

Abstract
Background
The most recognizable phenotype of severe asthma comprises people who are blood eosinophil and FeNO‐high, driven by type 2 (T2) cytokine biology, which responds to targeted biological therapies. However, in many people with severe asthma, these T2 biomarkers are suppressed but poorly controlled asthma persists. The mechanisms driving asthma in the absence of T2 biology are poorly understood.
Objectives
To explore airway pathology in T2 biomarker‐high and ‐low severe asthma.
Methods
T2 biomarker‐high severe asthma (T2‐high, n = 17) was compared with biomarker‐intermediate (T2‐intermediate, n = 21) and biomarker‐low (T2‐low, n = 20) severe asthma and healthy controls (n = 28). Bronchoscopy samples were processed for immunohistochemistry, and sputum for cytokines, PGD2 and LTE4 measurements.
Results
Tissue eosinophil, neutrophil and mast cell counts were similar across severe asthma phenotypes and not increased when compared to healthy controls. In contrast, the remodelling features of airway smooth muscle mass and MUC5AC expression were increased in all asthma groups compared with health, but similar across asthma subgroups. Submucosal glands were increased in T2‐intermediate and T2‐low asthma. In spite of similar tissue cellular inflammation, sputum IL‐4, IL‐5 and CCL26 were increased in T2‐high versus T2‐low asthma, and several further T2‐associated cytokines, PGD2 and LTE4, were increased in T2‐high and T2‐intermediate asthma compared with healthy controls.
Conclusions
Eosinophilic tissue inflammation within proximal airways is suppressed in T2 biomarker‐high and T2‐low severe asthma, but inflammatory and structural cell activation is present, with sputum T2‐associated cytokines highest in T2 biomarker‐high patients. Airway remodelling persists and may be important for residual disease expression beyond eosinophilic exacerbations.
Registered at ClincialTrials.gov: NCT02883530.
Keywords: cytokine, eosinophil, FeNO, severe asthma, Th2
People with T2‐high and T2‐low severe asthma, and healthy controls, have similar airway tissue inflammatory cells counts, but sputum T2 cytokines persist in T2‐high asthma, likely driving eosinophilic exacerbations through recruitment of blood eosinophils. In both T2‐high and T2‐low asthma, extensive airway wall remodelling is present and likely contributes to residual disease expression beyond T2 exacerbations. As bronchoconstriction and mucus plugging are the predominant causes of airflow obstruction driving asthma symptoms, exacerbations and death, the factors that sustain these abnormal pathological features should be a priority for future research and drug development for severe asthma.Abbreviations: CCL, C‐C motif chemokine ligand; Eos, eosinophil; FeNO, fraction of exhaled nitric oxide; GC, goblet cell expressing MUC5AC; IL, interleukin; LTE4, leukotriene E4; MC, mast cell; Neu, neutrophil; T2, type2 cytokine; PGD2, prostaglandin D2; TSLP, thymic stromal lymphopoietin

Abbreviations
- ACQ
asthma control questionnaire
- AHR
airway hyperresponsiveness
- ASM
airway smooth muscle
- BTS
British Thoracic Society
- FEV1
forced expiratory volume
- GINA
Global Initiative for Asthma
- HVS
Healthy Volunteer Study
- ICS
inhaled corticosteroid
- IQR
interquartile range
- LT
leukotriene
- MBP
major basic protein
- MCT
tryptase only mast cell
- MCTC
tryptase and chymase mast cell
- PG
prostaglandin
- RBM
reticular basement membrane
- RTU
ready‐to‐use
- SEM
standard error mean
- T2
type 2 cytokines (IL‐4, IL‐5, IL‐13)
- T2‐high‐FNS
T2 biomarker‐high FeNO non‐suppressor
- T2‐intermediate
T2 biomarker‐intermediate
- T2‐low
T2 biomarker‐low
1. INTRODUCTION
Asthma is a common, chronic and persistent disorder that accounts for significant morbidity and mortality, costing the European Union ~17.7 billion euros/annum. 1 , 2 , 3 Approximately, 10% of patients have asthma which is resistant to current therapies. 2 , 3 This group consumes 50–60% of health care costs attributed to asthma, reflecting a considerable unmet clinical need.
Asthma is characterized by the presence of airway inflammation, airway remodelling and airway hyperresponsiveness (AHR). How these features interact and the extent of their interdependence is unclear. Furthermore, while many airway elements are implicated (epithelial dysfunction, mucous gland hyperplasia with mucus hypersecretion, airway smooth muscle [ASM] dysfunction and inflammatory cell activation), the relationships between these elements and molecular pathways are poorly understood.
Approximately, 80% of people with mild corticosteroid‐naïve asthma demonstrate evidence of a blood or airway eosinophilia, with a concomitant increase in the fraction of exhaled nitric oxide (FeNO), 4 , 5 although up to 95% of people with severe asthma have current or historical evidence of eosinophilia. 6 , 7 This eosinophilic phenotype is often characterized by increased expression of an airway gene expression signature driven by IL‐4 and IL‐13, 8 , 9 , 10 , 11 although tissue eosinophilia is also dependent on IL‐5. 12 Together, these cytokines are described as Th2 or type 2 (T2) cytokines. Monoclonal antibodies that specifically target the IL‐4Rα inhibiting IL‐4/−13, or which target IL‐5/IL‐5Rα, demonstrate the greatest therapeutic effect in people with severe asthma who are T2 biomarker‐high (FeNO and blood eosinophils for IL‐4/−13, blood eosinophils for anti‐IL‐5). 13 , 14 The introduction of these drugs has transformed the treatment of people with T2‐driven severe asthma, who by definition are relatively resistant to inhaled corticosteroids (ICS).
Little is known about the molecular pathways active in severe asthma that is not T2 cytokine/biomarker‐high. These patients respond poorly to ICS and T2 cytokine‐targeted treatments, and represent a major unmet clinical need. Transcriptomic analyses from several airway biopsy studies have demonstrated mutually exclusive expression of the prototypic T2 cytokine‐dependent gene signature and an IL‐17‐dependent gene signature in mild–severe asthma. 10 , 11 However, 50% of uncontrolled severe asthma patients on high‐dose corticosteroid treatment are neither T2‐high nor IL‐17‐high. 10 , 11 This clearly represents a major challenge as it is not known what drives asthma in the absence of T2 cytokine or IL‐17 signalling. Potential mechanisms include structural change (remodelling) with fixed airflow obstruction and/or intrinsic ASM dysfunction. Ongoing mast cell activation is evident across severe asthma phenotypes, 15 and although neutrophil‐driven disease has been proposed, neutrophil counts in the bronchial mucosa were no different between health and mild, moderate or severe asthma in many studies. 16 , 17 , 18 Some studies have also suggested there is over activity of interferon‐γ 19 , 20 , 21 which might aggravate AHR. 19
The aim of this study was to explore the airway immunopathology of people at the extremes of the T2 biomarker spectrum in order to define the key features that contribute to corticosteroid insensitive T2‐high severe asthma and T2‐low severe asthma.
2. METHODS
Detailed methods are available in the Appendix S1.
2.1. Ethics and consent
The RASP bronchoscopy study was a multi‐centre study, which recruited people with severe asthma prospectively using pre‐defined inclusion and exclusion criteria. It was approved by the East Midlands—Leicester South Research Ethics Committee (REC) (reference 16/EM/0260) and registered at ClinicalTrials.gov (NCT02883530). Biopsy samples collected using the same standard operating procedure were also used from the pre‐intervention arms of two other studies, i) a single‐centre bronchoscopy study assessing the effects of ICS on adult healthy volunteers (referred to as Leicester HVS from here) (NCT02476825, REC approval 15/EM/0313) (only pre‐intervention baseline biopsies were studied), and ii) a multi‐centre bronchoscopy study evaluating the effects of lebrikizumab on airway eosinophilic inflammation in uncontrolled asthma 22 (referred to as Genentech CLAVIER from here) (NCT02099656, independent ethics committee approval was obtained at all participating centres; only pre‐intervention baseline biopsies were studied). All participants gave written informed consent. The Leicester HVS ran contemporaneously with RASP and used the same standard operating procedure and the same bronchoscopist for both studies (PB).
2.2. Study population
Detailed inclusion and exclusion criteria are provided in the online repository text. People with asthma aged 18–70 were eligible, and all were current non‐smokers with a < 15 pack‐year smoking history.
Standard criteria for the diagnosis of asthma are summarized in the Appendix S1. Patients were deemed adherent in their asthma centre through the prior analysis of prescription refills, measurement of prednisolone and cortisol levels if appropriate, and/or FeNO suppression testing. 23 , 24 Current use of a biologic treatment was an exclusion criteria, and only one patient had used a biologic previously (omalizumab).
Participants underwent extensive evaluation at baseline including a full medical history, lung function testing, bronchial challenge using methacholine where appropriate, and induced sputum.
People with severe asthma were recruited prospectively and identified as follows: (i) patients with a previous FeNO ≥45 ppb and blood eosinophils ≥0.3 × 109/L, who did not suppress their FeNO during a FeNO suppression test 23 , 24 , 25 during routine clinical care, referred to as T2 biomarker‐high FeNO non‐suppressors (T2‐high‐FNS); (ii) patients with a FeNO ≤30 ppb and blood eosinophils ≤0.2 × 109/L identified in clinic, referred to as T2 biomarker‐low (T2‐low); this group was supplemented with 9 participants from the Genentech CLAVIER study. 22 (iii) patients who had exited the RASP T2‐biomarker (FeNO, blood eosinophils, periostin)‐driven treatment optimization study 6 with either intermediate biomarker measurements, referred to as T2 biomarker‐intermediate (T2‐intermediate) or low T2 biomarkers (as described for ii above). The rationale for the biomarker levels defining the T2‐low group is provided in the Appendix S1.
Healthy volunteers in Leicester HVS had no prior history or clinical evidence of lower respiratory disease and normal spirometry. Healthy volunteers with a history of rhinitis (perennial or seasonal) were required to have a PC20 methacholine >16 mg/ml.
2.3. Standard operating procedures
All centres were experienced in performing research bronchoscopies in asthma. RASP, Leicester HVS and CLAVIER used the same standard operating procedures for tissue collection and processing, and all immunohistochemical staining and analysis were performed in Leicester. These are summarized briefly as follows:
2.4. Bronchoscopy
Subjects underwent bronchoscopy conducted according to British Thoracic Society guidelines. 26 Mucosal biopsies were collected from 2nd to 5th generation bronchi under direct vision as per study procedure manual.
2.5. Immunohistochemistry
Biopsies were fixed in 4% neutral buffered formalin for 4 h at 4°C as described for the Clavier study, 22 then processed into paraffin wax, as per study procedure manual, with the same protocol used for all studies contributing to this analysis. Immunohistochemistry was performed in Leicester. All the laboratory procedures and processes were performed following the ISO9001‐2015 Quality Management System and GCP/GLP guidelines. Further immunostaining details are provided in the Appendix S1.
2.6. Assessment of immunopathology
High‐throughput morphologic analysis was performed on scanned sections using a Carl Zeiss Scanner Z1 and AxioCam HRc digital camera (Carl Zeiss, Germany) and ZEN desk 3.1 image analysis software. The following previously validated pathological features 17 were evaluated in whole sections as follows: (i) nucleated inflammatory cells (eosinophils, neutrophils, mast cells [tryptase+, chymase+]) in the airway epithelium, lamina propria, ASM bundles and airway glands, expressed as cells/mm; 2 (ii) the area occupied by epithelial, ASM and glandular structures expressed as a percentage of the total biopsy area; (iii) MUC5AC expressed as the percentage of airway epithelium staining positive; 16 (iv) reticular basement membrane (RBM) thickness expressed in microns. 27 The mean of two sections at least 18 μm apart was taken for each immunohistological analysis.
All pathological data were assessed by an observer blinded to the identity of the patient.
2.7. Sputum mediators
Induced sputum supernatants were collected in PBS. Cytokines (IL‐4/−5/−13/−31/−33, CCL17/26, TARC, TSLP, IFNγ and TNFα), prostaglandin (PG)D2 and leukotriene (LT)E4 were measured as described in the Appendix S1.
2.8. Statistical analysis
The approach to statistical analysis is provided in the Appendix S1.
3. RESULTS
3.1. Clinical characteristics
For bronchoscopy, we recruited 54 patients with asthma prospectively, with suitable tissue for immunohistochemical analysis available from 49, with an additional 9 T2‐low patients from the CLAVIER study and 28 healthy controls from the Leicester HVS. The clinical characteristics of the pre‐defined bronchoscopy study groups are presented in Table 1.
Table 1.
Baseline demographic data for the bronchoscopy cohort
| T2‐high FeNO‐NS [n = 17] | T2‐intermediate [n = 21] | T2‐low [n = 20] | Healthy (n = 28) | p value† | |
|---|---|---|---|---|---|
| Age—years | 57 (50–63)** | 56 (46.5–63)** | 51 (35.3–58.3) | 27 (22–50) | <0.0001 |
| Sex—M/F | 9/8 | 11/10 | 13/7 | 14/14 | 0.7596 |
| BMI (kg/m2) | 29.8 (24.9–32.6)** | 31.4 (26.8–38.7)**** | 30.3 (28.2–33.8)*** | 23.8 (21.4–27.3) | <0.0001 |
| Age onset—years | 28.8 ± 19.2 | 29.6 ± 19.7 | 18.4 ± 14.0 | N/A | 0.0935 |
| Ethnicity Caucasian—% | 100 | 90.5 | 89.5 | 78.6 | 0.1647 |
| Asthma duration—years | 27.2 ± 19.6 | 25.4 ± 14.8 | 29.4 ± 12.7 | N/A | 0.7166 |
| Atopic a —% | 86.7 | 61.1 | 82.4 | 32.1 | 0.0007 |
| Severe annual exacerbation frequency | 2.0 (1.0–3.5) | 1.0 (0.0–2.0) | 1.0 (1.0–5.0) | N/A | 0.1188 |
| ICS dose—BDP equivalent—mcg | 2000 (2000–2000) | 2000 (1000–2400) | 2000 (2000–2000) | N/A | 0.4632 |
| Maintenance oral corticosteroids—% | 29.4 | 33.3 | 20.0 | N/A | 0.6213 |
| Oral corticosteroid dose—mg |
10.0 (5–20) [n = 5] |
10.0 (5–20) [n = 7] |
8.75 (5–10) [n = 4] |
N/A | 0.7618 |
| Long‐acting β2‐agonist—% | 100 | 100 | 100 | N/A | 1.0000 |
| Long‐acting muscarinic antagonist—% | 52.9 | 38.1 | 45.0 | N/A | 0.6578 |
| Leukotriene receptor antagonist—% | 47.1 | 52.4 | 15.0 | N/A | 0.0313 |
| Theophylline—% | 41.2 | 14.3 | 5.0 | N/A | 0.0158 |
| Ex smoker—% | 29.4 | 33.3 | 35.0 | 14.3 | 0.3285 |
| Smoking—pack‐years | 0 (0.0–2.0) | 0.0 (0.0–4.5) | 0.0 (0.0–0.9) | 0.0 (0.0–0.0) | 0.3153 |
| FEV1 Pre‐BD—L | 2.02 (1.66–2.71)**** | 2.43 (2.02–2.79)*** | 2.55 (2.02–3.15) ** | 3.59 (2.89–4.08) | <0.0001 |
| FEV1 Pre‐BD—% predicted | 66.8 (55.1–80.7)**** | 78.3 (72.4–94.0)** | 71.4 (63.2–88.0)**** | 102.0 (94.5–109.8) | <0.0001 |
| FEV1/FVC—% | 59.4 (55.2–66.4)**** | 70.5 (62.6–78.3)** | 64.9 (56.1–73.5)**** | 81.0 (77.3–84.9) | <0.0001 |
| ACQ5 | 1.6 (0.8–2.3) | 1.2 (0.6–1.9) | 2.1 (1.7–3.1)¶ | N/A | 0.0352 |
| Total IgE—kU/L | 132 (94–274) | 69 (17–338) | 173 (73.8–350.3) | Not done | 0.4372 |
| FeNO—ppb | 71.0 (40.0–104.5) ****/ ##/§§§§ | 21 (15.0–31.0) | 15.0 (11.3–19.5) | 17.5 (12.3–25.3) | <0.0001 |
| Blood eosinophils (screening)—×109/L |
0.38 (0.23–0.58)****/ #/§§§§ |
0.17 (0.08–0.31) | 0.11 (0.07–0.17) | 0.09 (0.06–0.14) | <0.0001 |
| Blood eosinophils (highest recorded)—×109/L | 0.81 (0.60–1.12)# | 0.5 (0.24–0.66) | 0.45 (0.31–0.70) (n = 11) | Not available | 0.0228 |
| Sputum eosinophils—% | 10.7 (0.8–35.3) §§ [n = 8] | 1.2 (0.0–6.7) [n = 14] | 0.0 (0.0–0.3) [n = 15] | Not done | 0.0067 |
| Sputum neutrophils—% |
23.5 ± 27.9 [n = 8] |
47.4 ± 36.7 [n = 14] |
49.9 ± 27.7 [n = 15] | Not done | 0.1068 |
Continuous variables are presented as mean ± SD or median (interquartile range). BMI = Body Mass Index. BD = Bronchodilator. †All tests for continuous variables are ANOVA or Kruskal–Wallis across all groups unless indicated otherwise, with adjusted p‐values for between‐group comparisons obtained using Sidak's or Dunn's multiple comparison tests. For categorical variables, a Chi‐squared test was used across applicable groups.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with healthy control subjects. #p < 0.05, ##p < 0.01 compared with T2‐intermediate. §§§p < 0.001, §§§§p < 0.0001 compared with T2‐low. ¶p < 0.05 compared with T2‐intermediate.
Atopy refers to the presence of a positive skin test or the presence of a raised specific IgE to a common aeroallergen.
Both FeNO and blood eosinophils were markedly increased in the T2‐high‐FNS compared with the other groups, as expected from the entry requirements for this group. This phenotype, therefore, appears stable over time (Table 1). Sputum eosinophils were also highest in T2‐high‐FNS, but T2‐low patients were the most symptomatic (Table 1). Many T2‐intermediate and T2‐low patients had historical evidence of raised blood eosinophils (Table 1).
3.2. Tissue inflammatory cells
All subjects had lamina propria suitable for analysis of inflammatory cells, but there were fewer with suitable epithelium, ASM and submucosal glands for inflammatory cell analysis within these compartments, as shown in the corresponding figures. Immunostaining from one centre was unsuccessful for major basic protein and neutrophil elastase (n = 7 subjects), and these markers were excluded from the analysis.
Representative immunostaining is shown in Figure S1. Eosinophil counts in the airway lamina propria were similar in asthma and health (Figure S2A) and across asthma groups (Figure 1A). Eosinophils were rarely seen in the airway epithelium (Figure S2B,C), only present in the airway submucosal gland stroma in 3 asthmatics, and never seen within ASM.
FIGURE 1.
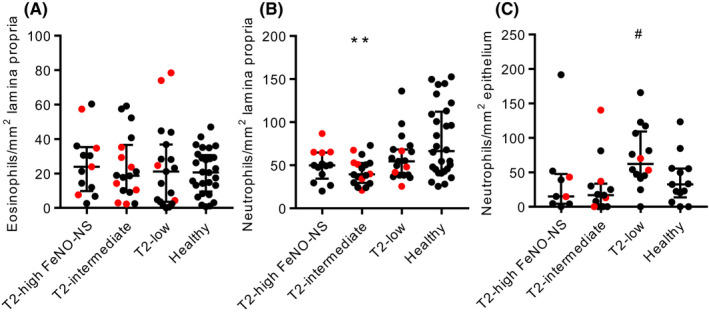
Eosinophil and neutrophil numbers in asthma subgroups and healthy controls. A) Eosinophil numbers in the airway lamina propria (Kruskal–Wallis p = 0.8444). B) Neutrophil numbers in the airway lamina propria (Kruskal–Wallis p = 0.0119). C) Neutrophil numbers in the airway epithelium (Kruskal–Wallis: p = 0.0197 all groups, p = 0.0105 asthma groups). **p = 0.0043 compared with healthy controls, #p = 0.0120 compared with T2‐intermediate (Dunn's multiple comparison test). Red points represent people taking oral corticosteroids
Neutrophil counts in the lamina propria were lower in asthma compared with health (p = 0.0052) (Figure S2D), with no differences between the asthma subgroups (Figure 1B). This difference between asthma and health was lost when the healthy control group was matched to those with asthma for age by removing people under the age of 25 (age did not influence any other immunohistological parameters—see online repository for further details). Neutrophil counts in the airway epithelium were similar between asthma and health (Figure S2E), but there was a significant difference across the asthma subgroups (Kruskal–Wallis p = 0.0105) (Figure 1C), with more epithelial neutrophils in T2‐low compared with T2‐intermediate asthma (p = 0.0120). Neutrophils were present within the ASM bundles in 2 asthmatics and 2 healthy controls and the airway gland stroma of 3 asthmatics.
Tryptase+ (total) and chymase+ mast cell counts were reduced in the airway lamina propria in severe asthma compared with health in keeping with previous studies 18 (p < 0.0001) (Figure S3A and B) and similar in the asthma subgroups (Figure 2A and B). Tryptase+ mast cell counts in the airway epithelium were similar in asthma and health (Figure S3C) and similar in the asthma subgroups (Figure S3D). Chymase+ mast cells were also present in the airway epithelium but in much lower numbers than tryptase+ mast cells (Figure S3E and F).
FIGURE 2.
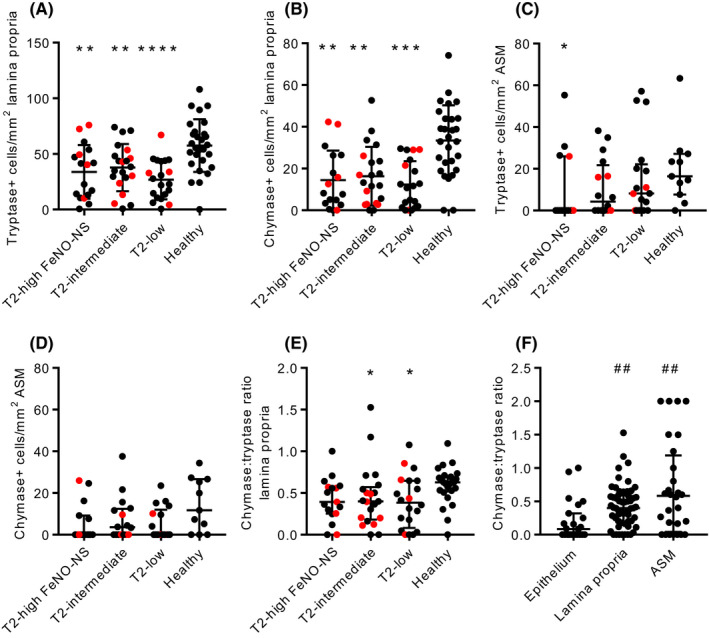
Mast cell numbers in asthma subgroups and healthy controls. A) Tryptase+ mast cell numbers in the airway lamina propria (ANOVA p < 0.0001). B) Chymase+ mast cell numbers in the airway lamina propria (Kruskal–Wallis p < 0.0001). C) Tryptase+ mast cell numbers within the airway smooth muscle (ASM) (Kruskal Wallis p = 0.0923). D) Chymase+ mast cell numbers within the ASM (Kruskal–Wallis p = 0.1346). E) The chymase:tryptase ratio in the lamina propria (Kruskal–Wallis p = 0.0280). F) The chymase:tryptase ratio in the airway epithelium, lamina propria and ASM in people with asthma (Kruskal–Wallis, p = 0.0018) (where chymase+ cells were present but there were no tryptase+ cells, values of 2 were given). For A‐E) *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with healthy controls (Dunnett's or Dunn's multiple comparison tests as appropriate). For F) ##p < 0.01 compared with airway epithelium (Dunn's multiple comparison test). Red points represent people taking oral corticosteroids
Tryptase+ mast cells were present in the airway glands in 9/19 asthmatics who had glandular tissue present and 2/3 healthy controls. These cells rarely contained chymase, with chymase present in only 3 patients. Tryptase+ and chymase+ mast cells were often present within the ASM with reduced chymase+ numbers in asthma compared with health (p = 0.0514 and p = 0.0263, respectively) (Figure S3G and H). There were no differences between the asthma groups (Figure 2C and D).
The ratio of chymase+:tryptase+ mast cells was significantly lower in asthmatic lamina propria compared with healthy lamina propria (p = 0.0022) (Figure S3I), but similar across asthma subgroups (Figure 2E), and demonstrated a gradient from the airway epithelium, through the lamina propria to the ASM in asthma (Kruskal–Wallis p = 0.0018) (Figure 2F).
3.3. Tissue remodelling
Airway smooth muscle area expressed as a percentage of total biopsy area was increased in asthma versus health (p = 0.0006) (Figure S4) and to a similar extent across the different asthma subgroups (Figure 3A). Airway submucosal glands expressed as a percentage of total biopsy area were also increased in asthma versus health (p = 0.0150) (Figure S4), accounted for by increases in the T2‐intermediate and T2‐low asthma groups (Figure 3B). There was a significant difference across the asthma subgroups in submucosal glandular tissue area (Kruskal–Wallis p = 0.0386), but no significant post hoc difference between asthma subgroups. MUC5AC expressed as the percentage of airway epithelium staining was elevated in asthma compared with healthy controls (p < 0.0001) (Figure S4), with similar increases in all asthma subgroups (Figure 3C).
FIGURE 3.
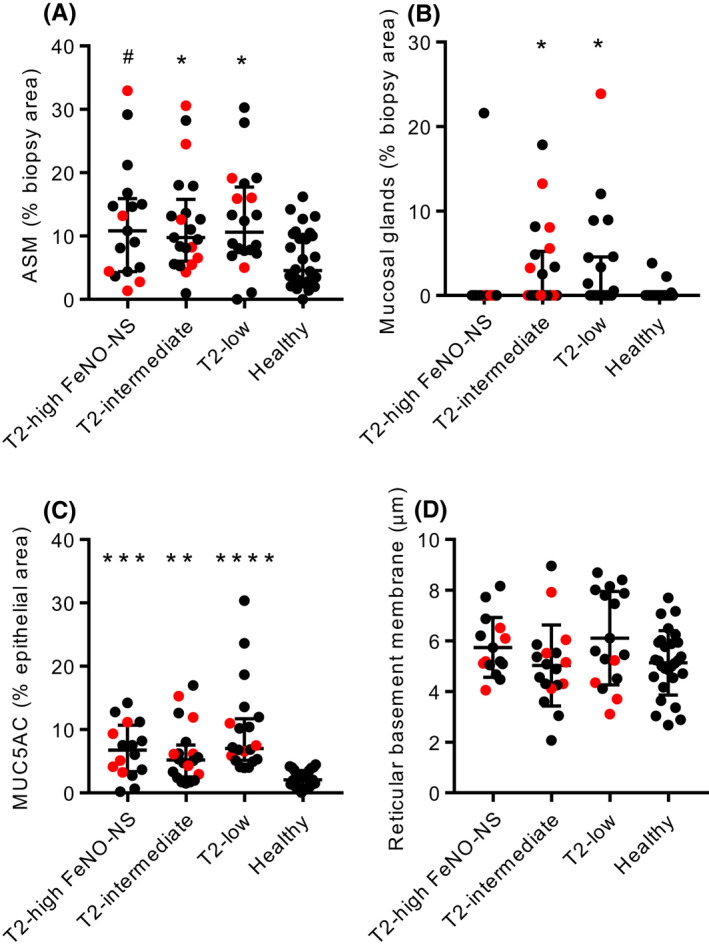
Remodelling features in asthma subgroups and healthy controls. A) Airway smooth muscle (ASM) mass expressed as a percentage of total biopsy area (Kruskal–Wallis p = 0.0098). B) Mucosal gland mass expressed as a percentage of total biopsy area (Kruskal–Wallis p = 0.0026). C) MUC5AC immunostaining expressed as a percentage of epithelial area (Kruskal–Wallis p < 0.0001). D) Reticular basement membrane (RBM) thickness (ANOVA p = 0.0910). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with healthy controls (Dunn's multiple comparison test). For A), #p = 0.0506 compared with healthy (Dunn's multiple comparison test). Red points represent people taking oral corticosteroids
There were no differences between asthma and health or across the asthma groups for epithelial area expressed as percentage of airway biopsy total area (Figure S4), or for reticular basement membrane thickness (Figure 3D and Figure S4).
Post hoc re‐analysis of the data restricting the T2‐low group to blood eosinophils ≤0.15x109/L and FeNO ≤25 ppb produced similar results for both inflammatory cell infiltration and remodelling features (Figure S9).
3.4. Sputum cytokines, PGD2 , LTE4
Sputum supernatants were available from 34/54 of the bronchoscopy cohort described above, 2 people without bronchoscopic samples collected for immunohistochemistry, and 7 participants who passed screening but did not proceed to bronchoscopy, commonly because they withdrew or exacerbated. The clinical characteristics of these 43 patients and 6 healthy controls are summarized in Appendix S2 and are similar to the primary bronchoscopy cohort. A subset of these subjects have been described previously. 28
IL‐5, CCL17, CCL26 and TSLP concentrations were elevated significantly in asthma overall compared with health (Figure S5). IL‐4, IL‐5 and CCL26 were significantly higher in T2‐high‐FNS compared with both T2‐low asthma and healthy controls (Figure 4). CCL17 and TSLP concentrations were increased in T2‐high‐FNS and T2‐intermediate asthma compared with healthy controls, but similar across the asthma groups (Figure 4). IL‐33 was increased in T2‐high‐FNS compared with healthy controls (Figure 4). There were no differences for IFNγ, IL‐31 or TNFα evident (Figure S5). Within the asthma patients (n = 43), there were strong positive correlations between all T2‐associated cytokines (Figure S6).
FIGURE 4.
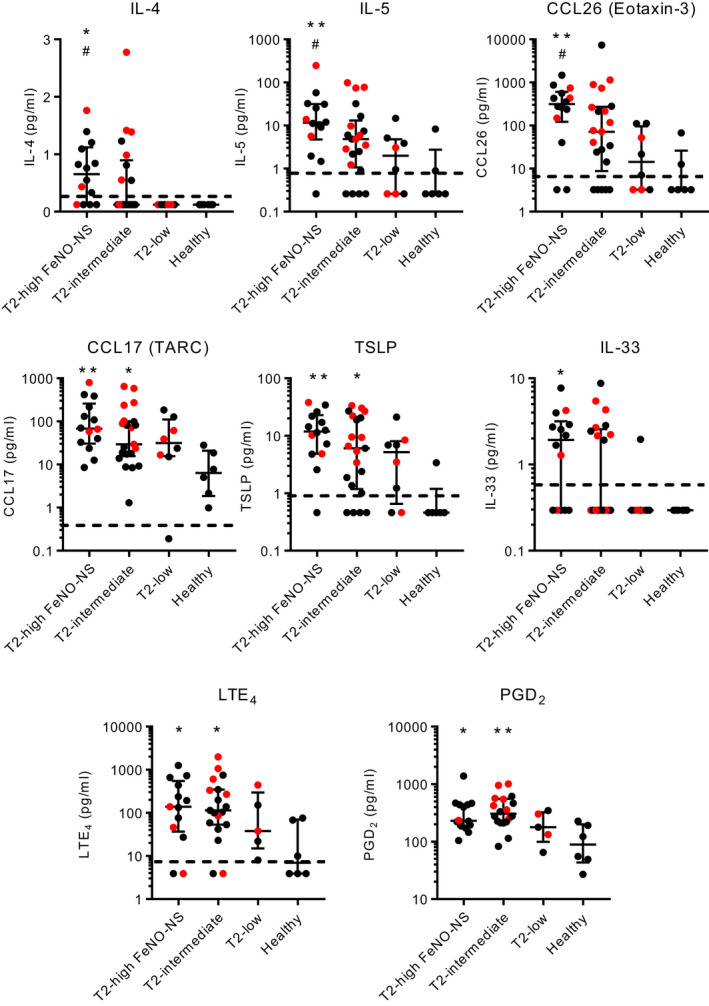
T2‐associated cytokine/chemokine, LTE4 and PGD2 concentrations in induced sputum supernatants in asthma subgroups and healthy controls. *p < 0.05, **p < 0.01, compared with healthy controls (Dunn's multiple comparison test). #p < 0.05 compared with T2‐low (Dunn's multiple comparison test). Red points represent people taking oral corticosteroids. Dotted lines represent the lower limit of detection
FeNO correlated with all T2 cytokines and most strongly with IL‐4 (r s = 0.661, p < 0.0001). Sputum eosinophils correlated with all T2 cytokines except IL‐13 and most strongly with IL‐5 (r s = 0.644, p < 0.0001). Blood eosinophils correlated weakly with most sputum T2 cytokines. FeNO correlated with blood eosinophils (r s = 0.589, p < 0.0001) and sputum eosinophils (r s = 0.487, p = 0.003), but the correlation between sputum and blood eosinophils was weak (r s = 0.331, p = 0.045, n = 37) (Figure S6).
For PGD2 and LTE4, samples were available for only 5 T2‐low patients. Both PGD2 and LTE4 were elevated in asthma compared with healthy controls (Figure S7) and elevated in both T2‐high‐FNS and T2‐intermediate patients compared with health (Figure 4). Levels of PGD2 in T2‐low asthma were similar to those in T2‐high‐FNS, and there were no significant differences between the asthma groups (Figure 4).
4. DISCUSSION
We have compared the immunopathology of highly polarized severe asthma phenotypes, namely people who have persistently raised T2‐biomarkers despite electronically monitored high‐dose ICS treatment (T2‐high‐FNS), people who have had their treatment titrated against T2 biomarkers (T2‐intermediate), and people with low T2 biomarkers (T2‐low). Our aim was to explore the airway immunopathological factors that might be contributing to disease expression in these T2‐high versus T2‐low cohorts.
These asthma groups were clearly separated by their expression of two widely accepted biomarkers of T2 activity, FeNO and blood eosinophils, in spite of taking similar doses of ICS and oral corticosteroids. Both the T2‐intermediate and T2‐low groups had significantly higher blood eosinophil counts in the past, suggesting that persistent T2‐biomarker‐low asthma is rare as described. 6 , 7 Nevertheless, these T2‐low patients remain very symptomatic and prone to exacerbations (Table 1).
The definition for T2‐low we used was planned before the results of more recent phase 3 trials of biologic therapies became available. T2 activity is not all‐or‐nothing, but graded, and there is debate about what thresholds define T2‐low. The UK severe asthma registry now defines T2‐low as blood eosinophils <0.15 × 109/L and FeNO ≤25 ppb. However, while a blood eosinophil count of (0.15 × 109/L) is the quoted cut for response to IL‐5‐targeted therapies, the clinical response just above this is minimal and of doubtful clinical significance, 29 as reflected in the UK National Institute for Clinical Excellence (NICE) prescribing criteria where the cut for treatment eligibility is 0.30 × 109/L. Furthermore, the upper 95% CI for blood eosinophils in healthy controls is at least 0.2 × 109/L. 30 In our T2‐low cohort, only one patient had a FeNO >25 ppb, and that was 27 ppb, while 75% of patients had blood eos ≤0.15. The T2‐low blood eosinophil counts and FeNO levels were similar to the healthy control group. However, a post hoc analysis of the data limiting the T2‐low group to those with blood eosinophils <0.15 × 109/L and FeNO ≤25 ppb does not alter the conclusions of this study, and it is clear from the sputum data that this T2‐low group had suppressed T2 cytokine expression in the airways and that our original thresholds were reasonable.
In keeping with raised FeNO and blood eosinophils in T2‐high‐FNS, there were increased concentrations of many T2‐associated cytokines in their induced sputum supernatant compared with T2‐low patients and healthy controls. These were strongly inter‐correlated and correlated with FeNO and sputum eosinophils, indicating a rich T2‐environment. However, tissue eosinophil numbers were not elevated in any of the asthma groups compared with healthy controls and were similar across the asthma groups, despite raised sputum eosinophils in the T2‐high‐FNS. These data may appear counterintuitive, but are consistent with the U‐BIOPRED cohort where there was no evidence of tissue eosinophilic inflammation in severe asthma compared with health, despite increased sputum eosinophils in severe asthma overall. 18 Several other 16 , 31 but not all 17 , 32 studies of severe asthma concur that there is no tissue eosinophilia. This is unlikely to be because bronchoscopic sampling is insensitive as studies of mild steroid‐naïve asthma show a consistent increase in tissue eosinophils (epithelium and lamina propria), and this is reduced consistently by inhaled corticosteroids. 33 , 34 The discrepancy with sputum eosinophil counts in T2‐high patients suggests there is ongoing eosinophil trafficking, although we might have expected to see increased eosinophils in the airway epithelium if that is the case. Nevertheless, the high levels of eosinophil chemoattractants in sputum support this explanation, and thus, a heightened level of trafficking may explain their relatively reduced numbers in the tissue. In addition, it is possible that the survival of tissue‐resident eosinophils remains highly sensitive to the effects of ICS in stable‐state severe asthma, while production of epithelial chemokines and T2 cytokines by mast cells, ILC2 cells, Th2 T cells, and perhaps eosinophils that remain activated, 35 are relatively resistant to the effects of ICS in T2 biomarker‐high patients.
IL‐5 and anti‐IL‐4/13‐targeted therapies are most effective in severe asthmatics expressing high type 2 biomarkers, 14 , 36 and IL‐5‐targeted therapy likely works exclusively through its effects on eosinophil biology. These biologic therapies reduce the rate of severe asthma exacerbations to a greater extent than improving day‐to‐day symptoms and lung function. 36 , 37 As tissue eosinophil numbers were not increased in stable‐state T2‐high severe asthma, the efficacy of anti‐IL5/IL‐5Rα and anti‐IL‐4Rα therapy on exacerbations may be due to their effects on systemic rather than tissue eosinophils, preventing recruitment and further trafficking to the tissue during exacerbations. This interpretation is supported by the observations that blood eosinophils are a better predictor of response to mepolizumab than sputum eosinophils, 29 , 38 although not all studies have shown this. 39 However, activation of the remaining tissue eosinophils may also contribute to exacerbations. 35 Furthermore, as IL‐4 and IL‐13 have biological effects that extend well beyond an effect on eosinophils, it is likely that anti‐IL‐4Rα therapy impacts other components of IL‐4/13 biology in asthmatic airways that promote exacerbations, for example, by attenuating the effects of IL‐4/13 on AHR. 40 , 41
There are significant residual exacerbations in T2‐high severe asthma treated with T2 biologics, many of which are non‐eosinophilic in origin. 42 This, coupled with persisting symptomatology and airway dysfunction in both T2‐high and T2‐low asthma, requires explanation and represents a major unmet clinical need. Bronchoconstriction and AHR are defining features of asthma. Our study is cross‐sectional, but the presence of increased ASM mass within the airway wall across all severe asthma phenotypes when compared to healthy controls, despite intense treatment, provides further evidence that the ASM remains a key dysfunctional element driving severe asthma pathophysiology, 43 irrespective of the underlying inflammatory milieu. The ongoing production of potent bronchoconstrictors (LTE4 and PGD2) in the airways in both T2‐high and T2‐low asthma despite high‐dose ICS+/−oral corticosteroid therapy is likely important in the presence of this hyperresponsive pathologically remodelled ASM. It also suggests that everyone with severe asthma should receive a trial of a leukotriene receptor antagonist.
Mucus secretion is another major contributor to airflow obstruction in both mild and severe asthma, 44 , 45 , 46 , 47 , 48 particularly during exacerbations and in asthma deaths. 46 , 49 , 50 MUC5AC expression was increased in the airway epithelium and to a similar extent across the asthma cohorts. Many T2 and non‐T2 cytokines, growth factors and lipid mediators potentially increase MUC5AC expression and mucus secretion. 51 , 52 , 53 While T2 cytokines may make an important contribution to ongoing mucus production in T2‐high asthma and provide another mechanism through which anti‐IL4Rα therapy may work to reduce asthma exacerbations, further work is required to establish the many possible factors driving persistent MUC5AC expression in T2‐low asthma. In addition, the presence of submucosal glands was increased in asthma, although it is unclear why this signal was driven by T2‐low and T2‐intermediate asthma rather than T2‐high asthma.
Mast cells are activated by most stimuli considered important for the pathogenesis of asthma, its propagation and the development of asthma exacerbations. 54 Their vast array of mediators can account for many of the described pathological features of asthma and the disordered airway physiology. 54 Numerous cross‐sectional studies demonstrate that mast cells are continuously activated in asthmatic airways, irrespective of disease severity or phenotype. 15 , 54 , 55 The presence of similarly high sputum PGD2 concentrations across the asthma groups in our study is consistent with these previous studies. Mast cell numbers in the lamina propria are similar in mild steroid‐naïve asthma compared with health 16 , 17 , 56 and reduced by ICS. 57 Some studies found similar mast cell numbers in the lamina propria in severe asthma compared with health and others reduced numbers. 16 , 18 , 31 Our data are in keeping with the latter. Epithelial mast cells are increased in mild steroid‐naive asthma compared with healthy controls, 16 , 56 but reduced by ICS, 57 consistent with this and previous studies. 16 , 18 A role for mast cells in both T2‐high and T2‐low severe asthma is supported by a recent phase 3 trial of masitinib, which reduced the rate of severe asthma exacerbations in both T2 biomarker‐high and T2 biomarker‐low severe asthma. 58 , 59 Taken together, this suggests that it is the activity of mast cells rather than their numbers, which is important.
Mast cell infiltration of the ASM is a characteristic feature of mild asthma 54 , 60 and likely important physiologically as this places activated mast cells within the ASM bundles. 61 In our previous study investigating pathological heterogeneity, mast cell counts in the ASM were only elevated compared with healthy controls in mild–moderate asthma, but not in severe asthma. 17 U‐BIOPRED also found no difference in the number of mast cells in the ASM in severe asthma compared with healthy controls. 18 Our study is, therefore, in keeping with previous studies. However, mast cells within the ASM bundles in severe asthma may remain activated with important physiological consequences. Electron microscopy to assess the level of piecemeal degranulation as a feature of activation, as shown in milder asthma, 62 , 63 would help answer this question.
The term ‘neutrophilic asthma’ is often used to describe patients with high sputum neutrophil percentage counts without consideration of total cell counts. A sputum neutrophilia is common in people using ICS, as corticosteroids induce eosinophil apoptosis but prolong neutrophil survival, and an inverse correlation between sputum eosinophil and neutrophil counts is described. 64 A small proportion of severe asthmatics have a high total sputum neutrophil count and often complain of repeated episodes of purulent sputum. These patients can perhaps be accurately described as having a neutrophilic phenotype. 65 However, this and many previous studies have failed to find increased airway neutrophil infiltration in mild, moderate or severe asthma when compared to healthy controls. 16 , 17 , 18 , 31 , 66 That is not to say that neutrophils do not play a role, because like mast cells, their activation status may be more important than absolute numbers. Increased numbers of neutrophils expressing CEACAM6 in severe asthma suggest that neutrophil activation status may indeed be altered. 66
Our study has some limitations as it is cross‐sectional, and patients can only be bronchoscoped when clinically stable and not exacerbating. However, the remarkably similar pathology between T2‐high and T2‐low severe asthma suggests this does not fluctuate markedly over time. It is also not feasible to biopsy the distal small airways, and it is possible that the T2‐biomarker signals in breath and sputum arise from the distal airways where ICS may not reach. 67 However, the persisting T2 signals in people on oral corticosteroids argue against this. Furthermore, T2‐dependent gene signatures in proximal bronchial biopsies were associated with sputum eosinophilia and raised FeNO levels in previous studies. 10 Although there was no tissue inflammatory cell signal that distinguished T2‐high from T2‐low asthma, we believe that bronchoscopy as a research tool still has an important role to play in the further understanding of the pathology and molecular pathways contributing to steroid insensitivity and ongoing disease expression, exemplified by recent transcriptomic studies. 55 , 68 It will also continue to have an important role in understanding the efficacy on novel therapies on remodelling and inflammation and for demonstrating drug target engagement within the airways.
In summary, tissue eosinophilic inflammation is absent in both T2 biomarker‐high and T2 biomarker‐low severe asthma. Persisting T2 cytokine expression in people with T2 biomarker‐high severe asthma likely underpins the ability of T2‐targeted treatments to reduce eosinophilic asthma exacerbations, most likely by preventing the recruitment of eosinophils at the time of exacerbation. However, there are many clinical aspects of severe asthma that appear orthogonal to T2 inflammation (non‐eosinophilic exacerbations, impaired lung function and day‐to‐day symptoms), likely accounted for by persisting increases in ASM mass, glandular hyperplasia, enhanced mucus production and mast cell activation. Considering that bronchoconstriction and mucus plugging are the predominant causes of airflow obstruction driving asthma symptoms, exacerbations and death, the factors that sustain these abnormal pathological features remain an important area for future research and drug development.
CONFLICT OF INTEREST
SC has received non‐restricted research grants from the NIHR Oxford BRC, Sanofi‐Genzyme and the Quebec Respiratory Health Research Network; he is the holder of the Association Pulmonaire du Québec's Research Chair in Respiratory medicine; he received speaker honoraria from AstraZeneca, GlaxoSmithKline, Sanofi‐Regeneron and Valeo Pharma; he received consultancy fees for FirstThought; he has received sponsorship to attend international scientific meetings by AstraZeneca. He is an advisory board member for Biometry Inc—a company which is developing a FeNO device (myBiometry); the contract will be remunerated by stock options. RC has received lecture fees from GSK, AstraZeneca, Teva, Chiesi and Sanofi; honoraria for Advisory Board Meetings from GSK, AstraZeneca, Teva, Chiesi and Novarti; sponsorship to attend international scientific meetings from Chiesi, Napp, Sanofi, Boehringer, GSK and AstraZeneca and a research grant to her Institute from AstraZeneca for a UK multi‐centre study. AHM has received personal and institutional payment for talks, advisory board meetings, education and research funding from GSK, AstraZeneca, Teva, Chiesi, NAPP, Sanofi, Novartis, PI. TSCH has received grants from Pfizer Inc., the University of Oxford, the Wellcome Trust, The Guardians of the Beit Fellowship, the NIHR Oxford Biomedical Research Centre, Sensyne Health and Kymab during the conduct of the study; and personal fees from AstraZeneca, TEVA and Peer Voice outside the submitted work. IDP, In the last 5 years, IDP has received speaker's honoraria for speaking at sponsored meetings from Astra Zeneca, Boehringer Ingelheim, Aerocrine, Almirall, Novartis, Teva, Chiesi, Sanofi/Regeneron, Menarini and GSK, and payments for organizing educational events from AstraZeneca, GSK, Sanofi/Regeneron and Teva. He has received honoraria for attending advisory panels with Genentech, Sanofi/Regeneron, Astra Zeneca, Boehringer Ingelheim, GSK, Novartis, Teva, Merck, Circassia, Chiesi and Knopp, and payments to support FDA approval meetings from GSK. He has received sponsorship to attend international scientific meetings from Boehringer Ingelheim, GSK, AstraZeneca, Teva and Chiesi. He has received a grant from Chiesi to support a phase 2 clinical trial in Oxford. In 2014–5 and 2019–20, he was an expert witness for a patent dispute involving AstraZeneca and Teva. SJF has received grants from Boehringer Ingelheim and fees from AstraZeneca, Boehringer Ingelheim, Novartis, Teva and Chiesi. LPM declares Research funding from Chiesi and Merck; consultancies for Chiesi, Glaxo Smith Kline, Merck, Sanofi, Genentech and support to attend scientific meetings from Chiesi, Merck and Bayer. LGH reports grants from Genentech/Hoffman la Roche, during the conduct of the study; other from AstraZeneca, Boehringer Ingelheim, Chiesi, GSK and Napp Pharmaceuticals, personal fees from Novartis, Hoffman la Roche/Genentech Inc, Sanofi, Evelo Biosciences, Glaxo Smith Kline, Astra Zeneca, Teva, Theravance, Circassia, grants from Medimmune, Novartis UK, Roche/Genentech Inc and Glaxo Smith Kline, Amgen, Genentech/Hoffman la Roche, Astra Zeneca, Medimmune, Glaxo Smith Kline, Aerocrine and Vitalograph, outside the submitted work. CDA is an employee of Genentech. PHH is an employee of GSK. JRA is an employee of Genentech. DC is an employee of Genentech. PB has received research funding from Genentech via the University Hospitals of Leicester NHS Trust; consultancies for Boehringer Ingelheim, Genentech and Celldex Therapeutics via the University of Leicester. Support to attend scientific meetings from Chiesi, Teva and Sanofi‐Genzyme. BH, LC, FAS, SB, RS, VB no conflict of interest.
AUTHOR CONTRIBUTIONS
All authors reviewed the data and contributed to its interpretation, edited the manuscript and approved the final submitted version. LC and FAS: Performed immunohistochemistry and quantified and collated the data. SC: Performed cytokine measurements on induced sputum supernatants and quantified and collated the data. BH: Managed the study, contributed to patient recruitment and collated the demographic data. RC: Principal investigator in Glasgow. SB: Performed bronchoscopy in Glasgow. AHM: Principal investigator in Birmingham. RS: Recruited and bronchoscoped patients in Oxford. TSCH: Supervised sputum cytokine analysis. IDP: Principal investigator in Oxford. SJF: Principal investigator in Manchester, performed bronchoscopy. VB: Sample management and storage. LPM: Co‐Investigator in Belfast performed bronchoscopies in Belfast, on study progress committee. LGH: Principal investigator in Belfast, conceived and designed the study, academic lead for RASP‐UK programme, bronchoscopy study progress committee. CDA: Contributed to bronchoscopy protocol design and provided tissue samples from the CLAVIER cohort. PHH: Principal investigator in Southampton. JRA: Contributed to study design and provided tissue samples from the CLAVIER cohort. DC: Contributed to study design and manuscript drafting. PB: Conceived and designed the study, wrote the study protocol and ethical application, analyzed the data, drafted the manuscript and performed all bronchoscopies in Leicester. Chief investigator.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Appendix S1
Appendix S2
ACKNOWLEDGEMENTS
The authors thank the following people for their help in conducting the study—Paula McCourt, Gill Elliott (Leicester), Gareth Hynes, Katie Borg, Clare Connelly (Oxford) and Miriam Baca (Genentech). AHM acknowledges the MIDRU research and development team at Heartlands Hospital Birmingham UK, Mary Bellamy, Tamika Thompson, Dr Sarah Davies and Dr Anil Pillai. SJF acknowledges the support of the NIHR Manchester Biomedical Research Centre and the NIHR Manchester Clinical Research Facility.
Khalfaoui L, Symon FA, Couillard S, et al. Airway remodelling rather than cellular infiltration characterizes both type2 cytokine biomarker‐high and ‐low severe asthma. Allergy. 2022;77:2974‐2986. doi: 10.1111/all.15376
Funding information
This paper was supported by an MRC Stratified Medicine Grant MR/M016579/1, and in part by the National Institute for Health Research (NIHR) Leicester Biomedical Research Centre (Respiratory). Additional funding was received from Genentech to the University Hospitals of Leicester NHS Trust to support this study. SJF is supported by the NIHR Manchester Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
REFERENCES
- 1. Stock S, Redaelli M, Luengen M, Wendland G, Civello D, Lauterbach KW. Asthma: prevalence and cost of illness. Eur Respir J. 2005;25(1):47‐53. [DOI] [PubMed] [Google Scholar]
- 2. Wenzel S. Severe asthma in adults. Am J Respir Crit Care Med. 2005;172(2):149‐160. [DOI] [PubMed] [Google Scholar]
- 3. Proceedings of the ATS workshop on refractory asthma . Current understanding, recommendations, and unanswered questions. Am J Respir Crit Care Med. 2000;162(6):2341‐2351. [DOI] [PubMed] [Google Scholar]
- 4. Berry MA, Morgan A, Shaw DE, et al. Pathological features and inhaled corticosteroid response of eosinophilic and non‐eosinophilic asthma. Thorax. 2007;62:1043‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McGrath KW, Icitovic N, Boushey HA, et al. A large subgroup of mild‐to‐moderate asthma is persistently noneosinophilic. Am J Respir Crit Care Med. 2012;185(6):612‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heaney LG, Busby J, Hanratty CE, et al. Composite type‐2 biomarker strategy versus a symptom‐risk‐based algorithm to adjust corticosteroid dose in patients with severe asthma: a multicentre, single‐blind, parallel group, randomised controlled trial. Lancet Respir Med. 2020;9:57‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jackson DJ, Busby J, Pfeffer PE, et al. Characterisation of patients with severe asthma in the UKsevere asthma registry in the biologic era. Thorax. 2021;76(3):220‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woodruff PG, Modrek B, Choy DF, et al. T‐helper type 2‐driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choy DF, Modrek B, Abbas AR, et al. Gene expression patterns of Th2 inflammation and intercellular communication in asthmatic airways. J Immunol. 2010;186(3):1861‐1869. doi: 10.4049/jimmunol.1002568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choy DF, Hart KM, Borthwick LA, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7(301):301ra129. [DOI] [PubMed] [Google Scholar]
- 11. Ostling J, van Geest M, Schofield JPR, et al. IL‐17‐high asthma with features of a psoriasis immunophenotype. J Allergy Clin Immunol. 2019;144(5):1198‐1213. [DOI] [PubMed] [Google Scholar]
- 12. Flood‐Page P, Menzies‐Gow A, Phipps S, et al. Anti‐IL‐5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112(7):1029‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wenzel S, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455‐2466. [DOI] [PubMed] [Google Scholar]
- 14. Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360(10):973‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hinks TS, Zhou X, Staples KJ, et al. Innate and adaptive T cells in asthmatic patients: relationship to severity and disease mechanisms. J Allergy Clin Immunol. 2015;136(2):323‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shikotra A, Choy DF, Ohri CM, et al. Increased expression of immunoreactive thymic stromal lymphopoietin in patients with severe asthma. J Allergy Clin Immunol. 2011;129:104‐111. [DOI] [PubMed] [Google Scholar]
- 17. Siddiqui S, Shikotra A, Richardson M, et al. Airway pathological heterogeneity in asthma: visualization of disease microclusters using topological data analysis. J Allergy Clin Immunol. 2018;142(5):1457‐1468. [DOI] [PubMed] [Google Scholar]
- 18. Wilson SJ, Ward JA, Sousa AR, et al. Severe asthma exists despite suppressed tissue inflammation: findings of the U‐BIOPRED study. Eur Respir J. 2016;48(5):1307‐1319. [DOI] [PubMed] [Google Scholar]
- 19. Raundhal M, Morse C, Khare A, et al. High IFN‐gamma and low SLPI mark severe asthma in mice and humans. J Clin Invest. 2015;125(8):3037‐3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuo CS, Pavlidis S, Loza M, et al. T‐helper cell type 2 (Th2) and non‐Th2 molecular phenotypes of asthma using sputum transcriptomics in U‐BIOPRED. Eur Respir J. 2017;49(2):1602135. [DOI] [PubMed] [Google Scholar]
- 21. Seys SF, Grabowski M, Adriaensen W, et al. Sputum cytokine mapping reveals an 'IL‐5, IL‐17A, IL‐25‐high' pattern associated with poorly controlled asthma. Clin Exp Allergy. 2013;43(9):1009‐1017. [DOI] [PubMed] [Google Scholar]
- 22. Austin CD, Gonzalez Edick M, Ferrando RE, et al. A randomized, placebo‐controlled trial evaluating effects of lebrikizumab on airway eosinophilic inflammation and remodelling in uncontrolled asthma (CLAVIER). Clin Exp Allergy. 2020;50(12):1342‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heaney LG, Busby J, Bradding P, et al. Remotely monitored therapy and nitric oxide suppression identifies nonadherence in severe asthma. Am J Respir Crit Care Med. 2019;199(4):454‐464. [DOI] [PubMed] [Google Scholar]
- 24. Boddy CE, Naveed S, Craner M, Murphy AC, Siddiqui S, Bradding P. Clinical outcomes in people with difficult‐to‐control asthma using electronic monitoring to support medication adherence. J Allergy Clin Immunol Pract. 2020;9:1529‐1538. [DOI] [PubMed] [Google Scholar]
- 25. McNicholl DM, Stevenson M, McGarvey LP, Heaney LG. The utility of fractional exhaled nitric oxide suppression in the identification of nonadherence in difficult asthma. Am J Respir Crit Care Med. 2012;186(11):1102‐1108. [DOI] [PubMed] [Google Scholar]
- 26. Du Rand IA, Blaikley J, Booton R, et al. British Thoracic Society guideline for diagnostic flexible bronchoscopy in adults: accredited by NICE. Thorax. 2013;68(Suppl 1):i1‐i44. [DOI] [PubMed] [Google Scholar]
- 27. Sullivan P, Stephens D, Ansari T, Costello J, Jeffery P. Variation in the measurements of basement membrane thickness and inflammatory cell number in bronchial biopsies. Eur Respir J. 1998;12(4):811‐815. [DOI] [PubMed] [Google Scholar]
- 28. Couillard S, Shrimanker R, Chaudhuri R, et al. FeNO non‐suppression identifies corticosteroid‐resistant Type‐2 signaling in severe asthma. Am J Respir Crit Care Med. 2021;204:731‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double‐blind, placebo‐controlled trial. Lancet. 2012;380(9842):651‐659. [DOI] [PubMed] [Google Scholar]
- 30. Hartl S, Breyer MK, Burghuber OC, et al. Blood eosinophil count in the general population: typical values and potential confounders. Eur Respir J. 2020;55(5):1901874. [DOI] [PubMed] [Google Scholar]
- 31. Vrugt B, Wilson S, Underwood J, et al. Mucosal inflammation in severe glucocorticoid‐dependent asthma. Eur Respir J. 1999;13(6):1245‐1252. [DOI] [PubMed] [Google Scholar]
- 32. Wenzel SE, Schwartz LB, Langmack EL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respirat Crit Care Med. 1999;160(3):1001‐1008. [DOI] [PubMed] [Google Scholar]
- 33. Djukanovic R, Wilson JW, Britten KM, et al. Effect of an inhaled corticosteroid on airway inflammation and symptoms in asthma. Am Rev Respir Dis. 1992;145(3):669‐674. [DOI] [PubMed] [Google Scholar]
- 34. Laitinen LA, Laitinen A, Haahtela T. A comparative study of the effects of an inhaled corticosteroid, budesonide, and a beta 2‐agonist, terbutaline, on airway inflammation in newly diagnosed asthma: a randomized, double‐blind, parallel‐group controlled trial. J Allergy Clin Immunol. 1992;90(1):32‐42. [DOI] [PubMed] [Google Scholar]
- 35. Kelly EA, Esnault S, Liu LY, et al. Mepolizumab attenuates airway eosinophil numbers, but not their functional phenotype, in asthma. Am J Respir Crit Care Med. 2017;196(11):1385‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castro M, Corren J, Pavord ID, et al. Dupilumab efficacy and safety in moderate‐to‐severe uncontrolled asthma. N Engl J Med. 2018;378(26):2486‐2496. [DOI] [PubMed] [Google Scholar]
- 37. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198‐1207. [DOI] [PubMed] [Google Scholar]
- 38. Ortega HG, Yancey SW, Mayer B, et al. Severe eosinophilic asthma treated with mepolizumab stratified by baseline eosinophil thresholds: a secondary analysis of the DREAM and MENSA studies. Lancet Respir Med. 2016;4(7):549‐556. [DOI] [PubMed] [Google Scholar]
- 39. Mukherjee M, Aleman Paramo F, Kjarsgaard M, et al. Weight‐adjusted intravenous Reslizumab in severe asthma with inadequate response to fixed‐dose subcutaneous mepolizumab. Am J Respir Crit Care Med. 2018;197(1):38‐46. [DOI] [PubMed] [Google Scholar]
- 40. Venkayya R, Lam M, Willkom M, Grunig G, Corry DB, Erle DJ. The Th2 lymphocyte products IL‐4 and IL‐13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am J Respirat Cell Mol Biol. 2002;26(2):202‐208. [DOI] [PubMed] [Google Scholar]
- 41. Manson ML, Safholm J, James A, et al. IL‐13 and IL‐4, but not IL‐5 nor IL‐17A, induce hyperresponsiveness in isolated human small airways. J Allergy Clin Immunol. 2020;145(3):808‐817. e802. [DOI] [PubMed] [Google Scholar]
- 42. McDowell PJ, Diver S, Yang F, et al. The inflammatory profile of exacerbations in patients with severe refractory eosinophilic asthma receiving mepolizumab (the MEX study): a prospective observational study. Lancet Respir Med. 2021;9:1174‐1184. [DOI] [PubMed] [Google Scholar]
- 43. Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. 2003;167(10):1360‐1368. [DOI] [PubMed] [Google Scholar]
- 44. Cutz E, Levison H, Cooper DM. Ultrastructure of airways in children with asthma. Histopathology. 1978;2(6):407‐421. [DOI] [PubMed] [Google Scholar]
- 45. Huber HL, Koessler KK. The pathology of bronchial asthma. Arch Intern Med. 1922;30:689‐695. [Google Scholar]
- 46. Carroll NG, Mutavdzic S, James AL. Increased mast cells and neutrophils in submucosal mucous glands and mucus plugging in patients with asthma. Thorax. 2002;57(8):677‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dunican EM, Elicker BM, Gierada DS, et al. Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J Clin Invest. 2018;128(3):997‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ordonez CL, Khashayar R, Wong HH, et al. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med. 2001;163(2):517‐523. [DOI] [PubMed] [Google Scholar]
- 49. Kuyper LM, Pare PD, Hogg JC, et al. Characterization of airway plugging in fatal asthma. Am J Med. 2003;115(1):6‐11. [DOI] [PubMed] [Google Scholar]
- 50. Dunnill MS. The pathology of asthma with special reference to changes in the bronchial mucosa. J Clin Pathol. 1960;13:27‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thai P, Loukoianov A, Wachi S, Wu R. Regulation of airway mucin gene expression. Annu Rev Physiol. 2008;70:405‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Okumura S, Sagara H, Fukuda T, Saito H, Okayama Y. FcepsilonRI‐mediated amphiregulin production by human mast cells increases mucin gene expression in epithelial cells. J Allergy Clin Immunol. 2005;115(2):272‐279. [DOI] [PubMed] [Google Scholar]
- 53. Arthur GK, Duffy SM, Roach KM, et al. KCa3.1 K+ channel expression and function in human bronchial epithelial cells. PLoS One. 2015;10(12):e0145259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bradding P, Arthur G. Mast cells in asthma‐‐state of the art. Clin Exp Allergy. 2016;46(2):194‐263. [DOI] [PubMed] [Google Scholar]
- 55. Tiotiu A, Badi Y, Kermani NZ, et al. Association of differential mast cell activation to granulocytic inflammation in severe asthma. Am J Respir Crit Care Med. 2022;205(4):397‐411. [DOI] [PubMed] [Google Scholar]
- 56. Bradding P, Roberts JA, Britten KM, et al. Interleukin‐4, −5, and −6 and tumor necrosis factor‐alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol. 1994;10(5):471‐480. [DOI] [PubMed] [Google Scholar]
- 57. Wallin A, Sandstrom T, Soderberg M, et al. The effects of regular inhaled formoterol, budesonide, and placebo on mucosal inflammation and clinical indices in mild asthma. Am J Respir Crit Care Med. 1999;159(1):79‐86. [DOI] [PubMed] [Google Scholar]
- 58. Humbert M, de Blay F, Garcia G, et al. Masitinib, a c‐kit/PDGF receptor tyrosine kinase inhibitor, improves disease control in severe corticosteroid‐dependent asthmatics. Allergy. 2009;64(8):1194‐1201. [DOI] [PubMed] [Google Scholar]
- 59. Davidescu L, Chanez P, Ursol G, et al. Late Breaking Abstract ‐ Masitinib in severe asthma: Results from a randomized, phase 3 trial. Eur Respir J. 2020;56:4612. [Google Scholar]
- 60. Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346:1699‐1705. [DOI] [PubMed] [Google Scholar]
- 61. Begueret H, Berger P, Vernejoux JM, Dubuisson L, Marthan R, Tunon‐De‐Lara JM. Inflammation of bronchial smooth muscle in allergic asthma. Thorax. 2007;62(1):8‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Djukanovic R, Wilson JW, Britten KM, et al. Quantitation of mast cells and eosinophils in the bronchial mucosa of symptomatic atopic asthmatics and healthy control subjects using immunohistochemistry. Am Rev Respir Dis. 1990;142(4):863‐871. [DOI] [PubMed] [Google Scholar]
- 63. Roberts JA, Bradding P, Britten KM, et al. The long‐acting beta2‐agonist salmeterol xinafoate: effects on airway inflammation in asthma. Eur Respir J. 1999;14(2):275‐282. [DOI] [PubMed] [Google Scholar]
- 64. Arron JR, Choy DF, Laviolette M, et al. Disconnect between sputum neutrophils and other measures of airway inflammation in asthma. Eur Respir J. 2014;43(2):627‐629. [DOI] [PubMed] [Google Scholar]
- 65. Nair P, Surette MG, Virchow JC. Neutrophilic asthma: Misconception or misnomer? Lancet Respir Med. 2021;9:441‐443. [DOI] [PubMed] [Google Scholar]
- 66. Shikotra A, Choy DF, Siddiqui S, et al. A CEACAM6‐high airway neutrophil phenotype and CEACAM6‐high epithelial cells are features of severe asthma. J Immunol. 2017;198(8):3307‐3317. [DOI] [PubMed] [Google Scholar]
- 67. Singhania A, Rupani H, Jayasekera N, et al. Altered epithelial gene expression in peripheral airways of severe asthma. PLoS One. 2017;12(1):e0168680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kelsen SG, Agache IO, Soong W, et al. Astegolimab (anti‐ST2) efficacy and safety in adults with severe asthma: a randomized clinical trial. J Allergy Clin Immunol. 2021;148(3):790‐798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Appendix S1
Appendix S2