
Figure 4: General workflow for analysis of single-cell epigenomics datasets.
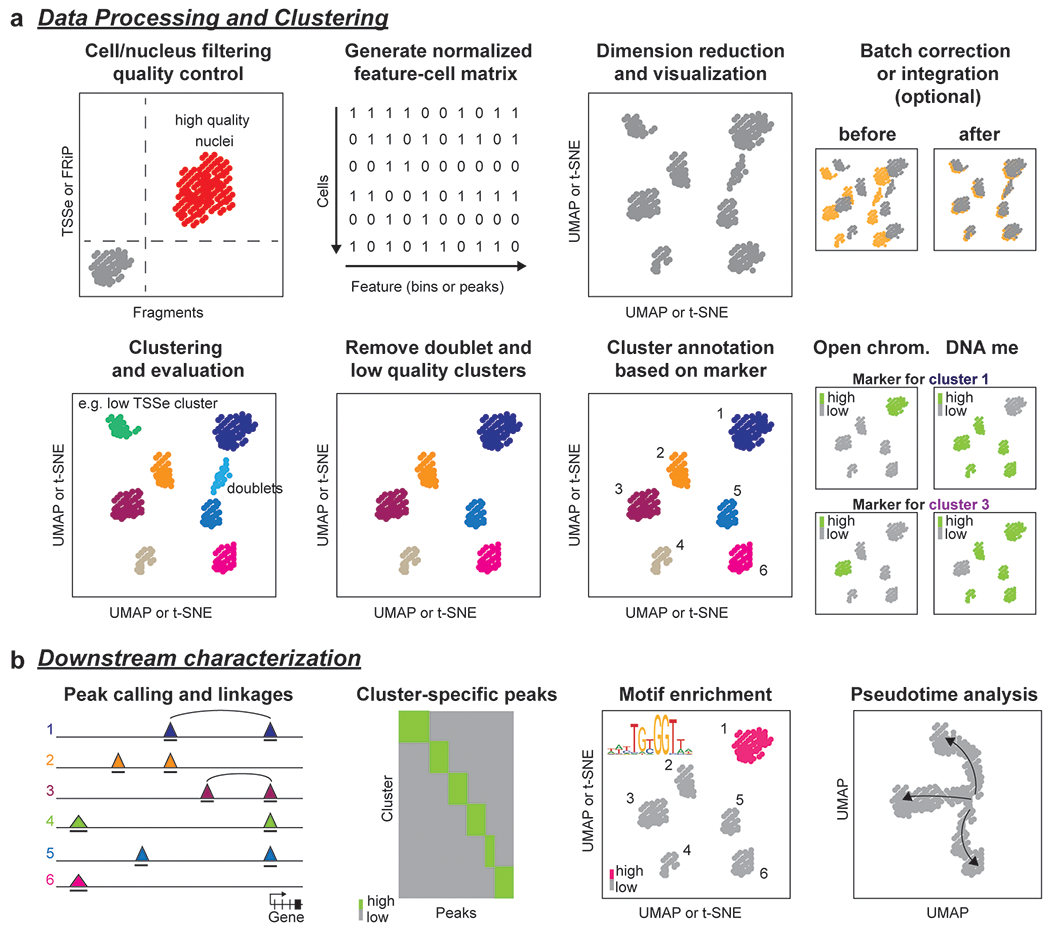
a After preprocessing and mapping, high quality nuclei or cells are detected using quality control criteria such as transcriptional start site enrichment (TSSe) for scATAC-seq, fraction of reads in peaks (FRiP) or the number of fragments/reads per nucleus. Next, a normalized cell-feature matrix is generated followed by dimension reduction and visualization in 2D space. Datasets from different modalities can be integrated to increase cell-type resolution and, if processing datasets from multiple experimental batches, batch correction might be necessary. b The nuclei are first grouped into clusters, then cell clusters with low quality or representing likely doublets are removed from downstream analysis. High quality clusters are annotated using, for example, high chromatin accessibility or low DNA methylation levels at marker gene loci. c Downstream analysis is exemplified for chromatin accessibility datasets. Reads from all nuclei from a cluster are combined to a cell-type-specific pseudobulk dataset to call peaks (triangles indicate signal pile-up and bold lines underneath tracks indicate peak regions) from scATAC-seq. Distal elements can be linked to target genes by assessing if two sites are accessible in the same cell (co-accessible sites are indicated by black arcs). If datasets were integrated with scRNA-seq data or data were generated using joint profiling of RNA and chromatin accessibility, accessibility of distal elements can be associated to putative target gene expression levels. To further characterize gene regulatory networks, cCREs are identified as peaks in each cell cluster followed by analysis of transcription factor motifs or footprints within the cCREs. Single-cell epigenomics data can also be used to generate pseudotime trajectories for analysis of developmental or cell state transitions. Here, computational integration or joint profiling of RNA and chromatin from the same cell can provide insight into the crosstalk and differences in timing between chromatin dynamics and gene expression changes.