

Abstract
Mass spectrometry (MS) has emerged at the forefront of quantitative proteomic techniques. Liquid chromatography‐mass spectrometry (LC‐MS) can be used to determine abundances of proteins and peptides in complex biological samples. Several methods have been developed and adapted for accurate quantification based on chemical isotopic labeling. Among various chemical isotopic labeling techniques, isobaric tagging approaches rely on the analysis of peptides from MS2‐based quantification rather than MS1‐based quantification. In this review, we will provide an overview of several isobaric tags along with some recent developments including complementary ion tags, improvements in sensitive quantitation of analytes with lower abundance, strategies to increase multiplexing capabilities, and targeted analysis strategies. We will also discuss limitations of isobaric tags and approaches to alleviate these restrictions through bioinformatic tools and data acquisition methods. This review will highlight several applications of isobaric tags, including biomarker discovery and validation, thermal proteome profiling, cross‐linking for structural investigations, single‐cell analysis, top‐down proteomics, along with applications to different molecules including neuropeptides, glycans, metabolites, and lipids, while providing considerations and evaluations to each application.
Keywords: isobaric tags, isotopic labeling, mass spectrometry, protein quantitation, quantitative proteomics, systems biology
Abbreviations
- Ac‐AG
acetyl‐alanine‐glycine
- AGC
automatic gain control
- aminoxyTMT
aminoxy reactive TMT
- AQUA
absolute abundance of target peptides via signal intensity
- BASIL
boosting to amplify signal with isobaric labeling
- biotin‐HPDP
N‐[6‐(biotinamidohexyl]‐3’‐(2’‐pyridyldithio propionamide
- CETSA
cellular thermal shift assay
- CILAT
cleavable isobaric labeled affinity tag
- CMT
combinatorial isobaric mass tag
- cPILOT
combined precursor isotopic labeling and isobaric tagging
- cysTMT
cysteine TMT
- DDA
data‐dependent acquisition
- DIA
data independent acquisition
- DiART
deuterium isobaric amine reactive tag
- DiLeu
N,N‐Dimethyl Leucine
- DiLeuPMP
dimethyl leucine containing pyrazolone analogue
- DLPTP
detection of ligand‐protein interactions from thermal profiles
- EASI
easily abstractable sulfoxide‐based isobaric tag
- ETD
electron‐transfer dissociation
- glycoTMT
carbonyl‐reactive tandem mass tag
- HOTMAQ
hybrid offset‐triggered multiplex absolute quantification
- iART
isobaric aldehyde reactive tag
- IBT
the isobaric tags
- iDiLeu
isotopic DiLeu
- iodoTMT
iodoacetyl TMT
- IPTL
Isobaric peptide termini labeling
- iqPIR
isobaric quantitative protein interaction reporter
- IT
injection time
- iTRAQH
isobaric tag for absolute and relative quantification hydrazide
- LFQ
label free quantitation
- MBR
match between runs
- mdDiLeu
mass defect DiLeu
- mTRAQ
mass tags for absolute and relative quantification
- NETLOP
NHS‐ester tandem labeling in a one‐pot
- NeuCode
neutron encoding
- NHS
N‐hydroxysuccinimide
- NPARC
nonparametric analysis of response curves procedure
- phospho‐TPP
phosphoproteomics combined with TPP
- PISA
proteome integral solubility alteration
- PRM
parallel reaction monitoring
- QUANTITY
quaternary amine containing isobaric tag for glycan
- qXL‐MS
quantitative cross‐linking with mass spectrometry
- RP
resolving power
- SCoPE‐MS
single cell proteomics by mass spectrometry
- SCX
strong cation exchange
- SPS
synchronous precursor scan
- SRM
selected reaction monitoring
- SUGAR
isobaric multiplex labeling reagents for carbonyl‐containing compound
- SWATH‐MS
sequential windowed acquisition of all theoretical fragment ion mass spectra
- TMT
tandem mass tag
- TMTC
complement TMT
- TMTpro
proline‐based tandem mass tag
- TOMAHAQ
triggered by offset, multiplexed, accurate‐mass, high‐resolution and absolute quantification
- TPP
thermal proteome profiling
- TSTU
1,1,3,3‐tetramethyl‐O‐(N‐succinimidyl uronium tetrafluoroborate
- XL‐MS
cross‐linking mass spectrometry
1. INTRODUCTION
Mass spectrometry (MS) is a highly sensitive analytical tool that can be used to determine abundance levels of proteins and peptides in complex biological mixtures. Quantification results can be used to understand different biological and pathological processes. Quantitative proteomics combined with liquid chromatography‐mass spectrometry (LC‐MS) has been a leading method to quantify proteins and peptides. Existing proteome‐wide quantification methods can be classified into label‐free proteomics and label‐based proteomics [1, 2, 3, 4].
Label‐free proteomics does not involve any derivatization with chemical isotopic labeling of a sample and requires each sample to be separated in individual LC‐MS or LC/LC‐MS/MS runs [5, 6]. Relative quantification of proteins is performed either by comparison of chromatographic peak areas of peptide peak or via spectral counting of identified protein spectra [7]. Label‐free quantification (LFQ) has been found to be effective for high identification of proteins and peptides based on advancements in data processing and acquisition strategies such as delayed normalization, maximizing peptide ratio extraction, utilization of programs like MaxQuant or OpenMS [8, 9]. LFQ does inherently suffer from several limitations compared to chemical labeling methods due to variability in reproducibility between technical replicate injections. Such variation can arise from differing sample preparation between samples, variation in sample injection, and time limitations due to running only one sample at a time, thus creating long run times for larger quantities of samples. This may lead to sample degradation, thus artifactually affecting quantitation results. It also is prone to sample loss, ionization efficiency variation, and retention time shifts between runs.
Chemical isotope labeling was first introduced through the isotope‐coded affinity tag (ICAT) strategy and expanded to other methods, including stable isotope labeling by amino acids in cell culture (SILAC), dimethyl labeling, 18O labeling, and neutron encoding (NeuCode) SILAC [10, 11, 12, 13]. These methods introduce small mass differences via heavy isotopologues either at the protein or peptide level that can be distinguished from one another at the MS1 precursor spectrum. Multiple samples can be simultaneously analyzed as respectively labeled peptides can be resolved via MS. Furthermore, pooling of samples prior to LC‐MS decreases sample variation across the workflow, signal variation, and overall analysis time [6, 14, 15]. To determine abundance changes, ratios of peptide signal intensities are compared between heavy/light peptide pairs with further protein statistical evaluation. Chemical isotope labeling has limitations in multiplexing capabilities as the higher the number of labeled samples analyzed at the MS1 level; spectral complexity increases due to each analyte contributing multiple peaks [16]. Different methods generally also require a minimum mass shift of 4 Da to prevent overlap of isotopic envelopes, thereby requiring a tag with larger mass that could inherently reduce the overall identification numbers or requiring a high‐resolution mass spectrometer that can distinguish small mass differences [17, 18].
MS2‐based isobaric quantification overcomes some of the limitations to the MS1‐based quantification methods by labeling the same peptides with isobaric tags. Instead of labeling via mass difference methods such as SILAC and dimethyl labeling, isobaric tags are structures that have similar physical and chemical properties and identical masses with different isotopic configurations in each channel to label peptides from various samples. A standard design for an isobaric tag consists of three elements: (1) a reactive group to target specific functional groups or residues on peptides, (2) a balance group that contains isotopes to ensure the same overall mass of the tag based on the reporter group ion mass, and (3) a reporter group that contains discrete isotopes for different channels that allows for relative quantification between samples. Peptides of the same m/z values at the MS1 level will appear as one composite peak, which upon fragmentation will produce peptide fragment ion peaks and reporter ion peaks. Relative intensities of reporter ions are distinguished at small m/z values to not interfere with peptide backbone fragment ions at higher mass and allow for determination of relative quantitative information. The peptide fragment ion peaks are utilized to resolve amino acid sequences, which are then assigned to a protein identification based on sequences. A typical workflow for isobaric labeling is illustrated in Figure 1. Isobaric tags allow for high multiplexing capabilities that increase throughput and reproducibility due to pooling of samples prior to running on LC‐MS and decreasing variability with no increase of spectral complexity [19]. We discuss notable examples of isobaric tags in the following sections and summarize their key features in Table 1.
FIGURE 1.
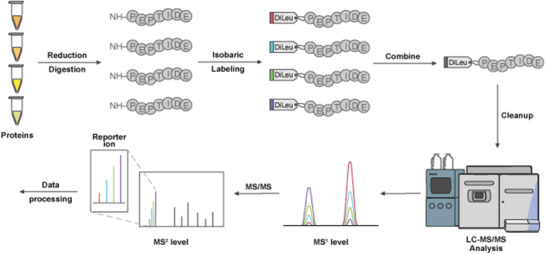
Overview of Isobaric Tag Workflow. The standard protocol involves proteins that undergo reduction, alkylation, and digestion to generate peptides. For the case of DiLeu 4‐plex, up to four samples are labeled with different channels, and are combined at equal concentrations prior to clean up steps. DiLeu‐labeled samples are analyzed via LC‐MS/MS where at the MS1 level, peptides from the pooled samples will appear as a single composite peak, which after fragmentation will show distinct reporter ion masses between m/z 114 and 118. The intensity of the reporter ion will indicate the relative amount of peptide in the mixture. All LC‐MS/MS data will undergo further data processing for downstream analysis
TABLE 1.
Advantages/disadvantages of isobaric tags comparison of major pros and cons with related references
| Isobaric tag | Multiplexing capacity | ETD‐compatible | Reporter ion yield | Deuterium | Cost (channel) | References |
|---|---|---|---|---|---|---|
| iTRAQ | 2–8 plex | ✓ | ∼$120 | [20, 21, 22, 23, 24] | ||
| TMT |
2–11 plex 2–18 plex (TMTpro) |
✓ | ∼$120 | [17, 25, 26, 27, 29] | ||
| DiLeu | 2–21 plex | High (less ratio compression) | ✓ | ∼$3 | [30, 31, 32, 33, 34, 35] | |
| DiART | 2–6 plex | High (less ratio compression) | ✓ | ∼$3 | [38, 39, 40] | |
| IBT | 2–10 plex | High (less ratio compression) | ∼$3 | [41] |
2. TMT AND iTRAQ
The first example of an isobaric tag was proposed by Thompson et al., as Tandem Mass Tag (TMT) [20]. The concept of TMT was initially designed as a duplex tag with fragmentation occurring at the proline residue on the N‐terminus. This paradigm allowed for subsequent acquisition of peptide backbone and reporter ions via collision‐induced dissociation (CID) for relative quantification by labeling at free N‐termini of peptides along with ε‐amino functions of lysine residues. The 2‐plex tag had two different generations with the major difference including a proline enhancement group in the second version but maintaining the same reporter ion group and amine reactive group at the C‐terminus. The 2‐plex was increased to a 6‐plex by reducing the size of the reporter ion to a dimethylpiperidine and balance group to be more compact, while maintaining the same N‐hydroxysuccinimide (NHS) moiety as the reactive group [21]. The 6‐plex also removed the use of deuterium ions with 13C and 15N isotopes to remove the deuterium effect causing retention time shift. TMT was further extended to a 10‐plex system with NeuCode that took advantage of neutron binding energy differences between C and N isotopes to create mass shift differences of 6.3 mDa, which can be resolved at a resolution of 50k with reporter masses ranging from m/z 126 to 131 [22, 23], The 10‐plex was also modified to accommodate electron‐transfer dissociation (ETD) via substitution of heavy carbons for heavy nitrogens. 11‐plex is currently the highest commercially available multiplexing capacity due to synthetic capabilities and cost restrictions, but it would be possible for 18‐plex capabilities if every N and C in the structure are replaced with isotopic variants.
Recently, a 16‐plex proline‐based tandem mass tag (TMTpro) was established employing an isobutyl‐proline as the reporter ion along with two β‐alanine residues on the extended balance group instead of glycine‐based tags [23]. NeuCode was incorporated into the structure's initial nine tag channels to increase to a 16‐plex with 6.3 mDa differences and 1 Da differences between reporter ions. When compared to the 10/11‐plex, TMTpro had similar identification numbers for total proteins and peptides with high labeling efficiency, but TMTpro required less time for MS3 ion injection times. At lower collisional energies, TMT10/11‐plex outperformed TMTpro with quantitative accuracy, but at higher energies, TMTpro outperformed the former [24]. Currently TMTpro has the capacity of an 18‐plex with recent developments.
Isobaric tag for absolute and relative quantification (iTRAQ) was published not long after the first example of TMT with a 4‐plex tag design [19]. Similar to TMT, iTRAQ isobarically labels peptides at N‐termini and lysine side chains, with reporter ions being distinguishable after CID fragmentation at low m/z values. iTRAQ retains the same amine reactive NHS moiety as TMT but opts for a smaller carbonyl balance group of m/z 28 to 31 and a N‐methylpiperazine reporter ion group of m/z 114 to 117 for an overall total mass of 145. It also does not utilize any deuterium ions on the structure but does place 18O on the balance group compared to TMT and TMTpro which only utilizes 13C and 15N isotopes. Eventually iTRAQ was increased to an 8‐plex system with reporter ion masses ranging from m/z 113 to 121 [25]. Comparison later showed that lower identification numbers of protein and peptides result from the 8‐plex compared to the 4‐plex iTRAQ and the 6‐plex TMT, which may be due to internal fragmentation of the tag structure [26, 27, 28]. The findings of the initial study were questioned by Pottiez et al. This study compared 8‐plex iTRAQ to the 4‐plex iTRAQ with the 8‐plex providing more accurate quantitation compared to the 4‐plex version [29]. All three of the tags (4‐plex iTRAQ, 6‐plex TMT, and 8‐plex iTRAQ) though had similar dynamic ranges and precision with peptide‐spectrum matches. Figure 2 illustrates the structures of iTRAQ, TMT 10/11‐plex and TMTpro side by side.
FIGURE 2.
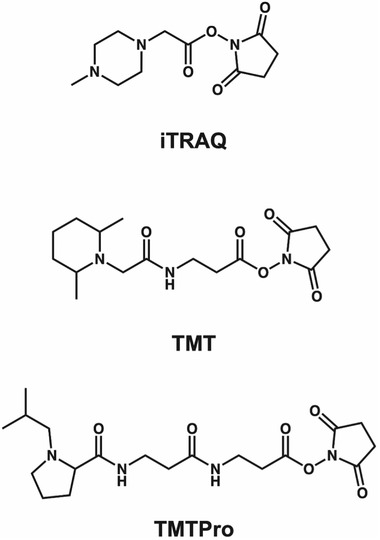
Chemical Structures of iTRAQ, TMT, and TMTPro. Each molecule consists of a reporter group, a mass balance group, and a peptide‐reactive group. iTRAQ contains a distribution of 13C, 15N, and 18O isotopes across the balance and reporter groups, while TMT and TMTPro consist of 13C and 15N only. iTRAQ consists of a N‐methylpiperazine reporter group, TMT with a dimethylpiperidine reporter and TMTPro contains an isobutyl‐proline reporter ion. Each tag carries an NHS reactive group, while mass normalization groups vary across each structure
3. DiLeu
N,N‐Dimethyl Leucine (DiLeu) is an isobaric tag developed by our lab as an alternative to the commercially available TMT and iTRAQ reagents. DiLeu was originally proposed as a 4‐plex tag by Xiang et al., for MS2 isobaric labeling with reporter ions ranging from m/z 115 to 118 and a mass shift of 145 [30]. The structure consists of a dimethylated leucine as the reporter group, a carbonyl balancing group and a triazine ester amine reactive group at the C‐terminus. One of the major differences compared to other isobaric tags is the inclusion of a triazine ester for the reactive group. DiLeu labeling has been reported to generate more intense reporter ions in comparison to iTRAQ‐labeled peptides due to the dimethylated reporter ion structure. DiLeu can also achieve better fragmentation of peptide backbones with reduced overall collision energy compared to other tags. Other benefits of DiLeu compared to TMT and iTRAQ include the lower cost to synthesize different channels, high yield percentage of tag over 80 to 90%, and do not require custom reagents to synthesize, while still maintaining high labeling efficiency and accurate quantification of proteins and peptides. A current limitation to DiLeu tagging strategy has been the relatively lower coverage due to sample loss from additional strong cation exchange (SCX) cleanup step for the need to remove excess tagging reagents. The leftover tagging reagents could also cause signal suppression of lower abundance proteins and peptides. The multiplexing capability of DiLeu was expanded upon via NeuCode to increase multiplexing from 4‐plex to 12‐plex with subtle mass differences of ∼6 mDa between different reporter ions [31]. These differences could be distinguished at baseline separation of resolving power (RP) 30K via Orbitrap HCD tandem MS, though the most accurate quantification occurred at RP of 60K and greater. Reducing the mass differences to 3 mDa via NeuCode and a stepwise mono‐methylation increased the multiplexing capabilities to a 21‐plex system as shown in Figure 3 [32]. All 21 reporter ions are resolvable at RP 60K (m/z 400) via HCD LC‐MS/MS with reporter ions ranging from m/z 115 to 118. A key difference with 21‐plex DiLeu is the implementation of a stepwise N‐monomethylation strategy to protect leucine with benzyl chloroformate to allow derivatization of a single N‐methyl group, rather than reductive dimethylation which was previously utilized. This strategy allows for incorporation of an odd number of deuterium isotopes onto the reporter ion to develop new channels. One feature of DiLeu tags is the incorporation of deuterium ions that affect retention time of labeled peptides. DiLeu, however, also takes advantage of placing deuterium proximal to the amines mitigating interactions with reversed‐phase stationary phases during LC separation.
FIGURE 3.
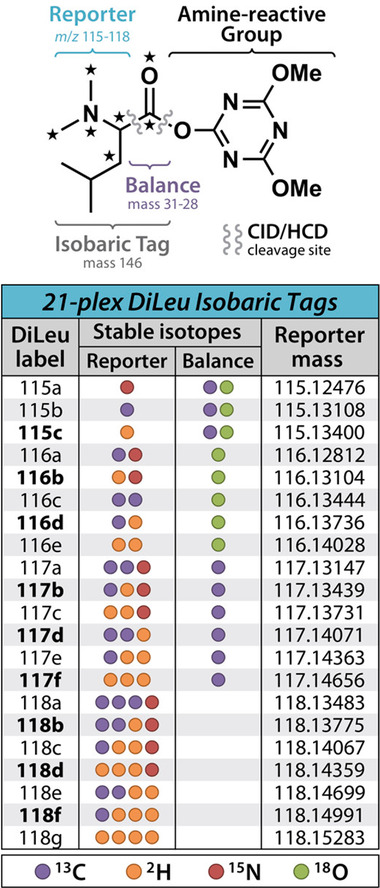
Chemical Structure of DiLeu and Multiplexing Chart. DiLeu isobaric tag structure with multiplexing capability chart illustrating the isotopic configurations across reporter ion and balance groups for each channel along with reporter ion masses
DiLeu multiplexing has also increased with incorporation of a β‐Alanine group in the balance group and 1 Da spacing between reporter groups, which increased multiplexing to an 8‐plex isobaric tag [33]. The 8‐plex DiLeu varied from the 4‐plex, 12‐plex and 21‐plex DiLeu tags due to the NHS moiety being more stable with the DiLeuAlaOH molecule [31, 34]. 8‐plex DiLeu exhibits greater retention time shifts from deuterium ions being placed on the balance group, in comparison to the more compact structures, but can be alleviated with utilization of non‐deuterated isotopologues of alanine [35]. DiAla and DiVal have also been synthesized by our lab as a variation on the dimethylated amino acid structure of DiLeu. We found that DiAla produced more abundant backbone peptide fragmentation allowing for higher protein identification and quantification numbers but resulted in lower reporter ion intensity compared to DiLeu [36, 37]. DiAla was still able to produce accurate quantitative analysis of peptides and complete labeling. The combinatorial usage of different demethylated amino acid tags may offer enhanced quantitation accuracy and protein identification coverage.
4. DiART AND IBT
Another alternative to commercially available isobaric tags is deuterium isobaric amine reactive tag (DiART) [38, 39]. DiART contains an NHS amine reactive group similar to TMT and iTRAQ, a β‐alanine balancer, and a N,N‐dimethyl leucine reporter group with m/z between 114 to 119 similar to DiLeu with the capability of 6‐plex. DiART has been compared to iTRAQ previously and reported to have a stronger reporter ion that enhanced signal to noise ratio (S/N) along with less ratio compression [40]. The isobaric tags (IBT) represent another alternative to other isobaric tags [41]. Structurally, IBT is similar to DiART, although the β‐Alanine balance group is replaced with alanine due to lower costs of isotopically labeled alanine. It is structurally identical to DiLeu, though it does not incorporate 2H and 18O isotopes to avoid deuterium chromatographic shifts. IBT also is activated in a different manner utilizing TSTU (1,1,3,3‐tetramethyl‐O‐(N‐succinimidyl) uronium tetrafluoroborate) for an NHS amine reactive moiety.
5. FACTORS/DISADVANTAGES
Isobaric tags offer many advantages compared to chemical isotopic labeling and label‐free quantification strategies through multiplexing, which increases sample throughput, quantitative accuracy, and reproducibility. Multiplexing also reduces run‐to‐run variability, instrument time, and missing values, although there are some disadvantages that must be considered. Ratio distortion is a common issue with isobaric tags when peptides of similar mass co‐isolate with each other in an isolation window that then distort the reporter ion intensity in the corresponding MS2 spectra [17]. Several methods have been developed with the intention of mitigating or removing this distortion from occurring including narrowing the precursor isolation window, addition of a gas‐phase reaction, delaying fragmentation at the apex of LC peaks, MS3 fragmentation, and estimation of redundant MS2 spectra from precursor ions [15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46].
Isotopic impurities cause contamination in reporter ion peaks due to isotopes contributing to adjacent reporter ions. Corrections must be made to account for this isotopic overlap to ensure accurate quantification. We recommend readers to view a previously published procedure that describes how to calculate peak areas that account for these contributions via reagent purity values [47].
The accuracy of quantification can also be affected by a mass detector's saturation point [48, 49]. Reporter ion intensities have upper intensity limits and intensities may be underestimated due to saturation effects of the detector, which is instrument dependent. Intensity can be influenced by increasing MS/MS acquisition while diminishing the number of possible identifications and also by spiking in samples of known concentration to confirm expected protein and peptide ratios [50].
Another important concern is that variability can occur from batch to batch in different isobaric tags. This was apparent when a large‐scale study was conducted on TMT isobaric labeling that identified significant missing values of proteins across different batches of tags, influencing the high precision of quantitation [51]. Although normalization methods could mitigate batch‐to‐batch variations, it is important to remember that variations could potentially occur from any isobaric tag, whether commercial or synthesized by a laboratory in‐house.
6. UNCONVENTIONAL LABELING METHODS
Besides the above‐mentioned tags, there are other novel isobaric tags that have been developed that could be called unconventional compared to the standard ideology of reporter, balance, and amine reactive groups. One such example is the combinatorial isobaric mass tag (CMT) [52]. The concept of CMT is that every isobaric tag will produce two reporter ions from fragmentation that are independent from one another with m/z values ranging from 126 to 128 and 172 to 175, respectively. This tag has the capacity of a 28‐plex utilizing mass shift differences of ∼6 mDa, even though the published results showed a 6‐plex, perhaps due to a constraint of RP or due to the amount of time and resources necessary to synthesize a set of 28‐plex tags.
Another set of examples involves peptide backbone fragmentation, where to circumvent the issue of reporter ion distortion, specific fragment ions are employed from the backbone to mitigate this effect. The first reported method was published in 2009 by Koehler et al., known as isobaric peptide termini labeling (IPTL) [53]. Derivatization occurs at lysine residues followed by succinylation with variants of succinic anhydride, which will generate different fragment ions that can be compared via b and y ion intensities from proteins and peptides. IPTL has since been expanded to several new strategies [54, 55, 56, 57]. Initial IPTL strategies are limited compared to labeling reagents due to minimal capacity to multiplex and complexity of MS2 spectra from multiplets of fragment ions. Since the initial report, new methods have been developed that increase multiplexing to a 7‐plex system and minimize spectral complexity via data software optimization [15, 42, 58–62].
Recent developments include chemical tagging strategy that will contain complete peptide sequences coupled to the balance and reporter ion groups referred to “peptide‐coupled reporter‐ion based quantification.” Instead of producing the same set of reporter ions for various peptides after fragmentation, reporter ions are peptide‐specific. The first example of this was complement TMT (TMTC) [15]. Designed as an alternative to replacing any MS3 scans or additional purification steps, TMTC relatively quantifies differences in samples from the complementary TMT fragment ion cluster rather than the reporter ions of the same masses. This method is limited due to the isotopic envelope of reporter ions from a precursor isolation window of 2 Th, and overall efficiency due to peptide ion charge state and sequence. To reduce these problems, a modified version was developed called TMTC+ that reduces the isolation window down to 0.4 Th, which also mitigates the long instrumental duty cycles from TMTC [42].
Another peptide‐coupled reporter ion tag was developed in 2018 by Winter et al., called easily abstractable sulfoxide‐based isobaric (EASI) tag [63]. The tag design includes an NHS amine reactive group, a balance group, and a neutral loss group that fragments at the sulfoxide group to enhance dissociation efficiency. This combined with an asymmetric isolation window to suppress adjacent peaks and lower collision energy needed for fragmentation makes it an attractive replacement for accurate and sensitive quantification as a 6‐plex. Similar to EASI tag, the acetyl‐alanine‐glycine (Ac‐AG) tag was developed as a new peptide‐coupled reporter‐ion‐based tag with improvement via ionization efficiency but is also versatile for DIA [64].
7. INCREASE IN MULTIPLEXING CAPABILITIES
Hybrid methods have been developed that combine MS1 and MS2 based quantification from isotopic mass differences and isobaric labeling to increase throughput and multiplexing capabilities. The initial idea of “hyperplexing” was developed by coupling 6‐plex TMT with triplex SILAC to achieve 18‐plex quantification [65, 66]. A recent method combined 16‐plex TMTpro along with 3‐plex mass tags for absolute and relative quantification (mTRAQ) to achieve 48‐plex capabilities for a novel NHS‐ester tandem labeling in a one‐pot (NETLOP) workflow [67]. A targeted proteomics approach has also been developed by combining 3 distinct TMT reagents and 6‐plex TMT tags with three mass variation of targeted peptides to achieve 54‐plex quantification [68]. Another technique made called combined precursor isotopic labeling and isobaric tagging (cPILOT) combining duplex stable isotope dimethyl labeling and 6‐plex TMT labeling permitting 12‐plex quantification of N‐termini and lysine residues [69]. We developed a similar approach known as DiLeu cPILOT combining stable isotope dimethyl labeling with 12‐plex DiLeu isobaric tags for 24‐plex quantification utilizing a synchronous precursor scan (SPS)‐MS3 acquisition method [70]. For more in‐depth discussions of higher order multiplexing, we refer the reader to an excellent review published elsewhere [71].
8. TARGETED ANALYSIS APPROACHES
Absolute quantification is generally accomplished with targeted mass spectrometry with methods such as selected reaction monitoring (SRM) and parallel reaction monitoring (PRM) [72, 73, 74, 75]. These methods combine stable‐isotope encoded peptides standards that are spiked in at known concentrations to determine absolute abundance of target peptides via signal intensity (AQUA) [76, 77]. A method known as triggered by offset, multiplexed, accurate‐mass, high‐resolution, and absolute quantification (TOMAHAQ) was developed with synthetic trigger peptides being utilized as an offset mass to trigger quantification [78]. These methods do depend on single point calibration, however, increasing the degree of potential inaccuracy due to the wide quantitative span of peptides across orders of magnitude and rely on expensive isotopically encoded peptide standards and isotopic labeling reagents. Recently our lab has developed a strategy similar to TOMAHAQ called hybrid offset‐triggered multiplex absolute quantification (HOTMAQ) strategy that combined isobaric 12‐plex DiLeu tags with 5‐plex isotopic DiLeu (iDiLeu) tags that utilize 3 Da mass differences between channels to enable accurate absolute quantification or targeted peptides in higher throughput [79, 80].
9. IMPROVEMENTS FOR IDENTIFICATION OF LOWER ABUNDANCE SPECIES
Efforts have been made to improve on identification of lower abundance peptides and proteins. Due to poor MS signal intensity and the suppression from higher abundant species, a recent influx of new methods has been developed to mitigate these effects [81, 82, 83]. Several of these techniques utilize match between runs (MBR) to align retention time measurements of lower abundant peptides to their corresponding m/z to a library or reference run to yield new identification. A recent method called BoxCar combines MBR with a new acquisition method to boost the precursor MS1 signal with a library to identify over 10 000+ proteins in a 100 min gradient LC‐MS/MS analysis [84]. One strategy that has caught the attention of the field is boosting to amplify signal with isobaric labeling (BASIL) [85]. This approach works by using isobaric tags with one of the channels being significantly higher in sample amount compared to other channels. This strategy enhances detectability of precursor signal intensities of peptides and identifiability from fragment spectra. The workflow for BASIL is illustrated in Figure 4.
FIGURE 4.
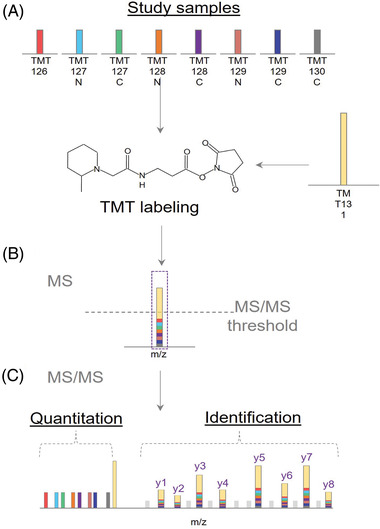
Illustration of BASIL Strategy. Adapted from Yi. L et al., (2019) with permission. (A) Study samples are labeled with a smaller amount of TMT tag, while the boosting sample is labeled with a larger amount of TMT tag. (B) Peptides will appear as a single composite peak at the MS1 level as a sum of all intensities from the study and boosting samples. (C) Tandem MS fragmentation of peptide backbones reveal the intensities of the TMT reporter ions along with quantification of the study samples
10. POST‐TRANSLATIONAL MODIFICATION (PTM) SPECIFIC TAGS
Isobaric tagging has been widely utilized in PTM quantitative analysis. For example, phosphoproteomics coupled with high‐specificity enrichments has been shown to be a well‐studied PTM with the use of isobaric tags [86]. In our lab, we have also developed a strategy to utilize boosting channels in 12‐plex DiLeu to enhance the analysis of intact glycoproteomics (under review). Besides isobaric tags targeting amine groups, other tags have been developed that are specifically designed to target sub‐classes of proteins and peptides. Initial concepts began with targeting cysteine residues with the cleavable isobaric labeled affinity tag (CILAT) [87]. CILAT takes advantage of an isobaric tag structure with affinity enrichment that utilizes a biotin affinity tag and acid‐labile linker. Another tag that targets cysteine residues is called cysteine TMT (cysTMT) [88, 89]. cysTMT was designed to be an alternative N‐[6‐(biotinamido)hexyl]‐3’‐(2’‐pyridyldithio) propionamide (biotin‐HPDP) to detect S‐nitrosylation with a similar Cys reactive group, a smaller balance group and a mass reporter. Subsequent development of iodoacetyl TMT (iodoTMT) has since replaced cysTMT with the principle remaining constant of quantifying protein S‐nitrosylation [90]. Compared to cysTMT, iodoTMT irreversibly labels Cys thiols that improves overall labeling efficiency.
Several tags have been developed to detect carbonyl groups for the purpose of quantification. The first of these tags developed was the carbonyl‐reactive tandem mass tag (glycoTMT) with a broader application of the tag being applied with aminoxy‐reactive TMT (aminoxyTMT) [91, 92]. Another tag known as quaternary amine containing isobaric tag for glycan (QUANTITY) was developed for quantification of N‐glycosylation [93]. Isobaric aldehyde reactive tag (iART) was created by Yang et al., as another alternative for glycan analysis [94]. Further development led to isobaric tag for absolute and relative quantification hydrazide (iTRAQH) [95]. Our lab has developed an alternative for analysis of carbonyl groups with isobaric multiplex labeling reagents for carbonyl‐containing compound (SUGAR) tags [96]. SUGAR features efficient multistep synthesis at a lower cost with high labeling efficiency of N‐glycans. Recently our lab developed the first isobaric tag aimed to quantify O‐glycans in the form of 4‐plex dimethyl leucine containing pyrazolone analogue (DiLeuPMP) [97].
11. APPLICATIONS OF ISOBARIC TAGS
11.1. Biomarker discovery
Quantitative proteomics has been combined in several facets to biomarker discovery in a variety of different diseases [98, 99]. One example used iTRAQ 8‐plex tags to label serum protein digests and identify biomarkers for acute myocardial infarction [100]. iTRAQ 8‐plex was also utilized to quantify targets for breast cancer from tissue samples [101]. Several examples have been used to identify cancer related targets including biomarkers for tumor metastasis and dysregulated proteins found in leukemic stem cells [102, 103, 104]. TMT has also been integrated into biomarker discovery by Tokuoka et al., to discover lipid biomarker candidates related to Alzheimer's disease [105]. A point of emphasis in biomarker studies to consider is potential variance when it comes to the type of sample being utilized and the type of quantification with tags. Isobaric tags can heavily underestimate fold changes of different disease states and, given that biomarkers need extensive orthogonal methods for verification and validation, this can be detrimental for potential false positives and negatives. Several strategies we have mentioned previously can be implemented to mitigate these effects. It is necessary, however, to have targeted approaches and absolute quantification methods for validation and verification of biomarkers.
11.2. Study of neuropeptides and isobaric tags
Neuropeptides comprise a large class of signaling molecules with prominent roles in various body functions including growth and metabolism [106]. Neuropeptides are very challenging to analyze due to their variety in functions, sequences, and sizes [107, 108, 109, 110]. They are also difficult to analyze due to their trace level in vivo. Several examples show it is possible to quantify neuropeptides at the MS1 level, however it is necessary to have MS2 quantification strategies to mitigate the spectral complexity at the precursor level [111, 112, 113, 114]. In one example, we quantified neuropeptides impacted from the gut microbiome with label‐free quantification and 10‐plex DiLeu isobaric tags with 282 labeled species exhibiting changes in regulation [114]. Another work performed relative quantification of neuropeptides from American lobsters using 4‐plex isobaric DiLeu tags at different stages of lobster brain development [115]. Most recently Sauer & Li performed relative quantification of neuropeptides in response to copper toxicity with 4‐plex DiLeu tags [116].
11.3. Isobaric tags and metabolomics
Metabolomics is a rapidly growing field of research in omics‐based studies. Metabolites are small molecules that are either intermediates or products of metabolic processes with typical molecular weights lower than 2000 Da. Challenges of metabolomics include the wide range of biological variance in metabolite levels and the structural variety of metabolite molecules that prevents the existence of a universal analysis method. Furthermore, some metabolites are very labile and may degrade quickly, thus making their analyses more difficult [117]. Typically, metabolomics can be studied in a targeted manner with MRM via tandem MS for an increase of sensitivity and selectivity of metabolites for absolute quantification, or from an untargeted approach for global detection of metabolites via relative quantification [118]. Several examples show absolute and relative quantification of metabolites utilizing isobaric tags [118, 119, 120, 121]. A study by Hao et al., featured the first use of mass defect DiLeu (mdDiLeu) tags to study metabolites from pancreatic cancer cells [121]. The 12‐plex DiLeu tags were also utilized to quantify urinary metabolites [122]. Furthermore, a software tool called Metandem has been developed to facilitate MS‐based isobaric labeling metabolomics [123].
11.4. Thermal proteome profiling with isobaric tags
Drug‐targeting methods to understand protein‐ligand interactions are becoming increasingly popular. Targeted approaches have proteins or peptides that are either activated or inhibited from chemical processes to measure different activities of large arrays of proteins at once. This is different compared to typical phenotypic approaches that rely on observable traits [124, 125, 126]. Approaches range from affinity purification, activity profiling, protein painting, studying of stabilities and enrichment strategies. [125, 126, 127, 128]. Many of these approaches are limited due to requiring chemical modifications and are limited to cell extracts, making them impractical for in vivo studies.
One approach that would not require chemical modifications is based on ligand binding. An increasingly popular method based on biophysical alterations to study protein abundance changes within recent years is from the cellular thermal shift assay (CETSA) [129], based on the principle that ligand binding will affect protein thermal stability. This method treats cells either with a control or drug of interest and perturbs them with subtle temperature differences via a thermal cycler to influence conformational state changes of protein‐ligand complexes. A thermal shift curve can then be built to distinguish differences in regulation of proteins based on drug‐ligand interactions.
TPP is based off CETSA where cells are treated either in vivo or extracted with subsequent heat perturbation and then labeled with isobaric tags for multiplexed quantitative mass spectrometry as illustrated in Figure 5 [130] Thermal shift curves are created from normalization of reporter ion intensities and complex programming to determine protein regulation differences. This method offers an unbiased approach towards studying drug target interactions in a high‐throughput manner. Nonetheless, it suffers from limited information related to the binding site.
FIGURE 5.
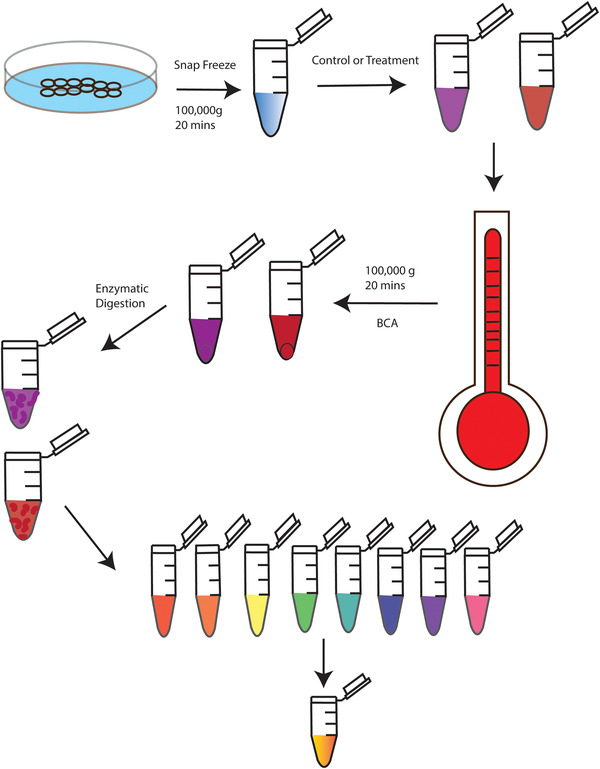
Thermal Proteome Profiling Workflow. Cells are snap frozen via liquid nitrogen for cell extraction, followed by ultra‐centrifugation. The supernatant is then subjected to treatment either with a control or drug with further exposure via a thermal cycler at varying temperatures. The heated samples are ultracentrifuged again with enzymatic digestion of the supernatant. Once peptides are formed, they are labeled with isobaric tags and combined at equal concentrations prior to cleanup steps and LC‐MS/MS analysis
Since TPP's inception, there have been several new methods developed to improve and expand its applicability [131]. TPP has been expanded to monitor membrane proteins, phosphorylated proteins and peptides via phosphoproteomics combined with TPP (phospho‐TPP), bacteria, plants, plasmodium, yeast, viruses, tissue samples, and plasma membrane proteins [132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145]. Other protocol‐based methods include a two‐dimensional approach that studies protein abundance and regulation changes simultaneously, a proteome integral solubility alteration (PISA) to increase throughput and reduction in data analysis, and utilization of a vacuum manifold to increase throughput [146, 147]. Data analysis approaches have been developed as well including nonparametric analysis of response curves procedure (NPARC) and detection of ligand‐protein interactions from thermal profiles (DLPTP) [148, 149].
11.5. Cross‐linking approaches
Cross‐linking mass spectrometry (XL‐MS) is a powerful tool to provide spatial details about proteins and protein‐protein interaction information complementary to current predominant techniques for protein structure determination, such as X‐ray crystallography. The advantages to this method include a requirement of small sample sizes, application to highly complex samples, and providing data on dynamic conformations and transient states in the solution [150].
The concept of quantitative cross‐linking with mass spectrometry (qXL‐MS) has emerged recently enabling the assessment of changes in proteins under different conditions. As in other quantitative proteomics studies, the application of isobaric labeling would be a major player in permitting large‐scale comparative analyses. An analytical platform proposed by Yu et al., involved an MS‐cleavable crosslinker DSSO and labeled cross‐linked peptides after enzymatic digestion [151]. In the MS2 scan, S‐C bonds in DSSO crosslinker were cleaved preferentially by CID to generate single peptide chains that still contain TMT tags. The peptide sequencing and quantification were then done at the MS3 level. The coupling of cleavable crosslinker and isobaric labeling not only facilitates the identification of cross‐linked peptides but also enables comparative analysis. An accurate quantitative result has been achieved to demonstrate the compatibility of isobaric labeling in cross‐linking. However, despite the use of TMT tags, only binary comparison has been conducted in this study where the advantage of the multiplexed capacity of isobaric tags was not presented.
Recently, a novel crosslinker has been introduced called isobaric quantitative protein interaction reporter (iqPIR), where stable isotopes were incorporated in different regions of crosslinker structures, eliminating the need for additional isobaric tagging [152]. Upon fragmentation, reporter ions were generated at the MS2 level with high sensitivity along with complementary fragment ions to provide additional quantitative information. This approach allows samples to be pooled once crosslinking has been done, therefore minimizing the variations which might be introduced during sample preparation. With the design using amino acids as building blocks, iqPIR can offer a higher multiplexing capacity, and has been extended to 6‐plex iqPIR recently.
11.6. Single‐cell analysis
The study of single cells enabling elucidation of many fundamental cellular processes and investigation of biological systems with single‐cell resolution offers tremendous value in biomedical research [153]. MS methods capable of analyzing thousands of proteins in a single experiment provide a promising tool towards this goal. However, despite the high sensitivity of MS, the losses arising from sample preparation and delivery of proteins to MS analyzers confined their development of single‐cell analysis [154]. Recently, advances in many aspects of workflows have favored analysis of small samples and made the profiling of considerable number of proteins from a single mammalian cell possible. Among those advances, isobaric labeling is one of the innovative strategies to alleviate several common difficulties in the single‐cell analysis field [155].
The first application of isobaric labeling to mammalian single‐cell analysis was published in 2018 [156]. Single Cell ProtEomics by Mass Spectrometry (SCoPE‐MS) strategy labeled single‐cell samples with distinct channels of TMT reagents and pooled them with carrier channels which labeled 200 cells with one of isobaric channels. As shown in Figure 6, the key feature of a second‐generation method (ScoPE2) is the isobaric carrier concept where different channels of TMT tags are employed to label proteins from a carrier sample, a reference sample, and single cells, with defined ratios [157]. For example, sample losses occurring throughout sample preparation due to nonspecific absorption over large surfaces have been reported to have a substantial impact on small samples, while the introduction of carrier samples significantly disperse the surface adhesion from single‐cell samples. Moreover, the signals of peptides that might fall below the detection limits would be detected with the enhancement from carrier samples and thus trigger MS2 fragmentation for sequencing peptides.
FIGURE 6.
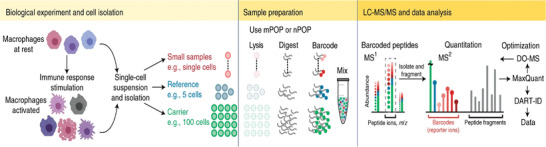
Single cell quantitative proteomics workflow via isobaric tagging. Adapted from Petelski A. et al., (2021) with permission. Individual cells are isolated from single‐cell suspensions, which are then lysed into proteins, digested into peptide chains, and labeled with isobaric tags for LC‐MS/MS analysis with subsequent data processing
This strategy has been further improved by Tsai et al., to achieve reliable and precise quantitation and optimal experimental conditions by evaluating different carrier‐to‐sample ratios and MS acquisition settings, such as automatic gain control (AGC) and injection time (IT) [158]. However, there are some potential pitfalls for using isobaric labeling in single‐cell analysis. According to the previous reports, experimental designs need to be carefully optimized for each instrument and sample type to achieve reliable quantification and maximum protein identification. Since additional fractionations and MS3 are not favorable to small samples, ratio compression presents a more serious problem for single‐cell analysis than bulk proteomics leading to skewed measurement. The choices of carrier samples that determine the sets of peptides to be enhanced and detected are also crucial for getting meaningful results.
11.7. DIA
DIA features higher reproducibility, fewer missing values, and better quantitative comparisons between runs compared to DDA [159]. The execution of fragmentation is not based on signals in MS1 scan, so DIA is more likely to detect proteins in low abundance. To achieve higher sample throughput, the applicability of isobaric labeling has been studied. Since DIA co‐isolates multiple precursor ions in a wider m/z window for MS2 fragmentation, all reporter ions from different precursor ions will be present in the same spectrum, meaning that those reporter ions no longer represent the abundance of specific peptides. Therefore, the unconventional mass tags that use NeuCode or produce complementary reporter ions are often employed for DIA analysis. We demonstrated a strategy using mdDiLeu to analyze human cerebrospinal fluid for biomarker discovery using DIA mode [160]. As shown in Figure 7, to ensure the labeled analytes to be isolated in the same DIA window and minimize MS1 spectral complexity, millidalton differences between channels were introduced to analytes, by which precursor ions remained to exhibit single ion peaks using a lower RP in the MS1 scan. Upon HCD fragmentation, multiplets of b‐ and y‐ions would be generated and detected under a higher RP for multiplexed quantification. This method showed a higher reproducibility and more quantifiable proteins compared to DDA while maintaining high quantitative accuracy.
FIGURE 7.
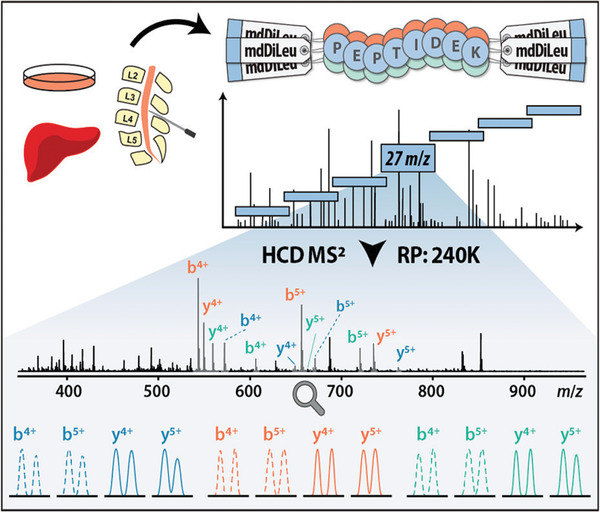
mdDiLeu DIA Workflow. Adapted from Zhong X. et al, (2020) with permission. Either cerebral spinal fluid or samples were labeled with mdDiLeu tags, with LC‐MS/MS analysis using a m/z 27 scan range for DIA isolation windows. These were analyzed using HCD tandem MS with further data processing to illustrate the different b and y ions for differentially labeled samples
The use of complementary reporter ions in DIA mode has been proposed to circumvent ratio distortion resulting from co‐isolation [18]. Although this method seems compatible with DIA mode, additional fragments originating from tag labeling further complicate DIA MS2 spectra which are already convoluted. The interpretation of spectra will become more challenging. On top of that, compared to high‐throughput methods without labeling empowered by simplifying sample complexities or comprehensive spectral libraries, the elevated spectral complexities and the requirement of ultra‐high RPs, which increases instrument cycle time, hinder the widespread use of isobaric labeling methods coupled with DIA [161, 162].
11.8. Top‐down proteomics
In contrast to bottom‐up proteomics, top‐down methods analyze intact proteins without using proteolytic digestion. This strategy preserves protein modifications to a higher extent and characterizes proteoforms that might be unexpected or not detectable using bottom‐up approaches. Owing to the nature of intact proteins, the development of chemical labeling encounters many challenges, including protein precipitation, incomplete labeling, and inherently lower signal‐to‐noise ratio [163]. Indeed, the earliest report of isobaric labeling of intact proteins from 2007 described the derivatization of standard proteins using iTRAQ, followed by gel electrophoresis separation and in‐gel tryptic digestion before MS analysis [164]. A later report took a similar approach using serum from patients who later developed pancreatic ductal adenocarcinoma. Following immunodepletion, serum proteins were labeled with 6‐plex TMT, separated using gel electrophoresis, then enzymatically digested [165].
Further pilot studies have demonstrated proof‐of‐principle for labeling‐based top‐down proteomics (without subsequent enzymatic digestion) using TMT reagents, albeit partially suffering from the previously mentioned limitations [166, 167]. IodoTMT reagents were applied to complex samples by Winkels et al. [168]. The use of thiol‐reactive TMT instead of amine‐reactive reagents, which is more sensitive to aqueous solutions, allowed a lower amount of organic solvent for labeling conditions, so that the depletion of larger proteins was not necessary to avoid protein precipitation. IodoTMT showed quantitative derivatization and accurate quantification in a 6‐plex analysis. This workflow performed isobaric labeling right after protein reduction, which minimized the variation between samples introduced during sample preparation. Although this method was restricted to cysteine‐containing proteoforms, simple and flexible experimental designs showed promising applicability for multiplexed top‐down proteomics.
12. CONCLUSIONS AND FUTURE DIRECTIONS
Relative quantitation via isobaric labeling is a powerful technique for quantitative proteomics studies because of the higher throughput and reproducibility from high multiplexing capabilities while mitigating variation from run to run. In this review, we have discussed a number of isobaric tagging reagents and their multiplexing and quantitation capabilities. We further explored a plethora of applications that isobaric tags can be applied to along with different molecules that can be studied. In recent years, the number of applications of isobaric labeling in MS‐based studies has skyrocketed. In parallel, the number of publications describing new labeling reagents and strategies, even beyond just proteomics, has also increased.
Based on recent trends in the field, we speculate that isobaric tagging technology will continue to grow with increasing multiplexing capabilities. The commercial TMTpro 16‐plex reagent set is currently available, though recent reports have described multiplexing from 21‐plex up to 48‐plex [32]. One limitation to increasing multiplexing capability is the necessity of higher MS2 resolution for ensuring quantitative accuracy, which could result in lower identification rates due to increased duty cycle. This limitation can be overcome with improving scanning speeds in newer high‐resolution instruments. For further increases to multiplexing, development of complementary ion tagging strategies will provide promising routes to higher sensitivity and numbers of quantified proteins in complex samples.
Concerning future isobaric labeling applications, we speculate that as single‐cell analyses become more widespread, so will isobaric labeling in these analyses. The advantages of sample pooling from multiplexing make quantitative comparisons between single cells more accessible [169, 170]. In studies of cancer for example, quantitative interrogation of cell‐to‐cell heterogeneity using isobaric labeling can elucidate changes that are masked when performing analyses from homogenized, bulk samples.
Quantitative proteomic information derived from studies using isobaric labeling have proven useful in guiding more targeted studies toward protein functions and disease pathologies. Given that technologies and strategies for isobaric labeling continue to evolve, it is certain that isobaric labeling will continue to remain at the forefront of quantitative proteomics.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
Preparation of this manuscript was funded in part by the National Institutes of Health through grants R01DK071801, RF1 AG052324, and P41GM108538. Lingjun Li acknowledges a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin‐Madison School of Pharmacy.
Sivanich, M. K. , Gu, T.‐J. , Tabang, D. N. , & Li, L. (2022). Recent advances in isobaric labeling and applications in quantitative proteomics. Proteomics, 22, e2100256. 10.1002/pmic.202100256
Michael K. Sivanich and Ting‐Jia Gu contributed equally to this work.
DATA AVAILABILITY STATEMENT
This is an invited review and there is no original data.
REFERENCES
- 1. Rauniyar, N. , & Yates, J. R. (2014). Isobaric labeling‐based relative quantification in shotgun proteomics. Journal of Proteome Research, 13(12), 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asara, J. M. , Christofk, H. R. , Freimark, L. M. , & Cantley, L. C. (2008). A label‐free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics, 8, 994–999. [DOI] [PubMed] [Google Scholar]
- 3. Bondarenko, P. V. , Chelius, D. , & Shaler, T. A. (2002). Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed‐phase liquid chromatography−tandem mass spectrometry. Analytical Chemistry, 74(18), 4741–4749. [DOI] [PubMed] [Google Scholar]
- 4. Gygi, S. P. , Rist, B. , Gerber, S. A. , Turecek, F. , Gelb, M. H. , & Aebersold, R. (1999). Quantitative analysis of complex protein mixtures using isotope‐coded affinity tags, Nature Biotechnology, 17, 994–999. [DOI] [PubMed] [Google Scholar]
- 5. Zhu, W. , Smith, J. W. , & Huang, C.‐M. (2010). Mass spectrometry‐based label‐free quantitative proteomics. Journal of Biomedical Biotechnology, 2010, 840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bantscheff, M. , Schirle, M. , Sweetman, G. , Rick, J. , & Kuster, B. (2007). Quantitative mass spectrometry in proteomics: A critical review. Analytical and Bioanalytical Chemistry, 389, 1017–1031. [DOI] [PubMed] [Google Scholar]
- 7. Liu, H. , Sadygov, R. G. , & Yates, J. R. (2004). A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Analytical Chemistry, 76, 4193–4201. [DOI] [PubMed] [Google Scholar]
- 8. Cox, J. , Hein, M. Y. , Luber, C. A. , Paron, I. , Nagaraj, N. , & Mann, M. (2014). Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed maxLFQ, Termed MaxLFQ. Molecular Cellular Proteomics, 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suits, F. , Hoekman, B. , Rosenling, T. , Bischoff, R. , & Horvatovich, P. (2011). Threshold‐avoiding proteomics pipeline. Analytical Chemistry, 83, 7786–7794. [DOI] [PubMed] [Google Scholar]
- 10. Ong, S.‐E. , Blagoev, B. , Kratchmarova, I. , Kristensen, D. B. , Steen, H. , Pandey, A. , & Mann, M. (2002). Stable isotope labeling by amino acids in cell culture, silac, as a simple and accurate approach to expression proteomics. Molecular Cellular Proteomics, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- 11. Hsu, J.‐L. , Huang, S.‐Y. , Chow, N.‐H. , & Chen, S.‐H. (2003). Stable‐isotope dimethyl labeling for quantitative proteomics. Analytical Chemistry, 75, 6843–6852. [DOI] [PubMed] [Google Scholar]
- 12. Yao, X. , Freas, A. , Ramirez, J. , Demirev, P. A. , & Fenselau, C. (2001). Proteolytic 180 labeling for comparative proteomics: model studies with two serotypes of adenovirus. Analytical Chemistry, 73, 2836–2842. [DOI] [PubMed] [Google Scholar]
- 13. Rose, C. M. , Merrill, A. E. , Bailey, D. J. , Hebert, A. S. , Westphall, M. S. , & Coon, J. J. (2013). Neutron encoded labeling for peptide identification. Analytical Chemistry, 85, 5129–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chahrour, O. , Cobice, D. , & Malone, J. (2015). Stable isotope labelling methods in mass spectrometry‐based quantitative proteomics. Journal of Pharmaceutical and Biomedical Analysis, 113, 2–20. [DOI] [PubMed] [Google Scholar]
- 15. Sonnett, M. , Yeung, E. , & Wühr, M. (2018). Accurate, sensitive, and precise multiplexed proteomics using the complement reporter ion cluster. Analytical Chemistry, 90, 5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boersema, P. J. , Aye, T. T. , Van Veen, T. A. B. , Heck, A. J. R. , & Mohammed, S. (2008). Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics, 8, 4624–4632. [DOI] [PubMed] [Google Scholar]
- 17. Ow, S. Y. , Salim, M. , Noirel, J. , Evans, C. , Rehman, I. , & Wright, P. C. (2009). iTRAQ underestimation in simple and complex mixtures: “The Good, the Bad and the Ugly”. Journal of Proteome Research, 8, 5347–5355. [DOI] [PubMed] [Google Scholar]
- 18. Pappireddi, N. , Martin, L. , & Wühr, M. (2019). A review on quantitative multiplexed proteomics. Chembiochem, 20, 1210–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ross, P. L. , Huang, Y. N. , Marchese, J. N. , Williamson, B. , Parker, K. , Hattan, S. , Khainovski, N. , Pillai, S. , Dey, S. , Daniels, S. , Purkayastha, S. , Juhasz, P. , Martin, S. , Bartlet‐Jones, M. , He, F. , Jacobson, A. , & Pappin, D. J. (2004). Multiplexed protein quantitation in Saccharomyces cerevisiae using amine‐reactive isobaric tagging reagents. Molecular and Cellular Proteomics, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- 20. Thompson, A. , Schäfer, J. , Kuhn, K. , Kienle, S. , Schwarz, J. , Schmidt, G. , Neumann, T. , & Hamon, C. (2003). Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- 21. Dayon, L. , Hainard, A. , Licker, V. , Turck, N. , Kuhn, K. , Hochstrasser, D. F. , Burkhard, P. R. , & Sanchez, J.‐C. (2008). Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6‐plex isobaric tags. Analytical Chemistry, 80, 2921–2931. [DOI] [PubMed] [Google Scholar]
- 22. Mcalister, G. C. , Huttlin, E. L. , Haas, W. , Ting, L. , Jedrychowski, M. P. , Rogers, J. C. , Kuhn, K. , Pike, I. , Grothe, R. A. , Blethrow, J. D. , & Gygi, S. P. (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Analytical Chemistry, 84, 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thompson, A. , Wölmer, N. , Koncarevic, S. , Selzer, S. , Böhm, G. , Legner, H. , Schmid, P. , Kienle, S. , Penning, P. , Höhle, C. , Berfelde, A. , Martinez‐Pinna, R. , Farztdinov, V. , Jung, S. , Kuhn, K. , & Pike, I. (2019). TMTpro: Design, synthesis, and initial evaluation of a proline‐based isobaric 16‐Plex tandem mass tag reagent set. Analytical Chemistry, 91, 15941–15950. [DOI] [PubMed] [Google Scholar]
- 24. Li, J. , Cai, Z. , Bomgarden, R. D. , Pike, I. , Kuhn, K. , Rogers, J. C. , Roberts, T. M. , Gygi, S. P. , & Paulo, J. A. (2021). TMTpro‐18plex: The expanded and complete set of TMTpro reagents for sample multiplexing. Journal of Proteome Research, 20, 2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choe, L. , D'ascenzo, M. , Relkin, N. R. , Pappin, D. , Ross, P. , Williamson, B. , Guertin, S. , Pribil, P. , & Lee, K. H. (2008). 8‐Plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer's disease. Proteomics, 7, 3651–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mertins, P. , Udeshi, N. D. , Clauser, K. R. , Mani, D. R. , Patel, J. , Ong, S.‐E. , Jaffe, J. D. , & Carr, S. A. (2012). iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Molecular and Cellular Proteomics, 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pichler, P. , Köcher, T. , Holzmann, J. , Mazanek, M. , Taus, T. , Ammerer, G. , & Mechtler, K. (2010). Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4‐Plex compared to TMT 6‐Plex and iTRAQ 8‐Plex on LTQ Orbitrap. Analytical Chemistry, 82, 6549–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Werner, T. , Becher, I. , Sweetman, G. , Doce, C. , Savitski, M. M. , & Bantscheff, M. (2012). High‐resolution enabled TMT 8‐plexing. Analytical Chemistry, 84, 7188–7194. [DOI] [PubMed] [Google Scholar]
- 29. Pottiez, G. , Wiederin, J. , Fox, H. S. , & Ciborowski, P. (2012). Comparison of 4‐plex to 8‐plex iTRAQ quantitative measurements of proteins in human plasma samples. Journal of Proteome Research, 11, 3774–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiang, F. , Ye, H. , Chen, R. , Fu, Q. , & et Li, L. (2010). N, N‐DiMethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Analytical Chemistry, 82, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frost, D. C. , Greer, T. , & Li, L. (2015). High‐resolution enabled 12‐Plex DiLeu isobaric tags for quantitative proteomics. Analytical Chemistry, 87(3), 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frost, D. C. , Feng, Y. , & Li, L. (2020). 21‐plex DiLeu isobaric tags for high‐throughput quantitative proteomics. Analytical Chemistry, 92, 8228–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frost, D. C. , Greer, T. , Xiang, F. , Liang, Z. , & Li, L. (2015). Development and characterization of novel 8‐plex DiLeu isobaric labels for quantitative proteomics and peptidomics. Rapid Communications in Mass Spectrometry, 29, 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang, R. , Sioma, C. S. , Wang, S. , & Regnier, F. E. (2001). Fractionation of isotopically labeled peptides in quantitative proteomics. Analytical Chemistry, 73, 5142–5149. [DOI] [PubMed] [Google Scholar]
- 35. Yu, Q. , Shi, X. , Greer, T. , Lietz, C. B. , Kent, K. C , & Li, L. (2016). Evaluation and application of dimethylated amino acids as isobaric tags for quantitative proteomics of the TGF‐Î2/Smad3 signaling pathway. Journal Proteome Research, 15(9), 3420–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gorman, G. S. , Speir, J. P , Turner, C. A. , & Amster, I. J. (1992). Proton affinities of the 20 common .alpha.‐amino acids. Journal of the American Chemical Society, 114(10), 3986–3988. [Google Scholar]
- 37. Harrison, A. G. (1997). The gas‐phase basicities and proton affinities of amino acids and peptides. Mass Spectrometry Review, 16(4), 201–217. [Google Scholar]
- 38. Zeng, D. , & Li, S. (2009). Revival of deuterium‐labeled reagents for protein quantitation. Chemical Communications, 23, 3369–3371. [DOI] [PubMed] [Google Scholar]
- 39. Zhang, J. , Wang, Y. , & Li, S. (2010). Deuterium isobaric amine‐reactive tags for quantitative proteomics. Analytical Chemistry, 82, 7588–7595. [DOI] [PubMed] [Google Scholar]
- 40. Chen, Z. , Wang, Q. , Lin, L. , Tang, Q. , Edwards, J. L. , Li, S. , & Liu, S. (2012). Comparative evaluation of two isobaric labeling tags, DiART and iTRAQ. Analytical Chemistry, 84, 2908–2915. [DOI] [PubMed] [Google Scholar]
- 41. Ren, Y. , He, Y. , Lin, Z. , Zi, J. , Yang, H. , Zhang, S. , Lou, X. , Wang, Q. , Li, S. , & Liu, S. (2018). Reagents for isobaric labeling peptides in quantitative proteomics. Analytical Chemistry, 90, 12366–12371. [DOI] [PubMed] [Google Scholar]
- 42. Wühr, M. , Haas, W. , Mcalister, G. C. , Peshkin, L. , Rad, R. , Kirschner, M. W. , & Gygi, S. P. (2012). Accurate multiplexed proteomics at the MS2 level using the complement reporter ion cluster. Analytical Chemistry, 84, 9214–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ting, L. , Rad, R. , Gygi, S. P. , & Haas, W. (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nature Methods, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wenger, C. D. , Lee, M. V. , Hebert, A. S. , Mcalister, G. C. , Phanstiel, D. H. , Westphall, M. S. , & Coon, J. J. (2011). Gas‐phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nature Methods, 8, 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iwasaki, M. , Tabata, T. , Kawahara, Y. , Ishihama, Y. , & Nakagawa, M. (2019). Removal of interference MS/MS spectra for accurate quantification in isobaric tag‐based proteomics. Journal of Proteome Research, 18, 2535–2544. [DOI] [PubMed] [Google Scholar]
- 46. Savitski, M. M. , Sweetman, G. , Askenazi, M. , Marto, J. A. , Lang, M. , Zinn, N. , & Bantscheff, M. (2011). Delayed fragmentation and optimized isolation width settings for improvement of protein identification and accuracy of isobaric mass tag quantification on orbitrap‐ type mass spectrometers. Analytical Chemistry, 83, 8959–8967. [DOI] [PubMed] [Google Scholar]
- 47. Shadforth, I. P. , Dunkley, T. , Lilley, K. , & Bessant, C. (2005). i‐Tracker: For quantitative proteomics using iTRAQTM . Bmc Genomics [Electronic Resource], 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin, W.‐T. , Hung, W.‐N. , Yian, Y.‐H. , Wu, K.‐P. , Han, C.‐L. , Chen, Y.‐R. , Chen, Y.‐J. , Sung, T.‐Y. , & Hsu, W.‐L. (2006). Multi‐Q: A fully automated tool for multiplexed protein quantitation. Journal of Proteome Research, 5, 2328–2338. [DOI] [PubMed] [Google Scholar]
- 49. Hu, J. , Qian, J. , Borisov, O. , Pan, S. , Li, Y. , Liu, T. , Deng, L. , Wannemacher, K. , Kurnellas, M. , Patterson, C. , Elkabes, S. , & Li, H. (2006). Optimized proteomic analysis of a mouse model of cerebellar dysfunction using amine‐specific isobaric tags. Proteomics, 6, 4321–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Burkhart, J. M. , Vaudel, M. , Zahedi, R. P. , Martens, L. , & Sickmann, A. (2011). iTRAQ protein quantification: A quality‐controlled workflow. Proteomics, 11, 1125–1134. [DOI] [PubMed] [Google Scholar]
- 51. Brenes, A. , Hukelmann, J. , Bensaddek, D. , & Lamond, A. I. (2019). Multibatch TMT reveals false positives, batch effects, and missing values. Molecular and Cellular Proteomics, 18, 1967–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Braun, C. R. , Bird, G. H. , Wühr, M. , Erickson, B. K. , Rad, R. , Walensky, L. D. , Gygi, S. P. , & Haas, W. (2015). Generation of multiple reporter ions from a single isobaric reagent increases multiplexing capacity for quantitative proteomics. Analytical Chemistry, 87, 9855–9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koehler, C. J. , Strozynski, M. , Kozielski, F. , Treumann, A. , & Thiede, B. (2009). Isobaric peptide termini labeling for MS/MS‐based quantitative proteomics. Journal of Proteome Research, 8, 4333–4341. [DOI] [PubMed] [Google Scholar]
- 54. Koehler, C. J. , Arntzen, M. Ø. , Strozynski, M. , Treumann, A. , & Thiede, B. (2011). Isobaric peptide termini labeling utilizing site‐specific N‐terminal succinylation. Analytical Chemistry, 83, 4775–4781. [DOI] [PubMed] [Google Scholar]
- 55. Jiang, H. , Yin, H. , Xie, L. , Zhang, Y. , Zhang, L. , Yang, P.‐Y. , & Lu, H. (2018). A novel triplex isobaric termini labeling quantitative approach for simultaneously supplying three quantitative sources. Analytica Chimica Acta, 1001, 70–77. [DOI] [PubMed] [Google Scholar]
- 56. Cao, T. , Zhang, L. , Zhang, Y. , Yan, G. , Fang, C. , Bao, H. , & Lu, H. (2017). Site‐specific quantification of protein ubiquitination on MS2 fragment ion level via isobaric peptide labeling. Analytical Chemistry, 89, 11468–11475. [DOI] [PubMed] [Google Scholar]
- 57. Zhang, S. , Chen, L. , Shan, Y. , Sui, Z. , Wu, Q. , Zhang, L. , Liang, Z. , & Zhang, Y. (2016). Pseudo isobaric peptide termini labelling for relative proteome quantification by SWATH MS acquisition. Analyst, 141, 4912–4918. [DOI] [PubMed] [Google Scholar]
- 58. Liu, J. , Zhou, Y. , Shan, Y. , Zhao, B. , Hu, Y. , Sui, Z. , Liang, Z. , Zhang, L. , & Zhang, Y. (2019). A multiplex fragment‐ion‐based method for accurate proteome quantification. Analytical Chemistry, 91, 3921–3928. [DOI] [PubMed] [Google Scholar]
- 59. Tian, X. , De Vries, M. P. , Visscher, S. W. J. , Permentier, H. P. , & Bischoff, R. (2020). Selective maleylation‐directed isobaric peptide termini labeling for accurate proteome quantification. Analytical Chemistry, 92, 7836–7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arntzen, M. Ø. , Koehler, C. J. , Barsnes, H. , Berven, F. S. , Treumann, A. , & Thiede, B. (2011). IsobariQ: Software for isobaric quantitative proteomics using IPTL, iTRAQ, and TMT. Journal of Proteome Research, 10, 913–920. [DOI] [PubMed] [Google Scholar]
- 61. Zhang, S. , Wu, Q. , Shan, Y. , Sui, Z. , Zhang, L. , & Zhang, Y. (2015). A paired ions scoring algorithm based on Morpheus for simultaneous identification and quantification of proteome samples prepared by isobaric peptide termini labeling strategies. Proteomics, 15, 1781–1788. [DOI] [PubMed] [Google Scholar]
- 62. Xie, L.‐Q. , Zhang, L. , Nie, A.‐Y. , Yan, G.‐Q. , Yao, J. , Zhang, Y. , Yang, P.‐Y. , & Lu, H.‐J. (2015). ITMSQ: A software tool for N‐ and C‐terminal fragment ion pairs based isobaric tandem mass spectrometry quantification. Proteomics, 15, 3755–3764. [DOI] [PubMed] [Google Scholar]
- 63. Virreira Winter, S. , Meier, F. , Wichmann, C. , Cox, J. , Mann, M. , & Meissner, F. (2018). EASI‐tag enables accurate multiplexed and interference‐free MS2‐based proteome quantification. Nature Methods, 15, 527–530. [DOI] [PubMed] [Google Scholar]
- 64. Tian, X. , De Vries, M. P. , Permentier, H. P. , & Bischoff, R. (2020). A versatile isobaric tag enables proteome quantification in data‐dependent and data‐independent acquisition modes. Analytical Chemistry, 12, 16149–16157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dephoure, N. , & Gygi, S. P. (2012). Hyperplexing: A method for higher‐order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Science Signal, 5(217), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Welle, K. A. , Zhang, T. , Hryhorenko, J. R. , Shen, S. , Qu, J. , & Ghaemmaghami, S. (2016). Time‐resolved analysis of proteome dynamics by tandem mass tags and stable isotope labeling in cell culture (TMT‐SILAC) hyperplexing. Molecular and Cellular Proteomics, 15(12), 3551–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xing, S. , Pai, A. , Wu, R. , & Lu, Y. (2021). NHS‐ester tandem labeling in one pot enables 48‐Plex quantitative proteomics. Analytical Chemistry, 93, 12827–12832. [DOI] [PubMed] [Google Scholar]
- 68. Everley, R. A. , Kunz, R. C. , Mcallister, F. E. , & Gygi, S. P. (2013). Increasing throughput in targeted proteomics assays: 54‐Plex quantitation in a single mass spectrometry run. Analytical Chemistry, 85, 5340–5346. [DOI] [PubMed] [Google Scholar]
- 69. Evans, A. R. , & Robinson, R. A. S. (2013). Global combined precursor isotopic labeling and isobaric tagging (cPILOT) approach with selective MS3 acquisition. Proteomics, 22, 3267–3272. [DOI] [PubMed] [Google Scholar]
- 70. Frost, D. C. , Rust, C. J. , Robinson, R. A. S. , & Li, L. (2018). Increased N, N‐Dimethyl leucine isobaric tag multiplexing by a combined precursor isotopic labeling and isobaric tagging approach. Analytical Chemistry, 90(18), 10664–10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Aggarwal, S. , Talukdar, N. C. , & Yadav, A. K. (2019). Advances in higher order multiplexing techniques in proteomics. Journal of Proteome Research, 18(6), 2360–2369. [DOI] [PubMed] [Google Scholar]
- 72. Gillette, M. A. , & Carr, S. A. (2013). Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nature Methods, 10(1), 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Doerr, A. (2013). Mass spectrometry‐based targeted proteomics. Nature Methods, 10(1), 23. [DOI] [PubMed] [Google Scholar]
- 74. Marx, V. (2013). Targeted proteomics. Nature Methods, 10(1), 19–223. [DOI] [PubMed] [Google Scholar]
- 75. Picotti, P. , & Aebersold, R. (2012). Selected reaction monitoring‐based proteomics: Workflows, potential, pitfalls and future directions. Nature Methods, 9(6), 555–566. [DOI] [PubMed] [Google Scholar]
- 76. Gerber, S. A. , Rush, J. , Steman, O. , Kirschner, M. W. , & Gygi, S. P. (2003). Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proceedings of the National Academy of Sciences of the United States of America, 100(12), 6940–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu, R. , Haas, W. , Dephoure, N. , Huttlin, E. L. , Zhai, B. , Sowa, M. E. , & Gygi, S. P. (2011). A large‐scale method to measure absolute protein phosphorylation stoichiometries. Nature Methods, 8(8), 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Erickson, B. K. , Rose, C. M. , Braun, C. R. , Erickson, A. R. , Knott, J. , Mcalister, G. C. , Wühr, M. , Paulo, J. A. , Everley, R. A. , & Gygi, S. P. (2017). A strategy to combine sample multiplexing with targeted proteomics assays for high‐throughput protein signature characterization. Molecular Cell, 65(2), 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhong, X. , Yu, Q. , Ma, F. , Frost, D. C. , Lu, L. , Chen, Z. , Zetterberg, H. , Carlsson, C. , Okonkwo, O. , & Li, L. (2019). HOTMAQ: A multiplexed absolute quantification method for targeted proteomics. Analytical Chemistry, 91(3), 2112–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Greer, T. , Lietz, C. B. , Xiang, F. , & Li, L. (2014). Novel isotopic N, N‐Dimethyl Leucine (iDiLeu) reagents enable absolute quantification of peptides and proteins using a standard curve approach. Journal of the American Society Mass Spectrometry, 26(1), 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tsou, C.‐C. , Tsai, C.‐F. , Tsui, Y.‐H. , Sudhir, P.‐R. , Wang, Y.‐T. , Chen, Y.‐J. , Chen, J.‐Y. , Sung, T.‐Y. , & Hsu, W.‐L. (2010). IDEAL‐Q, an automated tool for label‐free quantitation analysis using an efficient peptide alignment approach and spectral data validation. Molecular and Cellular Proteomics, 9(1), 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cox, J. , Hein, M. Y. , Luber, C. A. , Paron, I. , Nagaraj, N. , & Mann, M. (2014). Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed maxLFQ. Molecular and Cellular Proteomics, 13(9), 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Geyer, P. E. , Kulak, N. A. , Pichler, G. , Holdt, L. M. , Teupser, D. , & Mann, M. (2016). Plasma proteome profiling to assess human health and disease. Cell Systems, 2(3), 185–195. [DOI] [PubMed] [Google Scholar]
- 84. Meier, F. , Geyer, P. E. , Virreira Winter, S. , Cox, J. , & Mann, M. (2018). BoxCar acquisition method enables single‐shot proteomics at a depth of 10,000 proteins in 100 minutes. Nature Methods, 15(6), 440–448. [DOI] [PubMed] [Google Scholar]
- 85. Yi, L. , Tsai, C.‐F. , Dirice, E. , Swensen, A. C. , Chen, J. , Shi, T. , Gritsenko, M. A. , Chu, R. K. , Piehowski, P. D. , Smith, R. D. , Rodland, K. D. , Atkinson, M. A. , Mathews, C. E. , Kulkarni, R. N. , Liu, T. , & Qian, W.‐J. (2019). Boosting to Amplify Signal with Isobaric Labeling (BASIL) strategy for comprehensive quantitative phosphoproteomic characterization of small populations of cells. Analytical Chemistry, 91, 5794–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu, X. , Fields, R. , Schweppe, D. K. , & Paulo, J. A. (2021). Strategies for mass spectrometry‐based phosphoproteomics using isobaric tagging. Expert Review of Proteomics, 18(9), 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li, S. , & Zeng, D. (2007). CILAT ‐ A new reagent for quantitative proteomics. Chemical Communications, 21, 4193–4201. [DOI] [PubMed] [Google Scholar]
- 88. Murray, C. I. , Uhrigshardt, H. , O'meally, R. N. , Cole, R. N. , & Van Eyk, J. E. (2012). Identification and quantification of S‐nitrosylation by cysteine reactive tandem mass tag switch assay. Molecular and Cellular Proteomics, 11, M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Forrester, M. T. , Foster, M. W. , Benhar, M. , & Stamler, J. S. (2009). Detection of protein S‐nitrosylation with the biotin‐switch technique. Free Radical Biology and Medicine, 46, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Qu, Z. , Meng, F. , Bomgarden, R. , Viner, R. , Li, J. , Rogers, J. C. , Cheng, J. , Greenlief, C. M. , Cui, J. , Lubahn, D. B. , Sun, G. Y. , & Gu, Z. (2014). Proteomic quantification and site‐mapping of S‐Nitrosylated proteins using isobaric iodoTMT reagents. Journal of Proteome Research, 13, 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hahne, H. , Neubert, P. , Kuhn, K. , Etienne, C. , Bomgarden, R. , Rogers, J. C. , & Kuster, B. (2012). Carbonyl‐reactive tandem mass tags for the proteome‐wide quantification of N‐linked glycans. Analytical Chemistry, 84, 3716–3724. [DOI] [PubMed] [Google Scholar]
- 92. Zhou, S. , Hu, Y. , Veillon, L. , Snovida, S. I. , Rogers, J. C. , Saba, J. , & Mechref, Y. (2016). Quantitative LC‐MS/MS glycomic analysis of biological samples using aminoxyTMT. Analytical Chemistry, 88(15), 7515–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yang, S. , Wang, M. , Chen, L. , Yin, B. , Song, G. , Turko, I. V. , Phinney, K. W. , Betenbaugh, M. J. , Zhang, H. , & Li, S. (2015). QUANTITY: An isobaric tag for quantitative glycomics. Science Reports, 5, 8188–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yang, S. , Yuan, W. , Yang, W. , Zhou, J. , Harlan, R. , Edwards, J. , Li, S. , & Zhang, H. (2013). Glycan analysis by isobaric aldehyde reactive tags and mass spectrometry. Analytical Chemistry, 85(17), 8188–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Palmese, A. , De Rosa, C. , Chiappetta, G. , Marino, G. , & Amoresano, A. (2012). Novel method to investigate protein carbonylation by iTRAQ strategy. Analytical and bioanalytical chemistry, 404, 1631–1635. [DOI] [PubMed] [Google Scholar]
- 96. Feng, Y. , Chen, B. , Yu, Q. , Zhong, X. , Frost, D. C. , Ikonomidou, C. , & Li, L. (2019). Isobaric multiplex labeling reagents for carbonyl‐containing compound (SUGAR) tags: A probe for quantitative glycomic analysis. Analytical Chemistry, 91(4), 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Li, M. , Gu, T.‐J. , Lin, X. , & Li, L. (2021). DiLeuPMP: A multiplexed isobaric labeling method for quantitative analysis of O‐glycans. Analytical Chemistry, 93(28), 9845–9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cunningham, R. , Ma, D. , & Li, L. (2012). Mass spectrometry‐based proteomics and peptidomics for systems biology and biomarker discovery. Frontiers in Biology, 7(4), 313–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Westbrook, J. A. , Noirel, J. , Brown, J. E. , Wright, P. C. , & Evans, C. A. (2015). Quantitation with chemical tagging reagents in biomarker studies. Proteomics Clinical Applications, 9, 295–300. [DOI] [PubMed] [Google Scholar]
- 100. Du, C. , Weng, Y. , Lou, J. , Zeng, G. , Liu, X. , Jin, H. , Lin, S. , & Tang, L. (2019). Isobaric tags for relative and absolute quantification‐based proteomics reveals potential novel biomarkers for the early diagnosis of acute myocardial infarction within 3 h. International Journal of Molecular Medicine, 43, 1991–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wu, H. , Zhang, X.‐Y. , Niu, M. , Li, F.‐F. , Gao, S. , Wei, W. , Li, S.‐W. , Zhang, X.‐D. , Liu, S.‐L. , & Pang, D. (2020). Isobaric tags for relative and absolute quantitation in proteomic analysis of potential biomarkers in invasive cancer, ductal carcinoma in situ, and mammary fibroadenoma. Frontiers in Oncology, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chaerkady, R. , & Pandey, A. (2007). Quantitative proteomics for identification of cancer biomarkers. Proteomics Clinical Applications, 1, 1080–1089. [DOI] [PubMed] [Google Scholar]
- 103. Keshamouni, V. G. , Michailidis, G. , Grasso, C. S. , Anthwal, S. , Strahler, J. R. , Walker, A. , Arenberg, D. A. , Reddy, R. C. , Akulapalli, S. , Thannickal, V. J. , Standiford, T. J. , Andrews, P. C. , & Omenn, G. S. (2006). Differential protein expression profiling by iTRAQ−2DLC−MS/MS of lung cancer cells undergoing epithelial‐mesenchymal transition reveals a migratory/invasive phenotype. Journal of Proteome Research, 5, 1143–1154. [DOI] [PubMed] [Google Scholar]
- 104. Seshi, B. (2006). An integrated approach to mapping the proteome of the human bone marrow stromal cell. Proteomics, 6, 5169–5182. [DOI] [PubMed] [Google Scholar]
- 105. Tokuoka, S. M. , Kita, Y. , Shimizu, T. , & Oda, Y. (2019). Isobaric mass tagging and triple quadrupole mass spectrometry to determine lipid biomarker candidates for Alzheimer's disease. Plos One, 14(12), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Li, L. , & Sweedler, J. V. (2008). Peptides in the brain: Mass spectrometry‐based measurement approaches and challenges. Annual Review of Analytical Chemistry, 1, 451–483. [DOI] [PubMed] [Google Scholar]
- 107. Phetsanthad, A. , Vu, N. Q. , Yu, Q. , Buchberger, A. R. , Chen, Z. , Keller, C. , & Li, L. (2021). Recent advances in mass spectrometry analysis of neuropeptides. Mass Spectrometry Review, 24, e21734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Xie, F. , Romanova, E. , & Sweedler, J. (2011). Neuropeptidomics of the mammalian brain (pp. 229–242). Neuroproteomics. Humana Press. [Google Scholar]
- 109. Buchberger, A. , Yu, Q. , & Li, L. (2015). Advances in mass spectrometric tools for probing neuropeptides. Annual Review of Analytical Chemistry, 8, 485–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Buchberger, A. R. , Delaney, K. , Liu, Y. , Vu, N. Q. , Helfenbein, K. , & Li, L. (2020). Mass spectrometric profiling of neuropeptides in callinectes sapidus during hypoxia stress. Acs Chemical Neuroscience, 11, 3097–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Buchberger, A. R. , Vu, N. , Johnson, J. , DeLaney, K. , & Li, L. (2020). A simple and effective sample preparation strategy for MALDI‐MS imaging of neuropeptide changes in the crustacean brain due to hypoxia and hypercapnia stress. Journal of Proteome Research, 31, 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zhang, Y. , Buchberger, A. , Muthuvel, G. , & Li, L. (2015). Expression and distribution of neuropeptides in the nervous system of the crab Carcinus maenas and their roles in environmental stress. Proteomics, 15, 3969–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wilson, R. E. , Jaquins‐Gerstl, A. , & Weber, S. G. (2018). On‐column dimethylation with capillary liquid chromatography‐tandem mass spectrometry for online determination of neuropeptides in rat brain microdialysate. Analytical Chemistry, 90, 4561–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu, R. , Wei, P. , Keller, C. , Orefice, N. S. , Shi, Y. , Li, Z. , Huang, J. , Cui, Y. , Frost, D. C. , Han, S. , Cross, T‐W. L. , Rey, F. E. , & Li, L. (2020). Integrated label‐free and 10‐Plex DiLeu isobaric tag quantitative methods for profiling changes in the mouse hypothalamic neuropeptidome and proteome: Assessment of the impact of the gut microbiome. Analytical Chemistry, 92(20), 14021–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jiang, X. , Xiang, F. , Jia, C. , Buchberger, A. R. , & Li, L. (2018). Relative quantitation of neuropeptides at multiple developmental stages of the american lobster using N , N ‐dimethyl leucine isobaric tandem mass tags. ACS Chemical Neuroscience, 9, 2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sauer, C. S. , & Li, L. (2021). Mass spectrometric profiling of neuropeptides in response to copper toxicity via isobaric tagging. Chemical Research in Toxicology, 34, 1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zhao, S. , & Li, L. (2020). Chemical derivatization in LC‐MS‐based metabolomics study. Trends in Analytical Chemistry, 131, 1–11. [Google Scholar]
- 118. Yuan, W. , Anderson, K. W. , Li, S. , & Edwards, J. L. (2012). Subsecond absolute quantitation of amine metabolites using isobaric tags for discovery of pathway activation in mammalian cells. Analytical Chemistry, 84, 2892–2899. [DOI] [PubMed] [Google Scholar]
- 119. Murphy, J. P , Everley, R. A. , Coloff, J. L. , & Gygi, S. P. (2014). Combining amine metabolomics and quantitative proteomics of cancer cells using derivatization with isobaric tags. Analytical Chemistry, 86(7), 3585–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yuan, W. , Zhang, J. , Li, S. , & Edwards, J. L. (2011). Amine metabolomics of hyperglycemic endothelial cells using capillary LC‐MS with isobaric tagging. Journal of Proteome Research, 10(11), 5242–5250. [DOI] [PubMed] [Google Scholar]
- 121. Hao, L. , Johnson, J. , Lietz, C. B. , Buchberger, A. , Frost, D. , Kao, W. J , & Li, L. (2017). Mass defect‐based n,n‐dimethyl leucine labels for quantitative proteomics and amine metabolomics of pancreatic cancer cells. Analytical Chemistry, 89, 1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wei, P. , Hao, L. , Thomas, S. , Buchberger, A. R. , Steinke, L. , Marker, P. C. , Ricke, W. A. , & Li, L. (2020). Urinary amine metabolomics characterization with custom 12‐plex isobaric DiLeu labeling. Journal of the American Society of Mass Spectrometry, 31, 1854–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hao, L. , Zhu, Y. , Wei, P. , Johnson, J. , Buchberger, A. , Frost, D. , Kao, W. J , & Li, L. (2019). Metandem: An online software tool for mass spectrometry‐based isobaric labeling metabolomics. Analytica Chimica Acta, 1088, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Mateus, A. , Kurzawa, N. , Perrin, J. , Bergamini, G. , & Savitski, M. (2021). Drug target identification in tissues by thermal proteome profiling. Annual Review of Pharmacology and Toxicology, 62(14), 1–18. [DOI] [PubMed] [Google Scholar]
- 125. Ito, T. , Ando, H. , Suzuki, T. , Ogura, T. , Hotta, K. , Imamura, Y. , Yamaguchi, Y. , & Handa, H. (2010). Identification of a primary target of thalidomide teratogenicity. Science, 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- 126. Saghatelian, A. , Jessani, N. , Joseph, A. , Humphrey, M. , & Cravatt, B. (2004). Activity‐based probes for the proteomic profiling of metalloproteases. Proceedings of the National Academy of Sciences of the United States of America, 101(27), 10000–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. West, G. M. , Tucket, C. L. , Xu, T. , Park, S. K. , Han, X. , Yates, J. R. 3rd , & Fitzgerald, M. C. (2010). Quantitative proteomics approach for identifying protein‐drug interactions in complex mixtures using protein stability measurements. Proceedings of the National Academy of Sciences of the United States of America, 107, 9078–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Luchini, A. , Espina, V. , & Liotta, L. A. (2014). Protein painting reveals solvent‐excluded drug targets hidden within native protein‐protein interfaces. Nature Communication, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Molina, D. M. , Jafari, R. , Ignatushchenko, M. , Seki, T. , Larsson, E. A , Dan, C. , Sreekumar, L. , Cao, Y. , & Nordlund, P. (2013). Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science, 341(6141), 84–87. [DOI] [PubMed] [Google Scholar]
- 130. Savitski, M. M. , Reinhard, F. B. M. , Franken, H. , Werner, T. , Savitski, M. F. , Eberhard, D. , Molina, D. M. , Jafari, R. , Dovega, R. B. , Klaeger, S. , Kuster, B. , Nordlund, P. , Bantscheff, M. , & Drewes, G. (2014). Tracking cancer drugs in living cells by thermal profiling of the proteome. Science, 346(6205), 1–10. [DOI] [PubMed] [Google Scholar]
- 131. Mateus, A. , Kurzawa, N. , Becher, I. , Sridharan, S. , Helm, D. , Stein, F. , Typas, A. , & Savitski, M. M. (2020). Thermal proteome profiling for interrogating protein interactions. Molecular Systems Biology, 16, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Uhlén, M. , Fagerberg, L. , Hallström, B. M. , Lindskog, C. , Oksvold, P. , Mardinoglu, A. , Sivertsson, Å. , Kampf, C. , Sjöstedt, E. , Asplund, A. , Olsson, I. , Edlund, K. , Lundberg, E. , Navani, S. , Szigyarto, C. A.‐K. , Odeberg, J. , Djureinovic, D. , Takanen, J. O. , Hober, S. , … Pontén, F. (2015). Tissue‐based map of the human proteome. Science, 347(6220), 1260419. [DOI] [PubMed] [Google Scholar]
- 133. Mateus, A. , Hevler, J. , Bobonis, J. , Kurzawa, N. , Shah, M. , Mitosch, K. , Goemans, C. V. , Helm, D. , Stein, F. , Typas, A. , & Savitski, M. M. (2020). The functional proteome landscape of Escherichia coli. Nature, 588, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Mateus, A. , Bobonis, J. , Kurzawa, N. , Stein, F. , Helm, D. , Helver, J. , Typas, A. , & Savitski, M. (2018). Thermal proteome profiling in bacteria: Probing protein state in vivo. Molecular Systems Biology, 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Dziekan, J. M. , Wirjanata, G. , Dai, L. , Go, K. D. , Yu, H. , Lim, Y. T. , Chen, L. , Wang, L. C. , Puspita, B. , Prabhu, N. , Sobota, R. M. , Nordlund, P. , & Bozdech, Z. (2020). Cellular thermal shift assay for the identification of drug‐target interactions in the Plasmodium falciparum proteome. Nature Protocols, 15, 1881–1921. [DOI] [PubMed] [Google Scholar]
- 136. Dziekan, J. M. , Yu, H. , Chen, D. , Dai, L. , Wirjanata, G. , Larsson, A. , Prabhu, N. , Sobota, R. M. , Bozdech, Z. , & Nordlund, P. (2019). Identifying purine nucleoside phosphorylase as the target of quinine using cellular thermal shift assay. Science Translational Medicine, 11, eaau3174. [DOI] [PubMed] [Google Scholar]
- 137. Volkening, J. D. , Stecker, K. E. , & Sussman, M. R. (2019). Proteome‐wide analysis of protein thermal stability in the model higher plant arabidopsis thaliana. Molecular and Cellular Proteomics, 18, 308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Viéitez, C. , Busby, B. , Ochoa, D. , Mateus, A. , Galardini, M. , Jawed, A. , … Beltrao, P. (2019). Towards a systematic map of the functional role of protein phosphorylation. bioRxiv., 1–28. [Google Scholar]
- 139. Peck Justice, S. A. , Barron, M. P. , Qi, G. D. , Wijeratne, H. R. S , Victorino, J. F. , Simpson, E. R. , Vilseck, J. Z. , Wijeratne, A. B. , & Mosley, A. L. (2020). Mutant thermal proteome profiling for characterization of missense protein variants and their associated phenotypes within the proteome. Journal of Biological Chemistry, 295, 16219–16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Selkrig, J. , Stanifer, M. , Mateus, A. , Mitosch, K. , Barrio‐Hernandez, I. , Rettel, M. , Kim, H. , Voogdt, C. G. P. , Walch, P. , Kee, C. , Kurzawa, N. , Stein, F. , Potel, C. , Jarzab, A. , Kuster, B. , Bartenschlager, R. , Boulant, S. , Beltrao, P. , Typas, A. , & Savitski, M. M. (2021). SARS‐CoV‐2 infection remodels the host protein thermal stability landscape. Molecular Systems Biology, 17, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hasimoto, Y. , Sheng, X. , Murray‐Nerger, L. , & Cristea, I. (2020). Temporal dynamics of protein complex formation and dissociation during human cytomegalovirus infection. Nature Communication, 11, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Perrin, J. , Werner, T. , Kurzawa, N. , Rutkowska, A. , Childs, D. D. , Kalxdorf, M. , Poeckel, D. , Stonehouse, E. , Strohmer, K. , Heller, B. , Thomson, D. W. , Krause, J. , Becher, I. , Eberl, H. C , Vappiani, J. , Sevin, D. C. , Rau, C. E. , Franken, H. , Huber, W. , … Bergamini, G. (2020). Identifying drug targets in tissues and whole blood with thermal‐shift profiling. Nature Biotechnology, 38, 303–308. [DOI] [PubMed] [Google Scholar]
- 143. Becher, I. , Werner, T. , Doce, C. , Zaal, E. A. , Tögel, I. , Khan, C. A. , Rueger, A. , Muelbaier, M. , Salzer, E. , Berkers, C. R. , Fitzpatrick, P. F. , Bantscheff, M. , & Savitski, M. M. (2016). Thermal profiling reveals phenylalanine hydroxylase as an off‐target of panobinostat. Nature Chemical Biology, 12, 908–910. [DOI] [PubMed] [Google Scholar]
- 144. Gaetani, M. , Sabatier, P. , Saei, A. A. , Beusch, C. M. , Yang, Z. , Lundström, S. L. , & Zubarev, R. A. (2019). Proteome integral solubility alteration: A high‐throughput proteomics assay for target deconvolution. Journal of Proteome Research, 18, 4027–4037. [DOI] [PubMed] [Google Scholar]
- 145. Kalxdorf, M. , Günthner, I. , Becher, I. , Kurzawa, N. , Knecht, S. , Savitski, M. M. , Eberl, H. C , & Bantscheff, M. (2021). Cell surface thermal proteome profiling tracks perturbations and drug targets on the plasma membrane. Nature Methods, 18(1), 84–91. [DOI] [PubMed] [Google Scholar]
- 146. Li, J. , Van Vranken, J. G. , Paulo, J. A. , Huttlin, E. L. , & Gygi, S. P. (2020). Selection of heating temperatures improves the sensitivity of the proteome integral solubility alteration assay. Journal Proteome Research, 19, 2159–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Childs, D. , Bach, K. , Franken, H. , Anders, S. , Kurzawa, N. , Bantscheff, M. , Savitski, M. M. , & Huber, W. (2019). Nonparametric analysis of thermal proteome profiles reveals novel drug‐binding proteins*. Molecular and Cellular Proteomics, 18, 2506–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Kurzawa, N. , Becher, I. , Sridharan, S. , Franken, H. , Mateus, A. , Anders, S. , Bantscheff, M. , Huber, W. , & Savitski, M. M. (2020). A computational method for detection of ligand‐binding proteins from dose range thermal proteome profiles. Nature Communication, 11, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Saei, A. A. , Beusch, C. M. , Sabatier, P. , Wells, J. A. , Gharibi, H. , Meng, Z. , Chernobrovkin, A. , Rodin, S. , Näreoja, K. , Thorsell, A.‐G. , Karlberg, T. , Cheng, Q. , Lundström, S. L. , Gaetani, M. , Végvári, Á. , Arnér, E. S. J. , Schüler, H. , & Zubarev, R. A. (2021). System‐wide identification and prioritization of enzyme substrates by thermal analysis. Nature Communication, 12(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Piersimoni, L. , Kastritis, P. L. , Arlt, C. , & Sinz, A. (2021). Cross‐linking mass spectrometry for investigating protein conformations and protein‐protein interactions‐a method for all seasons. Chem. Rev., 122(8), 7500–7531. [DOI] [PubMed] [Google Scholar]
- 151. Yu, C. , Huszagh, A. , Viner, R. , Novitsky, E. J. , Rychnovsky, S. D. , & Huang, L. (2016). Developing a multiplexed quantitative cross‐linking mass spectrometry platform for comparative structural analysis of protein complexes. Analytical Chemistry, 88, 10301–10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chavez, J. D. , Keller, A. , Mohr, J. P. , & Bruce, J. E. (2020). Isobaric quantitative protein interaction reporter technology for comparative interactome studies. Analytical Chemistry, 92, 14094–14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kelly, R. T. (2020). Single‐cell proteomics: Progress and prospects. Molecular and Cellular Proteomics, 19, 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ctortecka, C. , & Mechtler, K. (2021). The rise of single‐cell proteomics. Analytical Science Adventures, 2, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Slavov, N. (2021). Single‐cell protein analysis by mass spectrometry. Current Opinion in Chemical Biology, 60, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Budnik, B. , Levy, E. , Harmange, G. , & Slavov, N. (2018). SCoPE‐MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome biology, 19, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Petelski, A. A. , Emmott, E. , Leduc, A. , Huffman, R. G , Specht, H. , Perlman, D. H. , & Slavov, N. (2021). Multiplexed single‐cell proteomics using SCoPE2. Nature Protocols, 16, 5398–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tsai, C.‐F. , Zhao, R. , Williams, S. M. , Moore, R. J. , Schultz, K. , Chrisler, W. B. , Pasa‐Tolic, L. , Rodland, K. D. , Smith, R. D. , Shi, T. , Zhu, Y. , & Liu, T. (2020). An improved boosting to amplify signal with isobaric labeling (iBASIL) strategy for precise quantitative single‐cell proteomics. Molecular and Cellular Proteomics, 19, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ludwig, C. , Gillet, L. , Rosenberger, G. , Amon, S. , Collins, B. C. , & Aebersold, R. (2018). Data‐independent acquisition‐based SWATH ‐ MS for quantitative proteomics: A tutorial. Molecular Systems Biology, 14, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhong, X. , Frost, D. C. , Yu, Q. , Li, M. , Gu, T.‐J. , & Li, L. (2020). Mass defect‐based DiLeu tagging for multiplexed data‐independent acquisition. Analytical Chemistry, 92, 11119–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lin, L. , Zheng, J. , Yu, Q. , Chen, W. , Xing, J. , Chen, C. , & Tian, R. (2018). High throughput and accurate serum proteome profiling by integrated sample preparation technology and single‐run data independent mass spectrometry analysis. Journal of Proteomics, 174, 9–16. [DOI] [PubMed] [Google Scholar]
- 162. Mc Ardle, A. , Binek, A. , Moradian, A. , Chazarin Orgel, B. , Rivas, A. , Washington, K. E. , Phebus, C. , Manalo, D.‐M. , Go, J. , Venkatraman, V. , Coutelin Johnson, C. W. , Fu, Q. , Cheng, S. , Raedschelders, K. , Fert‐Bober, J. , Pennington, S. R. , Murray, C. I. , & Van Eyk, J. E. (2022). Standardized workflow for precise mid‐ and high‐ throughput proteomics of blood biofluids. Clinical Chemistry, 68(3), 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Toby, T. K. , Fornelli, L. , & Kelleher, N. L. (2016). Progress in top‐down proteomics and the analysis of proteoforms. Annual Review of Analytical Chemistry, 9, 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wiese, S. , Reidegeld, K. A. , Meyer, H. E. , & Warscheid, B. (2007). Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics, 7(3), 340–350. [DOI] [PubMed] [Google Scholar]
- 165. Sinclair, J. , & Timms, J. F. (2011). Quantitative profiling of serum samples using TMT protein labelling, fractionation and LC‐MS/MS. Methods (San Diego, Calif.), 54(4), 361–369. [DOI] [PubMed] [Google Scholar]
- 166. Yu, D. , Wang, Z. , Cupp‐Sutton, K. A. , Guo, Y. , Kou, Q. , Smith, K. , Liu, X. , & Wu, S. (2021). Quantitative Top‐down proteomics in complex samples using protein‐level tandem mass tag labeling. Journal of the American Society of Mass Spectrometry, 32, 1336–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Hung, C.‐W. , & Tholey, A. (2012). Tandem mass tag protein labeling for top‐down identification and quantification. Analytical Chemistry, 84, 161–170. [DOI] [PubMed] [Google Scholar]
- 168. Winkels, K. , Koudelka, T. , & Tholey, A. (2021). Quantitative top‐down proteomics by isobaric labeling with thiol‐directed tandem mass tags. Journal of Proteome Research, 20, 4495–4506. [DOI] [PubMed] [Google Scholar]
- 169. Dou, M. , Clair, G. , Tsai, C.‐F. , Xu, K. , Chrisler, W. B. , Sontag, R. L. , Zhao, R. , Moore, R. J. , Liu, T. , Pasa‐Tolic, L. , Smith, R. D. , Shi, T. , Adkins, J. N. , Qian, W.‐J. , Kelly, R. T. , Ansong, C. , & Zhu, Y. (2019). High‐throughput single cell proteomics enabled by multiplex isobaric labeling in a nanodroplet sample preparation platform. Analytical Chemistry, 91(20), 13119–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Woo, J. , Williams, S. M. , Markillie, L. M. , Feng, S. , Tsai, C.‐F. , Aguilera‐Vazquez, V. , Sontag, R. L. , Moore, R. J. , Hu, D. , Mehta, H. S. , Cantlon‐Bruce, J. , Liu, T. , Adkins, J. N. , Smith, R. D. , Clair, G. C. , Pasa‐Tolic, L. , & Zhu, Y. (2021). High‐throughput and high‐efficiency sample preparation for single‐cell proteomics using a nested nanowell chip. Nature Communication, 12(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This is an invited review and there is no original data.