
Figure 4.
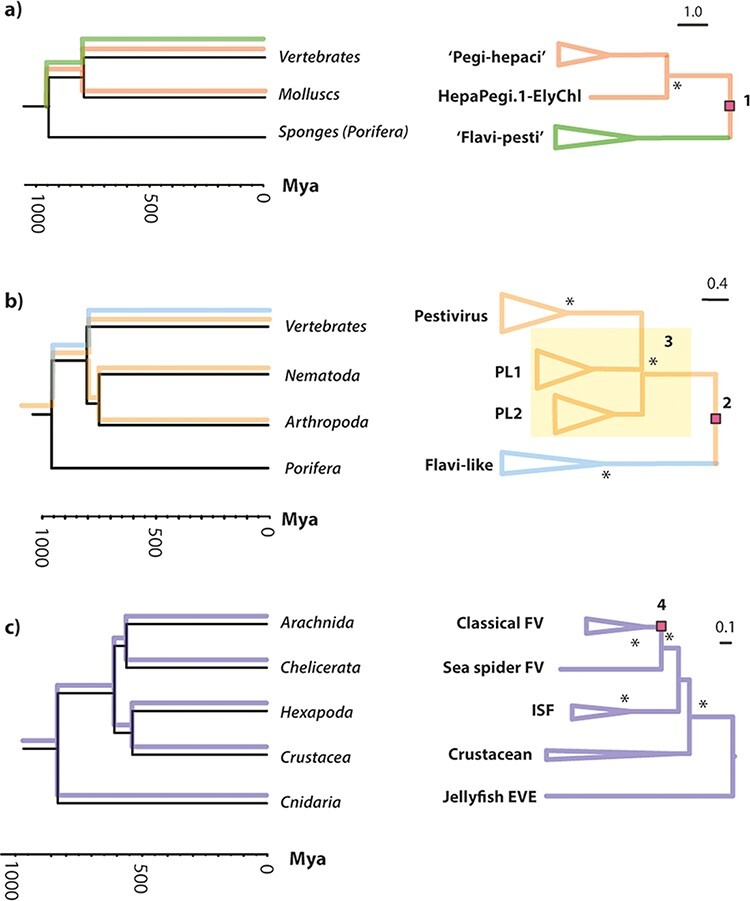
Putative codivergence of flavivirid groups and host phyla. ‘Tanglegrams’ illustrating matching topologies in animal (left) and flavivirid (right) phylogenies. Putative tracking of host lineages by viral lineages is indicated on the host phylogeny. Clades within which the branch lengths of virus phylogenies are correlated with divergence times in host animal lineages: the invertebrate and vertebrate splits in the (a) ‘HP’ lineage (R2 = 0.5); (b) ‘PL’ lineage (R2 = 0.7); (c) the cnidarian–arthropods, chelicerate–hexapoda, and crustacean–insect splits in the Flavivirus genus (R2 = 0.3). Note that, due to limited data, all comparisons rely on strong assumptions regarding virus phylogenies, indicated in the figure by numbers, as follows: (1) and (2) midpoint rooting was used, and the deepest divergence in the virus tree was approximately calibrated using the divergence date of porifera (the most basal animal lineage) in line with our hypothesis that major flavivirid lineages originated in early metazoans; (3) the splits between nematode and arthropod viruses in PL1 and PL2 clades—indicated by the yellow square—were poorly resolved in our phylogenies; (4) midpoint rooting was used, and it is assumed that the arthropod-borne classical flaviviruses originated in arachnids, as proposed in Fig. 5a.