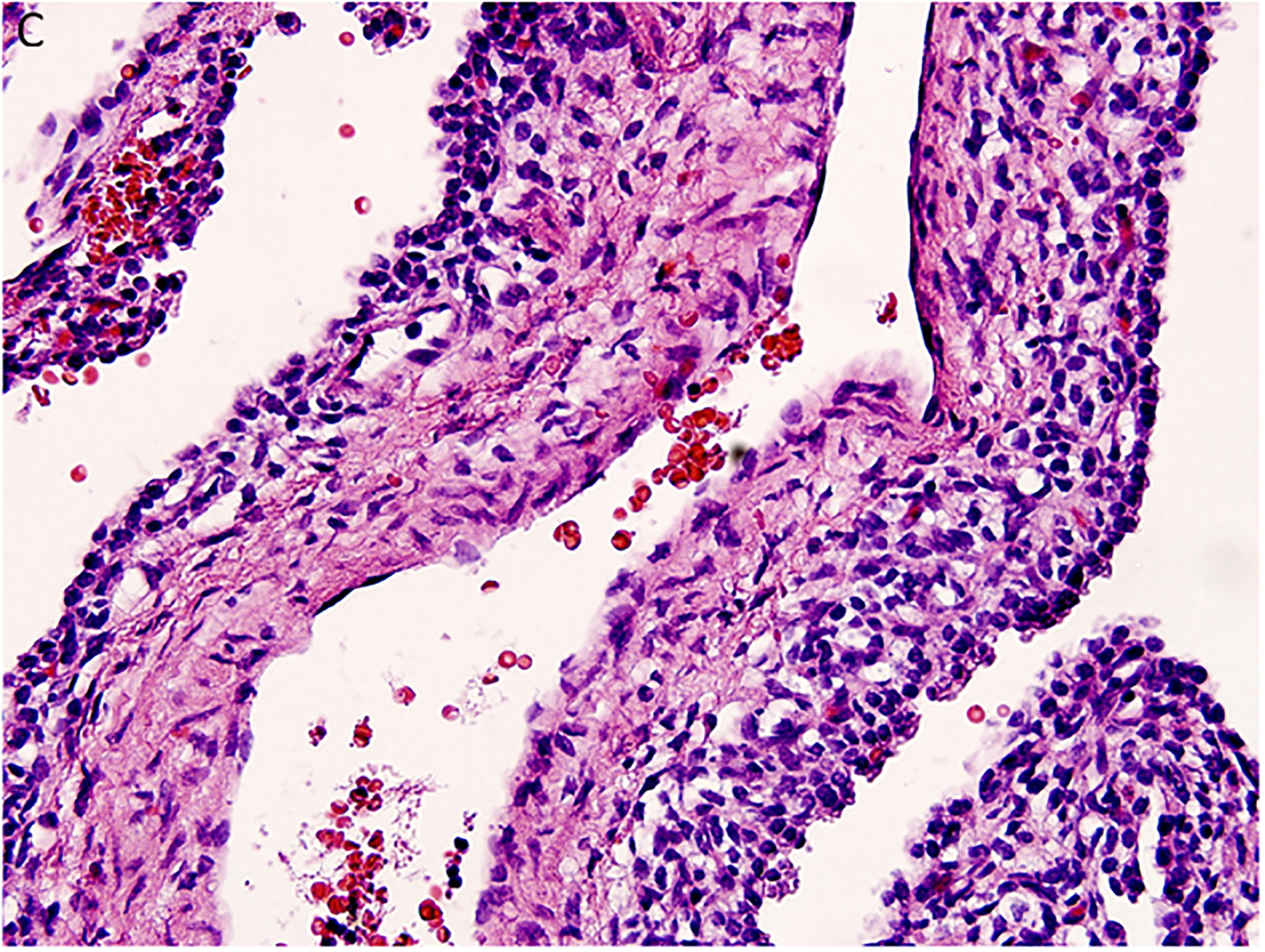
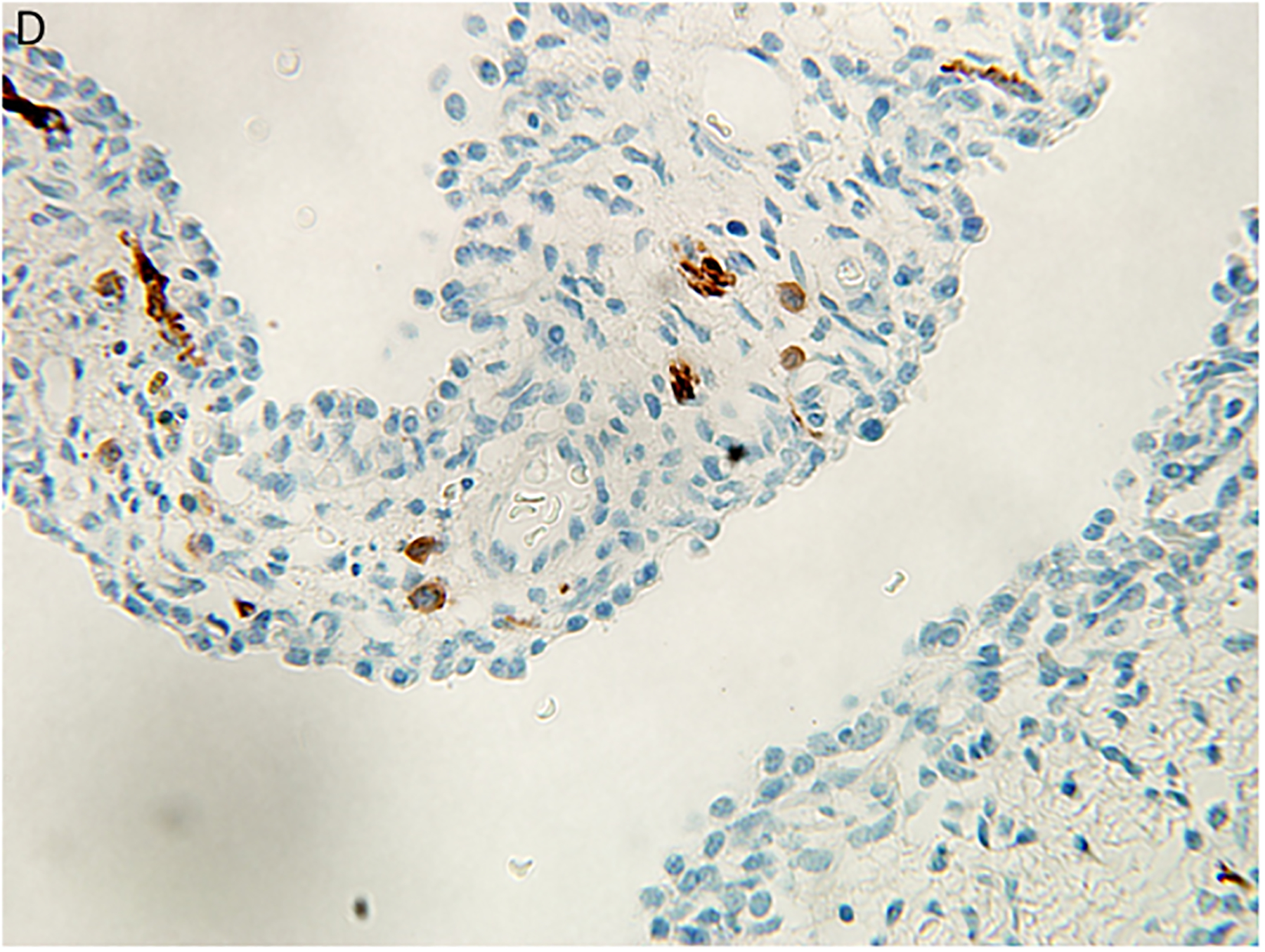
Pleuropulmonary blastoma type I presented with spontaneous pneumothorax in a 14-month-old girl. A cystic lesion was detected in the right lung. A. This low magnification demonstrates the typical multicystic pattern. Note that the cyst is covered with pleura along one edge (x40). B. The septal stroma is occupied by a uniform population of small primitive cells (x400). C. The septa demonstrates a cambium layer of small primitive cells and a fibrous stroma. Some septa only had the fibrous stroma (x400). D. Desmin immunostain shows the presence of individual positive cells in the septa. A similar pattern of immunostaining was obtained with myogenin (x400).
Histologically, the type I PPB can be a challenge if one is expecting to identify a uniform subepithelial cambium layer of small primitive round cells or evidence of rhabdomyoblastic differentiation in all cases (Figure 3C,D). A subepithelial fibrous stroma and prominent small blood vessels may be identified in many of the septa and alternating or interposed among those septa are other septae with immature small cells forming a cambium layer-like appearance. The septa are typically uniform in width, but some may be expanded by immature small cells and/or spindle cells; these hypercellular, expanded septa are thought to represent features of progression of a type I PPB. As long as there is no discernible grossly visible mass; however, the lesion is still regarded as a type I PPB.
Two categories of type I PPB are recognized and the above discussion is focused on type I PPB in the presence of immature mesenchymal cells with or without rhabdomyoblastic differentiation. The second category of type I PPB has the “r” appended to designate possible regression or failure to progress to a type II or III PPB (20). The stroma has a bland fibrovascular appearance without any foci of small primitive round cells after examination of the entire specimen. Small nodules of cartilage within the septa may be seen in both type I and type Ir PPBs. The median age at diagnosis of type Ir PPB was 47 months compared to 8 months in the case of type I PPB (16).
Regarding type I and type Ir PPB, it is our postulation that only some PPBs are fated to progress to the more aggressive and potentially lethal type II or type III PPB; this failure of tumor progression is also seen in Wilms tumor (nephrogenic rests) and neuroblastoma. Biallelic DICER1 mutations alone may be insufficient for malignant progression. Mutation and/or loss of TP53 and mutations of NRAS have been detected in PPBs in support of the notion that additional mutations are required for type I PPB to evolve into type II or III PPB with its lethal potential (21).
There are several lines of evidence in support of the progression of type I PPB to the tumefactive type II and III PPBs. The more compelling of these is the existence of type II PPB with its residual foci of type I PPB and solid areas of a multipatterned primitive sarcoma with rhabdomyoblastic and chondroid (not all cases) differentiation indistinguishable from type III PPB (Figure 4A–D) (8). In addition there are cases of type I PPB which have been diagnosed earlier in infancy as CCAM/CPAM 4 which recurred one or two years later as type II or III PPB (22). Importantly, these cases are seen less frequently today with the pathologic recognition of type I PPB. A third observation relates to the median ages at diagnosis of types I, II and III PPB at 8, 35 and 41 months, respectively which was noted in our earlier study of 50 cases and confirmed in our recent study of 350 cases (8,16).
Figure 4.
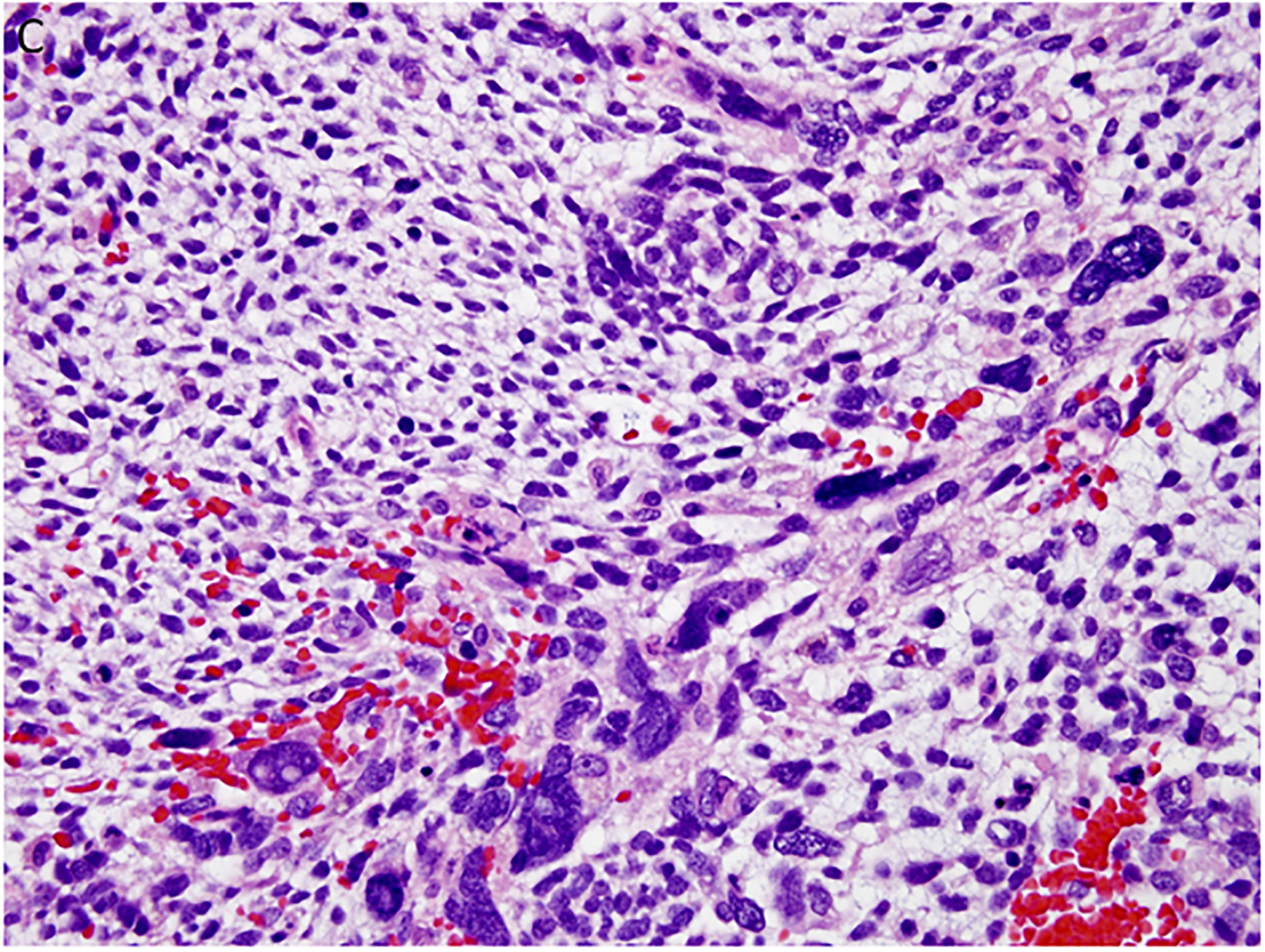
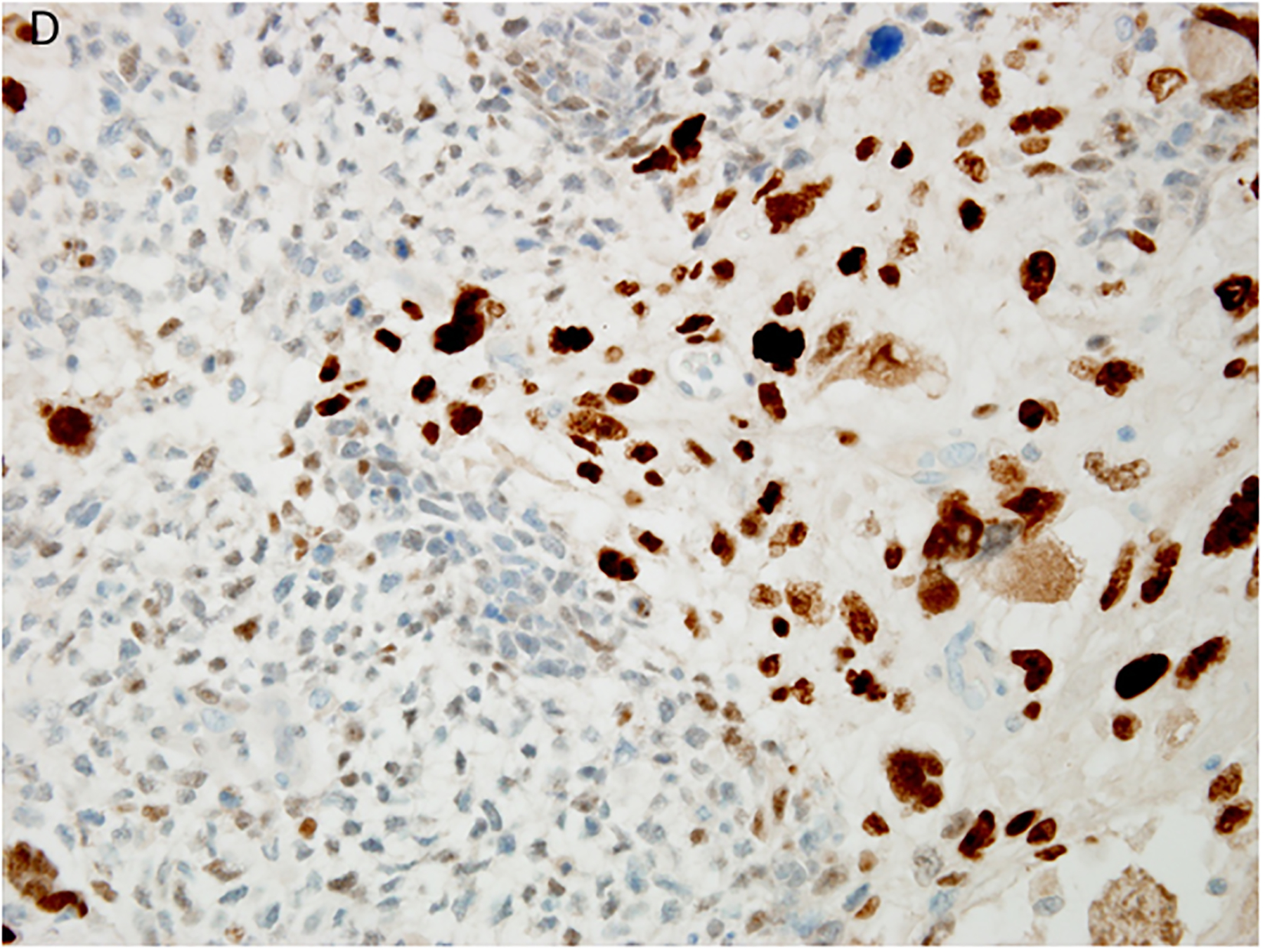
Pleuropulmonary blastoma type II presented in this 3-year-old female as a solid and cystic mass in the right chest. A. The only epithelium in a PPB is the remnant of the cyst lining (x400). B. This field shows the presence of a continuous epithelium of a residual cyst and an expanded stroma composed of uniform malignant small cells (x200). C. Focus of anaplastic cells are identified among the primitive malignant cells (x400). D. p53 immunostaining is especially intense in the nuclei of the anaplastic cells (x400).
Type II versus type III PPB and prognosis.
The earlier experience with type II and type III PPBs was attempt at primary resection with either a lobectomy or a debulking of the intrathoracic, friable mass (8). Our more recent experience is that a biopsy is performed after imaging studies have confirmed the presence of a mass. The biopsy demonstrates a primitive appearing, obviously malignant neoplasm with or without rhabdomyoblastic differentiation which has been confirmed by immunohistochemistry. Prior to the recognition of PPB, these cases were diagnosed as embryonal rhabdomyosarcoma (ERMS) and treated according to the former Intergroup Rhabdomyosarcoma Study Protocol. One recent study of ERMS and the rhabdomyosarcomatous component of PPB demonstrated an over-expression of insulin-like growth factor (IGF2) in ERMS and its low expression in PPB (23). Obviously it is not possible to perform IGF2 on a routine basis, but if there is a large intrathoracic or pulmonary-based mass combined with a biopsy demonstrating ERMS, then a diagnosis of PPB can be made in most cases or submitted to the IPPBR for consultation. In the course of our review, we have seen examples of Ewing sarcoma-primitive neuroectodermal tumor, pleuropulmonary synovial sarcoma, congenital-infantile fibrosarcoma and a recent example of malignant ectomesenchymoma (LPD).
Most children have a surgical resection of a type II or III PPB after receiving chemotherapy (16). Pathologic examination of the specimen is directed toward documenting the presence of residual tumor, assessment of any surgical margins and a determination, if possible, whether the PPB is type II or type III. There is prognostic importance to this distinction since by all measures children with type II PPB have a significant survival advantage over those with type III PPB (16). Even though the solid area of type II and type III PPBs are indistinguishable pathologically, the unavoidable conclusion is that type III PPB is clinically and biologically a more aggressive neoplasm. An explanation for this observation must await future studies.
Congenital lung cyst and PPB.
A congenital lung cyst of one suspected type or another, usually a CCAM/CPAM, is the clinical impression when such a case is submitted for pathologic examination. The diagnosis of type I PPB is established on the basis of the microscopic review. The existence of type I PPB has altered the landscape of congenital lung cysts and has resulted in the creation of a dilemma in the management of these cases, especially when the cyst is asymptomatic (24,25). Tagge, et al (26) were among the first in the pediatric surgery community to introduce a cautionary note in the nonoperative approach to the asymptomatic cyst because of the concern about a possible PPB. The resection of an asymptomatic lung cyst from an infant is not without its complications as reported in a study by Hall and associates (27). The latter authors reported that there were “no cases of PPB” among their 60 cases, but they had one example of “CCAM type 4,” which we have discussed above. An earlier study also from The Hospital for Sick Children, Toronto reported that 4% of resected lung cysts were PPBs (28).
Without a thorough histopathologic evaluation, there is no reliable approach to discriminate preoperatively one congenital pulmonary cyst from another including PPB (29). More than one cyst in the lung should alert to the possibility of PPB since most other congenital cysts are solitary, whereas type I PPB is considerably more likely to be multifocal. An accompanying multicystic lesion in the kidney representing CN establishes the nature of the lung cyst as a PPB. There is also the opportunity to perform genetic testing for the DICER1 germline mutation. Serum microRNA testing may become available in the future (30). Until that more reliable test is found, the controversy about the appropriate care of the child with an asymptomatic lung cyst is unlikely to abate (31–33).
DICER1-PPB familial tumor predisposition syndrome.
It was initially thought that 25% of PPB cases were familial; however, it now appears that 65%−70% of children with PPB have a heterozygous germline mutation in DICER1 and approximately 84% of those germline mutations were inherited from a parent (34). In terms of the other solid malignant neoplasms of childhood with an inherited component, PPB is only superseded by medullary thyroid carcinoma (Table 2). The vast majority of the other examples of PPB are also attributable to DICER1 mutations but in a mosaic or tumor specific cellular level (34). Mosaicism for RNase IIIb domain missense mutations appears to predict a severe phenotype with multiple primary tumors of various sites (34).
Table 2.
Hereditary Component of Various Solid Neoplasms Presenting in Childhood
| Tumor Type | Gene | Hereditary (%) |
|---|---|---|
| Medullary Thyroid Carcinoma | RET (10q11.2) | >90% |
| Pleuropulmonary Blastoma | DICER1 (14q32.13) | 65 – 70% |
| Adrenocortical Neoplasia | TP53 (17p13.1) | 50 – 80% |
| Optic Glioma | NF1 (17q11.2) | 45% |
| Retinoblastoma | RB1 (13q14.2) | 40% |
| Pheo-Paraganglioma | VHL, NF1, RET, SDHB, SDHD | 40% |
| ATRT/MRT | SMARCB1 (22q11.23) | 25% |
| Hepatoblastoma | APC (5q21-q22) | 8 – 9% |
| Wilms Tumor | WT1 (11p13) | 3 – 5% |
| Rhabdomyosarcoma | TP53, NF1, NBS1, ESC02, PTPH11, BUBIB, CREBBP, NF2, TSC1/2, WRN | 2 – 4% |
| Neuroblastoma | ALK1 (2p23) | 1 – 2% |
| CNS Tumors | PTCH1, RAS-NF1, NF2, WNT, RB1, TP53, VHL | <1 – 3% |
Abbreviations: ATRT/MRT = Atypical teratoid rhabdoid tumor tumor / malignant rhabdoid tumor; CNS = Central nervous system
Cystic nephroma of the pediatric kidney was the first documented extrapulmonary tumors in the setting of the DICER1 tumor predisposition syndrome; it is estimated that 9%−12% of children with PPB have a synchronous or metachromous CN (35). These multicystic lesions are also accompanied by a somatic DICER1 mutation in 70% of cases and are no longer regarded as histogenetically related to Wilms tumor or cystic partially differentiated nephroblastoma (36). DICER1-associated cystic nephroma, like type I PPB, has the potential to progress to a primitive sarcoma which is likely the same neoplasm previously reported as anaplastic sarcoma of the kidney (37). Notable also is the strong association between DICER1 and Sertoli-Leydig cell tumor (and gynandroblastoma) which suggests that patients with the latter tumors should be tested for germline or tumor DICER1 mutations (38–40). Overall nodular hyperplasia of the thyroid with or without papillary thyroid carcinoma may be the most commonly associated pathology in DICER1 germline mutation carriers. Though a somewhat disparate group of tumors, embryonal rhabdomyosarcoma of the cervix (not vagina), ciliary body medulloepithelioma, nasal chondromesenchymal hamartoma, hamartoma-like polyps of the small intestine and pituitary blastoma are the other DICER1-associated neoplasms (42–49). Like with a proportion of PPBs, some of these tumors occur in the absence of a germline or somatic DICER1 mutation, but rather have somatic mutations in the latter gene as in a recently reported case of an intraocular medulloepithelioma in an 18-year-old female (50). Any one of these tumors or any combination may be the presenting manifestation of the DICER1-related syndrome, so that it is particularly important for the clinician and/or pathologist to raise that concern and the suggestion for genetic counseling and testing.
PPB has served as an introduction to a unique set of seemingly unrelated neoplasms, all of which are manifestations of germline mutations in the DICER1 gene. The protein encoded by the DICER1 gene is a ribonuclease III enzyme with RNase IIIa and RNAase IIIb domains, which is essential for generation of miRNAs (51–53). Like most other tumor suppressor genes, DICER1 is critical in the regulation of cellular proliferation and differentiation during early development. Because DICER1 is intimately involved in branching morphology in the lung and elsewhere (10), it is not surprising that one of the earliest morphologic manifestations is the formation of cysts in the lungs and kidneys as two examples.
If there is anything that is apparent at this stage of the IPPBR understanding, it is that there is much more to be learned, but we have journeyed a long distance from those first 11 cases of PPB in 1988. PPB is today the most common primary malignant neoplasm of the lung in children (Table 3) (54).
Table 3.
Primary Lung Tumors in Children*
| No (%) | No (%) | ||
|---|---|---|---|
| 52 (44) | 71 (22) | ||
| 22 (19) | 51 (16) | ||
| 9 (8) | 51 (16) | ||
| 8 (7) | 41 (13) | ||
| 27 (23) | 29 (9) | ||
| 27 (8) | |||
| 23 (8) | |||
| 23 (8) | |||
| 118 (100) | 316 (100) | ||
Adapted from Dishop and Kuruvilla. Arch Pathol Lab Med 2008; 132:1079–1103
References:
- 1.Frances D, Jacobsen M. Pulmonary blastoma. Curr Top Pathol 1983; 73:265–294. [DOI] [PubMed] [Google Scholar]
- 2.Spencer H Pulmonary blastoma. J Pathol Bacteriol 1961; 82:161–165. [Google Scholar]
- 3.Travis WD, Brambilla E, Burke AP, Mars A, Nicholson AG. (Eds): World Health Organization Classification of Tumours of the Lung, Pleura, Thymus and Heart. Fourth edition. IARC Press: Lyon: 2015. [DOI] [PubMed] [Google Scholar]
- 4.Mechoulan A, Leclair MD, Yvinec M, Philippe HJ, Winer N. [Pleuropulmonary blastoma: a case of early neonatal diagnosis through antenatal scan screening]. Gynecol Obstet Fertil, 2007; 35:437–441. [DOI] [PubMed] [Google Scholar]
- 5.Miniati DN, Chintagumpala M, Langston C, et al. Prenatal presentation and outcome of children with pleuropulmonary blastoma. J Pediatr Surg 2006; 41:66–71. [DOI] [PubMed] [Google Scholar]
- 6.Manivel JC, Priest JR, Watterson J, et al. Pleuropulmonary blastoma. The so-called pulmonary blastoma of childhood. Cancer 1988; 62:1516–1526. [DOI] [PubMed] [Google Scholar]
- 7.Schneider DT, Brecht IB, Olson TA, Ferrari A. Rare tumors in children and adolescents. rare tumors: A different perspective on oncology. Pediatr Oncol 2012; 1:3–13. [Google Scholar]
- 8.Priest JR, McDermott MB, Bhatia S, Watterson J, Manivel JC, Dehner LP. Pleuropulmonary blastoma: a clinicopathologic study of 50 cases. Cancer, 1997; 80:147–161. [PubMed] [Google Scholar]
- 9.Priest JR, Watterson J, Strong L, et al. Pleuropulmonary blastoma: a marker for familial disease. J Pediatr 1996; 128:220–224. [DOI] [PubMed] [Google Scholar]
- 10.Hill DA, Ivanovich J, Priest JR, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science, 2009; 325:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris KS, Zhang Z, McManus MT, Harfe BD, Sun X. Dicer function is essential for lung epithelium morphogenesis. Proc Natl Acad Sci U S A, 2006; 103:2208–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bignold LP. The cell-type-specificity of inherited predispositions to tumours: review and hypothesis. Cancer Lett, 2004; 216:127–146. [DOI] [PubMed] [Google Scholar]
- 13.Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature, 2011;476:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin Y, Castro AM, Hoekstra M, et al. Fibroblast growth factor 9 regulation by microRNAs controls lung development and links DICER1 loss to the pathogenesis of pleuropulmonary blastoma. PLoS Genet, 2015; 11(5):e1005242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagh PK, Gardner MA, Ma X, et al. Cell- and developmental stage-specific DICER1 ablation in the lung epithelium models cystic pleuropulmonary blastoma. J Pathol 2015; 236:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messinger YH, Stewart DR, Priest JR, et al. Pleuropulmonary blastoma: a report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer, 2015; 121:276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stocker JT, Mani H, Husain AN. The respiratory tract. In: Stocker JT, Dehner LP, Husain AN, eds. Stocker and Dehner’s Pediatric Pathology. Third edition. Philadelphia: Lippincott Williams and Wilkins, 2011; 467–468. [Google Scholar]
- 18.MacSweeney F, Papagiannopoulos K, Goldstraw P, Sheppard MN, Corrin B, Nicholson AG. An assessment of the expanded classification of congenital cystic adenomatoid malformation and their relationship to malignant transformation. Am J Surg Pathol, 2003; 27:1139–1146. [DOI] [PubMed] [Google Scholar]
- 19.Hill DA, Dehner LP. A cautionary note about congenital cystic adenomatoid malformation (CCAM) type 4. Am J Surg Pathol, 2004; 28:554–555. [DOI] [PubMed] [Google Scholar]
- 20.Hill DA, Jarzembowski JA, Priest JR, Williams G, Schoettler P, Dehner LP. Type I pleuropulmonary blastoma: pathology and biology study of 51 cases from the International Pleuropulmonary Blastoma Registry. Am J Surg Pathol, 2008; 32:282–295. [DOI] [PubMed] [Google Scholar]
- 21.Pugh TJ, Yu W, Yang J, et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene, 2014; 33:5295–5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Libretti L, Ciriaco P, Casiraghi M, Arrigoni C, Zannini P. Pleuropulmonary blastoma in the area of a diagnosed congenital lung cyst. Ann Thorac Surg, 2008; 85:658–660. [DOI] [PubMed] [Google Scholar]
- 23.Venkatramani R, Triche TJ, Wang L, Shimada H, Mascarenhas L. Insulin-like growth factor 2 gene expression molecularly differentiates pleuropulmonary blastoma and embryonal rhabdomyosarcoma. J Pediatr Hematol Oncol, 2015; 37:e356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laberge JM, Bratu I, Flageole H. The management of asymptomatic congenital lung malformations. Paediatr Respir Rev, 2004; 5(Suppl A):S305–S312. [DOI] [PubMed] [Google Scholar]
- 25.Durell J, Lakhoo K. Congenital cystic lesions of the lung. Early Hum Dev, 2014; 90:935–939. [DOI] [PubMed] [Google Scholar]
- 26.Tagge EP, Mulvihill D, Chandler JC, Richardson M, Uflacker R, Othersen HD. Childhood pleuropulmonary blastoma: caution against nonoperative management of congenital lung cysts. J Pediatr Surg, 1996; 31:187–199. [DOI] [PubMed] [Google Scholar]
- 27.Hall NJ, Chiu PPL, Langer JC. Morbidity after elective resection and prenatally diagnosed asymptomatic congenital pulmonary airway malformations. Pediatr Pulmonol, 2015. July; doi: [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 28.Nasr A, Himidan S, Pastor AC, Taylor G, Kim PC. Is congenital cystic adenomatoid malformation a premalignant lesion for pleuropulmonary blastoma? J Pediatr Surg, 2010; 45:1086–1089. [DOI] [PubMed] [Google Scholar]
- 29.Oliveira C, Himidan S, Pastor AC, et al. Discriminating preoperative features of pleuropulmonary blastoma (PPB) from congenital cystic adenomatoid malformations (CCAM): a retrospective, age-matched study. Eur J Pediatr Surg, 2011; 21:2–7. [DOI] [PubMed] [Google Scholar]
- 30.Schultz KA, Harris A, Williams GM, et al. Reply: Serum microRNA screening for DICER1-associated pleuropulmonary blastoma. Pediatr Blood Cancer, 2014; 61:2331–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotecha S Should asymptomatic congenital cystic adenomatous malformations be removed? The case against. Paediatr Resp Rev, 2013; 14:171–172. [DOI] [PubMed] [Google Scholar]
- 32.Delacourt C, Hadchouel A, Dunlop NK. Shall all congenital cystic lung malformations be removed? The case in favour. Paediatr Resp Rev, 2013; 14:169–170. [DOI] [PubMed] [Google Scholar]
- 33.Baird R, Puligandla PS, Leberge JM. Congenital lung malformations: Informing best practice. Semin Pediatr Surg, 2014; 23:270–277. [DOI] [PubMed] [Google Scholar]
- 34.Brenneman M, Field A, Yang J, et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in DICER1 syndrome: a unique variant of the two-hit tumor suppression model [version 1; referees: 2 approved with reservations] F1000Research 2015, 4214 (doi: 10.126888/f1000research.6746.1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boman F, Hill DA, Williams GM, et al. Familial association of pleuropulmonary blastoma with cystic nephroma and other renal tumors: a report from the International Pleuropulmonary Blastoma Registry. J Pediatr, 2006; 149:850–854. [DOI] [PubMed] [Google Scholar]
- 36.Doros LA, Rossi CT, Yang J, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol, 2014; 27:1267–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vujanić GM, Kelsey A, Perlman EJ, Sandstedt B, Beckwith JB. Anaplastic sarcoma of the kidney: a clinicopathologic study of 20 cases of a new entity with polyphenotypic features. Am J Surg Pathol, 2007; 31:1459–1468. [DOI] [PubMed] [Google Scholar]
- 38.Schultz KA, Yang J, Doros L, et al. DICER1-pleuropulmonary blastoma familial tumor predisposition syndrome: a unique constellation of neoplastic conditions. Pathol Case Rev, 2014; 19:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heravi-Moussavi A, Anglesio MS, Cheng SWG, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med, 2012; 366:234–242. [DOI] [PubMed] [Google Scholar]
- 40.Schultz KA, Harris A, Messinger Y, et al. Ovarian Tumors related to intronic mutations in DICER1: a report from the International Ovarian and Testicular Stromal Tumor Registry. Fam Cancer, 2015. Aug 20 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doros L, Schultz KA, Stewart DR, et al. DICER1-related disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2015.2014. Apr 24. [Google Scholar]
- 42.Priest JR, Williams GM, Manera R, et al. Ciliary body medulloepithelioma: four cases associated with pleuropulmonary blastoma—a report from the International Pleuropulmonary Blastoma Registry. Br J Ophthalmol, 2011; 95:1001–1005. [DOI] [PubMed] [Google Scholar]
- 43.Kramer GD, Arepalli S, Shields CL, Shields JA. Ciliary body medulloepithelioma association with pleuropulmonary blastoma in a familial tumor predisposition syndrome. J Pediatr Ophthalmol Strabismus, 2014; 51:e48–50. [DOI] [PubMed] [Google Scholar]
- 44.Foulkes WD, Bahubeshi A, Hamel N, et al. Extending the phenotypes associated with DICER1 mutations. Hum Mutat, 2011; 32:1381–1384. [DOI] [PubMed] [Google Scholar]
- 45.Stewart DR, Messinger Y, Williams GM, et al. Nasal chondromesenchymal hamartomas arise secondary to germline and somatic mutations of DICER1 in the pleuropulmonary blastoma tumor predisposition disorder. Hum Genet, 2014; 133:1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mason KA, Navaratnam A, Theodorakopoulou E, Chokkalingam PG. Nasal Chondromesenchymal hamartoma (NCMH): a systematic review of the literature with a new case report. J Otolaryngol Head Neck Surg, 2015; 44:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Kock L, Sabbaghian N, Plourde F, et al. Pituitary blastoma: a pathognomonic feature of germline DICER1 mutations. Acta Neuropathol, 2014; 128:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahakitrungruang T, Srichomthong C, Pornkunwilai S, et al. Germline and somatic DICER1 mutations in a pituitary blastoma causing infantile-onset Cushing’s disease. J Clin Endocrinol Metab, 2014; 99:e1487–1492. [DOI] [PubMed] [Google Scholar]
- 49.Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol, 2012; 25:602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Durieux E, Descotes F, Nguyen AM, Grange JD, Devouassoux-Shisheboran M. Somatic DICER1 gene mutation in sporadic intraocular medulloepithelioma without pleuropulmonary blastoma syndrome. Hum Pathol, 2015; 46:783–787. [DOI] [PubMed] [Google Scholar]
- 51.Obsteter J, Dovc P, Kunej T. Genetic variability of microRNA regulome in human. Mol Genet Genomic Med. 2015; 3:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anglesio MS, Wang Y, Yang W, et al. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J Pathol, 2013; 229:400–409. [DOI] [PubMed] [Google Scholar]
- 53.Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer, 2014; 14:662–672. [DOI] [PubMed] [Google Scholar]
- 54.Dishop MK, Kuruvilla S. Primary and metastatic lung tumors in the pediatric population. Arch Pathol Lab Med, 2008; 132:1079–1103. [DOI] [PubMed] [Google Scholar]