

SUMMARY
Ductal carcinoma in situ (DCIS) is the most common precursor of invasive breast cancer (IBC), with variable propensity for progression. We perform multiscale, integrated molecular profiling of DCIS with clinical outcomes by analyzing 774 DCIS samples from 542 patients with 7.3 years median follow-up from the Translational Breast Cancer Research Consortium (TBCRC) 038 study and the Resource of Archival Breast Tissue (RAHBT) cohorts. We identify 812 genes associated with ipsilateral recurrence within five years from treatment and develop a classifier that predicts DCIS or IBC recurrence in both cohorts. Pathways associated with recurrence include proliferation, immune response, and metabolism. Distinct stromal expression patterns and immune cell compositions are identified. Our multiscale approach employed in situ methods to generate a spatially resolved atlas of breast precancers, where complementary modalities can be directly compared and correlated with conventional pathology findings, disease states, and clinical outcome.
eTOC blurb
Strand et al. have performed a large, comprehensive study to generate a spatially resolved breast precancer atlas. They present a prognostic classifier that predicts both precancer recurrence and invasive progression, which may form the basis for a future clinical test to guide breast precancer treatment.
Graphical Abstract
INTRODUCTION
As nonobligate precursors of invasive disease, precancers provide a unique vantage point to study molecular pathways and evolutionary dynamics leading to the development of life-threatening cancers. Breast ductal carcinoma in situ (DCIS) is one of the most common precancers across all tissues, with ~50,000 women diagnosed each year in the U.S.1. Current treatment of DCIS involves breast conserving surgery or mastectomy, with the goal of preventing invasive cancer. However, DCIS consists of a molecularly heterogeneous group of lesions, with highly variable risk of invasive progression. Improved understanding of which DCIS is likely to progress could spare a subgroup of women unnecessary treatment.
Identification of factors associated with disease progression has been studied extensively. Epidemiologic cancer progression models indicate that clinical features like age at diagnosis, tumor grade, and hormone receptor expression may have some prognostic value, but have limited ability to identify the biologic conditions that govern DCIS progression to invasive breast cancer (IBC). Previous molecular analyses of DCIS have studied either 1) cohorts of pure DCIS with known outcomes (e.g., disease-free vs recurrent), or 2) cross-sectional cohorts of DCIS with or without adjacent IBC2–10. These approaches have tested potentially divergent assumptions: recurrence of the DCIS as IBC may arise from neoplastic cells left behind when the DCIS was removed, be related to initial field effect, or develop independently. Longitudinal cohorts provide a perspective of cancer progression over time. Analysis of DCIS adjacent to IBC assumes these preinvasive areas are good models for pure DCIS and are ancestors of the invasive cancer cells, with synchronous lesions inferring progression. To date, these studies have not produced clear evidence for a common set of events associated with invasion.
Moreover, few genomic aberrations have been identified that can differentiate DCIS from IBC4,6,7,11–13 and microenvironmental processes, including collagen organization, myoepithelial changes, and immune suppression, may contribute to IBC development2,3,5. Presently, it remains unknown how these different molecular axes contribute to DCIS evolution.
Here, as part of the Human Tumor Atlas Network (HTAN) we present two DCIS cohorts, the Translational Breast Cancer Research Consortium (TBCRC) 038 study and the Resource of Archival Breast Tissue (RAHBT), for multimodal molecular analyses. We performed comprehensive integrated molecular profiling of these complementary, clinically annotated, longitudinally sampled cohorts, to understand the spectrum of molecular changes in DCIS and to identify both tumor and stromal predictors of subsequent events. We used multidimensional and multiparametric approaches to address central conceptual themes of cancer progression, ecology, and evolutionary biology. We hypothesize that the breast precancer atlas (PCA) presented here will facilitate phylogenetic analysis to reconstruct the relationship between DCIS and IBC, the natural history of DCIS, and factors that underlie progression to invasive disease.
RESULTS
Study Design and Cohorts
We generated two retrospective case-control cohorts of patients initially diagnosed with pure DCIS with or without a subsequent ipsilateral breast event (iBE, either DCIS or IBC) after surgical treatment. Identical eligibility criteria were used for outcome analysis in both cohorts. The RAHBT cohort used for outcome analysis has 97 cases with median diagnosis at age 53, and 40 months median time to recurrence. Over half (66.0%) had lumpectomy with radiation, 10.3% had lumpectomy without radiation, and 35% were identified as black. The TBCRC cohort included 216 patients with median diagnosis at age 52, and 48 months median time to recurrence. More than half (55.5%) had lumpectomy with radiation, 15.3% had lumpectomy without radiation, and 30.0% were identified as black (Table 1). Figure 1 shows an outline of cohorts and analyses in this study. Table 1 summarizes the RAHBT and TBCRC cohorts used for outcome analysis, Table S1 summarizes the RAHBT LCM cohort, and Table S2 summarizes the assays in this study by cohort.
Table 1.
Breast Pre-cancer Atlas Patient Cohorts with RNA-seq data and ipsilateral breast event used for outcome analysis.
| TBCRC | RAHBT | |||||
|---|---|---|---|---|---|---|
| DCIS without recurrence (N=95) | DCIS with DCIS Recurrence (N=66) | DCIS with Invasive Recurrence (N=55) | DCIS without recurrence (N=68) | DCIS with DCIS Recurrence (N=15) | DCIS with Invasive Recurrence (N=14) | |
| Year of Diagnosis | ||||||
| Median | 2009 | 2008 | 2006 | 2006 | 2008 | 2009 |
| Age at Diagnosis | ||||||
| Median | 54 | 54 | 50 | 52 | 53 | 52 |
| Mean (±SD) | 54.4 (±8.5) | 55.2 (±9.8) | 52.6 (±9.8) | 53.1 (±7.2) | 52.,5 (±6.0) | 55.1 (±11.1) |
| Grade | ||||||
| 1 | 5 [5.3%] | 6 [9.0%] | 3 [5.5%] | 18 [26.5%] | 4 [26.7%] | 3 [21.4%] |
| 2 | 37 [38.9%] | 26 [39.4%] | 19 [34.5%] | 28 [48.2%] | 4 [26.7%] | 8 [57.1%] |
| 3 | 53 [55.8%] | 34 [51.5%] | 33 [60.0%] | 22 [32.4%] | 7 [46.7%] | 2 [21.4%] |
| Pathologic Tumor Size | ||||||
| Median | 2.1 | 1.5 | 1.9 | |||
| Mean (±SD) | 2.7 (±1.9) | 2.2 (±2.0) | 2.8 (±2.6) | |||
| Marker Status | ||||||
| ER(+) | 60 [63.2%] | 41 [62.1%] | 37 [67.3%] | 55 [80.9%] | 8 [53.3%] | 12 [85.7%] |
| ER(−) | 35 [36.8%] | 25 [37.9%] | 18 [32.7%] | 13 [19.1%] | 7 [46.7%] | 2 [14.3%] |
| ER(+) Dx before 2000 | 0 | 2 [3.0%] | 4 [7.3%] | 3 [4.4%] | 0 | 3 [21.4%] |
| ER(+) Dx 2000 & after | 60 [63.2%] | 39 [59.1%] | 33 [60.0%] | 52 [76.5%] | 8 [53.3%] | 9 [64.3%] |
| ER(−) Dx before 2000 | 0 | 0 | 1 [1.8%] | 2 [2.9%] | 2 [13.3%] | 0 |
| ER(−) Dx 2000 & after | 35 [36.8%] | 25 [37.9%] | 17 [30.9%] | 11 [16.2%] | 5 [33.3%] | 2 [14.3%] |
| Treatment | ||||||
| Lumpectomy+Radiation | 58 [61.1%] | 40 [60.6%] | 22 [40.0%] | 6 [8.8%] | 2 [13.3%] | 2 [14.3%] |
| Lumpectomy−Radiation | 5 [5.3%] | 16 [25.2%] | 12 [21.8%] | 45 [66.2%] | 11 [73.3%] | 8 [57.1%] |
| Lumpectomy Radiation Unknown | 1 [1.1%] | 1 [1.5%] | 2 [3.6%] | 0 | 0 | 0 |
| Mastectomy | 31 [32.6%] | 9 [13.6%] | 19 [34.5%] | 17 [25.0%] | 2 [13.3%] | 4 [28.6%] |
| Time to Recurrence* (months) | ||||||
| Mean (±SD) | 105.7 (±37.0) | 52.7 (±39.9) | 71.2 (±43.9) | 139.8 (±52.7) | 54.9 (±40.4) | 73.4 (±68.4) |
| Median | 96 | 40 | 58 | 141 | 36 | 47 |
| Margins | ||||||
| Ink on tumor | 0 | 0 | 0 | 0 | 0 | 0 |
| <2mm | 27 [28.4%] | 28 [42.4%] | 17 [30.9%] | 15 [22.1%] | 4 [26.7%] | 6 [42.9%] |
| At least≥ 2mm | 37 [38.9%] | 25 [37.9%] | 21 [38.2%] | 11 [16.2%] | 4 [26.7%] | 1 [7.1%] |
| Clear, unknown mm | 31 [32.6%] | 13 [19.7%] | 17 [30.9%] | 42 [61.8%] | 7 [46.7%] | 7 [50.0%] |
| Race | ||||||
| White | 62 [65.2%] | 38 [57.6%] | 28 [50.9%] | 44 [64.7%] | 10 [66.7%] | 9 [64.3%] |
| Black | 22 [23.2%] | 21 [31.8%] | 22 [40.0%] | 24 [35.3%] | 5 [33.3%] | 5 [35.7%] |
| Asian | 2 [2.1%] | 1 [1.5%] | 2 [3.6%] | 0 | 0 | 0 |
| Pacific Islander | 0 | 1 [1.5%] | 0 | 0 | 0 | 0 |
| Other | 0 | 0 | 0 | 0 | 0 | 0 |
| Unknown | 9 [9.5%] | 5 [7.6%] | 3 [5.5%] | 0 | 0 | 0 |
To end of follow-up for no recurrence. See also Table S1.
Figure 1. Cohorts and methods outline.
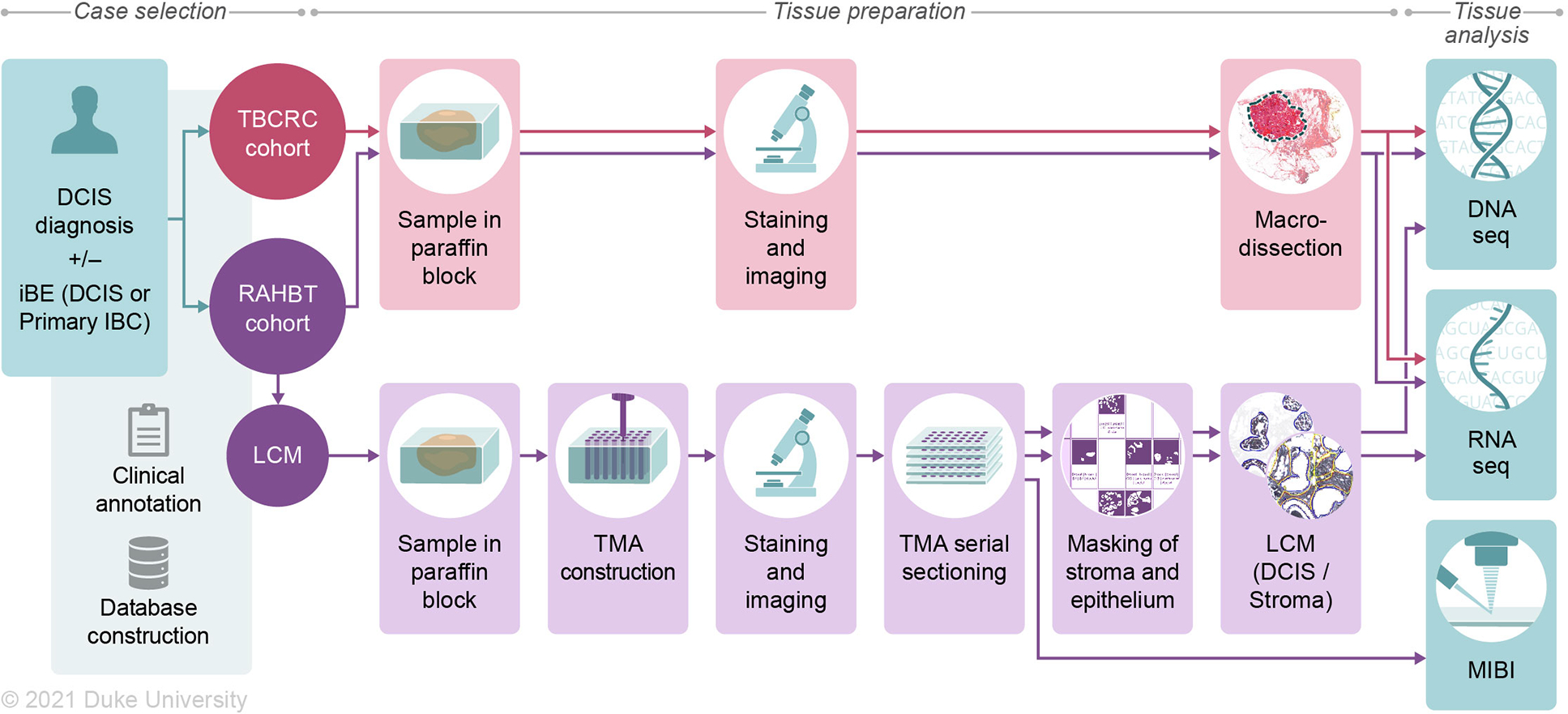
Two retrospective study cohorts were generated, consisting of ductal carcinoma in situ (DCIS) patients with either a subsequent ipsilateral breast event (iBE) or no later events after surgical treatment. TBCRC samples were macrodissected for downstream RNA and DNA analyses. RAHBT samples were 1) macrodissected like TBCRC, or 2) organized into a tissue microarray (TMA) from which serial sections were made for RNA, DNA, and protein (MIBI) analysis (RAHBT LCM cohort). TMA cores were laser capture microdissected to ensure pure epithelial and stromal components. See also Tables S1 and S2.
Prognostic classifier predicts early recurrence
The TBCRC and RAHBT cohorts were designed to investigate biological determinants of recurrence by matching patients with subsequent iBE to patients that did not have any events during long-term follow-up.
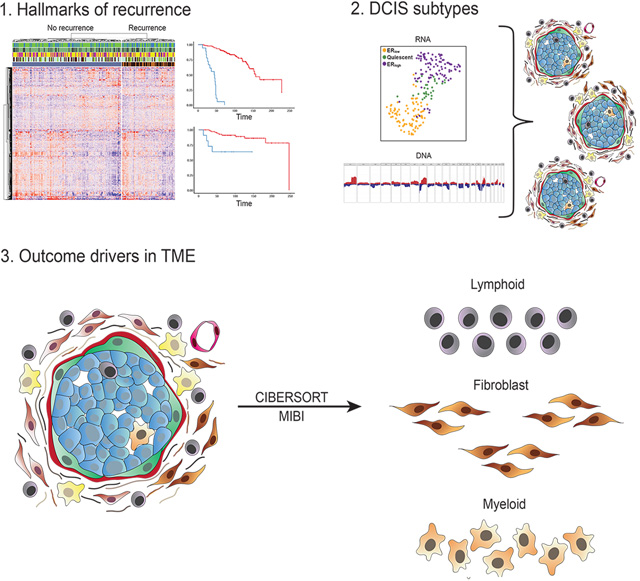
To identify gene expression patterns correlating with outcome, we analyzed RNA from primary DCIS with iBEs within 5 years vs the remaining samples in TBCRC, to avoid including non-clonal events that might be more common in later years. We identified 812 differentially expressed (DE) genes at 0.05 false discovery rate (FDR) (Figure 2A, Table S3).
Figure 2. Identification, training, and validation of 812 gene classifier.
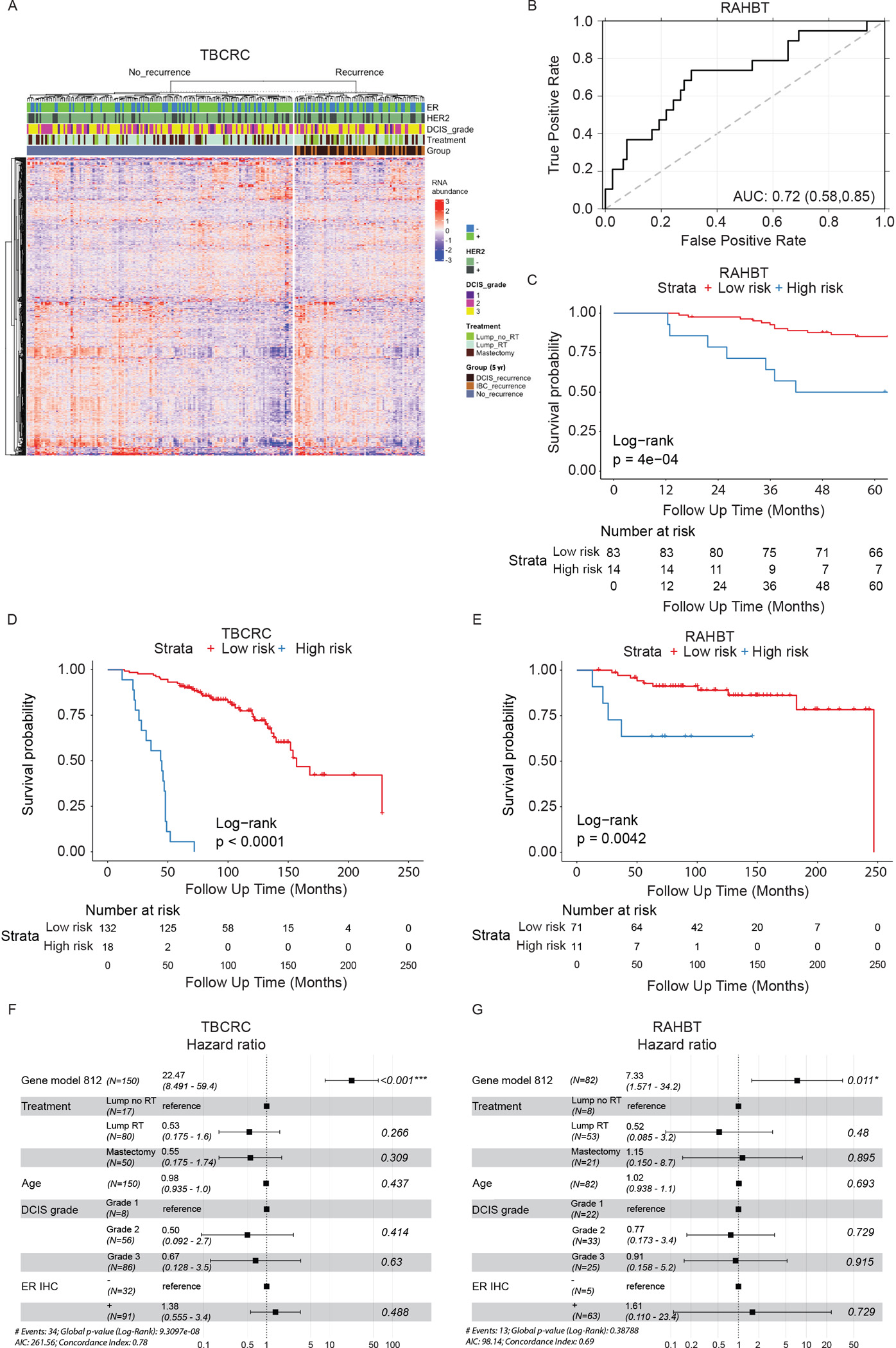
A) Heatmap of 812 differentially expressed (DE) genes from cases vs controls analysis (5-year outcome) in TBCRC. Covariates show ER and HER2 status, DCIS grade, treatment, and type of iBE/no iBE. B) ROC curve of the 812 gene classifier in RAHBT. C) Kaplan-Meier plot of time to iBE (5-year outcome) stratified by classifier risk groups in RAHBT. D-E) Kaplan-Meier plot of time to invasive progression (full follow-up) stratified by classifier risk groups in TBCRC (D) and RAHBT (E). C-E) P-values from log-rank tests. F-G) Forest plot of multivariable Cox regression analysis including classifier risk groups, treatment, age, DCIS grade, and ER status for invasive iBEs (full follow-up) in TBCRC (F) and RAHBT (G). See also Figure S1 and Table S3.
To identify copy number aberrations (CNAs) that correlate with outcome, we performed light-pass whole genome sequencing (WGS) on DNA from FFPE samples in both cohorts (n=228). We identified 29 recurrent CNAs across both cohorts, none of which were predictive of recurrence (Figure S1A). Given the absence of significant CNAs, we trained a Random Forest classifier in TBCRC using only the 812 DE genes. The classifier was validated in RAHBT, with an ROC AUC of 0.72 (Figure 2B), Precision 0.86, Recall 0.91, and F1 score 0.88, indicating that the classifier performed well also in the test cohort. The classifier significantly predicted any subsequent iBE in both cohorts (RAHBT P=0.0004, Figure 2C). Importantly it was also a significant predictor of invasive iBEs over the full follow-up time (TBCRC P<0.0001, RAHBT P=0.0042, Figure 2D–E), demonstrating the classifier could specifically identify DCIS that progress to IBC.
Next, we examined whether the 812 gene classifier remained an independent predictor of outcome when combined with clinical features. We performed multivariable Cox regression analysis including the classifier, treatment, age, clinical ER, and DCIS grade (Figure S1B–C). While multivariable analysis demonstrated a trend for treatment type and ER status for outcome, only the 812 gene classifier was significant in both cohorts (RAHBT HR=3.48, (95% CI: 1.14–10.6), P=0.028). Importantly, in multivariable analysis for invasive iBEs only, the classifier showed an even stronger prognostic value in both cohorts, with a hazard ratio of 7.33 in RAHBT (95% CI: 1.57–34.2, P=0.011, Figure 2F–G). While previous studies found association between ER status and DCIS outcome14–16, Kaplan-Meier analysis of clinical ER status (IHC-based) demonstrated a trend in RAHBT (P=0.053), but not in TBCRC (P=0.2, Figure S1D–E). Moreover, the 812 gene classifier showed no prognostic value for progression free disease or overall survival for 1064 IBCs from The Cancer Genome Atlas (TCGA17, Figure S1F–I), suggesting that the classifier is specific for the DCIS stage. To compare the 812 gene classifier to commercially available prognostic tests for DCIS, we calculated the Oncotype DCIS score as previously described18 using TBCRC and RAHBT RNA-sequencing data. We found that, in contrast to the 812 gene classifier, the DCIS Oncotype score did not differ between the outcome groups in either cohort (Figure S1J–K).
The 812 gene classifier likely represents several distinct biologic processes that promote recurrence and invasive progression. To further understand the biology and identify pathways involved in recurrence, we performed Gene Set Enrichment Analysis (GSEA) on DE genes between cases with 5-year recurrence vs the rest in TBCRC. We identified 11 Hallmark pathways significantly associated with early recurrence including those associated with proliferation, immune response, and metabolism (Figure S1L).
To further examine pathway activation status, we performed Gene Set Variation Analysis (GSVA) at the individual tumor level in 5-year outcome groups. Here, MYC and mTORc1 signaling were increased in cases vs controls and strongly correlated (Figure 3A–C). We also observed high correlation between cell cycle linked G2M and E2F pathways. Further, Glycolysis and Oxidative Phosphorylation were increased in cases, and the significant positive correlation between these two pathways indicated that metabolically active tumors use both pathways. Overall, this analysis confirmed the finding from the differential abundance and GSEA analysis of 5-year outcome groups.
Figure 3. Outcome-associated pathways in individual samples.
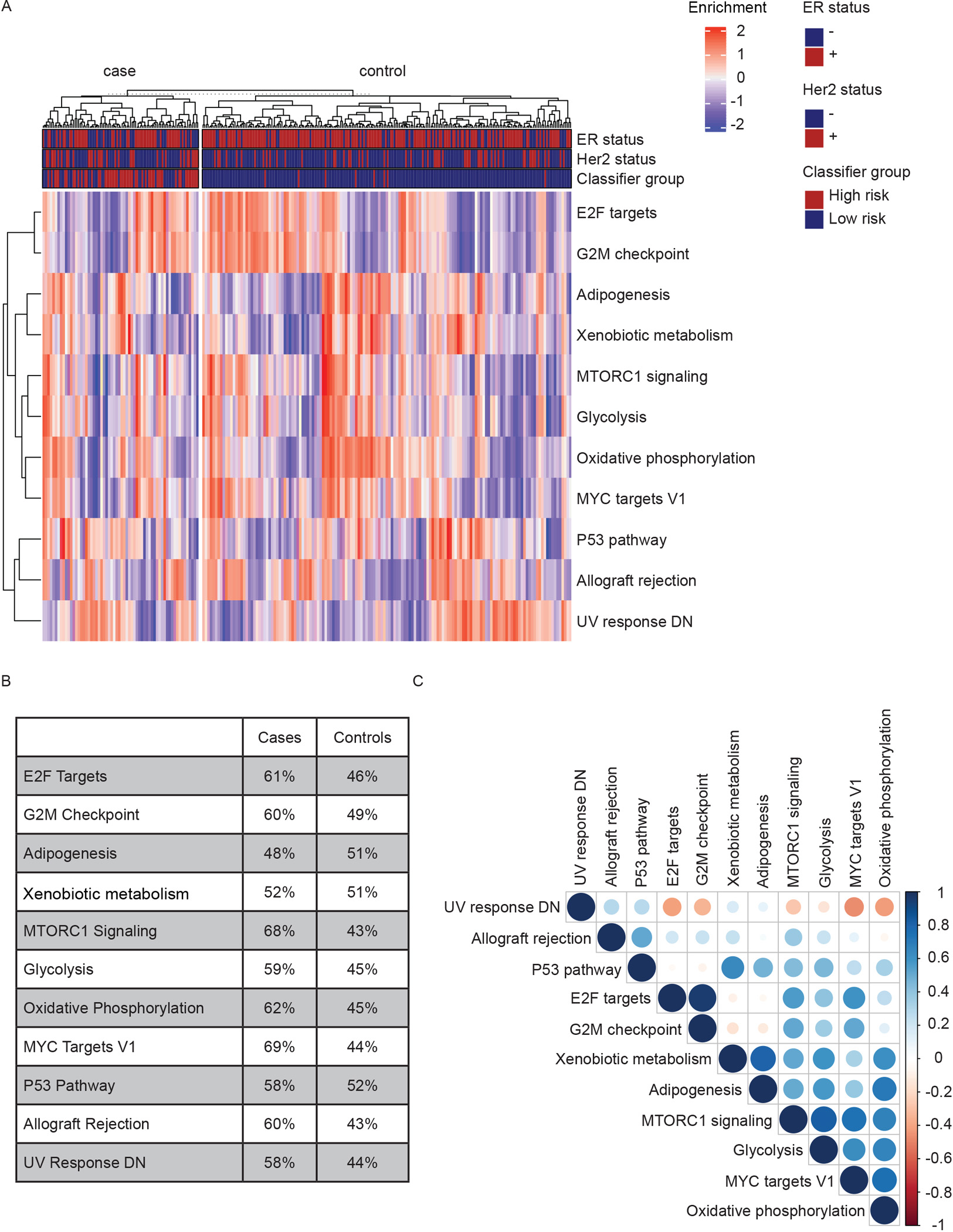
A) Heatmap of single-sample Gene Set Variation Analysis of 11 Hallmark pathways associated with recurrence. B) Percentage of samples in 5-year outcome groups enriched for each pathway in A). C) Plot of Pearson’s correlations between pathways in A). Blue: Positive. Red: Negative. White: P>0.05. Color intensity and circle size are proportional to correlation coefficients.
DCIS RNA clustering defines expression modules that drive outcome
Since proliferation and metabolism were identified as important pathways in recurrence, we next examined whether these pathways are driven by major DCIS phenotypes. Previous studies suggested that IBC subtypes do not fit well for DCIS 19. We hypothesized that a DCIS-specific classification scheme would better address DCIS biology. To investigate the biology behind the outcome analysis with emphasis on epithelial pathways, we performed unsupervised clustering of RNA-seq data from TBCRC (n=216) as well as an additional group of RAHBT cases (n=265, Table S1) where we generated epithelial-enriched samples by laser capture microdissection (LCM) to evaluate tumor cell expression patterns without contributions from the tumor microenvironment (TME, Figure S2A–F).
We performed non-negative matrix factorization (NMF) on all protein coding genes (GENCODE v33) with non-zero variance, evaluated the fit of 2–10 clusters, and selected a 3-cluster solution based on silhouette width, cophenetic value, maximizing cluster number, and replication in RAHBT (Figure S2G–J). The 3-cluster solution most reproducibly captured the biologic subgroups in both cohorts. To ensure the identified clusters were not an artifact of the clustering method, we ran consensus clustering in TBCRC, which rediscovered three clusters with high concordance with the NMF clusters (85.6%, Figure S2K). In both cohorts, cluster 1 had significantly higher ERBB2 and lower ESR1 expression compared to clusters 2 and 3 (Figure 4A, B), which both had increased ESR1 expression. We termed the three clusters ERlow, quiescent, and ERhigh respectively. To characterize these clusters, we conducted differential abundance analysis comparing each cluster individually to the other two combined (one-vs-rest). The deregulated pathways in each cluster were highly concordant across both cohorts, further supporting three transcriptional patterns in DCIS that are driven by the tumor cell compartment (PERlow=2.33×10−2; Pquiescent=8.37×10−2; PERhigh=9.20×10−10; hypergeometric test; Figure S2L).
Figure 4. Transcriptomic DCIS subtypes correlate with outcome pathways.
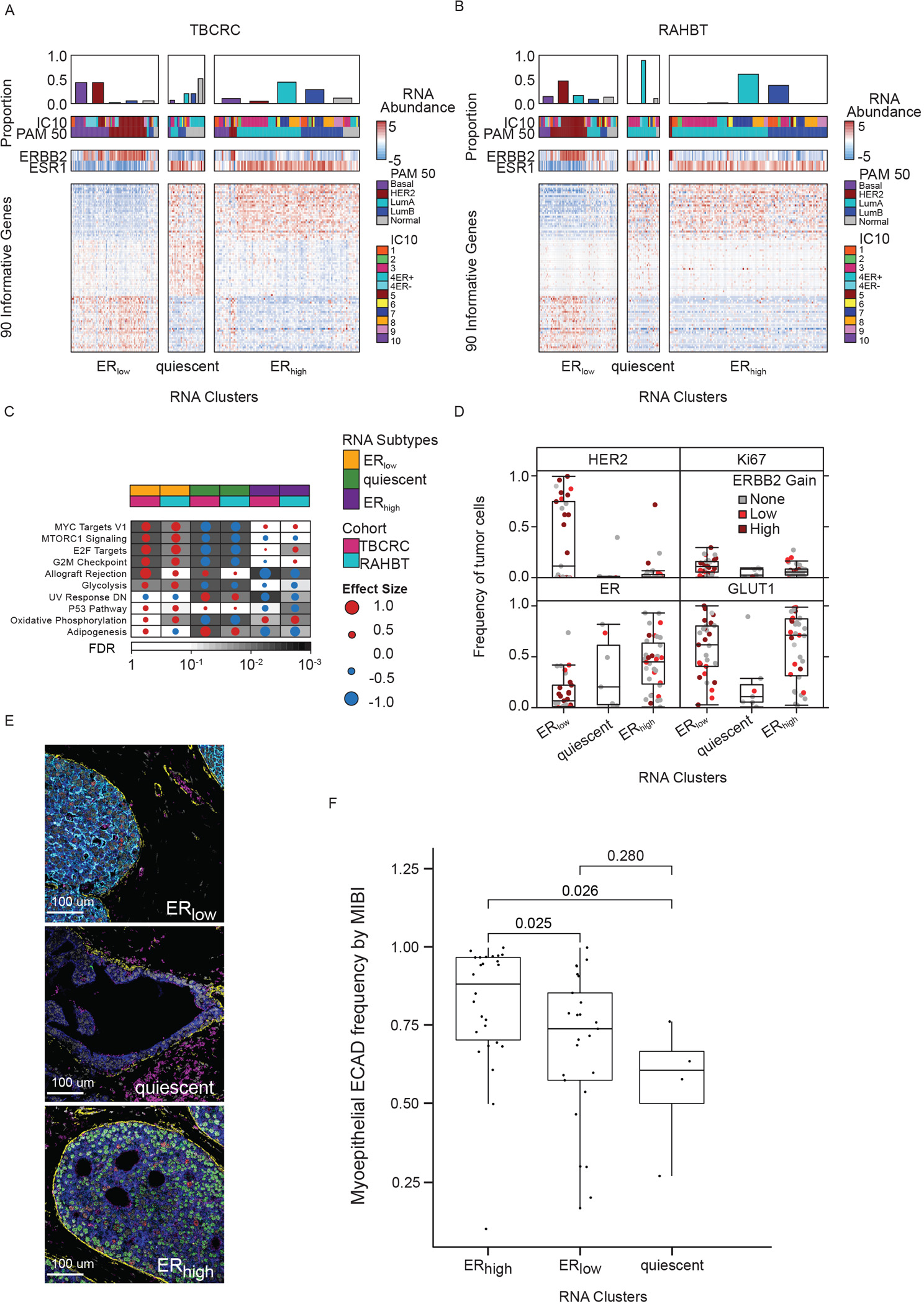
A) Heatmap of 90 informative genes, contributing to the three subtypes in TBCRC samples. Covariates indicate PAM50 and IC subtypes and ERBB2 and ESR1 mRNA abundance for each sample. B) Heatmap of DCIS subtypes in RAHBT. C) Gene Set Enrichment Analysis with Hallmark gene sets of each cluster vs rest for TBCRC and RAHBT LCM (outcome-associated pathways only). Dot size and color indicate magnitude and direction of pathway deregulation. Background shading indicates false discovery rate (FDR). Covariates indicate DCIS subtype and cohort. Effect size and FDR from GSEA algorithm. D) Box plots of HER2, ER, Ki67, and GLUT1 expression by MIBI in DCIS subtypes. Dot color indicates ERBB2 genomic amplification level. E) Representative MIBI images of the three subtypes. White=Nuc; Blue=PanKRT; Yellow=SMA; Pink=GLUT1; Cyan=HER2; Green=ER; Red=Ki67. F) Boxplot of myoepithelial ECAD frequency by MIBI in the three subtypes. P-values from Wilcoxon rank sum test. D, F): Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. See also Figure S2.
While we observed a differential expression of the estrogen response in the ERhigh cluster vs ERlow cluster, the most striking patterns involved pathways associated with DCIS recurrence (Figure 4C, Figure S2L). Pathways including MYC, mTOR signaling, and cell cycle pathways were enriched in ERlow and significantly depleted in the quiescent cluster. Moreover, the Allograft Rejection, p53 and Adipogenesis pathways were high in ERlow and low in ERhigh. Finally, ERhigh tumors were depleted for UV Response Down and enriched for Oxidative Phosphorylation pathways, both of which were associated with recurrence. None of the recurrence-associated pathways were enriched in the quiescent cluster. The presence of the Allograft Rejection pathway in RAHBT LCM epithelial samples, though not significant, suggests that immune cells have infiltrated the epithelial compartment in the involved samples. Thus, the 3-cluster solution identified pathways associated with recurrence.
Genomic and transcriptomic-based classifications of IBC20,21 have characterized the spectrum of invasive breast cancer subtypes, but it remains unclear whether these accurately describe the spectrum of DCIS. To investigate, we applied the PAM50 classification to TBCRC and RAHBT LCM epithelial DCIS samples and evaluated the correlation of each sample to the centroid of its assigned subtype. We compared this correlation to IBCs from TCGA through repeated downsampling of the TCGA. The median correlation was consistently lower in DCIS compared to IBC, with the most pronounced difference in the basal-like subtype (Figure S2M), as previously shown19. Significantly decreased correlation was also observed for luminal A (P=3.13×10−3) and normal-like subtypes (P=6.21×10−3). UMAP projection of the DCIS transcriptome revealed clear deviations from the PAM50 centroids (Figure S2N–O), and PAM50 failed to predict DCIS recurrence (Figure S2P–Q). These data suggest that while established IBC subtypes can be identified in DCIS, they do not fit DCIS as robustly as IBC, and are not prognostic in these premalignant lesions.
In support of the 3-cluster solution, we investigated MIBI protein expression for a subset of patients (n=71). The frequency of ER+ tumor cells was significantly higher in the quiescent and ERhigh subtypes compared to ERlow (log2FC=2.73; P=2.11×10−5; Wilcoxon rank sum test) while HER2+ tumor cells were significantly higher in the ERlow subtype (log2FC=4.88; P=3.74×10−2; Wilcoxon rank sum test; Figure 4D). Overall, the frequencies of ER+ and HER2+ tumor cells were well correlated with RNA abundance of ESR1 and ERBB2, respectively (Figure S2R–S). PGR levels were upregulated in quiescent and ERhigh compared to ERlow (Figure S2T). Based on MIBI data, quiescent lesions were depleted for Ki67 (log2FC=−1.46; P=8.08×10−2; Wilcoxon rank sum test) and GLUT1 (log2FC=−2.64; P=8.47×10−3) positive tumor cells, vs ERhigh and ERlow tumors, suggesting quiescent lesions are less proliferative and less metabolically active (Figure 4D–E).
In their analysis of DCIS tumors and TME by MIBI, Risom et al. identified myoepithelial E-cadherin expression as the most discriminative feature for risk of progression (Figure 6A-B in22). To investigate this in relation to the identified RNA clusters, we compared the distribution of myoepithelial E-cadherin frequency by MIBI in matched RAHBT LCM RNA samples. We found that ERhigh lesions had significantly higher myoepithelial E-cadherin frequency compared to ERlow and quiescent lesions (P≤0.026, Figure 4F). While most recurrence-associated pathways were enriched in ERlow lesions, this points to a feature associated with recurrence amongst ER+ DCIS tumors, and highlights that there are multiple paths to progression in DCIS.
Amplifications characteristic of high-risk of relapse IBC occur in DCIS
Next, we investigated how CNAs in DCIS contribute to pathways associated with DCIS recurrence. Amongst the 29 recurrent CNAs identified across both cohorts, we found 13 gains and 16 losses, occurring in 10.1–52.6% of DCIS samples (FDR<0.05; GISTIC2; Figure 5A). The identification of these common CNAs was not biased by depth of sequencing, but two were associated with cohort (1p21.3 and 10p15.3 deletions, Table S4). The most frequent alterations were gains of chromosomes 1q and 17q, including 17q12 where the ERBB2 oncogene is located, and loss of chromosome 17p, 16q, and 11q (Figure 5A), confirming prior findings5,9,12,23 and notably reflecting the CNA landscape of IBC20,24.
Figure 5. Characteristic IBC CNAs are present in DCIS.
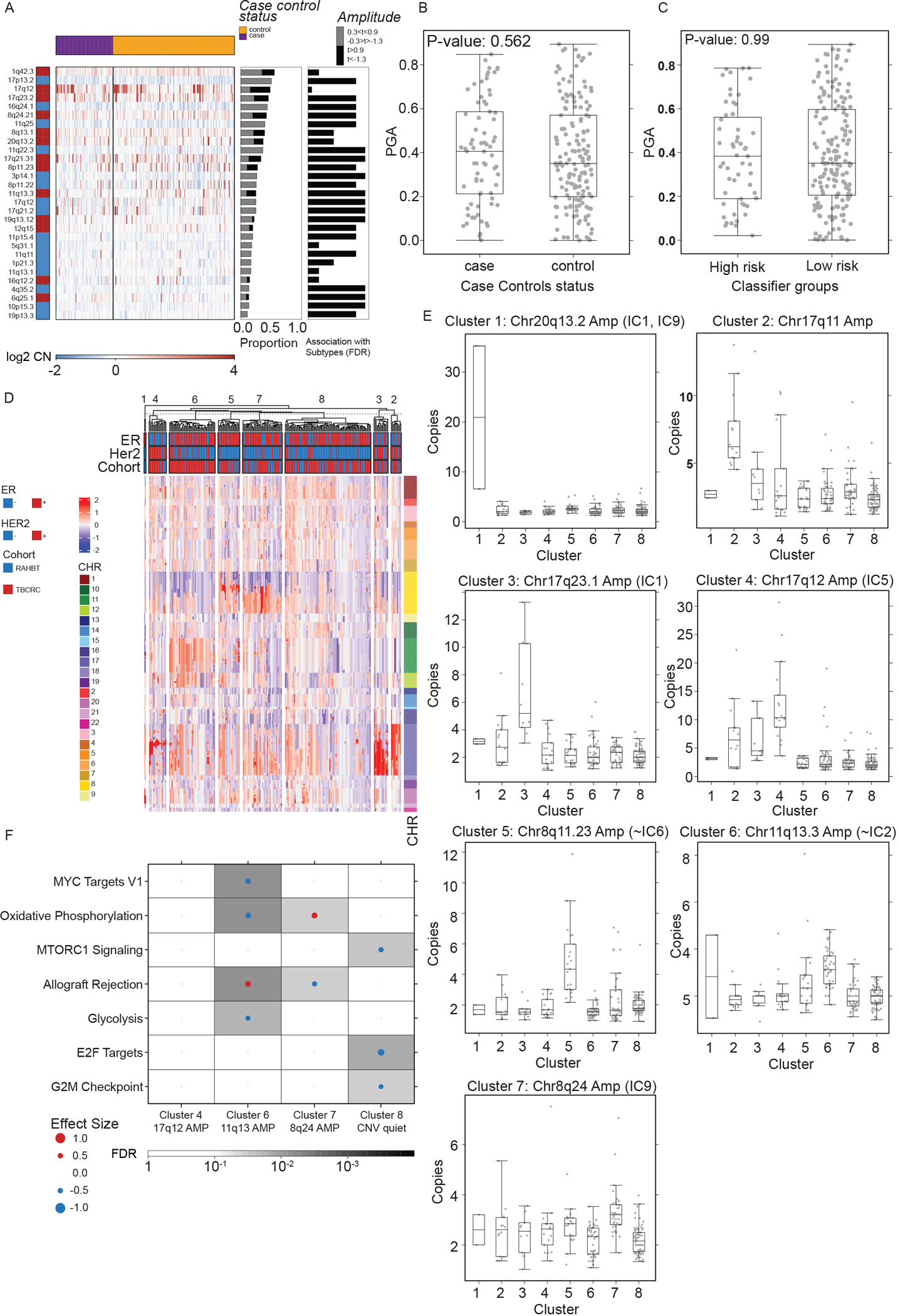
A) Heatmap (log2 copy number) of 29 recurrently altered copy number alterations (CNAs) in each sample grouped by 5-year outcome groups (top bar). Red = gain. Blue = loss. Middle barplot: Proportion of samples with each CNA. Right barplot: FDR from Kruskal-Wallis test of each CNA with outcome groups. B-C) Boxplot showing Proportion of the Genome copy number Altered (PGA) by 5-year outcome groups (B) and classifier risk groups (C). P-values from Kruskal-Wallis test. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. D) Unsupervised clustering of CNA landscape identified eight clusters. Heatmap of genomic segments (log2 copy number) in TBCRC and RAHBT samples. Covariates indicate ER and HER2 status (RNA-derived) and chromosomes for each segment. E) Boxplots of log2 copy numbers across the eight clusters, representing median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. F) GSEA Hallmark analysis of DE genes in matched RNA samples by DNA cluster for TBCRC and RAHBT, outcome-associated pathways only. Dot size and color represents the magnitude and direction of pathway deregulation. Background shading indicates FDR. Effect size and FDR from GSEA algorithm. See also Figure S3 and Table S4.
Next, we investigated if the distribution of Proportion of the Genome copy number Altered (PGA) was biased in the 5-year outcome groups or 812 gene classifier risk groups, but found no significant differential distribution (Figure 5B–C). PGA was not correlated to sequencing depth, nor predictive of iBEs (Figure S3A–B).
Early patterns of alterations may provide insight into the mechanisms of neoplastic lesion development and progression. To identify genomic subtypes in DCIS, we employed unsupervised NMF clustering of CNA segments on TBCRC and RAHBT jointly and identified eight clusters ranging in size from 2–98 samples (Figure 5D; Figure S3C–D) which were not biased by depth of sequencing (Figure S3E). CNA cluster 1 was characterized by chr20q13.2 amplification (Figure 5E). Three clusters were characterized by chr17q amplification (Cluster 2: 17q11, Cluster 3: chr17q23.1, Cluster 4: chr17q12). Cluster 5 was had chr8p11.23 amplification, Cluster 6 chr11q13.3 amplification, and Cluster 7 amplification of MYC on chr8q24. Cluster 8, the largest group (n=98), represented a CNA quiet subgroup, characterized by the absence or diminished signal of these CNAs.
Integrative subgroups (ICs) is an IBC classification scheme based on genomic copy number and expression profiles20. Intriguingly, despite the eight CNA clusters not being associated with recurrence (Figure S3F–G) several of these clusters were attributed to the presence or absence of CNAs characteristic of IC subtypes, namely the four high-risk of relapse ER+/HER2− subgroups (IC1,2,6,9) and the HER2-amplified (IC5) subgroup25 (Figure 5E). Of note, these four high-risk integrative subgroups (IC1,2,6,9) account for 25% of ER+/HER2− IBC and the majority of distant relapses25. Integrative subtypes are prognostic in IBC and improve the prediction of late relapse relative to clinical covariates. Understanding the clinical course of DCIS lesions harboring these high-risk invasive features is highly relevant in refining clinically meaningful risk associated with DCIS progression.
To identify enriched pathways in the eight CNA clusters, we investigated the differential abundance in matched RNA samples (DESeq2 one-vs-rest) and performed GSEA Hallmark analysis on the resulting gene lists. Clusters 6 (chr11q13 amplification) and 7 (chr8q24 (MYC) amplification) were enriched for pathways associated with recurrence (Allograft Rejection and Oxidative Phosphorylation, respectively), whereas Cluster 8 (CNA quiet) was depleted of recurrence associated pathways (Cell Cycle and mTORc1 signaling), and Cluster 6 was depleted of MYC targets (Figure 5F, Figure S3H). The remaining CNA clusters had no significant pathway enrichments. Thus, we identified a CNA-based cluster solution characterized by amplifications seen in high-risk IBC subtypes, including 17q12 (ERBB2) and 8q24 (MYC) amplification, some of which were significantly enriched or depleted for pathways associated with recurrence.
The DCIS TME reflects distinct immune and fibroblast states
The Hallmark pathways identified represent a diverse set of biologic events and may involve different components of the DCIS ecosystem including the cells within the TME. Accumulating evidence has shown that the TME is crucial for cancer development and progression26,27. To analyze the DCIS TME, we generated RAHBT LCM stromal samples by dissecting stromal tissue from the DCIS edge (Figure S2D–F).
To identify the contribution of epithelial and stromal components to the 812 gene classifier, we performed differential abundance analysis between stromal (n=196) and epithelial (n=265) samples from the RAHBT LCM cohort. We identified 9748 DE genes (FDR<0.05) between epithelium and stroma (5161 epithelial, 4587 stromal). An analysis of the 812 classifier genes showed that 20% were expressed primarily in stromal/TME cells, and 34% in epithelium (Table S3).
The MIBI method provides an orthogonal view of the TME and generates protein expression and identity of 16 different cell types including epithelial, fibroblasts, and immune cell types22. We used adjacent TMA sections to analyze RNA and MIBI expression on the same ducts. We compared MIBI-based cell type distribution across samples with the inferred cell type distribution from RNA expression data using CIBERSORTx (CSx, see Methods, Figure S4A–B), allowing us to cross-validate findings and extend observations on cell composition to DCIS samples without MIBI data, including the TBCRC cohort.
To define discrete TME phenotypes, we performed shared nearest neighborhood clustering of stromal RNA data and identified four distinct DCIS-associated stromal clusters (Figure 6A) and DE genes (DESeq2 each-vs-rest, Figure 6B). Pathway analyses (Figure 6C, S4C), MIBI protein expression and cell type distribution (Figure 6D), and CSx-inferred cell type distribution (Figure 6E, Figure S4D–G) were used to describe major characteristics of each cluster, which were termed Immune dense, Desmoplastic, Collagen-rich, and Normal-like. Figure 6F shows representative MIBI images of each cluster, with strong correlation with fibroblast states and immune cell density.
Figure 6. TME analysis.
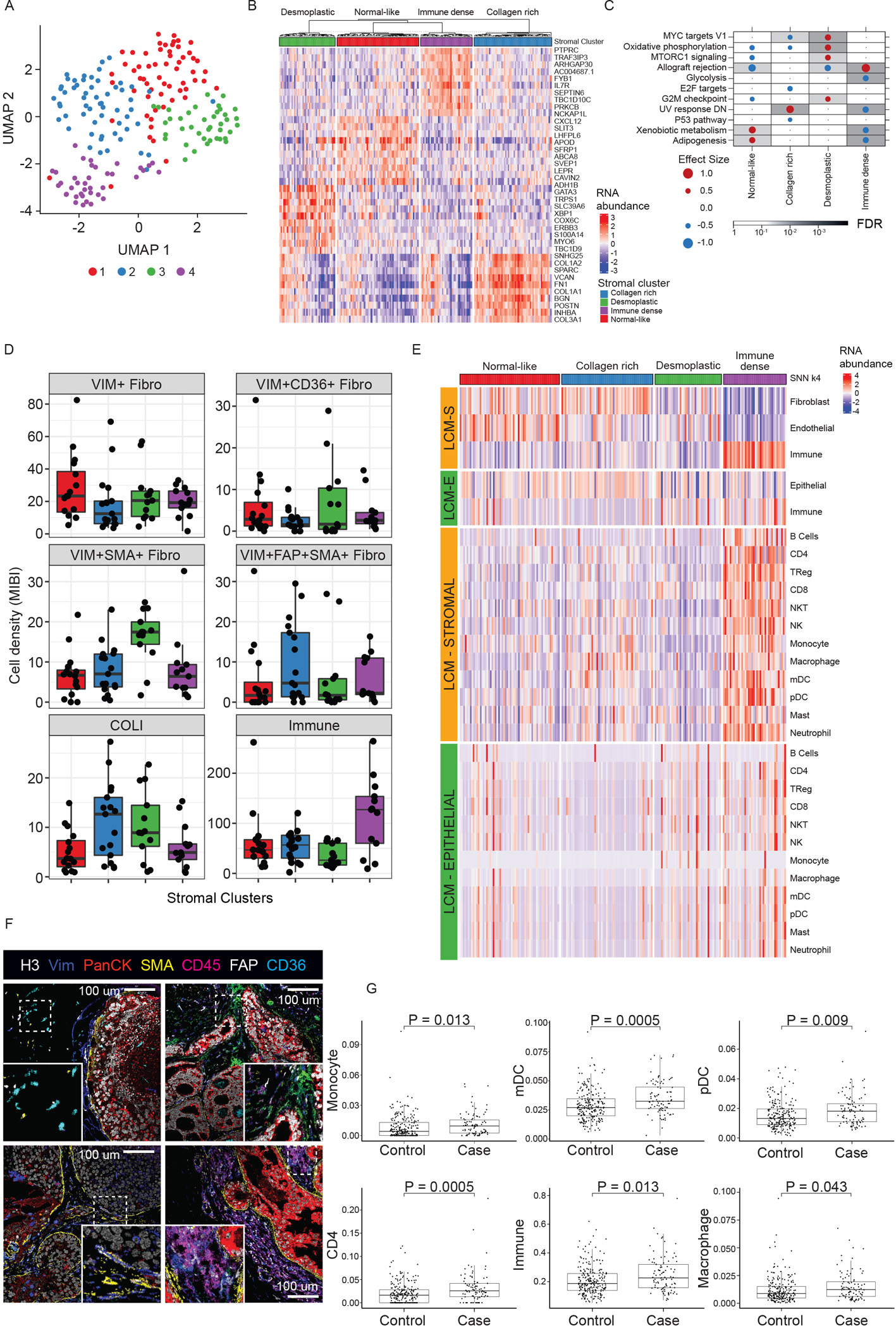
A) UMAP of DCIS stromal transcriptome colored by four identified clusters. B) Heatmap of top 10 up-regulated genes for each stromal cluster. C) GSEA Hallmark analysis of DE genes in each cluster vs rest, outcome-associated pathways only. Dot size and color represents the magnitude and direction of pathway deregulation. Background shading indicates FDR. Effect size and FDR from GSEA algorithm. D) MIBI-estimated cell density within clusters. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. E) Deconvolution analysis by CSx of epithelial and stromal LCM samples grouped by stromal clusters displaying immune cell and fibroblast abundance. F) Representative MIBI images of clusters reflecting different fibroblast states and total immune density. Top left: normal-like. Top right: Collagen rich (FAP+). Bottom left: Desmoplastic (SMA+). Bottom right: Immune dense (CD45 high). H3, histone 3; VIM, vimentin; panCK, pan cytokeratin; SMA, smooth muscle actin; FAP, fibroblast activated protein. G) CSx-inferred cell type distribution between cases with iBEs vs controls, TBCRC and RAHBT combined. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. Only cell types with FDR<0.05 shown (Wilcoxon rank sum test). See also Figures S4–6 and Table S5.
The Immune stromal cluster was the most distinct stromal subtype, with enrichment for the outcome-associated Allograft Rejection- and other immune activation pathways. MIBI and CSx data demonstrated a total abundance of immune cells more than twice that of any other cluster, with predominance of lymphoid over myeloid cells. A subgroup within this cluster was highly enriched for B cells, whereas another displayed overall balanced immune cell type composition. The Immune cluster also showed association with MIBI-identified T-cell and B-cell enriched neighborhoods (see22 for details), myoepithelial- and myeloid-enriched neighborhoods (Figure S5A), and was enriched for the ERlow subtype (Figure S5B).
The normal-like cluster was enriched for Gene Ontology pathways involved with ECM organization, Complement and Coagulation Cascades, Focal Adhesion, and PI3K-AKT signaling. The collagen-rich cluster was characterized by Collagen Metabolism, TGFb signaling, and Proteoglycans in Cancer, and Cell-Substrate and Focal Adhesion. This cluster had the highest fibroblast abundance and total myeloid cells, mostly associated with macrophages and myeloid dendritic cells (mDC). According to MIBI, this cluster was enriched in collagen and fibroblast associated protein positive (FAP+, VIM+, SMA+) myofibroblasts. The desmoplastic cluster was characterized by mammary gland development and fatty acid metabolism, high presence of VIM+, SMA+ myofibroblasts by MIBI, and higher levels of CD8+ T cells assessed by CSx vs the normal-like and collagen-rich clusters (Figure S5C).
These analyses indicate that the immune response is present in a discrete subset of cases. However, outcome analysis by stromal subtype demonstrated a modest outcome difference, without major contribution from the Immune subcluster (P=0.12, log-rank test, Figure S6A). We hypothesized that the outcome differences could be attributed to a subset of immune cells rather than the entire immune response, and analyzed CSx-inferred cell type distribution in 5-year outcome groups in TBCRC and RAHBT combined. We identified significantly higher levels of CD4+ T cells, myeloid- and plasmacytoid dendritic cells (pDC), monocytes, macrophages, and overall immune cells in cases vs. controls (Figure 6G). Furthermore, we found that several cell types, including CD4 T-cells, mDCs, and pDCs, were significant predictors of any iBE 5 years after treatment (univariable Cox regression analysis, Table S5). These differences in outcome groups were overall mirrored by CSx-inferred cell type distributions in the high- and low risk classifier groups (Figure S6B). Finally, we investigated the distribution of CSx-based cell types in 5-year outcome groups stratified by iBE type. The results overall reflected the analysis in cases vs. controls, with the largest differences observed between invasive iBEs and controls (Figure S6C).
Taken together, these results support the contributions of individual immune cells with high-risk outcomes. However, non-immune cell phenotypes are not well defined by this CSx approach but can still be identified as a biologic response. The desmoplastic cluster had the clearest and most favorable outcome (HR=0.23, P=0.06, Figure S6B), despite being enriched for several recurrence-associated pathways, including proliferative signals (MYC and G2M checkpoint) associated with poor outcome in the epithelial compartment. This highlights the complexity and differential contribution from the stromal and epithelial compartments.
DISCUSSION
The aims of the HTAN Breast Pre-Cancer Atlas are to 1) develop a resource of multi-modal spatially resolved data from breast pre-invasive samples that will facilitate discoveries by the scientific community regarding the natural history of DCIS and predictors of progression to life-threatening IBC; and 2) populate that platform with data from retrospective cohorts of patients with DCIS and demonstrate its use to construct an atlas to test novel biologic insights. Here, we examined two well-annotated, retrospective, longitudinal patient cohorts with or without a subsequent iBE. The two cohorts have important and distinct differences. They comprise subjects from diverse geographical sites, race/ethnicities, median years of diagnosis, and time to recurrence. There were no significant differences in age at diagnosis or treatment across cohorts. Together, these cohorts comprise a large series of matched case-control samples allowing great statistical power to perform the comprehensive studies reported here. A particular strength of the study is the complementary nature of the two cohorts, allowing for validation of our findings, as well as the capability to separately study the epithelial and stromal components in RAHBT LCM samples. Future observations on a DCIS cohort undergoing watchful waiting would provide outcome results that may be more aligned with emerging personalized treatment strategies of DCIS, that could include non-surgical options.
DCIS is a heterogeneous disease with variable prognosis but has defied attempts to identify molecular factors associated with future progression. Previous studies have evaluated the prognostic value of biomarkers associated with outcomes, with conflicting conclusions for virtually all markers tested, including ER, HER2, immune markers such as tumor infiltrating lymphocytes, and stromal characteristics. Many promising leads have not been reproducible due to multiple factors, including lack of endpoint standardization, differences between cohorts, small sample size, and limited datasets for validation with long-term outcomes.
Herein, we have developed and validated an 812 gene classifier which independently predicted risk of both overall recurrence and invasive progression. This classifier was highly associated with outcome in a multivariable model which included treatment, age, grade, and clinical ER status; the classifier had a HR of 22.5 (95% CI 8.5– 59.4) in the training set and 7.3 (95% CI 1.6– 34.2) in the validation set, over four-fold higher than has been previously reported for other prognostic markers for DCIS14.
Importantly, we found that this classifier was a stronger predictor of 5-year recurrence or progression than previously described clinical factors, including age at diagnosis, tumor grade, ER status, or treatment. The large dataset, with a high number of events, permitted an agnostic analysis of all genome-wide features and was thus less opportunistic than other, more limited studies. Further, since no a priori assumptions were made regarding whether to incorporate the molecular features of invasive cancer, we were able to construct a less biased predictor.
Our classifier is characterized by several Hallmark pathways including some related to cell cycle progression and growth factor signaling (E2F targets, G2M checkpoint, MYC targets, mTORc1 signaling) and metabolism (Glycolysis, Oxidative Phosphorylation). Examination of pathway activation status at the individual tumor level revealed the underlying complexity of the classifier. High correlation between cell cycle linked E2F and G2M pathways are consistent with a proliferation related signature. However, the strongest features of the classifier (distinguishing cases from controls) were MYC and MTORC1 signaling which are strongly correlated with each other but less so with the canonical proliferation pathways indicating that proliferation alone is not the central predictor. Interestingly, both Glycolysis and Oxidative Phosphorylation were increased in cases suggesting that heightened metabolic activity is associated with risk of progression regardless of whether it is anaerobic. Finally, Allograft Rejection, a broad immune pathway, was elevated in cases and in general appeared to be an independent component of the classifier. Overall, there are multiple components to this classifier that are elevated in different subsets of the tumors lending additional evidence that simplified predictors fail to capture the heterogeneity of the disease.
IBC has been genomically profiled with several approaches, including the PAM50 and IC classification schemes. While DCIS and IBC are part of the same neoplastic process, there are differences in the TME, evolutionary age, and inter-observer variability in diagnostic labeling at different stages of progression. This suggests that a DCIS-specific classification scheme would correlate better with biologic and clinical features of DCIS. Our analysis indicated the PAM50 subtypes are not apt for DCIS characterization, as previously described19,28. Instead, we identified three transcriptomic DCIS subgroups, characterized by ER signaling, proliferation and metabolism. These subtypes more accurately capture the spectrum of DCIS biology than IBC-derived subtypes, and represent the fundamental genomic organization at this early stage of breast neoplasia. They may represent the earliest variation in neoplasia transcriptome, potentially applicable to earlier stages such as hyperplasias.
There are several possible reasons why traditional IBC classifiers do not perform well on DCIS. HER2 expression is more common at the DCIS stage than at the IBC stage29, which may lead to a different transcriptomic distribution in DCIS vs IBC. Many ER− DCIS express HER2 without amplification, in contrast to IBC, where the HER2-amplified subtype is clearer. Moreover, DCIS cells are confined to the epithelial compartment and interact with myoepithelial cells and the basement membrane, thus presumably restricted by rules of differentiation that govern normal epithelial cells, which could constrain the transcriptomic variability of neoplastic cells and in turn possible subtypes. Finally, the evolutionary age of the neoplasm may influence classification differences in DCIS vs IBC. By comparing WGS data from DCIS and IBCs, we found that the same constellation of copy number changes was present in both, consistent with previous studies30–32. While DCIS had fewer genomic alterations than IBC, and a larger group of DCIS was classified as genomically quiescent, recurrent genomic events that drive the IBC-based IC scheme were evident at the DCIS stage.
A unique aspect of our study is the separate profiling of stromal and epithelial components through CSx analysis of LCM-derived RNA coupled with in situ MIBI protein expression. We identified four stromal subtypes characterized by distinct pathways, stromal-, and immune cell composition. Specific stromal patterns were correlated with epithelial expression patterns, and particularly HER2+/ER− DCIS were associated with a stronger immune response, potentially associated with co-amplification of ERBB2 (HER2) and chemokine encoding genes on the 17q12 chromosomal region3. A limitation of this study is that our CSx approach did not facilitate identification of non-immune stromal cell types.
Generating a DCIS atlas is similar to the effort of TCGA for IBC, but there are important differences. Working with DCIS samples is considerably more challenging; while IBC tumors are evident by gross exam, and can be easily obtained as fresh, fresh frozen, or archival material, this is not the case for pre-invasive lesions. DCIS can sometimes be recognized radiographically but is only precisely detailed by pathologic examination, making prospective tissue collection a challenge. Moreover, the transition from intraepithelial to invasive neoplasia is definitional for IBC. For DCIS, such a clear-cut definition does not exist. DCIS is broadly defined by cytologic and architectural changes compared to normal breast tissue by a growth of neoplastic cells in the inter-epithelial compartment.
One issue that should be noted is the genetic relationship between the primary DCIS and the subsequent ipsilateral cancer. Recent work33 on a large cohort indicates that 18% of ipsilateral invasive events may be unrelated to the primary DCIS based on mutations and CNAs. Non-clonal recurrences were more likely to be in a different breast quadrant and have discordant ER expression whereas time to recurrence and patient age were not significantly associated with clonality. While we did not examine the recurrences in the current study to determine clonality, it is likely that a similar fraction would be identified as “unrelated”. We anticipate that further refinement and validation of our classifier will be strengthened by eliminating non-clonal iBEs.
In conclusion, we have developed a genomic classifier that predicts both recurrence and invasive progression, using large, comprehensively annotated case-control data sets of primary DCIS. The classifier is comprised of both epithelial and stromal features. Our findings support that progression is a process that requires both invasive propensity among the DCIS cells and stromal permissiveness in the TME. We propose this classifier as the basis for a future clinical test to assess outcomes in patients with primary DCIS to guide a more individualized therapy, based on biologic risk. Future work will include further validation of the classifier and translation to clinical implementation. The Breast Pre-Cancer Atlas presented here provides a foundational advancement in the study of precancerous lesions and will be a valuable resource for years to come, with data available to the research community through the HTAN portal.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Robert West (rbwest@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA and DNA sequencing data, metadata, and MIBI and H&E imaging data, have been deposited at the HTAN portal and are publicly available as of the date of publication (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v1.p1). For further information see the key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | Abcam | ab212156 |
| CK7 | Spring | M3524.C |
| VIM | Cell Signaling Technologies | 5741BF |
| CD44 | Thermo Fisher Scientific | MA5-13890 |
| CK5 | Spring | M3274.C |
| PanCK | Thermo Fisher Scientific | MS-343_PABX |
| HIF1A | Abcam | ab210073 |
| CD45 | Cell Signaling Technologies | 13917BF |
| AR | Cell Signaling Technologies | 5153BF |
| HLADR/DP/DQ | Abcam | ab209968 |
| GLUT1 | Abcam | GR32744795 |
| ECAD | Abcam | ab201499 |
| CD20 | Cell Marque | 120M-8-Oem |
| MMP9 | Abcam | ab204850 |
| FAP | R&D Systems | AF3715 |
| CD11c | Abcam | ab216655 |
| HER2 | Millipore | 3013420 |
| CD3 | Cell Signaling Technologies | 85061BF |
| CD8 | Cell Marque | 107M-9-OEM |
| CD36 | Cell Signaling Technologies | 14347BF |
| MPO | R&D Systems | AF3667 |
| CD68 | Cell Signaling Technologies | 76437BF |
| pS6 | Cell Signaling Technologies | 4858BF |
| Granzyme B | Abcam | ab219803 |
| P63 | Cell Signaling Technologies | 39692BF |
| Ki67 | Cell Signaling Technologies | 9449BF |
| IDO1 | Spring | M5604.C |
| Anti-Biotin | BioLegend | 409002 |
| CD31 | Abcam | ab216459 |
| PD1 | Cell Signaling Technologies | 86163BF |
| CD14 | Cell Signaling Technologies | 56082BF |
| CD4 | Abcam | ab181724 |
| Anti-Alexa488 | Thermo Fisher Scientific | A11094 |
| Collagen 1 | Abcam | EPR7785 |
| SMA | Abcam | ab242395 |
| COX2 | Spring | M3214.C |
| Histone H3 | Cell Signaling Technologies | 4499BF |
| ER | Abcam | ab205850 |
| PDL1-biotin | Cell Signaling Technologies | 13684S |
| Chemicals, peptides, and recombinant proteins | ||
| SMARTScribe reverse transcriptase | Clontech | 639537 |
| SUPERase• In RNase inhibitor | Thermo Fisher Scientific | AM2694 |
| AMPure XP SPRI bead mix | Beckman Coulter | A63880 |
| Kapa HiFi HotStart ReadyMix | Kapa | KK2601 |
| Proteinase K, 20 mg/mL | NEB | P8107S |
| Proteinase K inhibitor | Millipore | 537470 |
| dNTP mix, 10 mM ea. | Thermo Fisher Scientific | R0191 |
| PhiX control library | Illumina | FC-110-3002 |
| TBS IHC Wash Buffer with Tween 20 | Cell Marque | Cat#935B-09 |
| PBS IHC Wash Buffer with Tween 20 | Cell Marque | Cat#934B-09 |
| Target Retrieval Solution, pH 9, (3:1) | Agilent (Dako) | Cat#S2375 |
| Avidin/Biotin Blocking Kit | Biolegend | Cat#927301 |
| Gelatin (cold water fish skin) | Sigma-Aldrich | Cat#G7765-250 |
| Xylene Histological grade | Sigma-Aldrich | Cat#534056-500 |
| Glutaraldehyde 8% Aqueous Solution EM Grade | EMS | Cat#16020 |
| Normal Donkey serum | Sigma-Aldrich | Cat#D9663-10ML |
| Bovine Albumin (BSA) | Fisher | Cat#BP1600-100 |
| Centrifugal filters (0.1 μm) | Millipore | Cat#UFC30VV00 |
| Biological samples | ||
| The Resource of Archival Breast Tissue (RAHBT) cohort, collected at Washington University in St. Louis. | HTAN portal | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v1.p1 |
| The Translational Breast Cancer Consortium (TBCRC) 038 cohort collected at 12 participating sites and administered by Duke University. | HTAN portal | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v1.p1 |
| Critical commercial assays | ||
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | 20024906 |
| KAPA HyperPlus Kit | Kapa Biosystems | #07962428001 |
| SeqCap Adapter Kit A | Kapa Biosystems | #7141530001 |
| Qubit® dsDNA HS Assay Kit (#) | Thermo Fisher Scientific | #Q32851 |
| PicoPure DNA Extraction kit | Thermo Fisher Scientific | #KIT0103 |
| MIBItag Conjugation Kit | IONpath | Cat#600XXX |
| ImmPRESS UNIVERSAL (Anti-Mouse/Anti-Rabbit) IgG KIT (HRP) | Vector Laboratories | Cat#M P-7500-15 |
| ImmPACT DAB (For HRP Substrate) | Vector Laboratories | Cat#SK-4105 |
| Deposited data | ||
| TBCRC & RAHBT RNA and DNA sequencing data | HTAN portal | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v1.p1 |
| TBCRC & RAHBT metadata | HTAN portal (Atlas name: HTAN Duke) | https://humantumoratlas.org (Atlas name: HTAN Duke) |
| RAHBT MIBI imaging data | HTAN portal (Atlas name: HTAN Duke) | https://www.humantumoratlas.org (Atlas name: HTAN Duke) |
| Software and algorithms | ||
| Data analysis using R | R | NA |
| Analysis code for R | Mendeley | https://data.mendeley.com/datasets/tbzv5hpvw5/1 |
All original code has been deposited at Mendeley Data and is publicly available as of the date of publication (https://data.mendeley.com/datasets/tbzv5hpvw5/1).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cohort collection and sample acquisition
RAHBT Cohort
The Resource of Archival Breast Tissue (RAHBT) is a data/tissue resource established by Drs. Allred and Colditz in 2008 focused on premalignant or benign breast disease. Uniform coding of premalignant lesions assures greater consistency and use of research. Follow-up through hospital record linkages documents subsequent breast lesions including IBC. The entire study population includes women ages 18 and older with documented cases of premalignant breast disease (including carcinoma in situ). The study was approved by the Washington University in St. Louis Institutional Review Board (IRB ID #: 201707090).
Women were identified as eligible through seven primary sources: Washington University School of Medicine Departmental databases (Surgery, Radiation Oncology, Pathology, and Radiology), and the Siteman Oncology Services Database (local tumor registry), the St. Louis Breast Tissue Repository, and the Women’s Health Repository. We reviewed all records, excluded women with cancer prior to qualifying premalignant lesions and identified 1831 unique women with DCIS or DCIS and subsequent recurrence. A common data set with pathologic details, risk factor data, treatment, and unique identifiers was created and used to follow these women for subsequent breast lesions. Centralized pathology review confirmed 174 cases of DCIS with recurrent lesions. For each case (with subsequent ipsilateral or contralateral breast events) we matched two controls who remained free from subsequent breast events based on race, year of diagnosis (+/− 5 years), age at diagnosis (+/− 5 years), and type of definitive surgery (mastectomy or lumpectomy). For each DCIS diagnosis we retrieved slides and blocks for pathology review, secured a whole slide image of each sample, marked for TMA cores, and prepared for laboratory processing. A total of 172 cases and 338 controls were cored for TMAs. Breast pathology review was completed by Drs. Allred, Warrick, DeSchryver, and Veis.
To define an external validation data set that used identical eligibility criteria to TBCR 038 including year of initial DCIS diagnosis, we identified an additional set of cases from RAHBT and used comparable laboratory procedures for RNA-seq.
For RAHBT, 97 patients were analyzed by RNA-seq (Table 1). The median age at diagnosis was 53, and median year of diagnosis 2006. Time to recurrence with ipsilateral IBC was 36 months, and to diagnosis of ipsilateral DCIS 46.9 months. For women in the cohort with no iBEs, median follow-up extended to 141 months. The total number of deaths by any cause was six. Treatment of initial DCIS ranged from lumpectomy with radiation (66.0%), and no radiation (10.3%) and mastectomy (23.7%). This subset of the RAHBT cohort was composed of 35.1% African American women.
For RAHBT LCM, 265 patients were analyzed by RNA-seq (Table S1). The median age at diagnosis was 53, and median year of diagnosis 2002. Time to recurrence with ipsilateral IBC was 80 months, and to diagnosis of ipsilateral DCIS 50 months. For women in the cohort with no iBEs, median follow-up extended to 111 months. Treatment of initial DCIS ranged from lumpectomy with radiation (52%), and no radiation (18%) and mastectomy (28%). This subset of the RAHBT cohort was composed of 25% African American women.
TBCRC 038 Cohort
TBCRC 038 is a retrospective multi-center study activated at 12 participating TBCRC (Translational Breast Cancer Consortium) sites, which identified women treated for ductal carcinoma in situ (DCIS) at one of the enrolling institutions between 01/01/1998 and 02/29/2016. The TBCRC and the Department of Defense (DOD) approved this study for the collection of archival tissues. Duke served as the initiating and central site for all data, samples, assays, and analysis. The study was approved by the Duke Health Institutional Review Board (Protocol ID: Pro00068646) as well as the IRB at each participating institution. Individual sites reviewed medical records to identify patients eligible for the study.
Study eligibility criteria included: Women aged 40–75 years at diagnosis of DCIS without invasion; no prior treatment for breast cancer; and definitive surgical excision with no ink on tumor margins and treated with mastectomy, lumpectomy with radiation, or lumpectomy. Cases (patients with subsequent iBEs) were matched 1:1 to controls with at least 5 years of follow-up without subsequent iBEs. Matching was based on year of diagnosis (+/−5 years), age at diagnosis (+/− 5 years), and DCIS nuclear grade (high grade vs. non-high grade). All cases consisted of initial diagnosis of pure DCIS, with ipsilateral recurrence occurring no less than 12 months from date of primary diagnosis. Clinical data, including treatment data, were collected at each site, and standardized data points were entered into a web-based portal. Tumor tissue was collected from FFPE blocks and cut into 5um sections. All slides were scanned and reviewed centrally by a breast pathologist (AH) to confirm the diagnosis. Tumor tissue marked by the pathologist was macrodissected for bulk analysis assays.
The 216 patients from the TBCRC cohort analyzed by RNA-seq (Table 1) includes 95 women without iBE after 5 or more years, 66 with DCIS iBEs, and 55 with IBC iBEs. Median time to IBC iBE for this subset was 58 months and 40 months to DCIS iBE. The total number of deaths by any cause was 12. 30% of this subset were African American.
METHOD DETAILS
TMA construction
Qualified DCIS or subsequent lesion slides were assembled for pathology review. The research breast pathologist marked the slides for best area to core (1mm) for the carcinoma in situ and later event. The TMAs were designed such that cases/controls were assigned randomly on the map. The Beecher Tissue Arrayer was used to take a core from the patient donor block and place it in the designated area of the recipient TMA block. Slides were then cut for research purposes, and stained H&E and unstained slides were prepared. The TMAs were stored in the St. Louis Breast Tissue Registry Lab at room temperature.
Slide cutting
A TMA cutting breakdown was established to include slides for laser capture microdissection (LCM PEN membrane glass slides) sequencing, multiplex protein (MIBI high-purity gold-coated slides) staining and charged glass slides for FISH analysis of the RAHBT TMAs. The order of the slides for the different assays was as follows:
Slide 1–3: FISH/routine IHC – 4 um slices on charged slides
Slide 4–6: RNA/DNA sequencing – 7 um slices on LCM membrane glass slides
Slide 7: MIBI analysis – 4 um slices on gold coated slides
Slide 8–10: FISH/routine IHC – 4 um slices on charged slides
Slide 11–13: RNA/DNA sequencing – 7 um slices on LCM membrane slides
Slide 14: MIBI analysis – 4 um slices on gold coated slides
Slide 15–17: FISH/routine IHC – 4 um slices on charged slides
Slide 18 H&E stained.
Digital H&E generation (scanners)
At Washington University School of Medicine, the H&E original slide and TMA slide for RAHBT was imaged (20x) by Aperio AT2 (Leica). ImageScope provides the software for viewing the slides. Images are stored on secure servers in the Dept of Pathology, Washington University School of Medicine.
Pathologic analysis and masking
For the TBCRC cohort, whole slide images of the H&E slide made from the block sourced for DNA and RNA was reviewed and scored for grade, presence of necrosis and architecture by a breast pathologist (AH). For the RAHBT LCM cohort, H&E images from the TMAs were used to score for grade, presence of necrosis and architecture by four breast pathologists (DJV, AH, SS, RBW). Areas of DCIS and normal tissue from the RAHBT TMAs were annotated and masked for LCM by two breast pathologists (SS and RBW).
Laser Capture microdissection
Consecutive sections of tissue microarray blocks were cut and mounted on PEN membrane slides. Slides were dissected immediately after staining on an Arcturus XT LCM System based on the masked areas. Epithelial and stromal sections were dissected separately (Figure S1). Each sample adhere to a CapSure HS LCM Cap (Thermo Fisher #LCM0215). After LCM, the cap was sealed in an 0.5 mL tube (Thermo Fisher #N8010611) and stored at −80°C until library preparation. The matching epithelial regions in consecutive slides were dissected for corresponding DNA libraries.
RNA-sequencing (smart-3seq)
Sequencing libraries were prepared according to the Smart-3SEQ method35 starting from dissected FFPE tissue on an Arcturus LCM HS Cap, except for the unique P5 index and universal P7 primers. Three control samples were added to each library preparation batch and sequence batch to allow batch effect analysis. Libraries were pooled together according to qPCR measurements and prepared according to the manufacturer’s instructions with a 1% spike-in of the PhiX control library (Illumina #FC-110–3002) and sequenced on an Illumina NextSeq 500 instrument with a High Output v2.5 reagent kit (Illumina # 20024906).
ER, HER2 status
Clinical ER status (by IHC) was available for 83.3% (180 of 216) of the TBCRC cohort, 83.5% (81 of 97) of the RAHBT cohort, and 46.8% (124 of 265) of the RAHBT LCM cohort.
Additionally, we called ER and HER2 positivity based on mRNA abundance levels of ESR1 and ERBB2, respectively. We applied a Gaussian mixture model with two components using the mclust R package (v5.4.7).
PAM50 and IC10
PAM50 subtypes were called using the genefu36 v2.22.1 R package. We compared the PAM50 subtypes called by genefu against subtypes called adjusting for the expected proportion of ER+ samples, as implemented in19. We found both methods to be highly concordant (>96% concordance). We compared the correlation of DCIS and IBC samples to the PAM50 centroids within the genefu R package using Spearman’s correlation. We also compared the silhouette widths based on Euclidean distances of the PAM50 subtypes to the de novo DCIS subtypes using the cluster R package (v2.1.1). IC10 subtypes were called using the iC10 (v1.5) R package. PAM50 subtypes were called in TBCRC and RAHBT separately, using the same protocols, given the differences in measurement techniques used in the two cohorts.
To compare PAM50 centroids in DCIS to TCGA: The TCGA cohort was downsampled to match the size of the DCIS cohort. The downsampling was repeated 1,000 times, and the median correlation for each of the 1,000 iterations was compared to the median DCIS correlations.
Differential abundance analyses
Differential abundance analysis was performed using the R package DESeq2 v1.30.137 with default options. P-values were adjusted for multiple testing using the Benjamini-Hochberg method. FDR<0.05 was considered significant for all DESeq2 analyses. Reads matrices were VST normalized for downstream analyses.
Unsupervised clustering: non-negative matrix factorization
We identified RNA and CNA based clusters by non-negative matrix factorization using the NMF R package v0.23.038. Each NMF rank was run 30 times to evaluate cluster stability. We comprehensively evaluated 2–10 clusters for each data type and evaluated cluster fit by cophenetic and silhouette values. RNA clusters were first discovered in TBCRC and replicated in RAHBT. We evaluated replication by quantifying the concordance of de novo clusters identified in RAHBT vs clusters determined from centroids identified in TBCRC.
CNA clusters were discovered in TBCRC and RAHBT jointly and compared against clusters identified in TBCRC and RAHBT individually to ensure robustness.
CIBERSORTx
Using single-cell RNA-seq datasets, a breast specific signature matrix was built to resolve proportions of tumor, fibroblasts, endothelial and immune cells from bulk RNA-seq data39. scRNAseq data was downloaded from Gene Expression Omnibus database (GEO data repository accession numbers GSE114727, GSE114725). Normalized counts were obtained using Seurat R package (v3.2.0), and used as single cell matrix input alongside with their cell type identities (code available: https://cibersortx.stanford.edu/, default parameters for “Create Signature Matrix/ scRNAseq input data”)40. The resultant signature matrix contained 3484 genes and allowed to resolve different immune cell types, including B, CD8 T, CD4 T, NKT, NK, mast cells, neutrophils, monocytes, macrophages and dendritic cells (code available https://cibersortx.stanford.edu/, “Impute Cell Fractions/Enable batch correction S-mode”, and default parameters). The signature matrix was first in-silico validated. In order to test the accuracy of the signature matrix, a set of samples (1/10 of each type) from the same scRNAseq dataset was reserved to build a synthetic matrix of bulk RNA-seq data. By mixing different proportions of single cell transcripts, the synthetic bulk was used to predict cell type proportions and subsequently correlated with the true proportions used to build the synthetic mix. Pearson’s coefficient was >0.75 in all the cases, and most >0.9. The aforementioned matrix was used to deconvolve the LCM RNA-seq samples and to compare CSx-estimated cell abundance with MIBI-identified cell types. Cell abundance between groups was compared by Wilcoxon rank sum test followed by Benjamini-Hochberg correction for multiple testing.
Shared Nearest Neighbor clustering
LCM stromal samples from RAHBT were classified using the Shared Nearest Neighbor clustering method implemented in the Seurat R package (v3.2.0). Data was normalized by negative binomial regression (sctransform R package, v0.3.2, variable.feature.n = “all.genes”). The first 15 principal components were used to identify the clusters and 16 different resolutions were compared, selecting resolution 0.75 and four clusters as the final solution. Positive markers were selected at a minimum fraction of 0.25 and the resultant gene list was used to further characterize each cluster by gene ontology and KEGG pathway analysis, implemented in clusterProfiler R package (version 3.18.1).
Pathway & Gene Set Enrichment Analyses
Gene set enrichment analyses were performed using fgsea R package (v1.12.0) based on the MSigDB Hallmark pathways v7.4, 41. All genes from differential abundance analyses were included and were ranked by their signed adjusted P-values. Pathways were considered enriched if adjusted P-values<0.05. We evaluated pathway concordance across the DCIS subtypes using a hypergeometric test.
Single sample gene set variation analysis was performed using the GSVA R package42 (v1.38.2) using default parameters.
Outcome analysis
Associations with time to event were quantified using Cox Proportional Hazard model correcting for treatment as indicated in the text. To standardize follow-up across TBCRC and RAHBT, we censored the follow-up time at 250 months, the maximum follow-up time in TBCRC. Kaplan-Meier plots as implemented in the R packages survival (v3.2.10) and survminer (v0.4.9) were used to visualize outcome differences.
The 812 gene classifier was built using the cforest implementation of Random Forest in the Caret (v6.0–91) R package using default parameters. The TBRCR cohort was used as the training cohort and the model was tested on the RAHBT cohort. Hyperparameters were tuned on the training cohort using four-fold cross validation. The mtry parameters 5, 20, 50, 100, 200, 500, and 800 were tested and the optimal mtry selected was 5. Accuracy of the classifier was assessed using ROC curve, Precision, Recall, and F1 score.
Breast cancer data (BRCA) from TCGA was downloaded from https://www.cancer.gov/tcga. A total of 1064 samples with available follow-up information was used to test the 812 gene classifier towards progression-free survival and overall survival as defined in the TCGA-BRCA metadata.
RNA for the TCGA samples was normalized using the same protocols as the DCIS RNA-sequencing (TBCRC and RAHBT cohorts, above). The accuracy of the classifier in the TCGA cohort was assessed using ROC curve, Precision, Recall, and F1 score.
DNA-sequencing
Genomic DNA was isolated from LCM FFPE cells using PicoPure DNA Extraction kit (Thermo Fisher Scientific # KIT0103). 50ul lysis buffer with Proteinase K were added to each sample and incubated at 65°C overnight. After inactivating proteinase K, the genomic DNA was cleaned up with AMPure XP beads at 3:1 ratio (Beckman Coulter# A63880) and eluted in the 10mM Tris-HCl (pH8.0).
DNA Libraries were constructed with KAPA HyperPlus Kit (Kapa Biosystems #07962428001). Barcode adapters were used for multiplexed sequencing of libraries with SeqCap Adapter Kit A (Kapa Biosystems #7141530001). DNA libraries were amplified by 19 PCR cycles. AMPure XP beads were used for the size selection and cleaning up. DNA libraries were eluted in the 30 μL 10mM Tris-HCl (pH8.0).
Library size distribution was assessed on an Agilent 2100 Bioanalyzer using the DNA 1000 assay and the concentration was measured by Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific # Q32851). For each lane, 12 samples were pooled and sequenced by Novogene (Sacramento, CA, US) on the Illumina HiSeq Platform, collecting 110G per 275M reads output of paired-end reads of 150 bp length.
Identification of recurrent CNAs (GISTIC)
Recurrent CNAs were identified from purity-adjusted segment CNA calls from QDNASeq for 228 DCIS samples using GISTIC2 v2.0.2343 run with the following parameters: -ta 0.3 -td 0.3 -qvt 0.05 -brlen 0.98 -conf 0.95 -armpeel 1 -res 0.01 -rx 0. To ensure CNAs were not biased by sequencing depth, recurrent CNAs significantly associated (FDR<0.05) with the number of uniquely mapped reads were filtered out. Associations were quantified by Mann-Whitney test. The number of uniquely mapped reads was determined from samtools flagstat (v1.9).
MIBI
We used a MIBI panel consisting of 37 metal-conjugated antibodies that capture 16 different cell types including epithelial, fibroblasts, and immune cell types. We took tissue sections from adjacent sections to those used for RNA-seq to spatially align the same ducts for both MIBI and RNA. For full details of the MIBI methods, see the companion paper22. Briefly, antibodies were conjugated to isotopic metal reporters. Tissues were sectioned (5μm section thickness) from tissue blocks on gold and tantalum-sputtered microscope slides. Imaging was performed using a MIBI-TOF instrument with a Hyperion ion source.
Multiplexed image sets were extracted, slide background-subtracted, denoised, and aggregate filtered. Nuclear segmentation was performed using an adapted version of the DeepCell CNN architecture. Single cell data was extracted for all cell objects and area normalized. The FlowSOM R package v1.22.044 was used to assign each cell to one of five major cell lineages (tumor, myoepithelial, fibroblast, endothelial, immune). Immune cells were subclustered to delineate B cells, CD4+ T cells, CD8+ T cells, monocytes, MonoDC cells, DC cells, macrophages, neutrophils, mast cells, double-negative CD4−CD8− T cells, and HLADR+ APC cells. Tumor and fibroblast cells were similarly sub clustered to reveal phenotypic subsets. A total of 16 cell populations were quantified and analyzed. For full details of the MIBI methods, see the companion paper22.
Data visualization
Boxplots, heatmaps, scatterplots and barplots were generated using the BoutrosLab.plotting.general R package v6.0.3 45, or the R packages ggplot2 (v3.3.3, boxplots), corrplot (v0.84, scatterplots), and ComplexHeatmap (v.2.6.2, heatmaps). UMAPs were generated using the umap (v0.2.7.0) R package with the number of genes indicated in the text. Mosaic plots were generated using the vcd (v1.4.8) R package.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq processing
RNA sequencing data was processed with 3SEQtools (https://github.com/jwfoley/3SEQtools). Single-end Illumina FASTQ files were generated from NextSeq BCL files with bcl2fastq (v2.20.0.422) and then aligned to reference hg38 with STAR aligner (v2.7.3a). Samples that did not meet a minimum threshold of uniquely aligned reads were filtered out. The samples in this study averaged 1.11 million uniquely aligned reads. Gene expression matrices of raw and normalized read counts were produced from BAM files with featureCounts (v1.6.4) of the Subread package (v2.4.2) and GENCODE Release 33.
Read counts were normalized using the variance stabilizing transformation (VST) implemented in the R package, DESeq2 (v1.30.1)37. The VST normalization procedure normalizes for library size and returns a matrix that is approximately homoscedastic. The same normalization method was used for both the TBCRC and RAHBT cohorts individually.
DNA-seq processing
Low-pass WGS data were preprocessed using the Nextflow-base pipeline Sarek46 v2.6.1 with BWA v0.7.17 for sequence alignment to the reference genome GRCh38/hg38 and GATK47 v4.1.7.0 to mark duplicates and calibration. The recalibrated reads were further processed and filtered for mappability, GC content using the R/Bioconductor quantitative DNA-sequencing (QDNAseq) v1.22.0 with R v3.6.0. For QDNAseq, 50-kb bins were generated from (http://doi.org/10.5281/zenodo.4274556). We kept only autosomal sequences after filtering due to low-depth mappability and GC correction. We used the QDNAseq corrected output and segmented for CN analysis using the circular binary segmentation (CBS) algorithm from DNAcopy R/Bioconductor package v1.60.0. Copy number aberrations were called using CGHcall v2.48.048. The R/Bioconductor package ACE v1.4.049 was used to estimate purity and ploidy. Proportion of the genome copy number altered (PGA) was calculated based on CNAs with |log2 ratio| > 0.3 based on the following:
Statistical analyses
We used Mann-Whitney U test to compare continuous distributions between two groups, as specified in the text. We used the Kruskal-Wallis test to compare continuous values between three groups. All statistical analyses were implemented in the R statistical language (v3.6.1). P-values were corrected for multiple hypothesis testing via Bonferroni (when <10 independent tests) or Benjamini & Hochberg (when >10 independent tests).
Supplementary Material
Table S3: 812 differentially expressed genes from DESeq2 analysis iBEs within 5 years vs. the rest in TBCRC. Related to Figure 2.
HIGHLIGHTS.
Development of a new classifier for DCIS recurrence or progression
Outcome associated pathways identified across multiple data types and compartments
Four stroma-specific signatures identified
CNAs characterize DCIS subgroups associated with high-risk invasive cancers
ACKNOWLEDGMENTS
This publication is part of the HTAN (Human Tumor Atlas Network) Consortium paper package. A list of HTAN members is available at humantumoratlas.org/htan-authors/. R01 CA185138-01 (ESH); U2C CA-17-035 Pre-Cancer Atlas (PCA) Research Centers (ESH, RBW, CM, KP, GAC, KO); UO1 CA214183 (JRM); DOD BC132057 (ESH, CM); BCRF 19–074 (ESH); BCRF 19-028 (GAC); PRECISION CRUK Grand Challenge (ESH); R01CA193694 (RBW, GAC), BCRF PPI-18-006 (RBW). AEI RYC2019-026576-I, “LaCaixa” Foundation LCF/PR/PR17/51120011 (JAS). SHS was supported by the Lundbeck Foundation (R288-2018-35) and the Danish Cancer Society (R229-A13616). KEH was supported by a CIHR Banting Postdoctoral Fellowship. TBCRC 038 was conducted by the TBCRC, which receives major funding support from The Breast Cancer Research Foundation and Susan G. Komen. Some results in this paper are based upon data generated by the TCGA Research Network34.
Footnotes
DECLARATION OF INTERESTS
CC serves on the Scientific Advisory Board and/or as consultant for GRAIL, Deepcell, Ravel, Viosera, NanoString, Genentech and holds equity in GRAIL, Deepcell, Ravel.
KP serves on the Scientific Advisory Board of Acrivon Therapeutics, Vividion Therapeutics, and Scorpion Therapeutics, holds equity in Scorpion Therapeutics and Vividion Therapeutics, is a consultant to Aria Pharmaceuticals, and received honorarium from Astra-Zeneca.
RMA is an inventor on patent US20150287578A, and board member and shareholder in IonPath Inc. TR and RBW have consulted for IonPath Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.American_Cancer_Society (2019). Breast Cancer Facts & Figures 2019–2020.
- 2.Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, et al. (2004). Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6, 17–32. 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 3.Gil Del Alcazar CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, Zi X, Kwak M, Bergholtz H, Su Y, et al. (2017). Immune Escape in Breast Cancer During In Situ to Invasive Carcinoma Transition. Cancer discovery 7, 1098–1115. 10.1158/2159-8290.Cd-17-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heselmeyer-Haddad K, Berroa Garcia LY, Bradley A, Ortiz-Melendez C, Lee WJ, Christensen R, Prindiville SA, Calzone KA, Soballe PW, Hu Y, et al. (2012). Single-cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumor heterogeneity yet conserved genomic imbalances and gain of MYC during progression. Am J Pathol 181, 1807–1822. 10.1016/j.ajpath.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesurf R, Aure MR, Mørk HH, Vitelli V, Lundgren S, Børresen-Dale AL, Kristensen V, Wärnberg F, Hallett M, and Sørlie T (2016). Molecular Features of Subtype-Specific Progression from Ductal Carcinoma In Situ to Invasive Breast Cancer. Cell reports 16, 1166–1179. 10.1016/j.celrep.2016.06.051. [DOI] [PubMed] [Google Scholar]
- 6.Newburger DE, Kashef-Haghighi D, Weng Z, Salari R, Sweeney RT, Brunner AL, Zhu SX, Guo X, Varma S, Troxell ML, et al. (2013). Genome evolution during progression to breast cancer. Genome Res 23, 1097–1108. 10.1101/gr.151670.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorringe KL, Hunter SM, Pang JM, Opeskin K, Hill P, Rowley SM, Choong DY, Thompson ER, Dobrovic A, Fox SB, et al. (2015). Copy number analysis of ductal carcinoma in situ with and without recurrence. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 28, 1174–1184. 10.1038/modpathol.2015.75. [DOI] [PubMed] [Google Scholar]
- 8.Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, Casasent T, Meric-Bernstam F, Edgerton ME, and Navin NE (2018). Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 172, 205–217 e212. 10.1016/j.cell.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abba MC, Gong T, Lu Y, Lee J, Zhong Y, Lacunza E, Butti M, Takata Y, Gaddis S, Shen J, et al. (2015). A Molecular Portrait of High-Grade Ductal Carcinoma In Situ. Cancer Res 75, 3980–3990. 10.1158/0008-5472.Can-15-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vincent-Salomon A, Bidard FC, and Pierga JY (2008). Bone marrow micrometastasis in breast cancer: review of detection methods, prognostic impact and biological issues. Journal of clinical pathology 61, 570–576. 10.1136/jcp.2007.046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pareja F, Brown DN, Lee JY, Da Cruz Paula A, Selenica P, Bi R, Geyer FC, Gazzo A, da Silva EM, Vahdatinia M, et al. (2020). Whole-Exome Sequencing Analysis of the Progression from Non-Low-Grade Ductal Carcinoma In Situ to Invasive Ductal Carcinoma. Clin Cancer Res 26, 3682–3693. 10.1158/1078-0432.Ccr-19-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao J, Weremowicz S, Feng B, Gentleman RC, Marks JR, Gelman R, Brennan C, and Polyak K (2006). Combined cDNA array comparative genomic hybridization and serial analysis of gene expression analysis of breast tumor progression. Cancer Res 66, 4065–4078. 10.1158/0008-5472.Can-05-4083. [DOI] [PubMed] [Google Scholar]
- 13.Johnson CE, Gorringe KL, Thompson ER, Opeskin K, Boyle SE, Wang Y, Hill P, Mann GB, and Campbell IG (2012). Identification of copy number alterations associated with the progression of DCIS to invasive ductal carcinoma. Breast cancer research and treatment 133, 889–898. 10.1007/s10549-011-1835-1. [DOI] [PubMed] [Google Scholar]
- 14.Kerlikowske K, Molinaro AM, Gauthier ML, Berman HK, Waldman F, Bennington J, Sanchez H, Jimenez C, Stewart K, Chew K, et al. (2010). Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst 102, 627–637. 10.1093/jnci/djq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ringberg A, Anagnostaki L, Anderson H, Idvall I, Ferno M, and South Sweden Breast Cancer, G. (2001). Cell biological factors in ductal carcinoma in situ (DCIS) of the breast-relationship to ipsilateral local recurrence and histopathological characteristics. European journal of cancer (Oxford, England : 1990) 37, 1514–1522. 10.1016/s0959-8049(01)00165-4. [DOI] [PubMed] [Google Scholar]
- 16.Roka S, Rudas M, Taucher S, Dubsky P, Bachleitner-Hofmann T, Kandioler D, Gnant M, and Jakesz R (2004). High nuclear grade and negative estrogen receptor are significant risk factors for recurrence in DCIS. Eur J Surg Oncol 30, 243–247. 10.1016/j.ejso.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416 e411. 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solin LJ, Gray R, Baehner FL, Butler SM, Hughes LL, Yoshizawa C, Cherbavaz DB, Shak S, Page DL, Sledge GW Jr., et al. (2013). A multigene expression assay to predict local recurrence risk for ductal carcinoma in situ of the breast. J Natl Cancer Inst 105, 701–710. 10.1093/jnci/djt067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergholtz H, Lien TG, Swanson DM, Frigessi A, Daidone MG, Tost J, Wärnberg F, and Sørlie T (2020). Contrasting DCIS and invasive breast cancer by subtype suggests basal-like DCIS as distinct lesions. NPJ breast cancer 6, 26. 10.1038/s41523-020-0167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. (2000). Molecular portraits of human breast tumours. Nature 406, 747–752. 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 22.Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, McCaffrey EF, Greenwald NF, Rivero-Gutierrez B, Strand SH, et al. (2022). Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 185, 299–310 e218. 10.1016/j.cell.2021.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trinh A, Gil Del Alcazar CR, Shukla SA, Chin K, Chang YH, Thibault G, Eng J, Jovanović B, Aldaz CM, Park SY, et al. (2021). Genomic Alterations during the In Situ to Invasive Ductal Breast Carcinoma Transition Shaped by the Immune System. Molecular cancer research : MCR 19, 623–635. 10.1158/1541-7786.Mcr-20-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russnes HG, Vollan HKM, Lingjærde OC, Krasnitz A, Lundin P, Naume B, Sørlie T, Borgen E, Rye IH, Langerød A, et al. (2010). Genomic architecture characterizes tumor progression paths and fate in breast cancer patients. Sci Transl Med 2, 38ra47. 10.1126/scitranslmed.3000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rueda OM, Sammut SJ, Seoane JA, Chin SF, Caswell-Jin JL, Callari M, Batra R, Pereira B, Bruna A, Ali HR, et al. (2019). Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 567, 399–404. 10.1038/s41586-019-1007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gil Del Alcazar CR, Alečković M, and Polyak K (2020). Immune Escape during Breast Tumor Progression. Cancer immunology research 8, 422–427. 10.1158/2326-6066.Cir-19-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinshaw DC, and Shevde LA (2019). The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res 79, 4557–4566. 10.1158/0008-5472.Can-18-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swanson DM, Lien T, Bergholtz H, Sørlie T, and Frigessi A (2019). A Bayesian two-way latent structure model for genomic data integration reveals few pan-genomic cluster subtypes in a breast cancer cohort. Bioinformatics 35, 4886–4897. 10.1093/bioinformatics/btz381. [DOI] [PubMed] [Google Scholar]
- 29.Allred DC, Clark GM, Tandon AK, Molina R, Tormey DC, Osborne CK, Gilchrist KW, Mansour EG, Abeloff M, Eudey L, and et al. (1992). HER-2/neu in node-negative breast cancer: prognostic significance of overexpression influenced by the presence of in situ carcinoma. J Clin Oncol 10, 599–605. 10.1200/jco.1992.10.4.599. [DOI] [PubMed] [Google Scholar]
- 30.Hwang ES, DeVries S, Chew KL, Moore DH 2nd, Kerlikowske K, Thor A, Ljung BM, and Waldman FM (2004). Patterns of chromosomal alterations in breast ductal carcinoma in situ. Clin Cancer Res 10, 5160–5167. 10.1158/1078-0432.Ccr-04-0165. [DOI] [PubMed] [Google Scholar]
- 31.Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, et al. (2003). Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A 100, 5974–5979. 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vincent-Salomon A, Lucchesi C, Gruel N, Raynal V, Pierron G, Goudefroye R, Reyal F, Radvanyi F, Salmon R, Thiery JP, et al. (2008). Integrated genomic and transcriptomic analysis of ductal carcinoma in situ of the breast. Clin Cancer Res 14, 1956–1965. 10.1158/1078-0432.Ccr-07-1465. [DOI] [PubMed] [Google Scholar]
- 33.Lips EH, Kumar T, Megalios A, Visser LL, Sheinman M, Fortunato A, Shah V, Hoogstraat M, Sei E, Mallo D, et al. (2021). Genomic profiling defines variable clonal relatedness between invasive breast cancer and primary ductal carcinoma in situ. medRxiv, 2021.2003.2022.21253209. 10.1101/2021.03.22.21253209. [DOI] [Google Scholar]
- 34.TCGA Research Network. https://www.cancer.gov/tcga. .
- 35.Foley JW, Zhu C, Jolivet P, Zhu SX, Lu P, Meaney MJ, and West RB (2019). Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Res 29, 1816–1825. 10.1101/gr.234807.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gendoo DM, Ratanasirigulchai N, Schröder MS, Paré L, Parker JS, Prat A, and Haibe-Kains B (2016). Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 32, 1097–1099. 10.1093/bioinformatics/btv693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brunet JP, Tamayo P, Golub TR, and Mesirov JP (2004). Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A 101, 4164–4169. 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308.e1236. 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. (2019). Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782. 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics 14, 7. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry. Part A : the journal of the International Society for Analytical Cytology 87, 636–645. 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 45.P’ng C, Green J, Chong LC, Waggott D, Prokopec SD, Shamsi M, Nguyen F, Mak DYF, Lam F, Albuquerque MA, et al. (2019). BPG: Seamless, automated and interactive visualization of scientific data. BMC bioinformatics 20, 42. 10.1186/s12859-019-2610-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia M, Juhos S, Larsson M, Olason PI, Martin M, Eisfeldt J, DiLorenzo S, Sandgren J, Díaz De Ståhl T, Ewels P, et al. (2020). Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Research 9, 63. 10.12688/f1000research.16665.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van de Wiel MA, Kim KI, Vosse SJ, van Wieringen WN, Wilting SM, and Ylstra B (2007). CGHcall: calling aberrations for array CGH tumor profiles. Bioinformatics 23, 892–894. 10.1093/bioinformatics/btm030. [DOI] [PubMed] [Google Scholar]
- 49.Poell JB, Mendeville M, Sie D, Brink A, Brakenhoff RH, and Ylstra B (2019). ACE: absolute copy number estimation from low-coverage whole-genome sequencing data. Bioinformatics 35, 2847–2849. 10.1093/bioinformatics/bty1055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3: 812 differentially expressed genes from DESeq2 analysis iBEs within 5 years vs. the rest in TBCRC. Related to Figure 2.
Data Availability Statement
RNA and DNA sequencing data, metadata, and MIBI and H&E imaging data, have been deposited at the HTAN portal and are publicly available as of the date of publication (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v1.p1). For further information see the key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | Abcam | ab212156 |
| CK7 | Spring | M3524.C |
| VIM | Cell Signaling Technologies | 5741BF |
| CD44 | Thermo Fisher Scientific | MA5-13890 |
| CK5 | Spring | M3274.C |
| PanCK | Thermo Fisher Scientific | MS-343_PABX |
| HIF1A | Abcam | ab210073 |
| CD45 | Cell Signaling Technologies | 13917BF |
| AR | Cell Signaling Technologies | 5153BF |
| HLADR/DP/DQ | Abcam | ab209968 |
| GLUT1 | Abcam | GR32744795 |
| ECAD | Abcam | ab201499 |
| CD20 | Cell Marque | 120M-8-Oem |
| MMP9 | Abcam | ab204850 |
| FAP | R&D Systems | AF3715 |
| CD11c | Abcam | ab216655 |
| HER2 | Millipore | 3013420 |
| CD3 | Cell Signaling Technologies | 85061BF |
| CD8 | Cell Marque | 107M-9-OEM |
| CD36 | Cell Signaling Technologies | 14347BF |
| MPO | R&D Systems | AF3667 |
| CD68 | Cell Signaling Technologies | 76437BF |
| pS6 | Cell Signaling Technologies | 4858BF |
| Granzyme B | Abcam | ab219803 |
| P63 | Cell Signaling Technologies | 39692BF |
| Ki67 | Cell Signaling Technologies | 9449BF |
| IDO1 | Spring | M5604.C |
| Anti-Biotin | BioLegend | 409002 |
| CD31 | Abcam | ab216459 |
| PD1 | Cell Signaling Technologies | 86163BF |
| CD14 | Cell Signaling Technologies | 56082BF |
| CD4 | Abcam | ab181724 |
| Anti-Alexa488 | Thermo Fisher Scientific | A11094 |
| Collagen 1 | Abcam | EPR7785 |
| SMA | Abcam | ab242395 |
| COX2 | Spring | M3214.C |
| Histone H3 | Cell Signaling Technologies | 4499BF |
| ER | Abcam | ab205850 |
| PDL1-biotin | Cell Signaling Technologies | 13684S |
| Chemicals, peptides, and recombinant proteins | ||
| SMARTScribe reverse transcriptase | Clontech | 639537 |
| SUPERase• In RNase inhibitor | Thermo Fisher Scientific | AM2694 |
| AMPure XP SPRI bead mix | Beckman Coulter | A63880 |
| Kapa HiFi HotStart ReadyMix | Kapa | KK2601 |
| Proteinase K, 20 mg/mL | NEB | P8107S |
| Proteinase K inhibitor | Millipore | 537470 |
| dNTP mix, 10 mM ea. | Thermo Fisher Scientific | R0191 |
| PhiX control library | Illumina | FC-110-3002 |
| TBS IHC Wash Buffer with Tween 20 | Cell Marque | Cat#935B-09 |
| PBS IHC Wash Buffer with Tween 20 | Cell Marque | Cat#934B-09 |
| Target Retrieval Solution, pH 9, (3:1) | Agilent (Dako) | Cat#S2375 |
| Avidin/Biotin Blocking Kit | Biolegend | Cat#927301 |
| Gelatin (cold water fish skin) | Sigma-Aldrich | Cat#G7765-250 |
| Xylene Histological grade | Sigma-Aldrich | Cat#534056-500 |
| Glutaraldehyde 8% Aqueous Solution EM Grade | EMS | Cat#16020 |
| Normal Donkey serum | Sigma-Aldrich | Cat#D9663-10ML |
| Bovine Albumin (BSA) | Fisher | Cat#BP1600-100 |
| Centrifugal filters (0.1 μm) | Millipore | Cat#UFC30VV00 |
| Biological samples | ||
| The Resource of Archival Breast Tissue (RAHBT) cohort, collected at Washington University in St. Louis. | HTAN portal | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v1.p1 |
| The Translational Breast Cancer Consortium (TBCRC) 038 cohort collected at 12 participating sites and administered by Duke University. | HTAN portal | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v1.p1 |
| Critical commercial assays | ||
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | 20024906 |
| KAPA HyperPlus Kit | Kapa Biosystems | #07962428001 |
| SeqCap Adapter Kit A | Kapa Biosystems | #7141530001 |
| Qubit® dsDNA HS Assay Kit (#) | Thermo Fisher Scientific | #Q32851 |
| PicoPure DNA Extraction kit | Thermo Fisher Scientific | #KIT0103 |
| MIBItag Conjugation Kit | IONpath | Cat#600XXX |
| ImmPRESS UNIVERSAL (Anti-Mouse/Anti-Rabbit) IgG KIT (HRP) | Vector Laboratories | Cat#M P-7500-15 |
| ImmPACT DAB (For HRP Substrate) | Vector Laboratories | Cat#SK-4105 |
| Deposited data | ||
| TBCRC & RAHBT RNA and DNA sequencing data | HTAN portal | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002371.v1.p1 |
| TBCRC & RAHBT metadata | HTAN portal (Atlas name: HTAN Duke) | https://humantumoratlas.org (Atlas name: HTAN Duke) |
| RAHBT MIBI imaging data | HTAN portal (Atlas name: HTAN Duke) | https://www.humantumoratlas.org (Atlas name: HTAN Duke) |
| Software and algorithms | ||
| Data analysis using R | R | NA |
| Analysis code for R | Mendeley | https://data.mendeley.com/datasets/tbzv5hpvw5/1 |
All original code has been deposited at Mendeley Data and is publicly available as of the date of publication (https://data.mendeley.com/datasets/tbzv5hpvw5/1).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.