

Summary
Metabarcoding approaches are exponentially increasing our understanding of soil biodiversity, with a major focus on the bacterial part of the microbiome. Part of the soil diversity are also eukaryotes that include fungi, algae, protists and Metazoa. Nowadays, soil eukaryotes are targeted with the same approaches developed for bacteria and archaea (prokaryotes). However, fundamental differences exist between domains. After providing a short historical overview of the developments of metabarcoding applied to environmental microbiology, we compile the most important differences between domains that prevent direct method transfers between prokaryotic and eukaryotic soil metabarcoding approaches, currently dominated by short‐read sequencing. These include the existence of divergent diversity concepts and the variations in eukaryotic morphology that affect sampling and DNA extraction. Furthermore, eukaryotes experienced much more variable evolutionary rates than prokaryotes, which prevent capturing the entire eukaryotic diversity in a soil with a single amplification protocol fit for short‐read sequencing. In the final part we focus on future potentials for optimization of eukaryotic metabarcoding that include superior possibility of functionally characterizing eukaryotes and to extend the current information obtained, such as by adding a real quantitative component. This review should optimize future metabarcoding approaches targeting soil eukaryotes and kickstart this promising research direction.
Introduction
The use of molecular approaches to study soil microorganisms catalysed the immense development of microbial ecology. While soil bacterial diversity has received attention since the late 80s, studies on microbial eukaryotes (fungi, protists and micro‐algae, but also soil microfauna) have been lagging behind. Yet, they are currently gaining momentum, as large/global‐wide studies (Bates et al., 2013; Tedersoo et al., 2014; Mahé et al., 2017; Oliverio et al., 2020; Aslani et al., 2022) are generating immense amounts of data. The global understanding of diversity distribution and community assemblage rules (Aslani et al., 2022), as well as the discovery of novel environmental clades with high relevance for deep eukaryotic phylogeny (Burki et al., 2020), has considerably increased our understanding of soil eukaryotic diversity. Major breakthroughs were made, such as showing that eukaryotes in soils are more diverse than in aquatic systems (Singer et al., 2021), the most dominant eukaryotes are phagotrophs with small protists dominating (Oliverio et al., 2020) and that phototrophs are major contributors of the global carbon cycle (Jassey et al., 2022).
The precursory nature of ecological environmental studies focusing on prokaryotes has led to the fact that eukaryote‐focused environmental diversity studies use largely the same methodology. Nevertheless, the last 10 years saw the development of new curated genetic databases dedicated to eukaryotes (Guillou et al., 2012) and the expansion of existing ones (Quast et al., 2013). However, sampling designs, laboratory protocols, data analyses tools and concepts that are used by most soil ecologists interested in eukaryotes are still mostly based on – or even are identical to – the approaches used to study prokaryotes.
Our aim in this perspective is to highlight fundamental differences between prokaryotes and eukaryotes, and the resulting consequences for environmental metabarcoding studies. Indeed, the immense morphological diversity of eukaryotes as well as their fast and inhomogeneous evolutionary rates prevent the application of protocols for eukaryotic metabarcoding that were developed to study prokaryotes. Indeed, unified protocols for all eukaryotes can only provide partial images of the domain's diversity in soils. In addition to this comparative list of warnings, we highlight avenues to take to optimize the methodology to study soil eukaryotes.
Historical context; why did the study of eukaryotes lag behind prokaryotic studies?
Environmental microbiology with a focus on an in toto nucleic acid extraction, the current standard to assess microbial biodiversity, has started to reveal the immense diversity of planktonic bacteria in the Sargasso Sea (Lane et al., 1985; Giovannoni et al., 1990). Similar approaches followed shortly afterwards for Archaea (DeLong, 1992). Protocols were developed at that time to extract environmental DNA from soil (Tsai and Olson, 1992). These approaches led to the discovery of many major lineages composed exclusively of uncultured organisms, called ‘environmental clades’ (for a review encompassing all domains of life, see López‐García and Moreira (2008)). While prokaryotic microbiology developed considerably, microbial eukaryotic environmental diversity remained unstudied for more than a decade. Casually, the first study aimed at characterizing changes in communities in artificial systems after virus‐induced lysis in filamentous cyanobacteria (van Hannen et al., 1999). Eukaryotes gained more attention in aquatic systems, and studies revealed novel eukaryotic clades in marine (López‐García et al., 2001; Moon‐van der Staay et al., 2001) and freshwater hyperacidic systems (Amaral‐Zettler et al., 2002). Early environmental molecular explorations of soil eukaryotic diversity were still hampered by the low sequencing depth provided by amplicon cloning/Sanger sequencing approaches used by then, which did not allow seeing beyond the over‐dominance of a few plant and fungal operational taxonomic units (OTUs). Most sequences of protists, fungi and animals could not be retrieved (Lesaulnier et al., 2008), thus overlooking most eukaryotic diversity. Taxon‐specific primers circumvented this problem and were applied to investigate certain eukaryotic groups such as among protists (Bass and Cavalier‐Smith, 2004; Lara et al., 2007). In the meanwhile, soil bacteria studies were flourishing (Borneman et al., 1996; Chelius and Triplett, 2001; Sessitsch et al., 2001; Zhou et al., 2002). Later, the development and application of high‐throughput sequencing (HTS) technologies allowed comprehensive insights into microbial diversity in soils, such as of bacteria (Roesch et al., 2007), leading eventually to a better understanding of the distribution of bacterial diversity across global ecosystems (Delgado‐Baquerizo et al., 2018).
These methodological developments also benefited eukaryotic diversity surveys, as they allow overcoming the obstacle represented by the few over‐dominant OTUs and make possible the retrieval of a more complete picture of the eukaryotic biodiversity in soils. Eventually, the development of HTS in the last decade saw the considerable development of large soil metabarcoding initiatives, including global diversity surveys (Bates et al., 2013; Oliverio et al., 2020; Aslani et al., 2022). All these eukaryote‐focused approaches are mainly transferred from prokaryotic examples, often even using the same samples taken for prokaryotes for which the sampling has been optimized. The question remains how well transferable these approaches are and how accurately these data reflect the true eukaryotic diversity found in soils.
Two different diversity concepts
Species (i.e. independent evolutionary units) are the basic unit of biological diversity. While this concept has largely been developed based on macroscopic and sexual eukaryotes such as plants and animals, species are more challenging to delimit in the asexual prokaryotes. Some researchers even doubt the meaningfulness of the notion of species in prokaryotes, where genes are commonly exchanged between organisms (Doolittle and Zhaxybayeva, 2009). Indeed, bacterial and archaeal genomes possess a relatively restricted number of specific genes (‘core genome’), while many functional genes related to adaptations to the environment are freely transferred horizontally through plasmid exchanges and conjugation (Paquola et al., 2018; Kloub et al., 2021) (Fig. 1). Diversity units are therefore constituted by these core genomes (Bobay, 2020) which also include relatively variable genes that can be used to differentiate between close related clinical strains (Bourdin et al., 2021) or to observe geographical isolation (Qin et al., 2022). Yet, prokaryotic environmental studies aim at a more complete overview of the prokaryotic diversity than the few taxa for which genomes can be compiled as metagenome‐assembled genomes (Howe et al., 2014). These studies still need to apply metabarcoding based on arbitrary criteria such as genetic distances between strains to delimit species, such as the 97% 16S rRNA gene identity ‘rule’ (Stackebrandt et al., 2002) or amplicon sequence variants (Callahan et al., 2017). The delimitation of independent evolutionary units still remains unclear in bacteria, therefore the link between OTUs and bacterial biological species is not obvious. An example is provided by the human pathogen Salmonella flexneri, which shares 99.9% similarity with Escherichia coli on the 16S rRNA gene (Fukushima et al., 2002), with even E.coli variants ranging from pathogenic to mutualistic (Dethlefsen et al., 2007). A recent study showed that more than hundred thousand E. coli and Shigella genomes varied in their pathogenicity depending on genomic features which are invisible at the 16S rRNA gene level (Abram et al., 2021).
Fig. 1.
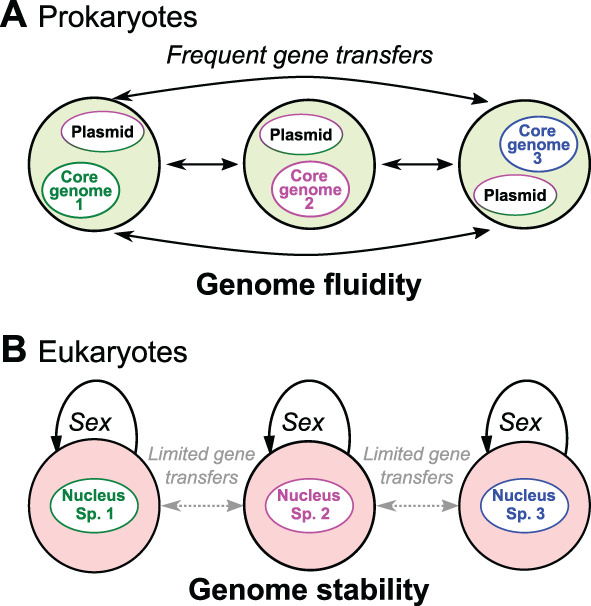
Differences between prokaryotic and eukaryotic diversity. While the first exchange functional genes frequently between taxa (A), eukaryotic genomes are much more stable and functions can be related to taxonomic affiliation (B).
In eukaryotes, biodiversity has been studied first on organisms that are easy to observe, that is, plants and animals, where genetic cohesion can essentially be characterized by sexual compatibility and population structure. These rules can also be applied to eukaryotic microbes, as most are considered sexual (Hofstatter and Lahr, 2019). Therefore, scientists interested in soil eukaryotes mostly benefit from the concept of species and all its developments (De Queiroz, 2007) as a benchmark for diversity. Genomes are much more stable in the eukaryotic world, and even if horizontal gene transfers have played a role in evolutionary histories, they remain extremely rare and would not bias diversity estimations (Van Etten and Bhattacharya, 2020) (Fig. 1). The term ‘core genome’ previously mentioned for bacteria does not apply here, as all genes are transmitted mostly within species, therefore, the OTUs obtained in environmental diversity studies are representative of the diversity. It must be noted that in microbial eukaryotes, organisms form temporal to permanent associations, most often composed by a consumer and a phototrophic organism (the consumer is then called ‘mixotroph’ by protistologists). Metabarcoding studies will retrieve sequences of both organisms, which we also consider as separate, like bacteria in a consortium.
Also in eukaryotes, the identity of species is not directly translatable from the commonly used 18S rRNA gene focused on in metabarcoding approaches. The reason is that this gene is simply not variable enough in most taxa. We illustrate this caveat in Fig. 2, showing that humans share exactly the same full 18S rRNA gene sequence with hominid primates (Fig. 2). Eukaryotic environmental metabarcoding targets even shorter parts of this gene, mostly the ‘hypervariable’ regions v4 and v9 of the 18S (Vaulot et al., 2022). Therefore, the reduced amount of phylogenetic information retrieved in current metabarcoding approaches would classify humans within the same taxonomic unit as elephants (Fig. 2). This vision would obfuscate a tremendous amount of diversity as the genetic information would be systematically pooled. This could lead to the fact that rare and potentially endangered species could be considered abundant if classified the same as some common species. For this reason, while 18S rRNA gene metabarcoding may provide a general overview of eukaryotic diversity in a soil, OTUs cannot be considered as equivalent to species, the basic unit of diversity. In order to allow species delimitation in eukaryotic metabarcoding studies by gaining taxonomic resolution, several variable genetic markers have been developed for the different groups and tested for specific resolution. The nuclear internal transcribed spacer (ITS), the mitochondrial cytochrome oxidase (COI) and the chloroplastic 23S rRNA in photosynthetic organisms are more variable than the 18S rRNA gene (Pawlowski et al., 2012) and are (or could be) used in group‐specific metabarcoding (see Section Potential for species‐level resolution in eukaryotic metabarcoding).
Fig. 2.
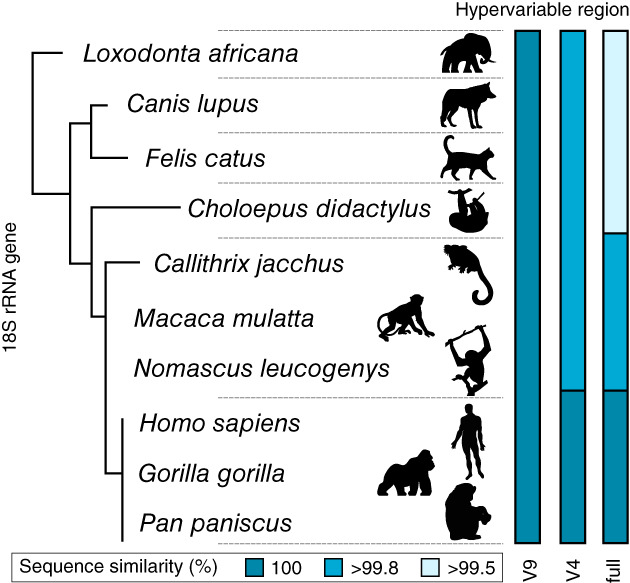
Phylogenetic tree illustrating percentages of genetic similarity between humans and different mammals based on some commonly used markers. v4 and v9 correspond to variable regions of the 18S rRNA gene.
Different morphologies change sampling protocols
Eukaryotes are characterized by an immense morphological variation (in terms of size, shape and many other traits) that greatly exceeds that of bacteria and archaea (Fig. 3). These variations affect how sampling for metabarcoding experiments in soil needs to be conducted. As soil eukaryotes range from few micrometres (several flagellated and amoeboid protists) to the centimetre or even meter scale (earthworms, burrowing animals and fungal mycelia), sample size and distribution need to be increased compared to studies focusing on the purely microscopic prokaryotes (Jurburg et al., 2021; Potapov et al., 2022). Sampling for the smallest and most abundant eukaryotic consumers – nanoflagellates and ‘naked’ amoebae might be the same as for bacteria. These eukaryotic organisms are considered the main drivers of nutrient cycling in soils (Rønn et al., 2002). However, even larger unicells such as Arcellinida (testate amoebae) would then need further upscaling or possibly filtering for concentrating cells (Kosakyan et al., 2015). These microbial top predators can dominate microbial biomass in certain systems such as peat bogs (Jassey et al., 2013; Marcisz et al., 2014). Also, microscopic nematodes, the most abundant animals on Earth that can exceed the importance of protists for nutrient cycling in some soils (Coleman et al., 1984; Griffiths, 1990), cannot be sampled with the prokaryotic protocols as sample sizes exceeding 100 g of soil are needed for reliable diversity analyses (Wiesel et al., 2015). Due to the functional overlap of the key microbiome predators protists and nematodes, as well as other fauna that perform similar functions (Thakur and Geisen, 2019), a separation between metazoans and unicellular organisms would be artificial and provide a biased vision of eukaryotic functional diversity. Like for large protists, nematodes and certainly also rotifers are extracted from the soil matrix before molecular work (Kawanobe et al., 2021). In order to retrieve all microbial‐sized eukaryotic diversity in metabarcoding analyses, huge amounts of soil should be taken. This would increase dramatically complicate and increase costs of sampling, and setting up controlled experiments such as mesocosms. At the same time, spatial heterogeneity of smaller organisms would be shadowed by sampling large amounts. A solution may come from applying different sampling strategies that target different organisms size classes and pooling nucleic acids extractions together. Sampling protocols to be applied depend therefore on the research question (and mostly on the target taxa), but also on the type of soil investigated.
Fig. 3.
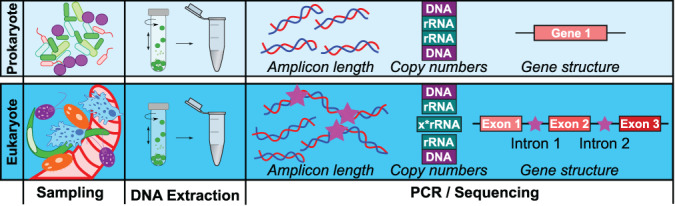
Schematic workflow comparison between prokaryotes and eukaryotes. Sampling and DNA extraction steps will be performed according to the targeted organisms. Fundamental differences between prokaryotes and eukaryotes (amplicon length, gene copy numbers and presence of intron in the eukaryote gene) can be observed in the PCR/Sequencing steps.
After sampling, nucleic acids need to be extracted in order to continue the flowchain in environmental metabarcoding. Some bacterial and archaeal taxa are recalcitrant to generic extraction protocols, as for instance the highly resistant spore‐forming bacteria (Dineen et al., 2010). In eukaryotes, differences between taxa exist also given their variability in morphology. In soil, most protists have the capacity to produce resistant dormant life stages or cysts (Ekelund and Rønn, 1994; Geisen et al., 2018). Some of these cysts are extremely resistant, and can withstand wet heat up to 50°C and dry heat to 120°C (Fenchel, 1987). This resistance is shown by successful resurrection of protist taxa after tenths of thousands of years from Arctic permafrost (Shmakova and Rivkina, 2015; Shmakova et al., 2016). The particularly recalcitrant cysts of Acanthamoeba spp. are also highly resistant to a variety of chemical agents (Turner et al., 2000). In turn, soil protist diversity includes highly fragile and network forming amoeboid organisms, which are abundant and widespread, such as foraminiferans (Holzmann et al., 2021) and variosean amoebae (Berney et al., 2015). These organisms, which can reach sizes of more than 1 mm, are disrupted when soil is ground or sieved, and their DNA can get lost. These differences make it impossible to assess eukaryotic diversity in an unbiased way using a single nucleic acid extraction approach. As it is mostly not feasible to combine many different protocols and it is also not always the goal to uncover the entire eukaryotic diversity, we propose to use a single extraction approach (Santos et al., 2017). Ideally, biases in extraction should be known to reliably discuss potential presence, absence and abundance of eukaryotic taxa present in a sample, and although certain trends are identifiable (Santos et al., 2017), taxon oriented‐research would be needed to adapt protocols to target eukaryotic groups.
Consequences of rRNA evolutionary rates on eukaryotic PCR‐based surveys
After sampling and extracting nucleic acids, the amplification of DNA extractions for environmental DNA surveys is also a source of bias that differs between prokaryotes and eukaryotes. One of the premises of environmental metabarcoding surveys is that taxa are equally amplified in all clades, as the sequences of the designed primers are highly conserved through evolution. For prokaryotes, protocols have been developed to obtain an optimized coverage of the whole diversity using primers flanking the v3–v5 variable regions of the 16S rRNA gene, with a size that is suited for short, max 550 bp targeting Illumina sequencing. For instance, the primer pair 515f/806r 16S SSU rRNA gene has been designed to match perfectly with flanking regions of more than 90% of all bacterial and archaeal 16S barcoding regions (Walters et al., 2011), making these oligonucleotides excellent broad‐range primers to cover prokaryotes (Knight et al., 2018). A recent test (03/2022) performed on the SILVA database (Klindworth et al., 2012) still shows that, despite the upload of most recent data, these primers still accommodate over 86% of all bacterial and archaeal sequences.
In comparison to their bacterial counterparts, eukaryotic ribosomal 18S rRNA genes present profoundly more heterogeneity in the regions flanking the most variable barcoding regions. Indeed, eukaryotes are famous for their fast‐evolving ribosomal genes that changed at different paces between clades. As an illustration, diverging evolution paces between eukaryotic 18S rRNA genes caused artefacts that disrupted the topology of the first eukaryotic trees (‘long branch attraction’) and caused misinterpretations on the evolutionary history of the whole domain (Philippe and Germot, 2000). These 18S rRNA gene‐related differences still cause issues in metabarcoding studies. The first consequence is that a universal PCR protocol cannot be designed for the whole domain Eukarya, because of a lack of universally conserved regions for primer design. Therefore, eukaryotic environmental molecular diversity studies based on PCRs performed on the 18S rRNA gene are systematically biased against certain organisms (Vaulot et al., 2022). Some common and diverse soil groups, such as the protistan Amoebozoa and Heterolobosea (Discoba) usually have sequences that are too variable in even conserved regions to design perfectly matching primers (Vaulot et al., 2022). Unfortunately, primers do not only ‘eliminate’ certain large clades from metabarcoding data. Evolutionary paces vary within classes, orders and even families, and supergroups tend to be partially eliminated. Often factors of these differences remain elusive, but organisms with mutualistic or parasitic lifestyles that are also common in soil animals and plants tend to evolve faster, as exemplified with the intracellular rhizarian parasite Microcytos (Hartikainen et al., 2014), or, living within terrestrial organisms, Microsporidia (Brinkmann et al., 2005).
The fast and heterogeneously evolving eukaryotic ribosomal genes also incorporate often introns of many hundreds of base pairs and supplementary loops that render the retrieval of certain sequences using modern HTS technology, such as Illumina's MiSeq, impossible. The presence of these insertions varies quickly in evolution, and closely related taxa can gain or lose them as illustrated in Heterolobosea (Geisen et al., 2015a), diatoms (Han et al., 2018) and in Arcellinid testate amoebae (Lara et al., 2008).
Among the two most commonly used variable regions of the 18S rRNA gene (v4 and v9), v4 is particularly prone to amplification biases (Vaulot et al., 2022). On the other hand, v4 is longer and provides more phylogenetic information (Fig. 2). The use of one region or the other depends of course on the research question, and experimental designs need to consider these limitations. In sum, there is no silver bullet solution to track all soil eukaryotic molecular diversity in metabarcoding studies. For eukaryotes, an homogenization of PCR protocols as recommended in the Earth Microbiome Project (Thompson, 2017) would systematically bias our vision of diversity against ‘non‐standard’ taxa. These do represent a substantial amount of the eukaryotic diversity, which might be largely lost in environmental eukaryotic DNA diversity studies.
Future perspectives on eukaryotic metabarcoding in soils
Despite the fact that eukaryotic metabarcoding approaches in soils are still in their infancy compared to prokaryotic studies, many methodological developments should overcome the current shortcomings. Here we list some potentials and recommendations to improve soil eukaryotic metabarcoding, which we summarize in Fig. 4:
Different diversity concepts need to be considered. While prokaryotic 16S rRNA OTUs can be considered as diversity proxies, in eukaryotes 18S rRNA OTUs correspond to real taxa that most of the time can be annotated to genus or family level or above, but rarely to species level. The level of taxonomic definition depends on the group considered and the targeted gene.
Sampling needs to be optimized when larger protists and metazoa are targeted by upscaling sample sizes or possibly concentrating organisms that cover also the local heterogeneity in organismal distribution. Holistic eukaryotic analyses covering protists and at least the most abundant ‘microbial sized’ animals (e.g. nematodes and rotifers) could be conducted by pooling nucleic acid extractions obtained from different protocols dimensioned for different organisms size classes. Including also the larger soil eukaryotes can be of fundamental value in ecological studies through their role as major microbiome predators, which influence its composition and functioning.
Environmental nucleic acids extraction and PCR will invariably bias the true information on eukaryotic diversity. Consensus approaches need to be applied by using the most generalist protocols (Santos et al., 2017; Vaulot et al., 2022), which allow cross‐sample comparisons. Still, it must be kept in mind that taxon‐specific protocols are needed for a substantial part of all soil eukaryotic clades if the entity of eukaryotic diversity is meant to be studied.
Next, we list a series of developments that will potentially open new research avenues in eukaryotic metabarcoding.
Fig. 4.
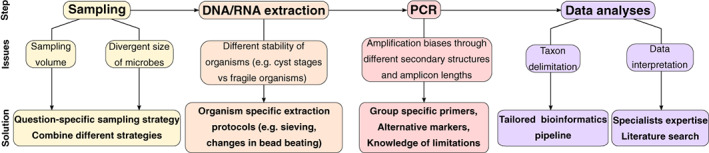
Illustration of the workflow followed in a eukaryotic metabarcoding experiment. Issues specific to eukaryotic metabarcoding are illustrated specifically, as well as solutions proposed to overcome these problems.
Functional annotations of eukaryotic metabarcoding data are readily possible compared to bacterial metabarcoding data
A major opportunity offered by eukaryotic environmental DNA surveys is the possibility to infer functions from taxonomic barcoding sequences. Eukaryotes keep to a large extent the inherited functions, like for instance photosynthesis in diatoms or active phagocytosis in Amoebozoa. This characteristic, which derives from the stability of eukaryotic genomes, allows a relatively reliable functional annotation of OTUs (Singer et al., 2021). Functions are conserved along clades at different taxonomic depths. For instance, in some protist groups, like in ciliates, function is conserved within classes to a certain extent, and a finer taxonomical resolution that can be reached with 18S rRNA sequences may refine even more the functional assignment (Mieczan, 2009). Thus, annotation based on ribosomal sequences can be relatively precise (Lara et al., 2007; Lara and Acosta‐Mercado, 2012). Other groups can be more challenging to characterize, like chrysophytes, where photosynthetic capacity was lost many times during the groups evolutionary history (Boenigk et al., 2005). This characteristic makes functional inference based on ribosomal sequences difficult in this group known to include both phagotrophic and phototrophic organisms. This situation is probably going to change as more species will be functionally characterized in the future. Therefore, functional annotations based on relatively easily obtainable 18S rRNA gene reads can be of major additional benefit to normal taxonomic information as the ecological role of distinct and entire communities of eukaryotes can be studied. It is a matter of time to implement an automated functional annotation tool for all soil eukaryotes.
Bacteria can generally not be reliably assigned a function in soils. Although functions can be related to some OTUs in bacteria and archaea (e.g. cyanobacteria, methanogens, etc.), the immense often unknown diversity in soils along with the plasticity of their genomes renders this prediction perilous. Some bacterial functional prediction pipelines like FAPROTAX have been used with some success in soil (Sansupa et al., 2021). However, this approach is based on the assumption that if two organisms share the same 16S rRNA gene sequence, then their genome (and thus their function) should be identical; an assumption that does not take into account bacterial genomic plasticity. In this sense, an approach through direct sequencing of functional genes retrieved from environmental samples has been favoured for years (see e.g. Gremion et al., 2004). In addition, recent deep‐sequencing efforts that reconstitute full genomes from metagenomes, metagenome‐assembled genomes (MAGs), are starting to be applied and reveal reliable potential gene functions of soil bacteria (Crits‐Christoph et al., 2018). Yet, this approach is only possible for a few numerically dominant taxa as many tens of thousands of bacterial taxa constitute the immensely large metagenome of a fraction of a gram of soil. Therefore, most bacteria remain functionally unknown in soils. It must be noted that, even worse than for bacteria, MAGs of soil eukaryotes cannot be assembled. This is caused by larger genomes and eukaryotic rarity compared to bacteria and archaea (Xiong et al., 2021), which diminishes eukaryotic sequence information in metagenomic data. Furthermore, eukaryotic functional genes can often not even be assigned to known functions (Sibbald and Archibald, 2017). Yet, and until new developments of sequencing technologies allow increasing even more sequencing depth to the point that eukaryotic genomes are assembled from metagenomes, the possibility of characterizing eukaryotic functional diversity directly from metabarcodes provides great insights into terrestrial ecosystems functioning.
The quantitative aspect of eukaryotic metabarcoding
Quantitative aspects are important information in any ecological study, and it would be desirable to include them in environmental metabarcoding surveys. Quantitative data such as abundance and especially biomass information are needed to estimate, for example, the importance of biodiversity in the global carbon, nitrogen and phosphorus cycles through food chains and across ecosystems. The question on how to interpret the number of reads obtained per OTU has been subject to a heated debate. Numbers of reads result approximately from the number of single cells times the number of copies per cell. These numbers vary between and within bacterial and archaeal taxa, spanning one order of magnitude, which prevents establishing a direct relationship between numbers of cells and numbers of reads (Stoddard et al., 2015). In eukaryotes, these numbers may vary by up to six orders of magnitude (Lavrinienko et al., 2021). As a consequence, studies show discrepancies between numbers of individuals and proportions of eukaryotic phylotypes in environmental metabarcoding studies (Gong and Marchetti, 2019). Ciliates, for instance, tend to be overrepresented, possibly due to their highly multiploid macronuclei (Geisen et al., 2015b). These numerical biases are combined with all potential flaws mentioned above (DNA extraction, PCR and sequencing). Therefore, Jurburg et al. (2021) even suggested to handle eukaryotic metabarcoding data as presence/absence, without any quantitative interpretation. This approach, however, needs to be very carefully considered for most ecological studies as numerical comparisons can provide vastly extended information compared to presence/absence data; in this sense, numerical data are far more important than richness information.
While copy numbers per genome do vary, total biovolumes of the organisms represented by different phylotypes can correlate with numbers of reads per OTU. Gonzalez‐de‐Salceda and Garcia‐Pichel (2021) recently found an allometric relationship, where cell biovolume and ribosomal operon copy numbers in microorganisms (including bacteria and archaea) are correlated well. A similar relationship exists also for multicellular organisms, namely, nematodes (Schenk et al., 2019). Correlating eukaryote biovolumes with numbers of reads in metabarcoding studies is in our view the way to bring quantitative aspects into the currently at best semi‐quantitative, qualitative metabarcoding data of soil eukaryotes. These biovolumes can be converted into C‐biomass equivalents, such as done for nematodes (van den Hoogen et al., 2019), which can be instrumental in following nutrient flows in ecosystems (Gilbert et al., 1998). However, calibration studies are needed to evaluate the biases inherent to a given eukaryotic taxon. A large database of individual species biovolumes is needed for broader use in ecological studies. In the meanwhile, sequence read numbers can still be considered as semi‐quantitative, but should be used with caution when applying numerically sensitive analyses as in correlation networks.
Potential for species‐level resolution in eukaryotic metabarcoding
As stated above, the 18S rRNA gene often provides a rather coarse taxonomic resolution for most eukaryotic groups. However, other molecular markers can increase taxonomic resolution. Due to the closer comparability to animals and plants, a species‐level taxonomic resolution can be used to apply the ecological theoretical background developed for plants and animals during the past decades and centuries. For that purpose, several barcoding genes have been tested for their taxonomic definition in isolated organisms and on pure cultures. Primers have been designed in order to cover the largest possible range of organisms within their target groups.
While short‐read metabarcoding approaches have been standard since its implementation almost a decade ago to target soil eukaryotes (Bates et al., 2013), long‐read HTS approaches, such as PacBio and Nanopore, are rapidly developing. As their error rate per base pair is decreasing, they can potentially allow the retrieval of full‐length ribosomal operon, combining species or even population‐resolved taxonomic information based on the ITS region with the phylogenetic information provided by 18S and 28S rRNA genes (Jamy et al., 2020; Tedersoo et al., 2021).
Another promising gene for metabarcoding is the COI. This gene is conserved across most animals (Hebert et al., 2003) and is used for rotifers, where it largely outperforms the 18S rRNA gene in terms of taxonomic resolution (Fontaneto et al., 2019). COI is also a reference for soil microarthropods (Porter et al., 2019). Although priming regions are too variable to be used to scan all nematode diversity (Ahmed et al., 2019), COI is a promising marker to delimit species within nematode families (Bai et al., 2020). In soil protist metabarcoding, this marker was applied to Arcellinida (lobose testate amoebae) revealing niche differentiation between closely related species (Singer et al., 2018). Together, metabarcoding focusing on genes other than the 18s rRNA gene and long‐read sequencing provide ultra‐high taxonomic resolution, which can be used for in‐depth studies on taxonomic profiles, species‐level biogeographic analyses or bioindication/tracking invasive species.
Concluding remarks
We here provide an overview of how the major differences that exist between prokaryotes and eukaryotes need to be taken into consideration when performing metabarcoding studies, from the conceptualization of experiments to data interpretation. Particularly in terrestrial systems, methods have first been optimized for prokaryotes, which leads to suboptimal experimental designs and misinterpretations of results in studies aiming at studying soil eukaryotes. Furthermore, we provide perspectives on ecologically relevant data that can be obtained through eukaryotic metabarcoding approaches, like functional information on soil eukaryotes. Methodological developments, thorough calibration and optimization efforts are still needed to refine quantitative interpretation of sequence read numbers; however, it can be foreseen that many methodological gaps will be filled in the years to come. Then, the possibility of applying fast evolving markers used for barcoding opens the door to many applications in academic (phylogeography, community ecology, etc.) and applied fields (bioindication, pathogen monitoring, etc.). Taken together, all these approaches will reveal the great power of eukaryotic environmental microbiology. Still, until then, it is of crucial importance to involve experts in studies; and, furthermore to train students in all steps of a given study, from design to interpretation.
Acknowledgements
Our research was funded by: FEDER/Ministerio de Ciencia e Innovación – Agencia Estatal de Investigación (Project PGC2018‐094660‐B‐I00) to E.L. and NWO‐VENI from the Netherlands Organization for Scientific Research (016.Veni.181.078) to S.G.
References
- Abram, K. , Udaondo, Z. , Bleker, C. , Wanchai, V. , Wassenaar, T.M. , Robeson, M.S. , and Ussery, D.W. (2021) Mash‐based analyses of Escherichia coli genomes reveal 14 distinct phylogroups. Commun Biol 4: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, M. , Back, M.A. , Prior, T. , Karssen, G. , Lawson, R. , Adams, I. , and Sapp, M. (2019) Metabarcoding of soil nematodes: the importance of taxonomic coverage and availability of reference sequences in choosing suitable marker(s). MBMG 3: e36408. [Google Scholar]
- Amaral‐Zettler, L.A. , Gómez, F. , Zettler, E. , Keenan, B.G. , Amils, R. , and Sogin, M.L. (2002) Eukaryotic diversity in Spain's River of Fire. Nature 417: 137. [DOI] [PubMed] [Google Scholar]
- Aslani, F. , Geisen, S. , Ning, D. , Tedersoo, L. , and Bahram, M. (2022) Towards revealing the global diversity and community assembly of soil eukaryotes. Ecol Lett 25: 65–76. [DOI] [PubMed] [Google Scholar]
- Bai, M. , Qing, X. , Qiao, K. , Qiao, K. , Ning, X. , Xiao, S. , et al. (2020) Mitochondrial COI gene is valid to delimitate Tylenchidae (Nematoda: Tylenchomorpha) species. J Nematol 52: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass, D. , and Cavalier‐Smith, T. (2004) Phylum‐specific environmental DNA analysis reveals remarkably high global biodiversity of Cercozoa (Protozoa). Int J Syst Evol Microbiol 54: 2393–2404. [DOI] [PubMed] [Google Scholar]
- Bates, S.T. , Clemente, J.C. , Flores, G.E. , Walters, W.A. , Parfrey, L.W. , Knight, R. , and Fierer, N. (2013) Global biogeography of highly diverse protistan communities in soil. ISME J 7: 652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney, C. , Geisen, S. , Van Wichelen, J. , Nitsche, F. , Vanormelingen, P. , Bonkowski, M. , and Bass, D. (2015) Expansion of the ‘Reticulosphere’: diversity of novel branching and network‐forming amoebae helps to define Variosea (Amoebozoa). Protist 166: 271–295. [DOI] [PubMed] [Google Scholar]
- Bobay, L.‐M. (2020) The prokaryotic species concept and challenges. In The Pangenome: Diversity, Dynamics and Evolution of Genomes, Tettelin, H. , and Medini, D. (eds). Cham: Springer International Publishing, pp. 21–49. [Google Scholar]
- Boenigk, J. , Pfandl, K. , Stadler, P. , and Chatzinotas, A. (2005) High diversity of the ‘Spumella‐like’ flagellates: an investigation based on the SSU rRNA gene sequences of isolates from habitats located in six different geographic regions. Environ Microbiol 7: 685–697. [DOI] [PubMed] [Google Scholar]
- Borneman, J. , Skroch, P.W. , O'Sullivan, K.M. , Palus, J.A. , Rumjanek, N.G. , Jansen, J.L. , et al. (1996) Molecular microbial diversity of an agricultural soil in Wisconsin. Appl Environ Microbiol 62: 1935–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdin, T. , Monnier, A. , Benoit, M.‐È. , Bédard, E. , Prévost, M. , Quach, C. , et al. (2021) A high‐throughput short sequence typing scheme for Serratia marcescens pure culture and environmental DNA. Appl Environ Microbiol 87: e01399‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann, H. , van der Giezen, M. , Zhou, Y. , de Raucourt, G.P. , and Philippe, H. (2005) An empirical assessment of long‐branch attraction artefacts in deep eukaryotic phylogenomics. Syst Biol 54: 743–757. [DOI] [PubMed] [Google Scholar]
- Burki, F. , Roger, A.J. , Brown, M.W. , and Simpson, A.G.B. (2020) The new tree of eukaryotes. Trends Ecol Evol 35: 43–55. [DOI] [PubMed] [Google Scholar]
- Callahan, B.J. , McMurdie, P.J. , and Holmes, S.P. (2017) Exact sequence variants should replace operational taxonomic units in marker‐gene data analysis. ISME J 11: 2639–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelius, M.K. , and Triplett, E.W. (2001) The diversity of archaea and bacteria in association with the roots of Zea mays L . Microb Ecol 41: 252–263. [DOI] [PubMed] [Google Scholar]
- Coleman, D.C. , Anderson, R.V. , Cole, C.V. , Mc Clellan, J.F. , Woods, L.E. , Trofymow, J.A. , and Elliott, E.T. (1984) Roles of protozoa and nematodes in nutrient cycling. In Microbial‐Plant Interactions. Madison, WI: John Wiley & Sons, pp. 17–28. [Google Scholar]
- Crits‐Christoph, A. , Diamond, S. , Butterfield, C.N. , Thomas, B.C. , and Banfield, J.F. (2018) Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 558: 440–444. [DOI] [PubMed] [Google Scholar]
- De Queiroz, K. (2007) Species concepts and species delimitation. Syst Biol 56: 879–886. [DOI] [PubMed] [Google Scholar]
- Delgado‐Baquerizo, M. , Oliverio, A.M. , Brewer, T.E. , Benavent‐González, A. , Eldridge, D.J. , Bardgett, R.D. , et al. (2018) A global atlas of the dominant bacteria found in soil. Science 359: 320–325. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. (1992) Archaea in coastal marine environments. Proc Natl Acad Sci U S A 89: 5685–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen, L. , McFall‐Ngai, M. , and Relman, D.A. (2007) An ecological and evolutionary perspective on human–microbe mutualism and disease. Nature 449: 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineen, S.M. , Aranda, R., IV , Anders, D.L. , and Robertson, J.M. (2010) An evaluation of commercial DNA extraction kits for the isolation of bacterial spore DNA from soil. J Appl Microbiol 109: 1886–1896. [DOI] [PubMed] [Google Scholar]
- Doolittle, W.F. , and Zhaxybayeva, O. (2009) On the origin of prokaryotic species. Genome Res 19: 744–756. [DOI] [PubMed] [Google Scholar]
- Ekelund, F. , and Rønn, R. (1994) Notes on protozoa in agricultural soil with emphasis on heterotrophic flagellates and naked amoebae and their ecology. FEMS Microbiol Rev 15: 321–353. [DOI] [PubMed] [Google Scholar]
- Fenchel, T. (1987) Ecology of Soil Protozoa. Madison, Wis: Science Technical. [Google Scholar]
- Fontaneto, D. , Eckert, E.M. , Anicic, N. , Lara, E. , and Mitchell, E.A.D. (2019) We are ready for faunistic surveys of bdelloid rotifers through DNA barcoding: the example of Sphagnum bogs of the Swiss Jura Mountains. Limnetica 38: 213–225. [Google Scholar]
- Fukushima, M. , Kakinuma, K. , and Kawaguchi, R. (2002) Phylogenetic analysis of Salmonella, Shigella, and Escherichia coli strains on the basis of the gyrB gene sequence. J Clin Microbiol 40: 2779–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisen, S. , Bonkowski, M. , Zhang, J. , and De Jonckheere, J.F. (2015a) Heterogeneity in the genus Allovahlkampfia and the description of the new genus Parafumarolamoeba (Vahlkampfiidae; Heterolobosea). Eur J Protistol 51: 335–349. [DOI] [PubMed] [Google Scholar]
- Geisen, S. , Laros, I. , Vizcaíno, A. , Bonkowski, M. , and De Groot, G.A. (2015b) Not all are free‐living: high‐throughput DNA metabarcoding reveals a diverse community of protists parasitizing soil metazoa. Mol Ecol 24: 4556–4569. [DOI] [PubMed] [Google Scholar]
- Geisen, S. , Mitchell, E.A.D. , Adl, S. , Bonkowski, M. , Dunthorn, M. , Ekelund, F. , et al. (2018) Soil protists: a fertile frontier in soil biology research. FEMS Microbiol Rev 42: 293–323. [DOI] [PubMed] [Google Scholar]
- Gilbert, D. , Amblard, C. , Bourdier, G. , and Francez, A.‐J. (1998) The microbial loop at the surface of a peatland: structure, function, and impact of nutrient input. Microb Ecol 35: 83–93. [DOI] [PubMed] [Google Scholar]
- Giovannoni, S.J. , Britschgi, T.B. , Moyer, C.L. , and Field, K.G. (1990) Genetic diversity in Sargasso Sea bacterioplankton. Nature 345: 60–63. [DOI] [PubMed] [Google Scholar]
- Gong, W. , and Marchetti, A. (2019) Estimation of 18S gene copy number in marine eukaryotic plankton using a next‐generation sequencing approach. Front Mar Sci 6: 6. [Google Scholar]
- Gonzalez‐de‐Salceda, L. , and Garcia‐Pichel, F. (2021) The allometry of cellular DNA and ribosomal gene content among microbes and its use for the assessment of microbiome community structure. Microbiome 9: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremion, F. , Chatzinotas, A. , Kaufmann, K. , von Sigler, W. , and Harms, H. (2004) Impacts of heavy metal contamination and phytoremediation on a microbial community during a twelve‐month microcosm experiment. FEMS Microbiol Ecol 48: 273–283. [DOI] [PubMed] [Google Scholar]
- Griffiths, B.S. (1990) A comparison of microbial‐feeding nematodes and protozoa in the rhizosphere of different plants. Biol Fertil Soils 9: 83–88. [Google Scholar]
- Guillou, L. , Bachar, D. , Audic, S. , Bass, D. , Berney, C. , Bittner, L. , et al. (2012) The protist ribosomal reference database (PR2): a catalog of unicellular eukaryote small sub‐unit rRNA sequences with curated taxonomy. Nucleic Acids Res 41: D597–D604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J. , Zhang, L. , Wang, P. , Yang, G. , Wang, S. , Li, Y. , and Pan, K. (2018) Heterogeneity of intron presence/absence in Olifantiella sp. (Bacillariophyta) contributes to the understanding of intron loss. J Phycol 54: 105–113. [DOI] [PubMed] [Google Scholar]
- Hartikainen, H. , Stentiford, G.D. , Bateman, K.S. , Berney, C. , Feist, S.W. , Longshaw, M. , et al. (2014) Mikrocytids are a broadly distributed and divergent radiation of parasites in aquatic invertebrates. Curr Biol 24: 807–812. [DOI] [PubMed] [Google Scholar]
- Hebert, P.D.N. , Ratnasingham, S. , and de Waard, J.R. (2003) Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond Ser B: Biol Sci 270: S96–S99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstatter, P.G. , and Lahr, D.J.G. (2019) All eukaryotes are sexual, unless proven otherwise. Bioessays 41: 1800246. [DOI] [PubMed] [Google Scholar]
- Holzmann, M. , Gooday, A.J. , Siemensma, F. , and Pawlowski, J. (2021) Review: freshwater and soil foraminifera – a story of long‐forgotten relatives. J Foraminiferal Res 51: 318–331. [Google Scholar]
- Howe, A.C. , Jansson, J.K. , Malfatti, S.A. , Tringe, S.G. , Tiedje, J.M. , and Brown, C.T. (2014) Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci U S A 111: 4904–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamy, M. , Foster, R. , Barbera, P. , Czech, L. , Kozlov, A. , Stamatakis, A. , et al. (2020) Long‐read metabarcoding of the eukaryotic rDNA operon to phylogenetically and taxonomically resolve environmental diversity. Mol Ecol Resour 20: 429–443. [DOI] [PubMed] [Google Scholar]
- Jassey, V.E.J. , Meyer, C. , Dupuy, C. , Bernard, N. , Mitchell, E.A.D. , Toussaint, M.‐L. , et al. (2013) To what extent do food preferences explain the trophic position of heterotrophic and mixotrophic microbial consumers in a Sphagnum peatland? Microb Ecol 66: 571–580. [DOI] [PubMed] [Google Scholar]
- Jassey, V.E.J. , Walcker, R. , Kardol, P. , Geisen, S. , Heger, T. , Lamentowicz, M. , et al. (2022) Contribution of soil algae to the global carbon cycle. New Phytol 234: 64–76. [DOI] [PubMed] [Google Scholar]
- Jurburg, S.D. , Keil, P. , Singh, B.K. , and Chase, J.M. (2021) All together now: limitations and recommendations for the simultaneous analysis of all eukaryotic soil sequences. Mol Ecol Resour 21: 1759–1771. [DOI] [PubMed] [Google Scholar]
- Kawanobe, M. , Toyota, K. , and Ritz, K. (2021) Development and application of a DNA metabarcoding method for comprehensive analysis of soil nematode communities. Appl Soil Ecol 166: 103974. [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , and Glöckner, F.O. (2012) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Res 41: e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloub, L. , Gosselin, S. , Fullmer, M. , Graf, J. , Gogarten, J.P. , and Bansal, M.S. (2021) Systematic detection of large‐scale multigene horizontal transfer in prokaryotes. Mol Biol Evol 38: 2639–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight, R. , Vrbanac, A. , Taylor, B.C. , Aksenov, A. , Callewaert, C. , Debelius, J. , et al. (2018) Best practices for analysing microbiomes. Nat Rev Microbiol 16: 410–422. [DOI] [PubMed] [Google Scholar]
- Kosakyan, A. , Mulot, M. , Mitchell, E.A.D. , and Lara, E. (2015) Environmental DNA COI barcoding for quantitative analysis of protists communities: a test using the Nebela collaris complex (Amoebozoa; Arcellinida; Hyalospheniidae). Eur J Protistol 51: 311–320. [DOI] [PubMed] [Google Scholar]
- Lane, D.J. , Pace, B. , Olsen, G.J. , Stahl, D.A. , Sogin, M.L. , and Pace, N.R. (1985) Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A 82: 6955–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara, E. , and Acosta‐Mercado, D. (2012) A molecular perspective on ciliates as soil bioindicators. Eur J Soil Biol 49: 107–111. [Google Scholar]
- Lara, E. , Berney, C. , Harms, H. , and Chatzinotas, A. (2007) Cultivation‐independent analysis reveals a shift in ciliate 18S rRNA gene diversity in a polycyclic aromatic hydrocarbon‐polluted soil. FEMS Microbiol Ecol 62: 365–373. [DOI] [PubMed] [Google Scholar]
- Lara, E. , Heger, T.J. , Ekelund, F. , Lamentowicz, M. , and Mitchell, E.A.D. (2008) Ribosomal RNA genes challenge the monophyly of the Hyalospheniidae (Amoebozoa: Arcellinida). Protist 159: 165–176. [DOI] [PubMed] [Google Scholar]
- Lavrinienko, A. , Jernfors, T. , Koskimäki, J.J. , Pirttilä, A.M. , and Watts, P.C. (2021) Does intraspecific variation in rDNA copy number affect analysis of microbial communities? Trends Microbiol 29: 19–27. [DOI] [PubMed] [Google Scholar]
- Lesaulnier, C. , Papamichail, D. , McCorkle, S. , Ollivier, B. , Skiena, S. , Taghavi, S. , et al. (2008) Elevated atmospheric CO2 affects soil microbial diversity associated with trembling aspen. Environ Microbiol 10: 926–941. [DOI] [PubMed] [Google Scholar]
- López‐García, P. , and Moreira, D. (2008) Tracking microbial biodiversity through molecular and genomic ecology. Res Microbiol 159: 67–73. [DOI] [PubMed] [Google Scholar]
- López‐García, P. , Rodriguez‐Valera, F. , Pedrós‐Alió, C. , and Moreira, D. (2001) Unexpected diversity of small eukaryotes in deep‐sea Antarctic plankton. Nature 409: 603–607. [DOI] [PubMed] [Google Scholar]
- Mahé, F. , de Vargas, C. , Bass, D. , Czech, L. , Stamatakis, A. , Lara, E. , et al. (2017) Parasites dominate hyperdiverse soil protist communities in Neotropical rainforests. Nat Ecol Evol 1: 0091. [DOI] [PubMed] [Google Scholar]
- Marcisz, K. , Fournier, B. , Gilbert, D. , Lamentowicz, M. , and Mitchell, E.A.D. (2014) Response of Sphagnum peatland testate amoebae to a 1‐year transplantation experiment along an artificial hydrological gradient. Microb Ecol 67: 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczan, T. (2009) Ciliates in Sphagnum peatlands: vertical micro‐distribution, and relationships of species assemblages with environmental parameters. Zool Stud 48: 33–48. [Google Scholar]
- Moon‐van der Staay, S.Y. , De Wachter, R. , and Vaulot, D. (2001) Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature 409: 607–610. [DOI] [PubMed] [Google Scholar]
- Oliverio, A.M. , Geisen, S. , Delgado‐Baquerizo, M. , Maestre, F.T. , Turner, B.L. , and Fierer, N. (2020) The global‐scale distributions of soil protists and their contributions to belowground systems. Sci Adv 6: eaax8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquola, A.C.M. , Asif, H. , Pereira, C.A.d.B. , Feltes, B.C. , Bonatto, D. , Lima, W.C. , and Menck, C.F.M. (2018) Horizontal gene transfer building prokaryote genomes: genes related to exchange between cell and environment are frequently transferred. J Mol Evol 86: 190–203. [DOI] [PubMed] [Google Scholar]
- Pawlowski, J. , Audic, S. , Adl, S. , Bass, D. , Belbahri, L. , Berney, C. , et al. (2012) CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol 10: e1001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe, H. , and Germot, A. (2000) Phylogeny of eukaryotes based on ribosomal RNA: long‐branch attraction and models of sequence evolution. Mol Biol Evol 17: 830–834. [DOI] [PubMed] [Google Scholar]
- Porter, T.M. , Morris, D.M. , Basiliko, N. , Hajibabaei, M. , Doucet, D. , Bowman, S. , et al. (2019) Variations in terrestrial arthropod DNA metabarcoding methods recovers robust beta diversity but variable richness and site indicators. Sci Rep 9: 18218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapov, A.M. , Beaulieu, F. , Birkhofer, K. , Bluhm, S.L. , Degtyarev, M.I. , Devetter, M. , et al. (2022) Feeding habits and multifunctional classification of soil‐associated consumers from protists to vertebrates. Biol Rev. (In press). [DOI] [PubMed] [Google Scholar]
- Qin, Q.‐L. , Wang, Z.‐B. , Cha, Q.‐Q. , Liu, S.‐S. , Ren, X.‐B. , Fu, H.‐H. , et al. (2022) Biogeography of culturable marine bacteria from both poles reveals that ‘everything is not everywhere’ at the genomic level. Environ Microbiol 24: 98–109. [DOI] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch, L.F.W. , Fulthorpe, R.R. , Riva, A. , Casella, G. , Hadwin, A.K.M. , Kent, A.D. , et al. (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1: 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rønn, R. , McCaig, A.E. , Griffiths, B.S. , and Prosser, J.I. (2002) Impact of protozoan grazing on bacterial community structure in soil microcosms. Appl Environ Microbiol 68: 6094–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansupa, C. , Wahdan, S.F. , Hossen, S. , Disayathanoowat, T. , Wubet, T. , and Purahong, W. (2021) Can we use functional annotation of prokaryotic taxa (FAPROTAX) to assign the ecological functions of soil bacteria? Appl Sci 11: 688. [Google Scholar]
- Santos, S.S. , Nunes, I. , Nielsen, T.K. , Jacquiod, S. , Hansen, L.H. , and Winding, A. (2017) Soil DNA extraction procedure influences protist 18S rRNA gene community profiling outcome. Protist 168: 283–293. [DOI] [PubMed] [Google Scholar]
- Schenk, J. , Geisen, S. , Kleinboelting, N. , and Traunspurger, W. (2019) Metabarcoding data allow for reliable biomass estimates in the most abundant animals on earth. MBMG 3: e46704. [Google Scholar]
- Sessitsch, A. , Weilharter, A. , Gerzabek, M.H. , Kirchmann, H. , and Kandeler, E. (2001) Microbial population structures in soil particle size fractions of a long‐term fertilizer field experiment. Appl Environ Microbiol 67: 4215–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakova, L. , Bondarenko, N. , and Smirnov, A. (2016) Viable species of Flamella (Amoebozoa: Variosea) isolated from ancient Arctic permafrost sediments. Protist 167: 13–30. [DOI] [PubMed] [Google Scholar]
- Shmakova, L.A. , and Rivkina, E.M. (2015) Viable eukaryotes of the phylum Amoebozoa from the Arctic permafrost. Paleontol J 49: 572–577. [Google Scholar]
- Sibbald, S.J. , and Archibald, J.M. (2017) More protist genomes needed. Nat Ecol Evol 1: 0145. [DOI] [PubMed] [Google Scholar]
- Singer, D. , Kosakyan, A. , Seppey, C.V.W. , Pillonel, A. , Fernández, L.D. , Fontaneto, D. , et al. (2018) Environmental filtering and phylogenetic clustering correlate with the distribution patterns of cryptic protist species. Ecology 99: 904–914. [DOI] [PubMed] [Google Scholar]
- Singer, D. , Seppey, C.V.W. , Lentendu, G. , Dunthorn, M. , Bass, D. , Belbahri, L. , et al. (2021) Protist taxonomic and functional diversity in soil, freshwater and marine ecosystems. Environ Int 146: 106262. [DOI] [PubMed] [Google Scholar]
- Stackebrandt, E. , Frederiksen, W. , Garrity, G.M. , Grimont, P.A. , Kämpfer, P. , Maiden, M.C. , et al. (2002) Report of the ad hoc committee for the re‐evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52: 1043–1047. [DOI] [PubMed] [Google Scholar]
- Stoddard, S.F. , Smith, B.J. , Hein, R. , Roller, B.R.K. , and Schmidt, T.M. (2015) rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res 43: D593–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedersoo, L. , Albertsen, M. , Anslan, S. , Callahan, B. , and Druzhinina, I.S. (2021) Perspectives and benefits of high‐throughput long‐read sequencing in microbial ecology. Appl Environ Microbiol 87: e00626‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedersoo, L. , Bahram, M. , Põlme, S. , Kõljalg, U. , Yorou, N.S. , Wijesundera, R. , et al. (2014) Global diversity and geography of soil fungi. Science 346: 1256688. [DOI] [PubMed] [Google Scholar]
- Thakur, M.P. , and Geisen, S. (2019) Trophic regulations of the soil microbiome. Trends Microbiol 27: 771–780. [DOI] [PubMed] [Google Scholar]
- Thompson, P.B. (2017) The Spirit of the Soil: Agriculture and Environmental Ethics. New York: Routledge. [Google Scholar]
- Tsai, Y.L. , and Olson, B.H. (1992) Detection of low numbers of bacterial cells in soils and sediments by polymerase chain reaction. Appl Environ Microbiol 58: 754–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, N.A. , Russell, A.D. , Furr, J.R. , and Lloyd, D. (2000) Emergence of resistance to biocides during differentiation of Acanthamoeba castellanii . J Antimicrob Chemother 46: 27–34. [DOI] [PubMed] [Google Scholar]
- van den Hoogen, J. , Geisen, S. , Routh, D. , Ferris, H. , Traunspurger, W. , Wardle, D.A. , et al. (2019) Soil nematode abundance and functional group composition at a global scale. Nature 572: 194–198. [DOI] [PubMed] [Google Scholar]
- Van Etten, J. , and Bhattacharya, D. (2020) Horizontal gene transfer in eukaryotes: not if, but how much? Trends Genet 36: 915–925. [DOI] [PubMed] [Google Scholar]
- van Hannen, E.J. , Zwart, G. , van Agterveld, M.P. , Gons, H.J. , Ebert, J. , and Laanbroek, H.J. (1999) Changes in bacterial and eukaryotic community structure after mass lysis of filamentous cyanobacteria associated with viruses. Appl Environ Microbiol 65: 795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaulot, D. , Geisen, S. , Mahé, F. , and Bass, D. (2022) pr2‐primers: an 18S rRNA primer database for protists. Mol Ecol Resour 22: 168–179. [DOI] [PubMed] [Google Scholar]
- Walters, W.A. , Caporaso, J.G. , Lauber, C.L. , Berg‐Lyons, D. , Fierer, N. , and Knight, R. (2011) PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 27: 1159–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel, L. , Daniell, T.J. , King, D. , and Neilson, R. (2015) Determination of the optimal soil sample size to accurately characterise nematode communities in soil. Soil Biol Biochem 80: 89–91. [Google Scholar]
- Xiong, W. , Jousset, A. , Li, R. , Delgado‐Baquerizo, M. , Bahram, M. , Logares, R. , et al. (2021) A global overview of the trophic structure within microbiomes across ecosystems. Environ Int 151: 106438. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Xia, B. , Treves, D.S. , Wu, L.‐Y. , Marsh, T.L. , O'Neill, R.V. , et al. (2002) Spatial and resource factors influencing high microbial diversity in soil. Appl Environ Microbiol 68: 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]