

Summary
Ambient RNA contamination in single-cell and single-nuclei RNA sequencing (snRNA-seq) is a significant problem, but its consequences are poorly understood. Here, we show that ambient RNAs in brain snRNA-seq datasets have a nuclear or non-nuclear origin with distinct gene set signatures. Both ambient RNA signatures are predominantly neuronal and we find that some previously annotated neuronal cell types are distinguished by ambient RNA contamination. We detect pervasive neuronal ambient RNA contamination in all glial cell types unless glia and neurons are physically separated prior to sequencing. We demonstrate that this contamination can be removed in silico and show that a previous single-nuclei RNA-seq based annotation of immature oligodendrocytes are glial nuclei contaminated with ambient RNAs. After ambient RNA removal, we detect rare, committed oligodendrocyte progenitor cells not annotated in most previous adult human brain datasets. Together, these results provide an in-depth analysis of ambient RNA contamination in brain single-nuclei datasets.
Graphical Abstract
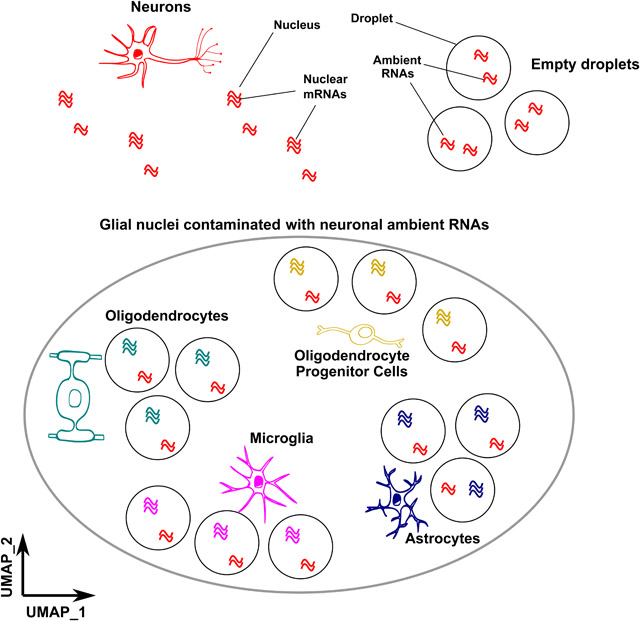
In brief
Caglayan et al. examine brain single nuclei datasets and uncover signatures of neuronal contamination in glia that mask rare cell types and lead to improper cell annotation. The authors show how to reduce this contamination by using either in silico approaches or physical separation of cell types.
Introduction
Single-nuclei RNA-sequencing (snRNA-seq) experiments involve nuclei isolation and subsequent capture of each nucleus in a single droplet containing a unique cell barcode. However, this capture process can also encapsulate freely floating transcripts, resulting in contamination of the endogenous expression profile. These extraneous transcripts have been previously referred to as ‘ambient RNAs’ (Luecken and Theis, 2019; Young and Behjati, 2020). Since ambient RNAs are expected to be primarily derived from more abundant cell types, ambient RNA contamination in less abundant cell types can result in a considerably skewed endogenous expression profile. Thus, failure to account for ambient RNA contamination can result in biological misinterpretation, especially in cell types with less abundant transcripts. Many previous studies have not removed ambient RNA contamination from the endogenous expression profiles of their datasets; therefore, it is possible that ambient RNA contamination constitutes an important problem that has led to misinterpretations in the downstream analyses. In our study, we address this possibility be reanalyzing several previously published datasets.
In addition to contaminating the endogenous expression profiles of nuclei, ambient RNAs are also captured in droplets that do not capture nuclei. These droplets are referred to as ‘empty droplets’ (Lun et al., 2019; Macosko et al., 2015). Interestingly, empty droplets do not always show a clear separation from the non-empty droplets in terms of unique read counts, underscoring the high levels of ambient transcripts that are captured in most single-nuclei (and single-cell) preparations (Lun et al., 2019). The lack of clear separation between empty and non-empty droplets based on read counts makes it difficult to justify using a read count based cutoff (also called UMI -unique molecular identifier- cutoff). A UMI cutoff can lead to mis-calling of empty droplets as non-empty droplets or mis-calling of certain cell types that contain fewer transcripts than others as empty droplets (Luecken and Theis, 2019). Recent tools have addressed this problem by distinguishing non-empty and empty droplets by using other metrics such as expression profile and nuclear fraction (Heiser et al., 2021; Lun et al., 2019; Muskovic and Powell, 2021). However, the composition of empty droplets is tissue-dependent and the specific composition of empty droplets for a given tissue is often not explored. Without proper understanding of the transcriptional composition of empty droplets, it can be difficult to decide whether a given cluster of cell barcodes are empty droplets or non-empty droplets that captured real nuclei / cells.
Taken together, ambient RNAs pose two challenges: contamination of the endogenous profile of real nuclei /cells and ambiguous separation of empty and non-empty droplets. With respect to these two issues, contamination in real nuclei / cells has received less attention. Several recently developed tools are specifically designed to remove ambient RNA contamination from real nuclei / cells; however, their utilization has been limited (Fleming et al., 2019; Yang et al., 2020; Young and Behjati, 2020). Additionally, while these tools aim to be effective across different tissues, the genetic signatures of ambient RNA contamination differ between tissue types. Thus, it is also important to characterize the overrepresented genes in the ambient RNA population of a given tissue type. This will aid both the interpretation of previously published datasets as well as assess the effects of ambient RNA contamination removal tools for specific tissues by evaluating the levels of ambient RNA population before and after the removal of contamination. In this study, we focus on snRNA-seq studies from brain tissue, one of the most frequently profiled tissues using this technique due to its high cellular heterogeneity (Bakken et al., 2021; Jakel et al., 2019; Nagy et al., 2020; Velmeshev et al., 2019). Brain tissue is also a good model to understand the effects of ambient RNA contamination since neurons are more abundant and contain more transcripts than glia in the adult mammalian cortex (Ruzicka et al., 2020). Therefore, ambient RNA profiles from snRNA-seq studies of brain should be biased for neurons. We hypothesize that this bias may contribute to both empty droplets with neuronal signatures as well as distinctive neuronal read contamination in non-neuronal cell types.
Here, we analyzed ambient RNA signatures from human brain snRNA-seq datasets (Table S1) by retaining additional cell barcodes that are typically removed due to low UMI counts. We found two types of ambient RNAs separated by their intronic read ratio: non-nuclear ambient RNAs with low intronic reads and nuclear ambient RNAs with high intronic reads. Comparisons with nuclei-sorted datasets revealed that non-nuclear ambient RNA can be cleared by physical nuclei sorting. We show that the ambient RNA signature is predominantly neuronal in origin, and all glia are contaminated with ambient RNAs. Ambient RNA contamination in glia was removed in datasets depleted of neurons (NeuN-sorted datasets; NeuN-SDs) before droplet capture (Hodge et al., 2019). As an in-silico alternative, we used CellBender (Fleming et al., 2019) and subsequent subcluster cleaning, which also removed detectable ambient RNA contamination from glia. Re-analysis of the oligodendrocyte lineage trajectory after ambient RNA removal revealed that previously annotated immature oligodendrocytes are likely glial nuclei contaminated with ambient RNA. Instead, we found a rare, transient cell type named COPs (committed oligodendrocyte progenitor cells), which were previously described in an adolescent mouse dataset (Marques et al., 2016) but not described in most human snRNA-seq datasets with few exceptions (Jakel et al., 2019; Perlman et al., 2020). Together, our results provide an in-depth analysis of ambient RNA contamination in single-nuclei brain datasets and reveal misinterpreted results that can be explained by ambient RNA contamination.
Results
Both nuclear and non-nuclear ambient RNAs confound cell type annotation
Studies of adult brain tissue have repeatedly shown greater number of transcripts present in neurons compared to glia (Ruzicka et al., 2020). Interestingly, several snRNA-seq studies reported some neuronal cell types (e.g. Neu-NRGN and Neu-mat in Figure 1A) that have fewer transcript counts than other neurons (Ruzicka et al., 2020; Velmeshev et al., 2019). We also noticed that Neu-NRGNs had higher mitochondrial reads than other neuronal cell types (Velmeshev et al., 2019) (Figure 1B). Since this dataset was generated by nuclei isolation, but not nuclei sorting (purification of DAPI+ nuclei with flow cytometry), we refer to it as non-sorted dataset 1 (NSD1). Considering the possibility of non-nuclear transcript contamination in nuclei-based sequencing, cell barcodes with high non-nuclear contamination should contain lower intronic read ratios since intronic reads will not be present in non-nuclear transcripts. We thus hypothesized that Neu-NRGNs and Neu-mat may be represented by cell barcodes with a high amount of such non-nuclear transcript contamination in NSD1. To test this hypothesis, we calculated the intronic read ratio per cell barcode (see methods) and found that Neu-NRGN but not Neu-mat displayed markedly lower intronic read ratios compared to other cell types (Figure 1C). To test whether these cell types are similar to normally discarded cell barcodes with low UMI counts, we then clustered an excess number of cell barcodes that contained low UMI counts together with the annotated cell barcodes from the original publication. To identify annotated cell barcodes that cluster with low UMI cell barcodes, we focused on the clusters that were largely, but not fully, filtered out (>75% filtered) in the original publication and named them “ambient clusters” (Figure 1D–E). We found that Neu-NRGN cell barcodes predominantly clustered with the ambient clusters while other cell types were almost absent in ambient clusters, except for Neu-mat (Figure 1F). If Neu-NRGN cell barcodes are indeed highly contaminated with non-nuclear ambient RNA, they should also be depleted of long non-coding RNAs (lncRNAs), which are retained in the nucleus (Guo et al., 2020). Indeed, Neu-NRGN barcodes contained fewer lncRNAs than other neurons (Figure 1G). An interesting example is MALAT1, which has elevated expression in the brain (Bernard et al., 2010) and was depleted among Neu-NRGN cell barcodes relative to other cell types (Figure 1H). Together these results indicate that Neu-NRGN cell barcodes contain high non-nuclear ambient RNA contamination and are unlikely to represent intact nuclei.
Figure 1. Neu-NRGNs are comprised of non-nuclear ambient RNAs.
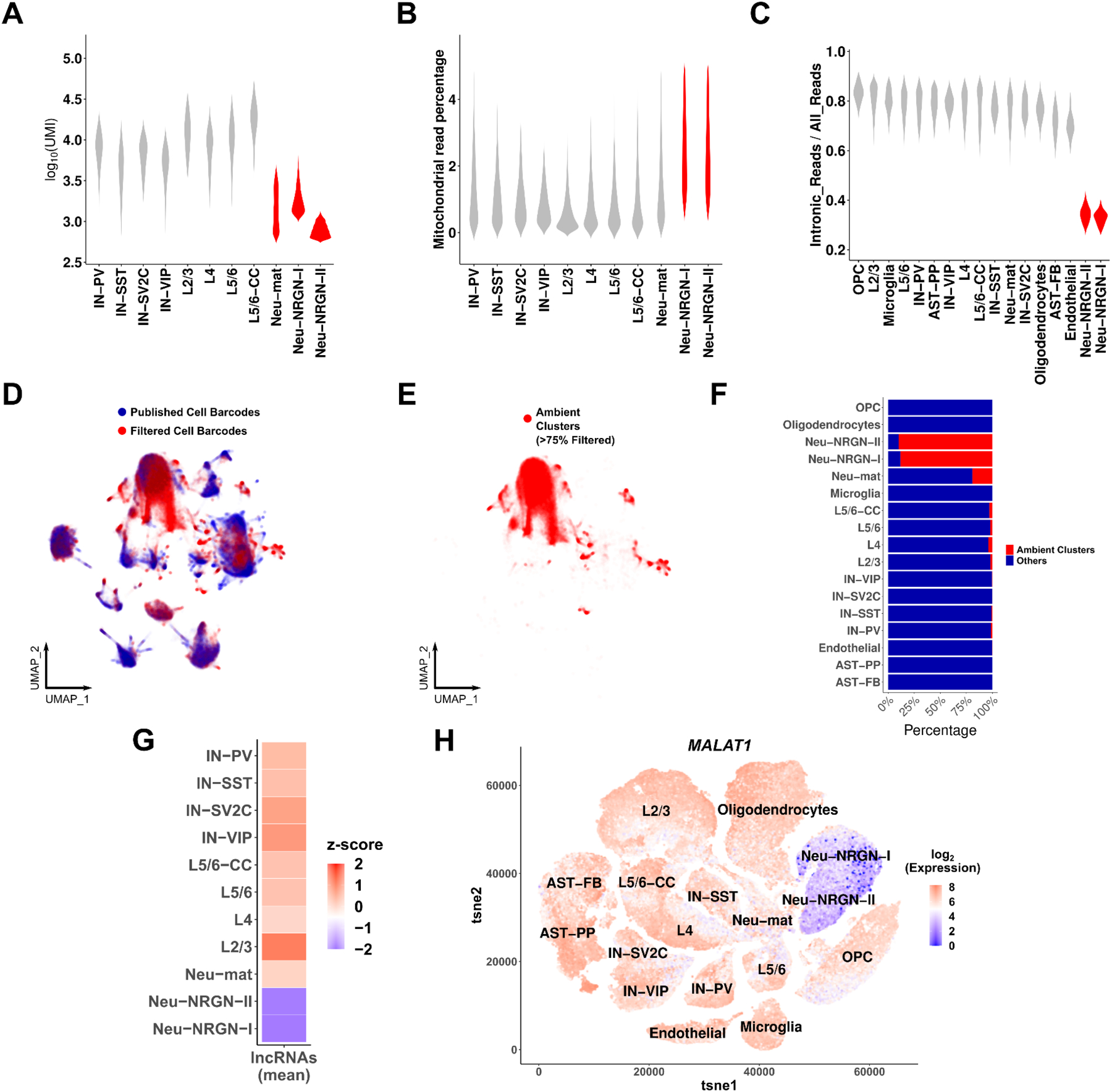
(A) Log10 transformed UMI counts per neuronal cell type in NSD1. Cell types with unusually low UMI count are shown in red. (B) Mitochondrial read percentage per neuronal cell type in NSD1. Cell types with significantly higher mitochondrial read percentage are shown in red (Wilcoxon rank sum test. P-value < 0.05). (C) Intronic read ratios of all cell types in NSD1. (D) UMAP representation after co-embedding of same numbers of published (blue) and filtered cell barcodes (red) (dataset: NSD1). (E) Clusters that are >75% composed of filtered cell barcodes are highlighted and named ambient clusters (dataset: NSD1). (F) Bar plot of the percentage of cell barcodes in ambient clusters per cell type. Red: ambient clusters, blue: other clusters. (G) Heatmap of normalized, log-transformed and z-scored expression levels of lncRNAs across cell types. The means of all lncRNAs were taken before calculating z-scores. (H) tSNE plot of the normalized and log transformed expression level of MALAT1 in all nuclei in NSD1.
We hypothesized that non-nuclear ambient RNA can be removed by fluorescence activated nuclei sorting (FANS). To test this, we analyzed another cortical snRNA-seq dataset, named sorted dataset 1 (SD1), in which the authors performed nuclei sorting (purification of DAPI+ nuclei with flow cytometry) (Lake et al., 2018). This sample preparation contrasts with the previous dataset (NSD1) we discussed that performed nuclei isolation but not nuclei sorting by flow cytometry (Figure 1). Since a low intronic read ratio indicates the presence of non-nuclear transcripts, we then determined the intronic read ratio in both datasets to assess whether non-nuclear transcripts are removed by nuclei sorting. As expected, NSD1 displayed a low intronic read ratio in cell barcodes with low UMI counts since these cell barcodes are primarily associated with ambient RNAs (Figure 2A). In contrast, we found that the intronic read ratio did not markedly change with increasing UMI counts in SD1 (Figure 2B). We then assessed ambient RNA signatures after the removal of non-nuclear ambient RNA and found ambient clusters in SD1 (Figure 2C–D). Interestingly, SD1 ambient cluster markers were highly and distinctly enriched in the Neu-mat markers from NSD1 (Figure 2E). We also observed significant enrichment of SD1 ambient cluster markers in other neuronal cell type markers although it was markedly less compared to Neu-mat. Co-clustering of SD1 ambient clusters and NSD1 cell types also grouped SD1 ambient clusters and Neu-mat cell barcodes together (Figure 2F). Overall, these results indicate that Neu-mat cell barcodes carry an unusually high nuclear ambient RNA signature and Neu-NRGN cell barcodes are distinctly contaminated with non-nuclear ambient RNA. These results also reveal that nuclei sorting, but not nuclei isolation alone, effectively removes non-nuclear ambient RNA; however, nuclei sorting cannot remove nuclear ambient RNA. Therefore, we named NSD1 ambient RNA markers as ‘non-nuclear ambient markers’, and SD1 ambient RNA markers as ‘nuclear ambient markers’ (Table S2). We selected a combination of the top 500 nuclear ambient markers and the top 500 non-nuclear ambient markers (logFC > 1 & FDR < 0.05) for the subsequent enrichment analyses.
Figure 2. Non-nuclear and nuclear ambient RNAs are distinct from each other.
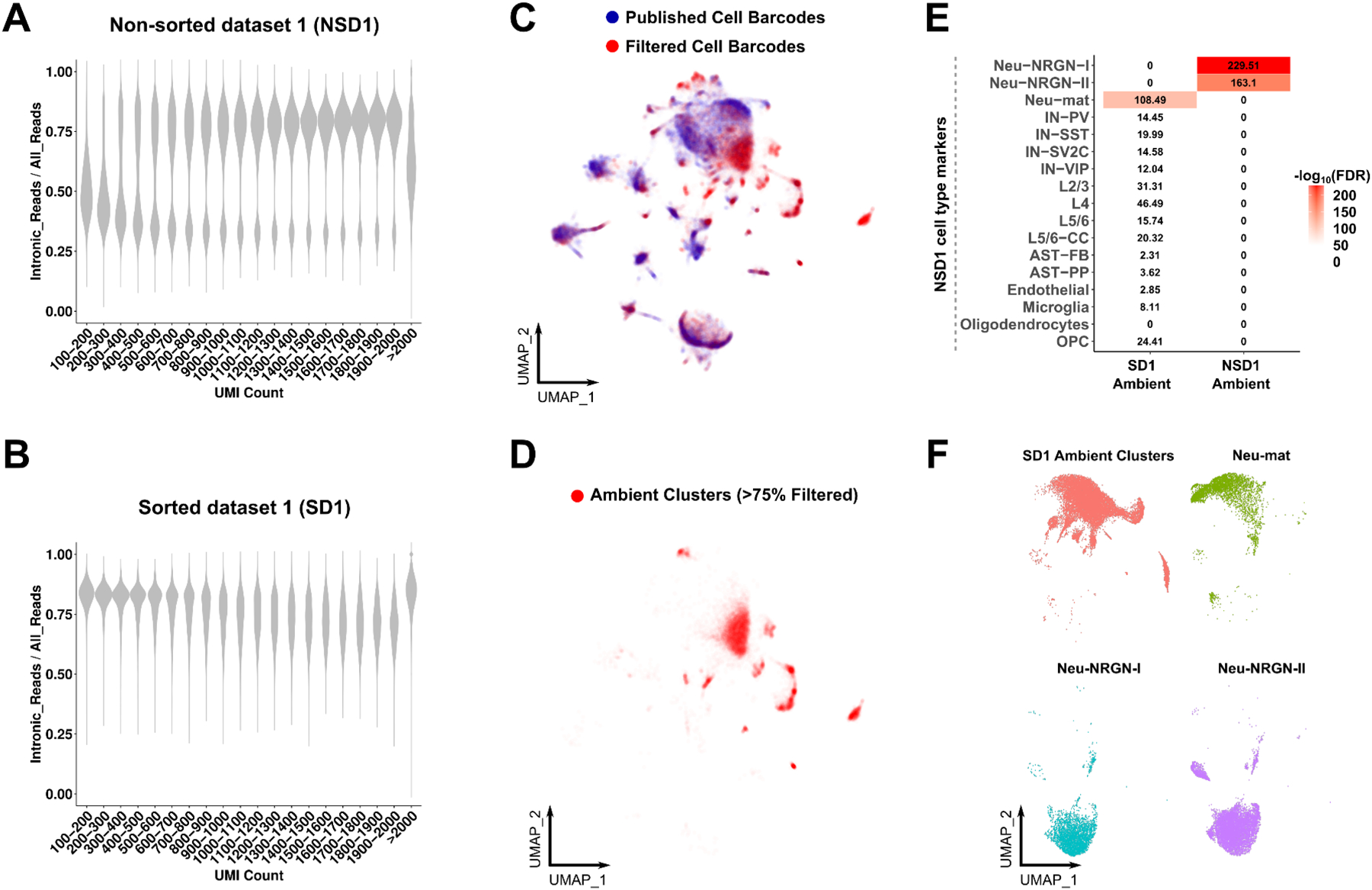
(A-B) Intronic read ratio across increasing UMI count in (A) NSD1 and (B) SD1. UMI counts are divided into intervals of 100 from 100–2000. (C) UMAP representation after co-embedding of the same numbers of published (blue) and filtered cell barcodes (red) (dataset: SD1). (D) Clusters that are >75% composed of filtered cell barcodes are highlighted and named ambient clusters (dataset: SD1). (E) Heatmap of enrichments between ambient RNA markers and Neu-mat or Neu-NRGN cell types in NSD1 (Fisher’s exact test, log10(FDR)). (F) Co-embedding of Neu-NRGNs, Neu-mat and SD1 ambient clusters. See also Figures S1–2 and Tables S2–3.
To provide independent support for these results, we then analyzed a second snRNA-seq cortical human dataset that did not include nuclei sorting (non-sorted dataset 2, NSD2, see Methods and Table S1) and a cortical human dataset that was generated after nuclei sorting (sorted dataset 2, SD2) (Tran et al., 2021). We reproduced a similar pattern of increasing intronic read ratio with increasing UMI in the NSD2, whereas this was not observed in SD2 (Figure S1A–B). We then similarly identified ambient clusters from the NSD2 dataset (Figure S1C). The intronic read ratio distribution in ambient clusters was bimodal and we divided the clusters into two categories: cell barcodes with high intronic read ratio (High-Intron-CB) and cell barcodes with low intronic read ratio (Low-Intron-CB) (Figure S1D). In line with the previous results, genes overrepresented in Low-Intron-CB were highly enriched in non-nuclear ambient RNA markers whereas High-Intron-CB were enriched in nuclear ambient RNA markers (Figure S1E).
Signatures and sources of non-nuclear and nuclear ambient RNAs
To better understand the ambient RNA marker genes, we performed gene ontology enrichment and found that non-nuclear ambient RNA markers are enriched for genes involved in ribosomal, mitochondrial, and synaptic functions, whereas nuclear ambient RNA markers are enriched for genes related to synaptic function (Figure S2A–B; Table S3). As previously hypothesized (Thrupp et al., 2020), we asked whether non-nuclear ambient RNAs are enriched in genes comprising synaptosomes, which are also marked by ribosomal, mitochondrial and synaptic activity (Hafner et al., 2019). Using the top 500 markers in each ambient RNA group, we found that non-nuclear ambient RNAs are more enriched than nuclear ambient RNAs for transcripts of both vGLUT1-depleted (originating from postsynapse + soma) and vGLUT1-enriched (originating from presynapse) synaptosome markers (Hafner et al., 2019) (Figure S2C). Since nuclear ambient RNAs also showed significant association and were enriched in overall synaptic function but not ribosomal and mitochondrial function (Table S3), we then hypothesized that nuclear ambient RNAs are derived from highly expressed genes captured in neuronal nuclei. Indeed, nuclear ambient RNAs largely overlapped with genes that are highly expressed in neurons (Figure S2C). We note that this also explains the significant enrichments between neuronal cell type markers and nuclear ambient RNA markers that we observed (Figure 2E). We also observed that the top non-nuclear ambient RNA and nuclear ambient RNA markers were also distinct, further underscoring that ambient RNAs are derived from different sources (Figure S2C).
Ambient RNA contamination of glial nuclei can be removed in silico
Given that neuronal genes are overrepresented in both ambient RNA types, we then hypothesized that ambient RNA contamination can make the transcriptomic profile of glial cell types appear more neuronal-like. As sorting for nuclei that do not express the neuronal marker NeuN (referred to as NeuN-) prior to droplet capture should remove neuronal ambient RNAs, we compared sorted datasets (SDs) and non-sorted datasets (NSDs) with three NeuN- sorted snRNA-seq datasets (NeuN- SD) (Bakken et al., 2021; Hodge et al., 2019; Sadick et al., 2022). We identified genes significantly overrepresented in the four exemplar datasets (SD1, SD2, NSD1, NSD2) compared to the three NeuN- sorted datasets in 6 major cell types: excitatory and inhibitory neurons, oligodendrocytes, OPCs (oligodendrocyte progenitor cells), astrocytes and microglia (Table S4). We called these genes “NeuN- depleted genes”. For each comparison, we then selected the top 500 NeuN- depleted genes and performed enrichment for ambient RNA markers. Despite the neuronal signature of ambient RNA markers, their enrichments within the NeuN- depleted genes were consistently significant in all glial cell types across the studies (Figure 3A–B). Notably, the association of ambient RNA markers and NeuN- depleted genes was consistently less in neuronal cell types than in glia (Figure 3A–B). Together these results show that glial nuclei in cortical snRNA-seq are likely contaminated with neuronal ambient RNAs.
Figure 3. Ambient RNAs contaminate glia expression profiles.
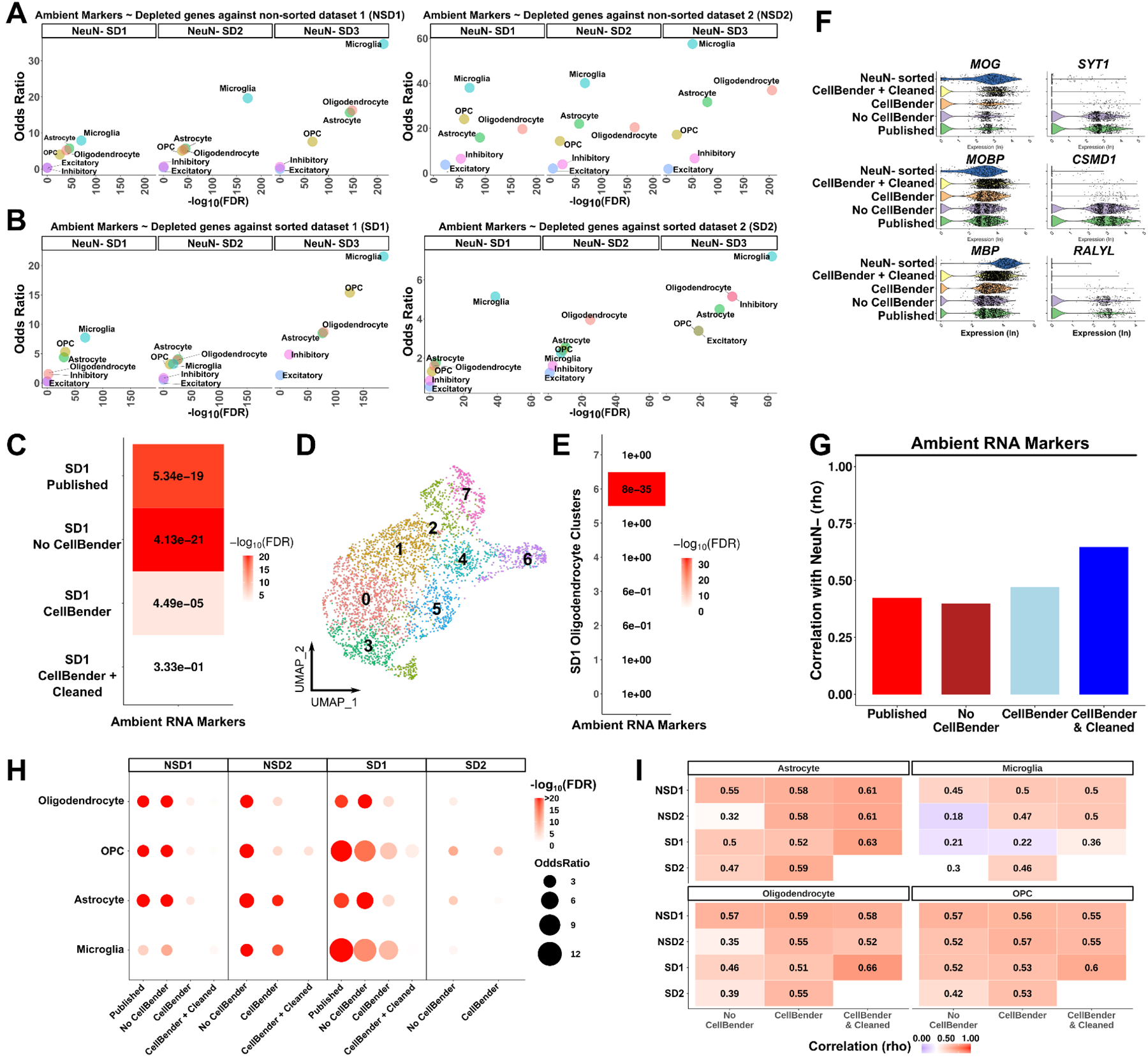
(A-B) Dot plots using odds ratio and FDR adjusted p-values as measurements of ambient RNA enrichment of genes depleted in NeuN- sorted datasets compared to other datasets that did not perform NeuN- sorting; comparisons include: (A) between NSDs and NeuN- SDs; (B) between SDs and NeuN- SDs (per major cell type using a Fisher’s exact test). (C) The same enrichment as in (A-B) after each analysis (in y-axis as rows) performed in oligodendrocytes from the SD1 dataset. Numbers: FDR value; colors scale: −log10(FDR). (D) UMAP plot of SD1 oligodendrocytes after CellBender. (E) Heatmap of enrichment between oligodendrocyte cluster markers and ambient RNA markers. (F) Violin plots of gene expression (log transformed) in oligodendrocytes after each analysis. Left column: oligodendrocyte markers, right column: ambient RNA markers. NeuN- sorted: NeuN- SD1. (G) Spearman rank correlations of all genes between SD1 oligodendrocytes and NeuN- sorted oligodendrocytes after each analysis (x-axis). (H) The same enrichment as in (C) performed in all datasets and glial cell types after each analysis. (I) Spearman rank correlations of all genes with the NeuN- sorted dataset. Correlations were performed per cell type per dataset (y-axis) after each analysis (x-axis). The numbers and color of the heatmaps indicate the correlation coefficient. See also Figures S3–9 and Table S4.
Our findings suggest that neuronal ambient RNAs contaminate glial nuclei unless samples are sorted to remove neuronal nuclei prior to droplet capture. To further test our finding, we used a dataset that employed two sorting strategies: one that depleted neurons (NeuN- and LHX2+ sorting also referred to as NeuN- SD3 in our comparisons) and one that did not deplete neurons (SOX9+ sorting) (Sadick et al., 2022) (Figure S3A–C). In line with our hypothesis, ambient RNA markers appeared less ‘expressed’ across glial nuclei in the dataset with neuron depletion (Figure S3D), and genes less represented in the neuron depleted dataset were significantly enriched in ambient RNA markers (Figure S3E). We note that two ambient RNA markers we exemplify (CSMD1 and RALYL) showed similar expression patterns between the two datasets. These genes are likely endogenously expressed in OPCs as high expression levels are detectable across the other NeuN- SDs (Bakken et al., 2021; Hodge et al., 2019), underscoring the importance of distinguishing ambient RNA contamination from endogenous transcripts per cell type. Together, these results provide further proof of neuronal ambient RNA contamination in the cortical snRNA-seq datasets.
Ambient RNA contamination within droplets that contain real nuclei is a general problem in snRNA-seq experiments and various tools exist to remove ambient RNA contamination (Fleming et al., 2019; Yang et al., 2020; Young and Behjati, 2020). To assess the performance of these tools in the analyzed datasets, we used NeuN- SDs as the ground truth and asked which tool will lower the percentage of reads explained by ambient RNA markers to the levels observed in NeuN- SDs in glial cell types. We applied SoupX (Young and Behjati, 2020), DecontX (Yang et al., 2020) and CellBender (Fleming et al., 2019) with default parameters on each dataset. Overall, we observed a lower percentage of ambient RNA in NeuN- SDs compared to other datasets where no removal was performed (Figure S4). Among the three ambient RNA removal tools, CellBender was consistently better at reducing the ambient RNA contamination levels across the datasets (Figure S4A–D). Comparison of all NeuN- SDs and ambient RNA removal tools in glial cell types showed that there was no significant difference between the NeuN- SDs and CellBender results in terms of the percentage of reads explained by ambient RNA markers (Figure S4E). We highlight this in oligodendrocytes from SD1 that show low levels of SYT1, CSMD1 and KCNIP4 after CellBender similar to NeuN- SDs, while DecontX and SoupX treated datasets display substantial levels of contamination (Figure S4F). Together, these results show that CellBender performs better than DecontX and SoupX to remove neuronal ambient RNA contamination from glial nuclei.
We next asked whether CellBender can fully remove ambient RNA contamination. For each dataset, we calculated the enrichments between ambient RNA markers and genes depleted in NeuN- SD1 (chosen as these genes have the lowest ambient RNA percentage in glial cell types among all NeuN- SDs) before and after applying CellBender (Figure S4A–D). Focusing on the oligodendrocytes (from SD1), we found that CellBender substantially reduced ambient RNA contamination (Figure 3C). However, enrichment was still significant, indicating that ambient RNA contamination was not fully removed. To investigate this, we next subclustered oligodendrocytes and found that markers of a small subcluster were highly enriched in ambient RNAs (Figure 3D–E). Removing this subcluster fully removed detectable ambient RNA contamination from oligodendrocytes (Figure 3C, F) and increased correlation with NeuN- sorted oligodendrocytes (Figure 3G). We then applied this procedure to each glial cell type per dataset and found that there was little to no contamination after CellBender and additional subcluster cleaning (Figure 3H). The removed subclusters had a consistently lower intronic read ratio in datasets that did not undergo nuclei-sorting, in line with the expectation that ambient RNA contamination contains non-nuclear reads unless nuclei are physically sorted (Figure S5A). Indeed, nuclei-sorted datasets contained similar intronic read ratios between removed subclusters and other nuclei. (Figure S5B). We note that SD2 contained fewer number of nuclei compared to the other datasets and did not demonstrate robust subclusters; thus, we omitted subcluster cleaning for SD2.
While neuronal nuclei are also expected to be contaminated with ambient RNAs, NeuN based sorting is not helpful in revealing ambient RNA contamination in neuronal nuclei since ambient RNAs are dominated by neuronal signatures. To assess the levels of ambient RNA contamination in neuronal nuclei, we leveraged the lower intronic read ratio of relatively more contaminated nuclei in the non-sorted datasets (Figure S5A). To reveal this association for all nuclei, we calculated a non-nuclear ambient RNA percentage and assessed its association with a non-intronic read ratio. Both measures are expected to be higher in ambient RNA contaminated nuclei. Indeed, we observed high correlations between the two metrics for all cell types (Figure S6A–C, E). In line with the previous results, ambient RNA contamination was decreased by CellBender and further removed by subcluster cleaning in all glial cell types (Figure S6A–B). Strikingly, CellBender did not reduce ambient RNA contamination from the neurons (Figure S6C, E). As expected, non-nuclear ambient RNA markers NRGN and CHN1 levels were higher in more contaminated neuronal nuclei whereas nuclear-retained MALAT1 levels were lower (Figure S6D, F). Contamination patterns were similar among the previously annotated neuronal subtypes of NSD1, indicating that ambient RNA contamination in neurons is a cell-type agnostic problem and, unlike glia, is not accounted for by CellBender (Figure S7A–B). Other ambient RNA contamination removal tools were also more effective in glia than neurons, indicating a general deficiency in the current methods to remove ambient RNA contamination from the dominant cell type in the tissue (Figure S7C–D).
To test the effect of ambient RNA removal on all genes, we then assessed the correlation of all expressed genes between a given dataset and a NeuN- sorted dataset. We found that ambient RNA removal consistently increased overall correlations, indicating that ambient RNA removal results in better reproducibility between datasets (Figure 3I). Neurons were also similarly correlated with the NeuN+ sorted dataset before and after CellBender (Figure S5C). These results indicate that ambient RNA contamination in glia can be effectively removed with CellBender and subcluster cleaning without undesired effects on the overall gene expression profile.
Ambient RNA contamination is also detected in a mouse brain snRNA-seq dataset
To assess if ambient RNA contamination is similar in mouse cortical snRNA-seq data, we generated snRNA-seq datasets from the frontal cortex of four P56 (postnatal day 56) mice. Similar to human datasets, the intronic read ratio was less in cell barcodes with low UMI counts (Figure S8A) and ambient clusters were distributed bimodally with Low-Intron-CB and High-Intron-CB (Figure S8B–C). Low-Intron-CB markers were also enriched in non-nuclear ambient RNAs whereas High-Intron-CB markers were enriched in nuclear ambient RNAs (Figure S8D). We similarly ran CellBender and performed subcluster cleaning on glial cell types. Both steps selectively removed the ambient RNA signature from all glial cell types (Figure S8E). Thus, we conclude that ambient RNA types and contamination of glial cell types by neuronal ambient RNAs are not specific to human brain datasets.
In-situ hybridization reveals no overlap of ambient RNA markers and glia
Since ambient RNA contamination arises from the RNAs released from dissociated cells and nuclei, we hypothesized that assays carried out using intact tissue should reveal considerably lower expression of neuronal ambient markers in glia. To test this, we used probes to an oligodendrocyte marker (Mog) and three ambient RNA markers (Rbfox1, Snap25, Syt1) and performed pairwise single molecule fluorescent in-situ hybridization (smFISH) on cortical slices from adult mice. Indeed, we found almost no overlap between any of the three ambient RNA markers and Mog (Figure S9A–D; Table S5). In contrast, the snRNA-seq from mouse frontal cortex indicated that >75% of nuclei contained reads from all three markers in the oligodendrocytes (Figure S9E). This prevalent contamination was abolished after the ambient RNA removal process (CellBender + subcluster cleaning) (Figure S9E). These results provide further support for the prevalence of neuronal ambient RNA contamination in glial snRNA-seq nuclei.
Previously annotated immature oligodendrocytes are glia contaminated with ambient RNAs
Glia can express genes that are typically associated with neuronal function. For example, OPCs can make synapse-like contacts with axons and express glutamatergic receptors that bind to neurotransmitters secreted by neurons, affecting oligodendrocyte maturation in vitro (Fields, 2015; Luse and Korey, 1959; Wake et al., 2011). We also found that glutamatergic receptors functionally studied in oligodendrocyte maturation (e.g. GRIA2, GRIA4, GRM5 in OPCs (Fields, 2015; Kougioumtzidou et al., 2017; Wake et al., 2011)) remain present in our analysis after ambient RNA removal (Figure S10A). However, such ambiguity in cell-type expression patterns raises the possibility that neuronal ambient RNA contamination in glia might have been implicated with biological function in previous snRNA-seq studies. For example, the snRNA-seq study that generated SD1 identified “immature oligodendrocytes”, which were marked by greater expression of many neuronal genes, but many marker genes of this cell type annotation were not independently validated (Lake et al., 2018). Based on our findings, we considered the alternative possibility that this excessive neuronal gene expression signature is ambient RNA contamination. In line with this interpretation, we found that ~80% (46 out of 58) of immature oligodendrocyte markers overlapped with the top 200 most abundant ambient RNA markers (Figure 4A). Using the gene-cell matrix from the original publication, we reconstructed the lineage trajectory between OPC-OL (Figure 4B). Cell barcodes between OPC and OL (‘transitioning cells’) showed high enrichment for both immature oligodendrocyte and ambient RNA markers (Figure 4B–C) and these cell barcodes were removed during our subcluster cleaning procedure (Figure 4D). These cell barcodes displayed similar UMI counts compared to OPC and OL indicating that they are also not glia-neuron doublets (Figure 4E). Since CellBender reduces ambient RNA contamination, we also assessed the OPC-OL trajectory after CellBender, which revealed a less continuous trajectory compared to the original dataset (Figure 4F). Similar to the original dataset, cell barcodes between OPC and OL were removed during subcluster cleaning (Figure 4G). We performed smFISH experiments to examine whether cells expressing the annotated immature oligodendrocyte markers (GRIN2A and SYT1) also express an oligodendrocyte lineage marker (OLIG2). We found essentially no overlap of expression of these genes in human cortical samples (Figure S9F–G; Table S5). In contrast, there was ~10% overlap of these markers in the SD1 snRNA-seq dataset (Figure S9H). Together, these results indicate that the OPC-OL pseudotime trajectory is driven by ambient RNA contamination rather than biological differentiation of oligodendrocytes.
Figure 4. Ambient RNA contamination causes misinterpretation of transitioning oligodendrocytes in the human brain.
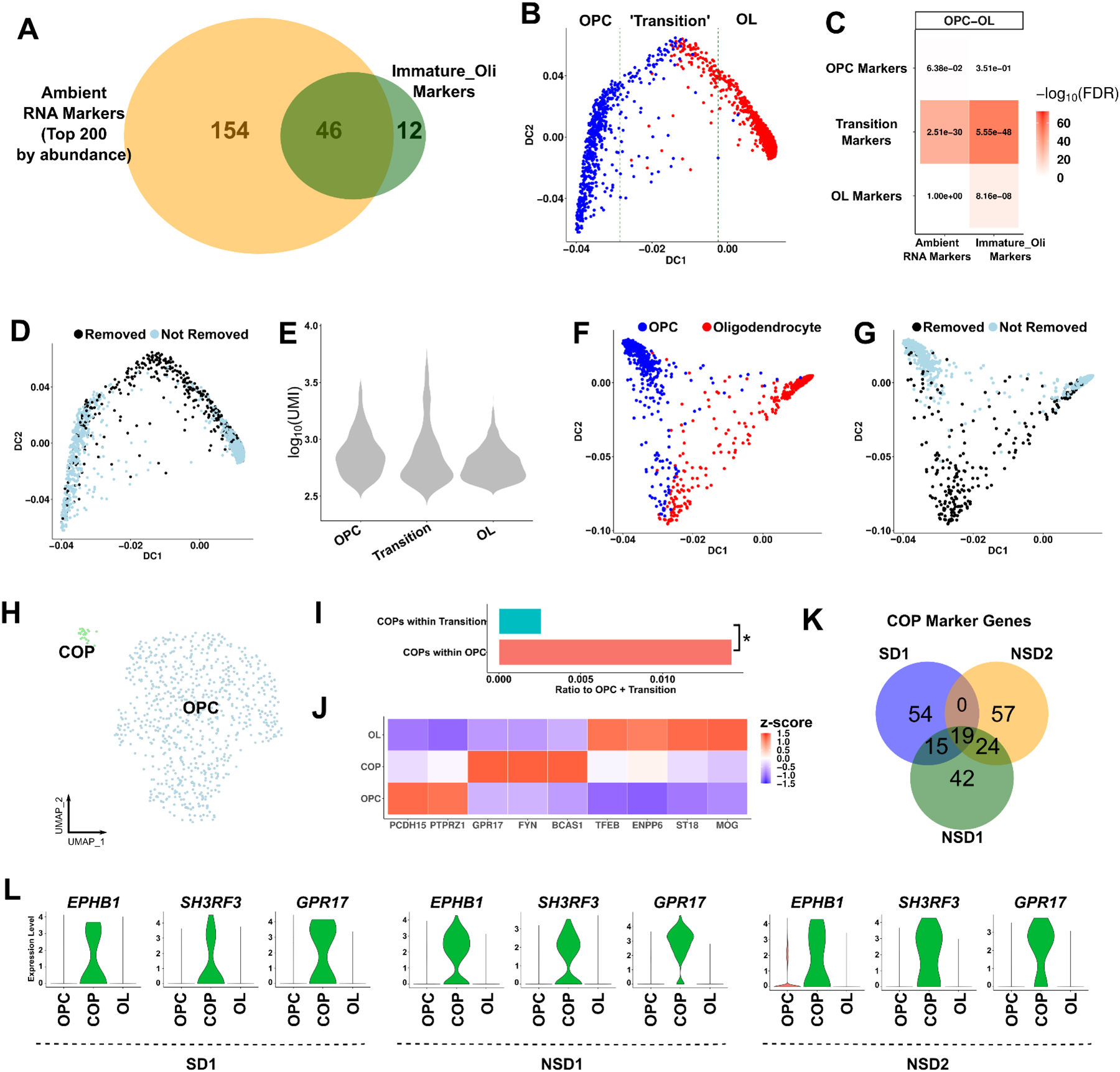
(A) Overlap of the immature oligodendrocyte markers in SD1 and the top 200 most abundant ambient RNA markers. (B) The oligodendrocyte lineage trajectory as reconstructed with destiny. The ‘transition’ zone: the 400 cell barcodes around the middle cell barcode based on DC1. (C) Heatmap enrichments between the trajectory zones (OPC, Transition, OL) and either ambient RNA or immature oligodendrocyte markers using a Fisher’s exact test. Numbers: FDR; color scale: −log10(FDR). (D) The same lineage trajectory as (B) with cell barcodes removed after subcluster cleaning highlighted. (E) UMI counts of cell barcodes within the OPC, Transition or OL zones. (F) The oligodendrocyte lineage trajectory after CellBender. (G) The same lineage trajectory as (F) with cell barcodes removed after subcluster cleaning highlighted. (H) UMAP of OPC subclustering. COP: committed OPCs. (I) The ratio of COPs within OPCs or within the transitioning cells to the total number of OPCs and transitioning cells. Asterisk: p-value < 0.05, Chi-square test. (J) Heatmap of oligodendrocyte lineage markers (z-scored across cell types per marker gene). (K) Overlap of COP markers (compared to OPCs) across datasets. The top 100 markers were selected (FDR < 0.05). (L) Violin plots of the expression levels of the top COP markers in three datasets. See also Figures S10–12 and Tables S5–6.
We hypothesized that ambient RNA contaminated nuclei can be detected as transitioning cells between any two glial cell types in pseudotime analysis as all glial nuclei are expected to contain neuronal ambient RNA contamination in a brain gray matter preparation. Therefore, we generated a pseudotime analysis between OPC and AST (astrocytes) and found similar ‘transitioning cells’ (Figure S10B–D). Since OPCs can achieve multipotency under certain conditions (Chamling et al., 2021; Sim et al., 2011; van Bruggen et al., 2017), we could not exclude the possibility that this could be a real biological function (i.e. OPCs differentiating into astrocytes). For a definitive answer, we tested two non-oligodendrocyte lineage cell types, AST and MIC (microglia), which also revealed similar ‘transitioning cells’ that were highly enriched in both ambient RNA and immature oligodendrocyte markers and were effectively removed by our ambient RNA removal process (Figure S10E–G). Given that immature oligodendrocytes also lack known markers of COPs or premyelinating oligodendrocytes (Pre-OL) (e.g. BCAS1, ENPP6, GPR17 (Hughes and Stockton, 2021)) (Figure S10H), our results indicate that nuclei previously annotated as immature oligodendrocytes in several snRNA-seq studies are not transitioning cells but rather glia with high contamination of neuronal ambient RNA.
Ambient RNA removal reveals rare cell type in adult human brain snRNA-seq datasets
Initial single cell studies on the adolescent mouse oligodendrocyte lineage identified COPs and NFOLs (newlyformed oligodendrocytes) as transitioning OL cells (Marques et al., 2016). This work established marker genes including Gpr17, which peaked in COPs, was reduced in NFOLs and was absent in mature OLs (Marques et al., 2016). A study in human induced pluripotent stem cell derived OPC culture also showed that GPR17 regulated OL maturation in human cells (Merten et al., 2018). However, few single-cell RNA-seq studies in adult human brain have identified these populations (Fernandes et al., 2021; Jakel et al., 2019). Since these studies used different annotation labels and marker genes, it is also unclear whether transitioning oligodendrocytes are consistent across human datasets. Robust annotation of these cells in human datasets is crucial to understand the role of the oligodendrocyte lineage in neurological diseases (Akay et al., 2021; Jakel et al., 2019; Nagy et al., 2020; Phan et al., 2020).
To determine whether we can identify COPs after ambient RNA removal, we subclustered OPCs. GPR17+ COPs were detectable and clustered separately than OPCs in NSD1, NSD2 and SD1 (Figure 4H, Figure S11A, C). In SD2, plotting of COP markers in the UMAP space revealed a small population of nuclei with high expression of COP markers, although they did not cluster separately due to the low number of nuclei in this dataset (Figure S11E). Importantly, COPs were significantly depleted within the transition zone of the pseudotime plot (Figure 4I), further indicating that previous pseudotime analyses were driven by ambient RNA contamination rather than demonstrating biological underpinnings of oligodendrocyte maturation. To validate the identity of COPs, we then plotted genes known to be associated with COPs or Pre-OLs (BCAS1 (Fard et al., 2017), GPR17 (Chen et al., 2009), FYN (Sperber and McMorris, 2001)) as well as genes that are upregulated in Pre-OLs but are also expressed in OLs (TFEB (Sun et al., 2018), ENPP6 (Xiao et al., 2016)). We found that BCAS1, GPR17 and FYN selectively marked COPs and TFEB and ENPP6 were upregulated in COPs compared to OPCs consistently across the datasets (Figure 4J, Figure S11B, D, E). Overall, 58 out of the 211 (27%) top markers of COPs were shared between at least two datasets (Figure 4K). To highlight previously undescribed markers for COPs in adult human brain, we then found the most specific COP markers compared to both OPCs and OLs which – in addition to GPR17, BCAS1, FYN – revealed TNS3 (Marques et al., 2016), SH3RF3, EPHB1, CRB1, SIRT2 and ARHGAP5 as additional potential markers for future studies of oligodendrocyte biology in human brain (Figure 4L; Table S6). Given that we could detect similar COP populations in all datasets, we also re-analyzed a previous study that identified COPs in adult human brain white matter (Jakel et al., 2019). Surprisingly, in the original annotation, COP markers did not have higher expression in COPs than OPCs (Figure S12A). Clustering OPCs and COPs revealed a subpopulation of nuclei that were very similar to COPs in other human datasets by their marker gene expression levels (‘COPs-New’) (Figure S12B–C). To assess whether previously annotated COPs (‘COPs-Old’) can be ambient RNA contamination, we also checked expression levels of neuronal genes. This revealed high expression of both ambient and non-ambient neuronal genes, indicating ‘COPs_Old’ might be OL-neuron doublets rather than ambient RNA contamination (Figure S12C). Indeed, COPs-Old displayed similar UMI count levels to neuronal cell types, in contrast to ambient RNA driven clusters which contained lower UMI count levels (Figure 1A, 4E, and S12D). These results provide further evidence of the extreme rarity of COPs in human brain datasets, which can be masked by technical artifacts.
Stepwise guideline for detection and removal of ambient RNAs
Our results show that a combination of existing tools and careful analysis can remove ambient RNA contamination and improve the biological relevance of results. To illustrate our approach in a more direct way, we present a stepwise guideline that outlines the major steps important in our analysis (Figure 5). While ambient RNA removal tools aim to be a one-step solution for this problem, we advise researchers to identify ambient RNA populations and their markers genes in their own dataset, which is achievable using common methods (Figure 5, Steps 1–4). This can then be used to assess whether a specific cell population is marked by high ambient RNA contamination which may not have been removed or cleaned of ambient RNAs by the specialized tools (e.g. CellBender, Figure 5, Steps 5–7). Taken together, we show pervasive contamination of glia by neuronal ambient RNAs, successfully remove them using available methods which reveals underappreciated biology of transitioning oligodendrocytes in adult human brain. We also provide a stepwise guideline outlining our integrated approach to tackle ambient RNA contamination in single-nuclei datasets from brain tissue.
Figure 5: Stepwise guidelines of ambient RNA marker detection and ambient RNA removal.
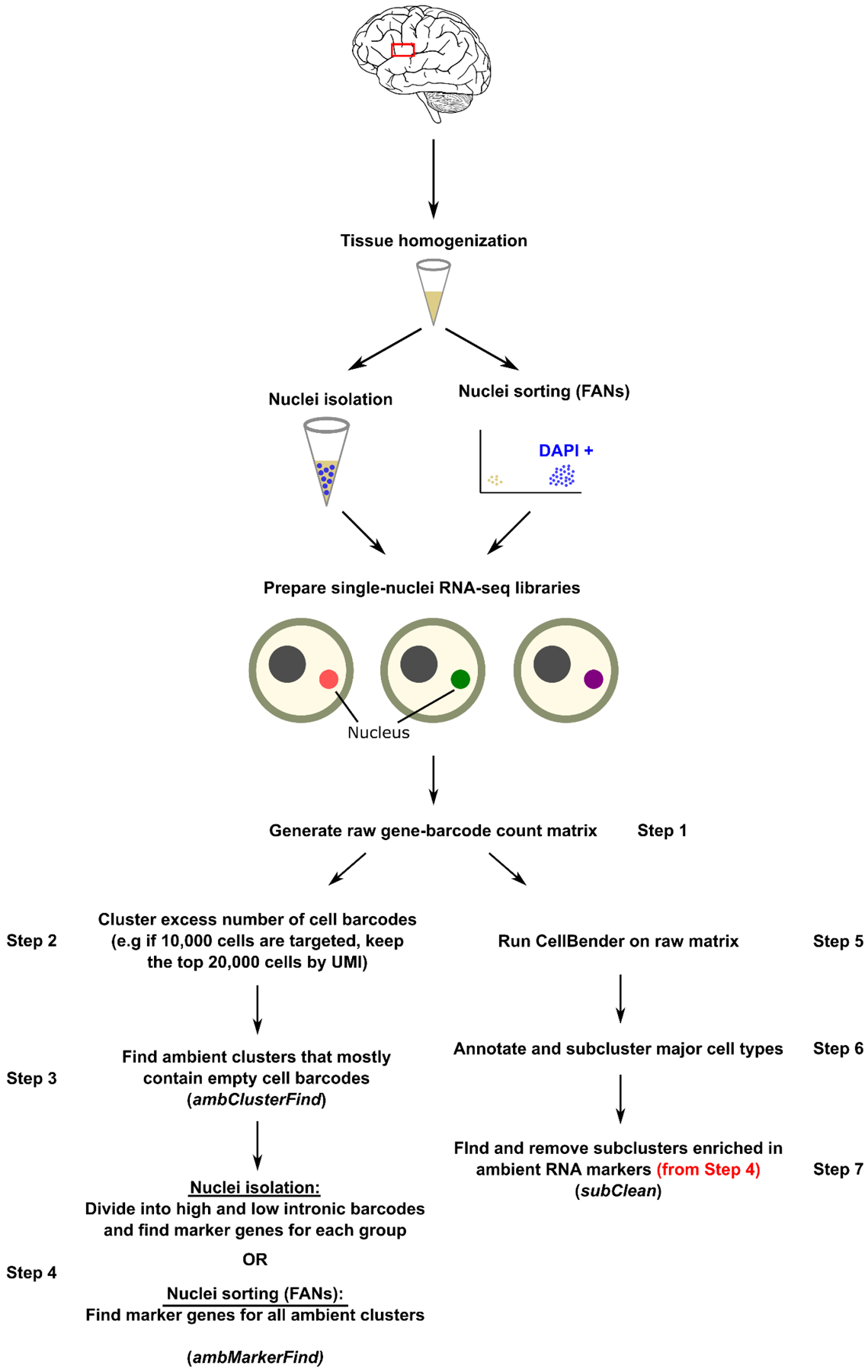
Steps 1–4 describe how to identify ambient RNA markers in the given dataset. Steps 5–7 describes how to use this information to further remove ambient RNA contaminated cell barcodes after a formal ambient RNA contamination removal tool such as CellBender is applied. Left: for non-sorted datasets, right: for nuclei-sorted datasets.
Discussion
Here, we provide an in-depth examination of ambient RNAs in brain snRNA-seq datasets. We identify nuclear and non-nuclear ambient RNAs with different gene signatures, and find that previously annotated neuronal cell types have high contamination of ambient RNAs (Ruzicka et al., 2020; Velmeshev et al., 2019). We then show that the high prevalence of neuronal reads in ambient RNAs contaminate glia, but can be effectively removed using CellBender and additional subcluster cleaning. These results are not unique to the human brain and are reproducible in mouse cortical snRNA-seq data. We also show that immature oligodendrocytes previously identified in snRNA-seq datasets are artifacts of neuronal ambient RNA contamination. After ambient RNA removal, we can identify populations of COPs in all human brain snRNA-seq datasets and highlight both known and previously undescribed markers of COPs. Finally, we provide a stepwise guideline of ambient RNA marker identification and removal.
Our findings suggest that single-nuclei isolation does not entirely remove non-nuclear reads. The presence of cell barcodes with high proportions of non-nuclear reads indicates that cytoplasmic mature RNAs also contribute to contamination during nuclei isolation. We found that marker genes of non-nuclear reads significantly overlap with mRNAs that localize to synaptosomes (Figure S2) and the non-nuclear ambient RNAs are largely abolished when the intact nuclei are physically sorted by FANS (Figure 2B and S1B). While these results indicate that non-nuclear reads are likely derived from all cell types, it is also possible that mature mRNAs can be carried over into droplets by the endoplasmic reticulum that is still attached to the nucleus after isolation or sorting. Therefore, some non-nuclear reads may be derived from the same cell as the captured nucleus.
We leveraged the intronic read ratio difference between the empty and non-empty droplets to reveal that previously annotated cell clusters (Neu-NRGNs) contain high levels of non-nuclear ambient RNA contamination and are likely empty droplets (Figure 1C). However, non-nuclear contamination measured by intronic read ratio is not sufficient to identify all healthy nuclei / cells. For example, a recent study highlighted the distinction of damaged cells and empty droplets that only contain ambient RNA (Muskovic and Powell, 2021). The authors noted that damaged cells contain similar intronic read ratio (i.e. nuclear fraction) to real cells, but they display lower UMI counts compared to other cells with similar annotation. Similarly, we find that the Neu-mat cluster has lower UMI counts compared to other neurons despite having a similar intronic read ratio, indicating that this cluster likely contains damaged nuclei (Figure 1A, C). Additionally, we observe that while empty droplets have lower intronic read ratios, these ratios are still substantially higher than zero, indicating that nuclear reads also contribute to ambient RNAs (Figure 1C, 2A). Finally, intronic read ratio is not an indicator of empty droplets in datasets that underwent nuclei sorting by flow cytometry since this procedure only removes non-nuclear ambient RNAs (Figure 2B). In nuclei-sorted datasets (e.g. SDs in this study), empty droplets can be better identified by assessing a given cluster’s enrichment for the ambient RNA markers (Figure 2E). We offer several functions to find ambient RNA markers for this purpose (Figure 5, steps 1–4).
In addition to empty droplets, ambient RNAs can contaminate all non-empty droplets. We focused on contamination in glial nuclei as they contain fewer transcripts than other cell types in the brain. We found that ambient RNA markers were underrepresented in the glial nuclei from studies that physically separated neurons and glia, indicating that some reads mapping to neuronal genes in datasets without neuron-glia separation are not representative of neuronal endogenous expression (Figure 3A–B). To remove the neuronal ambient RNA contamination in glia, we utilized CellBender (Fleming et al., 2019) and subsequent detection of subclusters with ambient RNA contamination. Together, these methods removed neuronal ambient RNA contamination from glial nuclei and improved correspondence with NeuN- sorted datasets (Figure 3C–I). Based on these results, we recommend two approaches for cortical snRNA-sequencing experiments: 1) physical separation of glia from nuclei (e.g. by FANS) or 2) in silico cleaning of neuronal ambient RNA contamination. Our approach for the in silico cleaning involves two steps: using a formal ambient RNA removal tool (CellBender) and subsequent removal of contaminated subclusters that are not successfully cleaned of ambient RNAs after CellBender (Figure 3C–E). We show that failure to remove ambient RNA contamination can have important consequences such as misannotation of contaminated glial nuclei as immature oligodendrocytes (Lake et al., 2018). Importantly, CellBender alone did not remove all ambient RNA contamination, and the remaining contaminated nuclei were positioned between OPCs and oligodendrocytes in the pseudotime trajectory (Figure 4B–G). We thus recommend utilizing both CellBender and subsequent subcluster cleaning to account for ambient RNA contamination. We provide a stepwise guideline for the in-silico approach we have taken to remove ambient RNA contamination from glial cell types (Figure 5).
Neuronal reads are abundant within the ambient RNA population, making ambient RNA contamination in glial cell types distinct from the endogenous gene expression of glial nuclei. In contrast, ambient RNA contamination in neurons is difficult to separate from the endogenous neuronal gene expression and cell barcodes with a higher percentage of ambient RNA markers may be biologically relevant. As an unbiased method, we used the positive correlation of non-intronic read ratio and ambient RNA percentage across cell barcodes as a measure of contamination in NSDs. This revealed that neither CellBender nor other tools (DecontX and SoupX) could substantially reduce ambient RNA contamination in neuronal cell types (Figure S6C–F, 7). While the cell barcodes with high contamination can be manually removed, determining a threshold for removal would be arbitrary and could reduce the number of nuclei retained for analysis. Currently, we suggest caution in interpreting “novel” neuronal cell types and cell states even if the common ambient RNA removal tools are applied. Our study shows that ambient RNA contamination in neurons can be assessed by using metrics such as intronic read ratio (if the dataset has non-nuclear ambient RNAs) and the percentage of ambient RNA markers identified from the same dataset or similarly prepared datasets from similar tissues.
We showed that analysis of nuclei from the oligodendrocyte lineage after ambient RNA removal revealed COPs in all independent datasets. While COPs have been identified before (Marques et al., 2016; Perlman et al., 2020), many studies did not annotate them (Nagy et al., 2020; Ruzicka et al., 2020; Sadick et al., 2022; Tran et al., 2021; Velmeshev et al., 2019). This could be hindered by both ambient RNA contamination and the rarity of COPs in adult brain. COPs are ~0.04% of cells in adult brain (NSD2 samples from 30–80 years old) and ~0.3% of cells in the adolescent brain ((NSD1 samples from 4–22 years old). We also showed that previously annotated COPs in adult human brain white matter are likely OL-neuron doublets and real COPs are detectable and similarly rare (~0.1% of all cells) (Figure S11). In line with this, live cell imaging in the mouse brain showed that ~80% of transitioning oligodendrocytes rapidly undergo cell death, which should result in a transient and rare cell population (Hughes et al., 2018). A carbon dating study of human genomic DNA in oligodendrocytes also showed low levels of oligodendrogenesis in adulthood, which further supports the rarity of transient cells in adult human brain (Yeung et al., 2014). Despite being rare, COPs are critical to examine since oligodendrocyte maturation is altered in both neurological diseases (de Faria et al., 2021; Phan et al., 2020) and human evolution (Miller et al., 2012; Zhu et al., 2018). Thus, ambient RNA removal is important for accurate analysis of underrepresented cell types. Another recent study also uncovered that glial cell types respond to enzymatic dissociation during single-cell and single-nucleus library preparation and confound the transcriptomic profile (Marsh et al., 2022). Here, we show that all glial cell types are also contaminated with neuronal ambient RNA transcripts, causing misinterpretation of glial single-cell analysis. Together, these results indicate that both data generation and data analysis of glial cell types should be revisited and updated.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further requests for resources should be directed to and will be fulfilled by the lead contact, Genevieve Konopka (genevieve.konopka@utsouthwestern.edu).
Materials availability
This study did not generate new unique reagents.
Data and Code Availability
Raw fastq files of mouse single-nuclei RNA-seq dataset are accessible in GEO with accession number: GSE198640. Re-analyzed processed matrices are accessible in GEO with accession number: GSE198951. All analysis codes are available in our github page: https://github.com/konopkalab/Ambient_RNA_In_Brain_snRNAseq
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human cortical tissue
Human postmortem posterior cingulate cortex samples were provided by the NIH NeuroBioBank. 17μm tissue sections were sectioned for the in-situ hybridization experiment detailed in the methods. Tissues from both adult (age range 45–75) males and females were used in the experiments. Demographic information of the samples is listed in Tables S1 and S5.
Mouse cortical tissue
All experiments were performed according to procedures approved by the UT Southwestern Institutional Animal Care and Use Committee. Mouse frontal cortex samples were collected for the single-nuclei RNA-sequencing and in-situ hybridization as detailed in the methods. Adult (postnatal day 56) male and female wildtype C57BL/6J mice were used in the experiments. Mice were maintained on a 12-hr light on/off schedule. Detailed information of the samples is listed in Table S1.
METHOD DETAILS
Preprocessing and count matrix generation
Datasets were downloaded from NCBI-GEO database (Table S1). Within the datasets, we only used the cells generated from cortical brain tissue. Specifically, NSD1 was from prefrontal and anterior cingulate cortex (n = 41 samples from anterior cingulate cortex and prefrontal cortex), NSD2 was from posterior cingulate cortex (n = 4 samples), SD1 was from prefrontal (n = 13) and visual cortex (n = 24), and SD2 was from anterior cingulate cortex (see below for final sample size). Barcode correction and filtering was done using umi_tools whitelist (retained top 20,000–80,000 cell barcodes per sample depending on the dataset to keep the ambient cell barcode population) and umi_tools extract (Smith et al., 2017). Alignment was done using STAR aligner (Dobin et al., 2013) with the reference genomes of GRCh38 (for the human datasets) or GRCm38 (for the mouse dataset). featureCount was used to count reads mapping to gene body only for uniquely mapping reads (Liao et al., 2014), and umi_tools count was used to create the count matrix. Count matrix using only intronic reads were similarly obtained using featureCount on a custom gtf that only contained introns (created using construct_introns from gread package in R (Srinivasan, 2016)). Intronic read ratios were then calculated per cell barcode by taking the ratio of the number of UMI counts mapping to introns and the number of UMI counts mapping to the gene body.
For DAPI+ sorted datasets, a Spearman’s rank correlation between UMI counts (log10) and intronic read ratio was computed for each sample. Only the samples with correlation lower than the correlation coefficient of 0.05 were considered sorted and used for further analysis. This criterion retained all samples in the SD1 study (Lake et al., 2018) and one sample (Br5400_sACC) in the SD2 study (Tran et al., 2021).
For ambient RNA cleanup, CellBender was used on the raw matrix of gene counts with default parameters (Fleming et al., 2019).
Single-nuclei library preparation
We processed 4 c57BL/6J P56 mice (2 males and 2 females). Mice were rapidly decapitated and brains were quickly removed. The isolated brain was quickly transferred to an ice-cold coronal brain section mold (Braintree Scientific, BS-A 5000) and washed with ice-cold 1X PBS (Cytiva, SH30256.01). The boundary of the olfactory bulb and frontal cortex was aligned to the first indentation where the first razor blade (Fisher Scientific, 12–640) was inserted to remove the olfactory bulb. The second razor blade was inserted into the third indentation. The coronal sections matched with coronal numbers 22–36 in the Allen Brain Atlas: Mouse Reference Atlas, Version 2 (2011). We then removed the subcortical region from this section and separated the left and right hemisphere samples into different Eppendorf tubes. The tubes were flash frozen in liquid nitrogen.
The nuclei isolation procedure was modified from our previous work (Ayhan et al., 2021). The frozen section from the left hemisphere was transferred to a Dounce homogenizer with 2ml of ice-cold Nuclei EZ lysis buffer (Sigma-Aldrich, NUC101). We then inserted pestle A for 23 strokes followed by pestle B for 23 strokes on ice. The homogenized sample was transferred to a 15ml conical tube. We added 2ml of ice-cold Nuclei EZ lysis buffer and incubated on ice for 5min. Nuclei were collected by centrifuging at 500 g for 5min at 4C. We discarded the supernatant and added 4ml of ice-cold Nuclei EZ lysis buffer to resuspend nuclei. We then repeated the incubation and centrifuge steps and resuspended the nuclei in 200ul of nuclei suspension buffer: 1X PBS, 1% BSA (ThermoFisher, AM2618), and 0.2 U/ul RNAse inhibitor (ThermoFisher, AM2696). Finally, the nuclei suspension was filtered twice through Flowmi Cell Strainers (Bel-Art, H13680–0040). We mixed 10ul of nuclei suspension with 10ul of 0.4% Trypan Blue (Gibco, 15-250-061) and loaded this suspension on a hemocytometer (SKC, DHC-N015) to determine the concentration. 10,000 nuclei/sample were used to prepare snRNA-seq libraries using 10X Genomics Single Cell 3′Reagent Kits v3 (Zheng et al., 2017). Libraries were sequenced by the McDermott Sequencing Core at UT Southwestern on a NovaSeq 6000.
Tissue processing, single-nuclei RNA-seq library preparation and sequencing for the NSD2 dataset was performed as previously described (Ayhan et al., 2021).
Ambient cluster analysis
To retain cell barcodes that predominantly contain ambient RNAs, we kept two times more cell barcodes than the original publication per sample. For datasets generated in this study, we retained two times more cell barcodes than the number of nuclei targeted. Therefore, the final count matrix included both the cell barcodes that mostly represented real nuclei (and were annotated as real cell types in the published datasets) and newly retained cell barcodes that mostly represented empty droplets. Since not all newly retained cell barcodes are predominantly ambient RNAs (e.g. they could be doublets, or low quality nuclei of various cell types), we then performed clustering to identify clusters that contained high numbers of newly retained cell barcodes and clustered distinctly compared to annotated cell types per dataset. The following methods from Seurat v3 (Stuart et al., 2019) were used to perform and visualize clustering: normalization (SCTransform), dimensionality reduction (RunPCA), batch correction (RunHarmony, default parameters), k-nearest neighbors (FindNeighbors) on batch corrected dimensions and clusters identification by shared nearest neighbors (FindClusters). UMAP embedding was then computed for visualization in 2D space (RunUMAP). Clusters that were largely composed of newly retained cell barcodes (>75%) were annotated as ambient clusters. We note that 75% is unusually high since only 50% of the newly retained barcodes were originally filtered out in the previous publication.
To identify ambient cluster marker genes, we ran DGE (differential gene expression) analysis using pseudobulk edgeR (Chen et al., 2016). Briefly, counts were aggregated per sample and pseudobulk DGE was run with pseudoBulkDGE function (method = ‘edgeR’) in the scran package (Lun et al., 2016). Ambient cluster markers were identified with logFC > 0.3 and FDR < 0.05 cutoffs.
Enrichment of ambient cluster markers with annotated cell types was done using a Fisher’s exact test from the GeneOverlap package (Shen L, 2020). The total number of expressed genes were used as background (we followed this strategy for all Fisher’s exact test enrichments).
Comparison with NeuN- datasets
To find differentially expressed genes in each dataset compared to the NeuN- sorted datasets, we first identified 6 major cell types in all datasets: excitatory neurons, inhibitory neurons, oligodendrocytes, OPCs, microglia and astrocytes. Using pseudobulk DGE (see above) in matched cell types, we then identified differentially expressed genes with significantly higher number of reads than in the given NeuN- sorted dataset. This was performed separately for each NeuN- sorted and other datasets. For the NeuN- sorted dataset and SD2 comparisons we used Wilcoxon rank sum test (FindMarkers function from Seurat) since we retained only one sample from this dataset (see Preprocessing and Count Matrix Generation).
For enrichment with ambient cluster markers, we selected the top 500 differentially expressed genes (ranked by logFC) among the NeuN- depleted genes in each comparison (logFC > 1 and FDR < 0.05). Similarly, the top 500 ambient RNA markers were selected from both nuclear ambient RNA and non-nuclear ambient RNA markers. Enrichment analyses were performed as above.
Ambient RNA removal with CellBender, DecontX and SoupX
All ambient RNA removal tools were run with the default parameters and according to the instructions. For CellBender (Fleming et al., 2019), the input was the raw gene – cell barcode count matrix. For DecontX (Yang et al., 2020), the input was the filtered matrix as recommended (Yang et al., 2020). For SoupX, both the filtered and raw matrices were given as input (Young and Behjati, 2020).
Subcluster cleaning of glia after CellBender
To subcluster glia after CellBender, we used the annotation provided in the original publication and processed each glia cell type separately per study. For the datasets generated in this study, we performed clustering as described and annotated glia based on established marker genes (e.g. MBP, PCDH15, APBB1IP, SLC1A3). Clustering was done similarly as above and marker genes of subclusters (identified using the default parameters in Seurat’s FindAllMarkers (Stuart et al., 2019) function were tested for enrichment of ambient RNA markers using a Fisher’s exact test. For this, we selected the top 500 (by logFC) ambient RNA markers from both nuclear and non-nuclear ambient RNA lists and combined them. We removed the subclusters with distinctly high levels of enrichment of ambient RNAs (FDR < 0.001 and odds ratio > 3) compared to other subclusters. All steps of ambient RNA contamination removal are outlined in Figure 5. We also showcase our mouse snRNA-seq dataset and provide analysis scripts that match each step in the stepwise guideline in our github page: https://github.com/konopkalab/Ambient_RNA_In_Brain_snRNAseq
Assessment of ambient RNA contamination signatures in glia
To compare ambient RNA contamination in glia after each type of analyses, we first found cell barcodes common between annotated cell barcodes in each original publication and the retained cell barcodes after CellBender. We then only retained common cell barcodes in all downstream analyses that compared the original dataset and analyses that included ambient RNA contamination removal (Figure 3). This was to ensure that only gene expression levels were different and the enrichments are not driven by cell barcode differences between analyses. However, we note that the analysis with CellBender + subcluster cleaning contained fewer cell barcodes as ambient RNA rich subclusters were removed after CellBender. To test ambient RNA marker enrichment, we first found differentially expressed genes with significantly greater number of reads than in the NeuN- sorted dataset per cell type per dataset (logFC > 1 and FDR < 0.05). These gene lists were then tested for enrichment of ambient RNA markers (the combined top 500 genes were used as before) using a Fisher’s exact test.
To test whether the global gene expression profile is altered after these different analysis methods, we found genes expressed in at least 5% of cells per cell type per dataset to remove lowly expressed genes. We then kept the genes that survive this threshold in all datasets and performed a Spearman rank correlation between each dataset and the NeuN- sorted dataset using the average log expression of genes in the normalized matrix.
In-situ hybridization and image quantification
Flash-frozen human postmortem cortical BA23 samples (n=3) and mouse whole brains (n=2) were embedded in Tissue-Tek CRYO-OCT Compound (#14-373-65, Thermo Fisher Scientific). We sectioned tissue at −20C to 17μm on Superfrost Plus Microscope slides (#12-550-15, Thermo Fisher Scientific). Fluorescent in situ hybridization (FISH) was performed using RNAScope® Multiplex Fluorescent Reagent Kit v2 assay for fresh frozen tissue (#323100, Advanced Cell Diagnostics) with the additional step of 0.05% Sudan Black B incubation at room temperature for 10 minutes after application of DAPI to quench autofluorescence. Species-specific probes were used for human: Hs-GRIN2A-C1 (485841), Hs-OLIG2-C2 (424191-C2), Hs-SYT1-C3 (525791-C3) and mouse: Mm-Syt1-C1 (491831), Mm-Rbfox1-C1 (519911), Mm-Snap25-C1 (516471), Mm-Mog-C2 (492981-C2) respectively. Opal fluorophores 520 (FP1487001KT, Akoya Biosciences), 570 (FP1488001KT, Akoya Biosciences) and 620 (FP1495001KT, Akoya Biosciences) were used to label C1, C2, and C3 channel respectively for the gene-specific probes after signal amplification.
We captured images from cortical areas of human and mouse by using a Zeiss LSM 710 at x20 magnification in the UT Southwestern Neuroscience Microscopy Facility. Maximum intensity projection images were generated from 13 slices of a Z stack. We randomly sampled 2–4 cortical areas (488×488 μm) from each brain section for both human and mouse to manually quantify the number of cells (DAPI 405 nm), neurons (GRIN2A, Syt1, Rbfox1, and Snap25, 488 nm; SYT1, 594 nm), and oligodendrocytes (OLIG2 and Mog, 555 nm). We then calculated the fraction of neurons, oligodendrocytes, and overlap between the two cell types.
Pseudotime analysis
To be consistent with SD1 (Lake et al., 2018), we used the DiffusionMap function from destiny (Angerer et al., 2016) only on the visual cortex samples to build pseudotime trajectories between OPC-OL or between other pairs of glial cell types using the matrix provided by the authors. Diffusion maps were created with parameters n_pcs=100 and k=100. The first two eigenvectors of diffusion maps were plotted for visualization. To identify markers of ‘transitioning’ cell barcodes, we found the middle cell barcode based on the first eigenvector (DM1) and labeled 200 cell barcodes around the middle cell barcode as ‘transitioning cells’. The remaining two groups of cell barcodes were labeled by their original annotation label (e.g. OPC). We then found marker genes for each of these pseudotime groups (FDR < 0.05 and logFC > 0.25 using FindMarkers in Seurat (Stuart et al., 2019)) and ran enrichment with ambient RNA markers and immature oligodendrocyte markers identified in SD1 using a Fisher’s exact test.
OPC subcluster analysis
To identify potential transitioning OPCs, we separately subclustered OPCs from three different datasets: SD1 (Lake et al., 2018), NSD1 (Velmeshev et al., 2019), and NSD2 (GEO accession: GSE198951) after CellBender and subcluster cleaning based on high ambient RNA contamination. We further removed subclusters with high expression of markers from two distinct major cell types as potential doublets. Committed oligodendrocyte progenitors (COPs) were identified by high expression of GPR17 (as previously established (Marques et al., 2016)) among other markers (e.g. BCAS1, FYN).
To identify subclusters of Jäkel et al. (Jakel et al., 2019), we performed dimensionality reduction and clustering on cells with the annotation of ‘OPCs’ and ‘COPs’ using Seurat v3 as described above. We then identified ‘COPs-New’ by the established marker genes (BCAS1, FYN, GPR17). For the heatmaps, all mature oligodendrocytes were combined and annotated as ‘OL’. Normalized and log transformed expression levels for each gene was then z-transformed across 4 cell type annotations (OPC, COP-New, COP-Old, OL). For the UMI counts plots, we retained the original labels for neuronal cell types. Both control and multiple sclerosis samples were used and no additional cell filtering (other than subsetting by annotation) was applied for all analyses.
Identification of COP Marker Genes
Genes upregulated in COPs compared to OPCs were identified using the FindMarkers function from Seurat. Significant genes (FDR < 0.05 and expressed in >10% of COPs) were ranked by their avg_logFC and the top 100 genes per dataset were selected.
To highlight genes specific to COPs compared to OPCs and OLs, we found the percentage of nuclei that expressed at least one read of each significant gene. We then computed the difference of percentages between both COPs-OPCs and COPs-OLs. We then took the intersection of the top 20 genes with the greatest difference in favor of COPs in both comparisons. This was repeated for all three datasets. Genes that marked COPs in at least two datasets were reported as COP markers within the oligodendrocyte lineage in human brain (Table S5)
Other enrichments
Gene ontology (GO) enrichment of ambient RNA signatures was done using the clusterProfiler package in R (Yu et al., 2012) with all expressed genes used as the background. The full table of GO results is available in Table S3.
To test enrichment of ambient RNA markers with vGLUT1-Depleted and vGLUT1-Enriched genes from Hafner et al. (Hafner et al., 2019), we first converted the mouse gene symbols to human gene symbols using SynGO (Koopmans et al., 2019). Fisher’s exact tests were performed as before.
To overlap ambient RNAs with highly represented genes in neurons in snRNA-seq datasets, we identified the top-represented genes among all neurons by taking the mean of each gene across all cell barcodes annotated as neurons separately in both SD1 and NSD1 (except for Neu-NRGNs and Neu-mat). The intersection of the top 500 genes in both datasets (403 genes) was used to overlap with ambient RNA markers.
Quantification and statistical analysis
All analysis-specific quantifications and statistics can be found in their corresponding method section. Individual statistics (e.g adjusted p-value, odds ratio, fold change) for each comparison can be found in the figure legends and on the figures. Sample sizes of the snRNA-seq dataset can be found in the methods. Unless otherwise stated, all samples from the associated publication were retained for the analyses of this study. Sample sizes of the in-situ hybridization experiment can also be found in the methods and the figure legends. We did not conduct a separate benchmarking for the selection of the statistical analyses, however we strived to select the most up to date and benchmarked methods (e.g we favored pseudobulk methods instead of the single-cell based methods for the differential gene expression analyses (Squair et al., 2021)).
Supplementary Material
Table S2 (Related to Figure 2): Statistics on the nuclear and non-nuclear ambient RNA markers. It also contains low-intronic ambient markers and high-intronic ambient markers for the non-sorted datasets (described in Figures S1,8).
Table S3 (Related to Figure S2): Statistic of the gene ontology enrichments of nuclear or non-nuclear ambient RNA markers.
Table S4 (Related to Figure 3): Statistics of the genes depleted in the NeuN- sorted datasets (NeuN- SDs) compared to SD1, SD2, NSD1 or NSD2 datasets. The list is ranked by the abundance of depleted genes (logCPM), all genes are depleted with FDR < 0.05.
Table S5 (Related to Figure S9): Quantification of the smFISH results.
Table S6 (Related to Figure 4): Statistics of the committed oligodendrocyte progenitor cell markers per dataset (SD1, NSD1, NSD2).
Key Resources Table
| Biological Samples | ||
| Adult mouse (P56) frontal cortex specimens | Table S1 | N/A |
| Adult human posterior cingulate cortex specimens | Table S1 | N/A |
| Deposited Data | ||
| Mouse snRNA-seq data | This paper | GEO: GSE198640 |
| Human nuclei-sorted snRNA-seq data (SD1) | Lake et al., 2018 | GEO: GSE97930 |
| Human nuclei-sorted snRNA-seq data (SD2) | Tran et al., 2021 | https://research.libd.org/globus (endpoint: jhpce#tran2021) |
| Human non-sorted snRNA-seq data (NSD1) | Velmeshev et al., 2019 | BioProject: PRJNA434002 |
| Human non-sorted snRNA-seq data (NSD2) | This paper | GEO: GSE198951 |
| NeuN- sorted dataset 1 | Hodge et al., 2019 | https://portal.brain-map.org/atlases-and-data/rnaseq/human-mtg-smart-seq |
| NeuN- sorted dataset 2 | Bakken et al., 2020 | https://assets.nemoarchive.org/dat-ek5dbmu |
| NeuN- sorted dataset 3 | Sadick et al., 2022 | GEO: GSE167494 |
| Human cortical oligodendrocyte data | Jäkel et al., 2019 | GEO: GSE118257 |
| Software and Algorithms | ||
| CellRanger v.3.0.2 | 10x Genomics | https://www.10xgenomics.com/products/single-cell-gene-expression/ |
| R version 4.1.2 | The R Project | https://www.r-project.org/ |
| Seurat_3.0.1 | Stuart et al., 2019 | https://github.com/satijalab/seurat |
| Scran_1.18.7 | Chen et al., 2016 | https://bioconductor.org/packages/release/bioc/html/scran.html |
| GeneOverlap_1.30.0 | Shen et al., 2021 | https://bioconductor.org/packages/release/bioc/html/GeneOverlap.html |
| CellBender_0.2.0 | Fleming et al., 2019 | https://github.com/broadinstitute/CellBender |
| DecontX | Yang et al., 2020 | https://github.com/campbio/celda |
| SoupX | Young & Behjati, 2020 | https://github.com/constantAmateur/SoupX |
| Destiny_3.8.1 | Angerer et al. 2015 | https://bioconductor.org/packages/release/bioc/html/destiny.html |
| clusterProfiler_4.2.2 | Yu et al., 2012 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| SynGO | Koopmans et al., 2019 | https://www.syngoportal.org/ |
| Umi-tools_1.1.1 | Smith et al., 2017 | https://github.com/CGATOxford/UMI-tools |
| STAR_2.7.10 | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Subread_2.0.1 | Liao et al., 2014 | https://sourceforge.net/projects/subread/files/ |
| Gread_0.99.3 | Srinivasan et al., 2016 | https://rdrr.io/github/asrinivasan-oa/gread/ |
| Critical commercial assays | ||
| RNAscope® Multiplex Fluorescent Reagent Kit v2 | ACD Bio-techne | Catalog #: 323100 |
| Chromium Single Cell 3′ v3 | 10x Genomics | Cat#1000153 |
| Chemicals, peptides and recombinant proteins | ||
| RNAscope® Probe-Hs-OLIG2-C2- mRNA | ACD Bio-techne | Catalog #: 424191-C2 |
| RNAscope® Probe Hs-SYT1-C3- mRNA | ACD Bio-techne | Catalog #: 525791-C3 |
| RNAscope® Probe-Hs-GRIN2A- mRNA | ACD Bio-techne | Catalog #: 485841 |
| RNAscope® Probe-Mm-Mog-C2 mRNA | ACD Bio-techne | Catalog #: 492981-C2 |
| RNAscope® Probe-Mm-Syt1- mRNA | ACD Bio-techne | Catalog #: 491831 |
| RNAscope® Probe-Mm-Rbfox1- mRNA | ACD Bio-techne | Catalog #: 519911 |
| RNAscope® Probe-Mm-Snap25- mRNA | ACD Bio-techne | Catalog #: 516471 |
Highlights.
Ambient RNA in single nuclei genomic data cause conspicuous contamination in glia
Ambient RNA contamination can be mitigated by physical sorting or in silico methods
Previously annotated immature oligodendrocytes are ambient RNA contaminated glia
Ambient RNA removal reveals unannotated COPs in all adult human brain datasets
Acknowledgments
The authors thank Dr. Dmitry Velmeshev, Dr. Arnold Kriegstein, Dr. Tao Wang and Dr. Lu Sun for their critical comments on the manuscript. We also thank Dr. Shin Yamazaki and the UTSW Neuroscience Microscopy Facility for their help with imaging, and Dr. Shane A. Liddelow and Michael O’Dea for additional information about their sorted dataset. The authors thank the NIH NeuroBioBank for providing human brain tissue. G.K. is a Jon Heighten Scholar in Autism Research and Townsend Distinguished Chair in Research on Autism Spectrum Disorders at UT Southwestern. E.C. is a Neural Scientist Training Program Fellow in the Peter O’Donnell Brain Institute at UT Southwestern. This work was partially supported by the James S. McDonnell Foundation 21st Century Science Initiative in Understanding Human Cognition Scholar Award, NHGRI (HG011641), NINDS (NS115821) and NIMH (MH126481, MH103517) to G.K. and an American Heart Association Postdoctoral Fellowship (915654) to Y.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Akay LA, Effenberger AH, and Tsai LH (2021). Cell of all trades: oligodendrocyte precursor cells in synaptic, vascular, and immune function. Genes Dev 35, 180–198. 10.1101/gad.344218.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer P, Haghverdi L, Buttner M, Theis FJ, Marr C, and Buettner F (2016). destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243. 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- Ayhan F, Douglas C, Lega BC, and Konopka G (2021). Nuclei isolation from surgically resected human hippocampus. STAR Protoc 2, 100844. 10.1016/j.xpro.2021.100844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TE, Jorstad NL, Hu Q, Lake BB, Tian W, Kalmbach BE, Crow M, Hodge RD, Krienen FM, Sorensen SA, et al. (2021). Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature 598, 111–119. 10.1038/s41586-021-03465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, Zhang MQ, Sedel F, Jourdren L, Coulpier F, et al. (2010). A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J 29, 3082–3093. 10.1038/emboj.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamling X, Kallman A, Fang W, Berlinicke CA, Mertz JL, Devkota P, Pantoja IEM, Smith MD, Ji Z, Chang C, et al. (2021). Single-cell transcriptomic reveals molecular diversity and developmental heterogeneity of human stem cell-derived oligodendrocyte lineage cells. Nat Commun 12, 652. 10.1038/s41467-021-20892-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Lun AT, and Smyth GK (2016). From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res 5, 1438. 10.12688/f1000research.8987.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu H, Wang S, Koito H, Li J, Ye F, Hoang J, Escobar SS, Gow A, Arnett HA, et al. (2009). The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat Neurosci 12, 1398–1406. 10.1038/nn.2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Faria O Jr., Pivonkova H, Varga B, Timmler S, Evans KA, and Karadottir RT (2021). Periods of synchronized myelin changes shape brain function and plasticity. Nat Neurosci 24, 1508–1521. 10.1038/s41593-021-00917-2. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fard MK, van der Meer F, Sanchez P, Cantuti-Castelvetri L, Mandad S, Jakel S, Fornasiero EF, Schmitt S, Ehrlich M, Starost L, et al. (2017). BCAS1 expression defines a population of early myelinating oligodendrocytes in multiple sclerosis lesions. Sci Transl Med 9. 10.1126/scitranslmed.aam7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes MGF, Luo JXX, Cui QL, Perlman K, Pernin F, Yaqubi M, Hall JA, Dudley R, Srour M, Couturier CP, et al. (2021). Age-related injury responses of human oligodendrocytes to metabolic insults: link to BCL-2 and autophagy pathways. Commun Biol 4, 20. 10.1038/s42003-020-01557-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD (2015). A new mechanism of nervous system plasticity: activity-dependent myelination. Nat Rev Neurosci 16, 756–767. 10.1038/nrn4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SJ, Marioni JC, and Babadi M (2019). CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. 791699. 10.1101/791699 %J bioRxiv. [DOI]
- Guo CJ, Xu G, and Chen LL (2020). Mechanisms of Long Noncoding RNA Nuclear Retention. Trends Biochem Sci 45, 947–960. 10.1016/j.tibs.2020.07.001. [DOI] [PubMed] [Google Scholar]
- Hafner AS, Donlin-Asp PG, Leitch B, Herzog E, and Schuman EM (2019). Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science 364. 10.1126/science.aau3644. [DOI] [PubMed] [Google Scholar]
- Heiser CN, Wang VM, Chen B, Hughey JJ, and Lau KS (2021). Automated quality control and cell identification of droplet-based single-cell data using dropkick. Genome Res 31, 1742–1752. 10.1101/gr.271908.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. 10.1038/s41586-019-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Orthmann-Murphy JL, Langseth AJ, and Bergles DE (2018). Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat Neurosci 21, 696–706. 10.1038/s41593-018-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, and Stockton ME (2021). Premyelinating Oligodendrocytes: Mechanisms Underlying Cell Survival and Integration. Front Cell Dev Biol 9, 714169. 10.3389/fcell.2021.714169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakel S, Agirre E, Mendanha Falcao A, van Bruggen D, Lee KW, Knuesel I, Malhotra D, Ffrench-Constant C, Williams A, and Castelo-Branco G (2019). Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547. 10.1038/s41586-019-0903-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, Cornelisse LN, Farrell RJ, Goldschmidt HL, Howrigan DP, et al. (2019). SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 103, 217–234 e214. 10.1016/j.neuron.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kougioumtzidou E, Shimizu T, Hamilton NB, Tohyama K, Sprengel R, Monyer H, Attwell D, and Richardson WD (2017). Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. Elife 6. 10.7554/eLife.28080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, and Zhang K (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80. 10.1038/nbt.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Luecken MD, and Theis FJ (2019). Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol 15, e8746. 10.15252/msb.20188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, McCarthy DJ, and Marioni JC (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res 5, 2122. 10.12688/f1000research.9501.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, participants in the 1st Human Cell Atlas, J., and Marioni JC (2019). EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol 20, 63. 10.1186/s13059-019-1662-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luse S, and Korey SJH-H, New York (1959). The Biology of Myelin. 59–81.
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L, Li H, Haring M, Hochgerner H, Romanov RA, et al. (2016). Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329. 10.1126/science.aaf6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SE, Walker AJ, Kamath T, Dissing-Olesen L, Hammond TR, de Soysa TY, Young AMH, Murphy S, Abdulraouf A, Nadaf N, et al. (2022). Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nature Neuroscience 25, 306–316. 10.1038/s41593-022-01022-8. [DOI] [PubMed] [Google Scholar]
- Merten N, Fischer J, Simon K, Zhang L, Schroder R, Peters L, Letombe AG, Hennen S, Schrage R, Bodefeld T, et al. (2018). Repurposing HAMI3379 to Block GPR17 and Promote Rodent and Human Oligodendrocyte Differentiation. Cell Chem Biol 25, 775–786 e775. 10.1016/j.chembiol.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DJ, Duka T, Stimpson CD, Schapiro SJ, Baze WB, McArthur MJ, Fobbs AJ, Sousa AM, Sestan N, Wildman DE, et al. (2012). Prolonged myelination in human neocortical evolution. Proc Natl Acad Sci U S A 109, 16480–16485. 10.1073/pnas.1117943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muskovic W, and Powell JE (2021). DropletQC: improved identification of empty droplets and damaged cells in single-cell RNA-seq data. Genome Biol 22, 329. 10.1186/s13059-021-02547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy C, Maitra M, Tanti A, Suderman M, Theroux JF, Davoli MA, Perlman K, Yerko V, Wang YC, Tripathy SJ, et al. (2020). Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat Neurosci 23, 771–781. 10.1038/s41593-020-0621-y. [DOI] [PubMed] [Google Scholar]
- Perlman K, Couturier CP, Yaqubi M, Tanti A, Cui QL, Pernin F, Stratton JA, Ragoussis J, Healy L, Petrecca K, et al. (2020). Developmental trajectory of oligodendrocyte progenitor cells in the human brain revealed by single cell RNA sequencing. Glia 68, 1291–1303. 10.1002/glia.23777. [DOI] [PubMed] [Google Scholar]
- Phan BN, Bohlen JF, Davis BA, Ye Z, Chen HY, Mayfield B, Sripathy SR, Cerceo Page S, Campbell MN, Smith HL, et al. (2020). A myelin-related transcriptomic profile is shared by Pitt-Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci 23, 375–385. 10.1038/s41593-019-0578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka WB, Mohammadi S, Davila-Velderrain J, Subburaju S, Tso DR, Hourihan M, and Kellis M (2020). Single-cell dissection of schizophrenia reveals neurodevelopmental-synaptic axis and transcriptional resilience. 2020.2011.2006.20225342. 10.1101/2020.11.06.20225342 %J medRxiv. [DOI] [Google Scholar]
- Sadick JS, O’Dea MR, Hasel P, Dykstra T, Faustin A, and Liddelow SA (2022). Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 110, 1788–1805 e1710. 10.1016/j.neuron.2022.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L SI (2020). GeneOverlap: Test and visualize gene overlaps. ttp://shenlab-sinai.github.io/shenlab-sinai/.
- Sim FJ, McClain CR, Schanz SJ, Protack TL, Windrem MS, and Goldman SA (2011). CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nat Biotechnol 29, 934–941. 10.1038/nbt.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber BR, and McMorris FA (2001). Fyn tyrosine kinase regulates oligodendroglial cell development but is not required for morphological differentiation of oligodendrocytes. J Neurosci Res 63, 303–312. . [DOI] [PubMed] [Google Scholar]
- Squair JW, Gautier M, Kathe C, Anderson MA, James ND, Hutson TH, Hudelle R, Qaiser T, Matson KJE, Barraud Q, et al. (2021). Confronting false discoveries in single-cell differential expression. Nat Commun 12, 5692. 10.1038/s41467-021-25960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan A (2016). gread: Fast Reading and Processing of Common Gene Annotation and Next Generation Sequencing Format Files. https://rdrr.io/github/asrinivasan-oa/gread/.
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LO, Mulinyawe SB, Collins HY, Ibrahim A, Li Q, Simon DJ, Tessier-Lavigne M, and Barres BA (2018). Spatiotemporal Control of CNS Myelination by Oligodendrocyte Programmed Cell Death through the TFEB-PUMA Axis. Cell 175, 1811–1826 e1821. 10.1016/j.cell.2018.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, Fourne Y, Matthews PM, Theys T, Mancuso R, et al. (2020). Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep 32, 108189. 10.1016/j.celrep.2020.108189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MN, Maynard KR, Spangler A, Huuki LA, Montgomery KD, Sadashivaiah V, Tippani M, Barry BK, Hancock DB, Hicks SC, et al. (2021). Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 109, 3088–3103 e3085. 10.1016/j.neuron.2021.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bruggen D, Agirre E, and Castelo-Branco G (2017). Single-cell transcriptomic analysis of oligodendrocyte lineage cells. Curr Opin Neurobiol 47, 168–175. 10.1016/j.conb.2017.10.005. [DOI] [PubMed] [Google Scholar]
- Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, and Kriegstein AR (2019). Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689. 10.1126/science.aav8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Lee PR, and Fields RD (2011). Control of local protein synthesis and initial events in myelination by action potentials. Science 333, 1647–1651. 10.1126/science.1206998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, Emery B, Li H, and Richardson WD (2016). Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci 19, 1210–1217. 10.1038/nn.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Corbett SE, Koga Y, Wang Z, Johnson WE, Yajima M, and Campbell JD (2020). Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol 21, 57. 10.1186/s13059-020-1950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung MS, Zdunek S, Bergmann O, Bernard S, Salehpour M, Alkass K, Perl S, Tisdale J, Possnert G, Brundin L, et al. (2014). Dynamics of oligodendrocyte generation and myelination in the human brain. Cell 159, 766–774. 10.1016/j.cell.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Young MD, and Behjati S (2020). SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 9. 10.1093/gigascience/giaa151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049. 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Sousa AMM, Gao T, Skarica M, Li M, Santpere G, Esteller-Cucala P, Juan D, Ferrandez-Peral L, Gulden FO, et al. (2018). Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362. 10.1126/science.aat8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2 (Related to Figure 2): Statistics on the nuclear and non-nuclear ambient RNA markers. It also contains low-intronic ambient markers and high-intronic ambient markers for the non-sorted datasets (described in Figures S1,8).
Table S3 (Related to Figure S2): Statistic of the gene ontology enrichments of nuclear or non-nuclear ambient RNA markers.
Table S4 (Related to Figure 3): Statistics of the genes depleted in the NeuN- sorted datasets (NeuN- SDs) compared to SD1, SD2, NSD1 or NSD2 datasets. The list is ranked by the abundance of depleted genes (logCPM), all genes are depleted with FDR < 0.05.
Table S5 (Related to Figure S9): Quantification of the smFISH results.
Table S6 (Related to Figure 4): Statistics of the committed oligodendrocyte progenitor cell markers per dataset (SD1, NSD1, NSD2).
Data Availability Statement
Raw fastq files of mouse single-nuclei RNA-seq dataset are accessible in GEO with accession number: GSE198640. Re-analyzed processed matrices are accessible in GEO with accession number: GSE198951. All analysis codes are available in our github page: https://github.com/konopkalab/Ambient_RNA_In_Brain_snRNAseq