

Abstract
Structural comparison reveals remote homology that often fails to be detected by sequence comparison. The DALI web server (http://ekhidna2.biocenter.helsinki.fi/dali) is a platform for structural analysis that provides database searches and interactive visualization, including structural alignments annotated with secondary structure, protein families and sequence logos, and 3D structure superimposition supported by color‐coded sequence and structure conservation. Here, we are using DALI to mine the AlphaFold Database version 1, which increased the structural coverage of protein families by 20%. We found 100 remote homologous relationships hitherto unreported in the current reference database for protein domains, Pfam 35.0. In particular, we linked 35 domains of unknown function (DUFs) to the previously characterized families, generating a functional hypothesis that can be explored downstream in structural biology studies. Other findings include gene fusions, tandem duplications, and adjustments to domain boundaries. The evidence for homology can be browsed interactively through live examples on DALI's website.
Keywords: AlphaFold Database, evolutionary classification, homology transfer of protein function, structural alignment
1. INTRODUCTION
The dropping cost of genome sequencing is closely followed, in inverse proportion, by a growing “ignorome”—genomic data that is incompletely annotated before being deposited in public databases (de Crécy‐Lagard et al., 2022; Stoeger et al., 2018). This is not surprising since common practices toward protein characterization are still hindered by expensive and time‐consuming methods, including reverse genetic studies, heterologous production of proteins including, or not, site‐directed mutagenesis, enzymatic assays, and protein structure determination by X‐ray crystallography, cryoelectron microscopy, or nuclear magnetic resonance, as well as research bias (Haynes et al., 2018). The concomitant increase of noncurated data calls for efficient tools that enable access to the underlying biochemical, biophysical, and evolutionary information retained in the yet unexplored huge amount of sequencing data. It is widely assumed that knowledge gained in one species can be transferred to another, even among species that are widely separated on the tree of life. This transfer is often performed at the level of proteins under the assumption that if two proteins are homologous, they will share conserved evolutionary domains typically represented by some structural and, although to a smaller extent, functional similarity (Ponting & Russell, 2002). For genomic studies carried out on nonmodel organisms, homology transfer of protein function is the key approach to annotate uncharacterized protein sequences. In favorable cases, the outcome of meta‐analysis of sequencing data might unravel ancient homologous relationships for genes previously thought to be lineage‐specific.
The most widely used tools for the analysis of protein families and evolutionary relationships described above are still based on sequence comparison (e.g., Altenhoff et al., 2021; Huerta‐Cepas et al., 2019; Mistry et al., 2020; Törönen et al., 2018). Here we seek to find deep phylogenetic relationships from proteins that share a similar three‐dimensional (3D) structure, despite their poor similarity in terms of primary structure. Given that tertiary structures are essential to gain understanding about protein function, structural features need to be examined in great detail to determine whether structural commonalities support a common functional role indicative of homology or not. “Protein families” encompass proteins that often share clear sequence similarity, accounting for cases with obvious high structural similarity. “Superfamilies,” also called clans, imply remote homology derived from a common ancestor. The superfamily classification ultimately relies on expert manual curation, integrating compelling evidence for homology from geometrical, sequence, and functional features that are unlikely to co‐occur by chance (Murzin et al., 1995). Many superfamilies have been discovered based on structural comparison. Until recently, this approach was limited by the sparsity of experimentally determined structures. However, the situation changed dramatically with the advent of accurate structure prediction based on deep learning (Jumper et al., 2021). The outcome of these studies generated roughly a million published structural models that are nowadays at our disposal (Varadi et al., 2022). To explore the path that was opened by the sudden availability of a plethora of structural models, tools for fast and efficient structural comparison are more needed than ever (Aderinwale et al., 2022; Bordin et al., 2022; van Kempen et al., 2002), and in this context, the DALI algorithm is one of the most sensitive (Holm, 2019).
In this work, we demonstrate the use of the DALI web server for protein structure comparisons in an effort to find evolutionary links between protein families that are represented in the Protein Data Bank (PDB) and those that are not. Protein families were defined by Pfam 35.0, a state‐of‐the‐art reference in protein domains classification (Mistry et al., 2020). We use the Pfam database as a baseline to target evolutionary relationships that often fail to be detected by standard approaches. Taking advantage of the AlphaFold Database version 1 (AFDB; Varadi et al., 2022), we selected a representative AlphaFold model for each protein family in Pfam 35.0 lacking experimental structural information. We then used the DALI web server to search for the closest structural match in the PDB and to assess the biological significance of the match. Interactive analysis tools, together with one‐click navigation links, allowed the rejection of poor candidates in seconds, whilst for interesting cases the time to assess evidence for homology ranged from 1 min up to an hour. Altogether, our results show that the DALI web server provides a powerful interface to identify remote structural homologs, as showcased here by 100 specific examples.
2. METHODS
2.1. Selection of targets
The representative set of proteins used in this study was selected from Pfam 35.0, which contains a total of 19,632 families. Among these, 7770 families are grouped into 655 clans, corresponding to superfamilies in other classifications (Mistry et al., 2020). A total of 59% of the Pfam families are represented in AlphaFold Database version 1 (AFDB1), which covers the human proteome as well as the proteomes of other important model organisms (Varadi et al., 2022). Many Pfam families represent domains, covering only part of a protein. For each Pfam family, we selected a representative member of median length for that family. On the basis that AlphaFold models cover UniProt sequences end‐to‐end, Pfam domain boundaries directly match the expected residue number present in the coordinates of AlphaFold models. Taking advantage of this premise, the 3D coordinates for the family representative were extracted from the AlphaFold model of the full‐length protein. Since not all the segments of AlphaFold models are modeled with equal confidence, segments that showed low‐confidence modeling were excluded from the analysis. The cut‐off pLDDT <70 removed ~17% of the residues, resulting in cropped models.
Furthermore, given that not all AlphaFold models are compact and globular, two filtering criteria were implemented. These removed many ill‐folded models from the analysis as confirmed by visual inspection. First, the contact order (CO) had to meet the range of native proteins (CO > 0.05) (Plaxco et al., 1998) and, second, in order to ensure globularity, the model had to exhibit at least 60 confidently modeled residues represented by at least four secondary structure elements, that is, either alpha helices or beta strands (Kabsch & Sander, 1983). The filter on the secondary structure will miss small domains stabilized by disulfide bonds or metal binding. On the other hand, it eliminates a much larger number of models with 1–3 helices, which have a limited set of packing arrangements, making physical convergence a likely explanation for fold similarities.
Finally, the analysis focused exclusively on protein families for which no structural information existed. Pfam provides public tables that link (i) PDB structures to protein families, and (ii) distantly related families to clans. These tables were useful to exclude either families that have a member assigned with a PDB structure (pdbmap set) and/or families that are indirectly linked to a PDB structure by having a clan member with a PDB structure (clanpdb set). Upon restricting the dataset to protein families/clans with no structural information in Pfam 35.0 (nopdb set), 1,412 families remained for downstream analysis (Figure 1).
FIGURE 1.
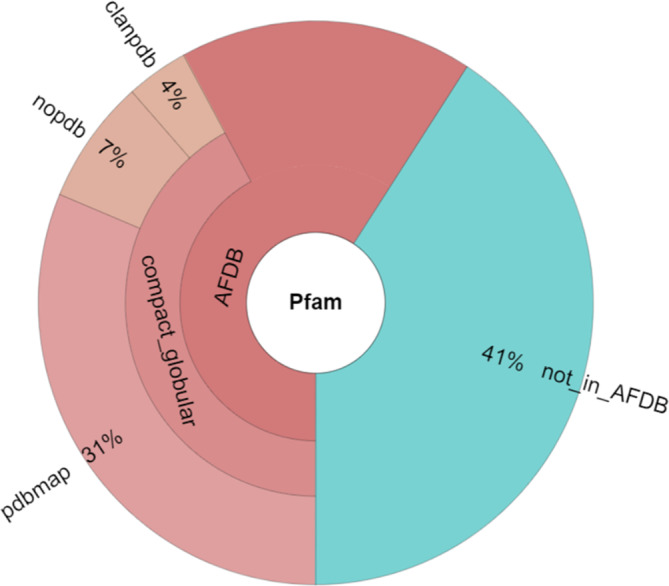
Structural coverage of Pfam 35.0 families by AlphaFold Database version 1.
2.2. Structural classification
The nopdb set was further analyzed by DALI searches of the cropped models against the PDB (data from March 2022). Cropped models were used because AlphaFold typically models low confidence segments as unstructured wide loops, which only would slow down the search with no reward in informative matches. The top hit was extracted and a pairwise structural alignment was generated with the full, noncropped model. The DALI web server has an option to interactively generate stacked sequence logos of selected structurally aligned proteins. To save time, we appended precomputed stacked sequence logos of the top pair to the HTML result page of the pairwise alignments. These modified web pages were screened visually on the DALI website. Tables 1, 2, 3, 4 provide links to each pairwise comparison. The examples can also be accessed via the web page http://ekhidna2.biocenter.helsinki.fi/dali/Tables_examples.html.
TABLE 1.
AlphaFold models validated after the release of Pfam 35.0
| pfamid | pfam_description | Accession | From | To | PDB hit | Z | RMSD | lali | %ide |
|---|---|---|---|---|---|---|---|---|---|
| PF03080 | Neprosin | O64859 | 184 | 395 | 7zvaA | 28.1 | 1.3 | 208 | 51 |
| PF03706 | Lysylphosphatidylglycerol synthase TM region | P75770 | 14 | 296 | 7duwA | 26.6 | 2.4 | 239 | 26 |
| PF03802 | Apo‐citrate lyase phosphoribosyl‐dephospho‐CoA transferase | P0A6G5 | 13 | 177 | 7dcmA | 27.6 | 0.9 | 165 | 99 |
| PF04113 | Gpi16 subunit, GPI transamidase component | E9QH65 | 77 | 628 | 7w72T | 52.5 | 7.8 | 520 | 69 |
| PF04114 | Gaa1‐like, GPI transamidase component | Q6AYM8 | 125 | 615 | 7w72A | 42.3 | 1.1 | 465 | 93 |
| PF04573 | Signal peptidase subunit | I1JRE6 | 1 | 165 | 7p2pB | 12.3 | 2.4 | 162 | 30 |
| PF06703 | Microsomal signal peptidase 25 kDa subunit (SPC25) | P58684 | 23 | 181 | 7p2qC | 13.5 | 2.5 | 156 | 27 |
| PF08014 | Domain of unknown function (DUF1704) | Q7TQE7 | 265 | 511 | 7z5gB | 33.9 | 0.7 | 247 | 66 |
| PF09773 | Meckelin (transmembrane protein 67) | E9QB24 | 156 | 982 | 7fh1A | 44.6 | 2.0 | 730 | 62 |
| PF10510 | Phosphatidylinositol‐glycan biosynthesis class S protein | Q96S52 | 22 | 547 | 7w72S | 44.4 | 2.7 | 511 | 92 |
| PF11380 | Stealth protein CR2, conserved region 2 | Q54UF6 | 90 | 198 | 7dxiB | 8.7 | 1.8 | 108 | 38 |
| PF11744 | Aluminum activated malate transporter | K7VD86 | 88 | 494 | 7vojA | 35.0 | 2.0 | 338 | 47 |
| PF12810 | Glycine rich protein | Q9UM73 | 726 | 987 | 7ls0B | 38.3 | 0.6 | 225 | 100 |
| PF15778 | UNC80 N‐terminal | Q8N2C7 | 16 | 236 | 7sx3E | 11.4 | 4.9 | 239 | 89 |
| PF17517 | IgGFc binding protein | Q9Y6R7 | 129 | 419 | 7wpqE | 13.8 | 3.7 | 252 | 33 |
Note: AlphaFold Database models are identified by UniProt accession and residue range. These examples have sequence identity >25% and query coverage >80% in structural alignment.
TABLE 2.
Statistically significant sequence conservation in binomial test.
| pfamid | Bonf. p | pfam_description | PDB hit's family | PDB hit | Z | %ide |
|---|---|---|---|---|---|---|
| PF03016 | 0.00141 | Exostosin family with glycosyltransferase activities | PF05686 Glycosyltransferase family 90 | 5f84A | 11.7 | 14 |
| PF03690 | 0.0221 | Uncharacterized protein family (UPF0160); DHH motif | PF01368 DHH family phosphoesterase (CL0137) | 6mtzB | 8.3 | 24 |
| PF03942 | 0.00417 | DTW domain | PF04034 RIbosome biogenesis protein, C‐terminal | 5apgA | 9.8 | 21 |
| PF04250 | 0.000649 | Protein of unknown function (DUF429) | PF02075 Crossover junction endodeoxyribonuclease RuvC (CL0219) | 6s16B | 9.5 | 15 |
| PF04293 | 0.00291 | SpoVR like protein | PF05960 Bacterial protein of unknown function (DUF885) | 3o0yA | 13.2 | 14 |
| PF04632 | 1.08 E‐10 | Fusaric acid resistance protein family | PF11744 Aluminum activated malate transporter | 7vq3A | 12.7 | 19 |
| PF06356 | 0.0330 | Protein of unknown function (DUF1064) | PF08722 (CL0236) TnsA‐like endonuclease | 1t0fA | 8.3 | 13 |
| PF07759 | 1.94 E‐07 | Protein of unknown function (DUF1615) | PF01464 Transglycosylase SLT domain (CL0037) | 6cf9A | 12.3 | 18 |
| PF08889 | 7.61 E‐05 | WbqC‐like protein family | PF10079 Bacillithiol biosynthesis BshC | 4wbdA | 12.1 | 12 |
| PF10070 | 9.41 E‐10 | Probable inorganic carbon transporter subunit DabA | PF08936 Carboxysome shell carbonic anhydrase | 2fgyA | 13.5 | 13 |
| PF10307 | 0.00428 | HAD domain family 1 in Swiss Army Knife RNA repair proteins | PNKP1, component of bacterial RNA repair complex | 4xruA | 10.2 | 14 |
| PF11001 | 0.0310 | Protein of unknown function (DUF2841) | PF04873 Ethylene insensitive 3 | 4zdsA | 12.0 | 20 |
| PF11070 | 0.000261 | Protein of unknown function (DUF2871) | PF00115 Cytochrome c and quinol oxidase polypeptide I (CL0714) | 6lh3B | 13.8 | 19 |
| PF11765 | 0.00648 | Hyphally regulated cell wall protein N‐terminal | Adhesin‐like wall protein 3b | 7o9oA | 28.6 | 18 |
| PF11875 | 0.0404 | DnaJ‐like protein C11, C‐terminal | PF02140 Galactose binding lectin domain | 2zx0A | 3.9 | 19 |
| PF11904 | 0.00762 | GPCR‐chaperone. Adjust domain boundaries. | LolA/B superfamily of lipoprotein localization factors (CL0048) | 2zf4F | 4.4 | 15 |
| PF12062 | 0.000201 | Heparan sulfate‐N‐deacetyse | Carbohydrate deacetylase Agd3 | 6nwzA | 18.5 | 20 |
| PF12222 | 1.64 E‐06 | Peptide N‐acetyl‐beta‐D‐glucosaminyl asparaginase amidase A | PF09112, PF09113 Peptide‐N‐glycosidase F (CL0612) | 4r4xA | 18.4 | 17 |
| PF12617 | 0.00701 | Iron–sulfur binding protein C terminal | PF01680 SOR/SNZ family (CL0036) | 4wy0G | 6.8 | 15 |
| PF13258 | 2.41 E‐05 | Domain of unknown function (DUF4049) | Tyrosine protein phosphatase WipA from Legionella | 5n6xA | 15.4 | 16 |
| PF13898 | 0.0374 | Deubiquitinating enzyme MINDY‐3/4, conserved domain | Deubiquitinase | 7bu0A | 3.8 | 9 |
| PF14033 | 0.0350 | Protein of unknown function (DUF4246) | PF13640 2OG‐Fe(i) oxygenase superfamily (CL0029) | 6n1fA | 13.0 | 9 |
| PF14130 | 0.0200 | Cap4 dsDNA endonuclease | N‐terminal endonuclease domain | 6vm6B | 11.8 | 10 |
| PF14646 | 4.99 E‐05 | MYCBP‐associated protein family | PapD‐like domain 7 (cell cycle protein) | 6fviA | 10.8 | 17 |
| PF14923 | 0.0211 | Coiled‐coil protein 142 | PF07393 Exocyst complex component Sec10 (CL0294) | 5h11A | 13.4 | 9 |
| PF14976 | 0.00943 | FAM72 protein | PF03226 Yippe zinc‐binding/DNA‐binding Mis18, centromere assembly (CL0080) | 5hj0C | 10.0 | 13 |
| PF15851 | 0.00453 | Domain of unknown function (DUF4723) | Glycosylphosphatidulinositol‐anchored high density lipoproterin‐binding protein 1 | 6e7kC | 11.1 | 24 |
| PF16062 | 4.79 E‐06 | Domain of unknown function (DUF4804) | Legionella effector protein MavL | 6omiB | 28.6 | 16 |
| PF17659 | 1.69 E‐08 | Family of unknown function (DUF5521) | PF16900 Replication protein A OB domain | 4gnxC | 13.1 | 18 |
| PF17720 | 6.78 E‐07 | Family of unknown function (DUF5565) | PF09414 RNA ligase (CL0078) | 5cotA | 7.3 | 18 |
| PF18168 | 0.0289 | Prim‐pol family 5 | PF01896 DNA primase small subunit (CL0243) | 5l2xA | 10.7 | 15 |
| PF18658 | 0.0274 | Spin‐doc zinc‐finger | PF19088 TUTase nucleotidyltransferase domain | 6iw6A | 4.2 | 14 |
Note: Query family has no structural information in Pfam 35.0; pfamid is hyperlinked to web page visualizing the structural comparison to PDB hit. Protein families of unknown function are highlighted in red.
TABLE 3.
Related known functions
| Pfamid | pfam_description | PDB hit's family | PDB hit | Z | %ide | Type |
|---|---|---|---|---|---|---|
| PF01548 | Transposase | PF02075 Crossover junction endodeoxyribonuclease RuvC (CL0219) | 4ld0B | 11.7 | 15 | E |
| PF01955 | Adenosylcobinamide amidohydrolase | PF03576 Peptidase family S58 (CL0635) | 5xyoA | 11.0 | 6 | E |
| PF03859 | CG‐1 domain | PF08549 SWI/SNF and RSC complexes subunit Ssr4 N‐terminal (Holm, 2020) | 7k7vA | 9.7 | 25 | B |
| PF04479 | RTA1 like protein. 7 transmembrane helices. | PF01036 Bacteriorhodopsin‐like protein (CL0192) | 1x0k1 | 13.0 | 10 | T |
| PF04666 | DUF659 Protein of unknown function (transposase‐like proteins with no known function) | DNA transposase | 2apcA | 18.2 | 14 | E |
| PF04709 | Anti‐Mullerian hormone, N terminal region | PF00688 TGF‐beta propeptide | 6sf2F | 6.0 | 20 | B |
| PF04724 | Glycosyltransferase family 17 | PF00535 Glycosyl transferase family 2 | 6e4qA | 10.7 | 10 | E |
| PF04765 | Protein of unknown function (DUF616). A number of the members are thought to be glycosyltransferases. | PF03414 Glycosyltransferase family 6 | 2vs3A | 10.6 | 7 | E |
| PF04833 | COBRA‐like protein | PF00553 Cellulose‐binding domain | 6qfsA | 8.4 | 21 | B |
| PF05123 | S‐layer like family, N‐terminal region | PF07752 S‐layer protein | 3u2gA | 3.2 | 12 | S |
| PF05124 | S‐layer like family, C‐terminal region | PDB protein's sequence assigned to the same Pfam family, but link is missing from pdbmap. | 6npsA | 6.5 | 16 | S |
| PF05428 | Corticotropin‐releasing factor binding protein (CRF‐BP) | PF00431 CUB domain (CL0164). Involved in binding interaction partners. | 6fzwD | 8.9 | 21 | B |
| PF05458 | Cd27 binding protein (Siva). Zinc finger. | PF02318 FYVE‐like zinc finger (CL0390) | 2cjsC | 6.1 | 10 | B |
| PF06081 | Aromatic acid exporter family member 1 | PF06081 Aromatic acid exporter family member 1 | 7vg3B | 13.3 | 25 | T |
| PF06189 | 5′‐nucleotidase | Polynucleotide kinase | 5ujoB | 9.6 | 14 | E |
| PF06420 | Mitochondrial genome maintenance MGM101 | PF04098 Rad52/22 family double‐strand break repair protein | 5xs0G | 12.4 | 16 | B |
| PF06524 | NOA36 protein (zinc finger protein 330) | PF00643 B‐box zinc finger | 6imqA | 4.5 | 17 | B |
| PF07906 | ShET2 enterotoxin, N‐terminal region. Is a cysteine protease (Pearson et al., 2017) | Ubiquitin‐specific protease (Schlieker et al., 2007) | 2j7qA | 9.6 | 8 | E |
| PF07958 | Protein of unknown function (DUF1688). Involved in uracil catabolism | PF09171 N‐glycosylase/DNA lyase | 1xqpA | 11.9 | 12 | E |
| PF09317 | Acyl‐CoA dehydrogenase, C‐terminal, bacterial type | C‐terminal domain of acyl‐CoA dehydrogenase | 3owaA | 11.6 | 12 | E |
| PF10113 | FeGP cofactor biosynthesis protein, fibrillarin family | PF04055 Radical SAM superfamily (CL0036) | 3t7vA | 19.8 | 16 | E |
| PF10238 | E2 transcription factor associated phosphoprotein | PF03228 Yippee zinc‐binding/DNA‐binding Mis18, centromere assembly | 5hj0C | 9.2 | 15 | B |
| PF10337 | Putative ER transporter, 6TM, N‐terminal | PF11447 Aluminum activated malate transporter | 7vojA | 16.3 | 11 | T |
| PF10343 | Potential Queuosine, Q, salvage protein family. DNA glycosidase activity has been suggested. | PF09171 N‐glycosylase/DNA lyase | 1xqoA | 12.8 | 9 | E |
| PF11726 | Inovirus Gp2. Involved in viral DNA replication via phospho‐Tyr mechanism. | HUH‐endonuclease superfamily | 2x3gA | 7.8 | 11 | E |
| PF12141 | Beta‐mannosyltransferases | PF04041 beta‐1,4‐mannooligosaccharide phosphorylase (CL0143) | 3tawA | 20.8 | 11 | E |
| PF14249 | Tocopherol cyclase | Diels‐Alderase (pericyclase) | 7dmnA | 21.7 | 8 | E |
| PF14616 | Transcription regulator Rua1, C‐terminal | Transcriptional regulator KAISO | 2lt7A | 3.0 | 18 | B |
| PF15051 | FAM198 protein. UniProt annotates query protein as Golgi associated kinase 1b. | PF06702 Golgi casein kinase, C‐terminal, Fam20 | 5yh3A | 18.3 | 18 | E |
| PF15083 | Colipase‐like | PF01114 Colipase, N‐terminal domain; PF02740 Colipase, C‐terminal domain (CL0621) | 1lpbA | 3.2 | 22 | B |
| PF15704 | Mitochondrial ATP synthase subunit | Mitochondrial ATP synthase associated protein ASA4 | 6rdq4 | 9.0 | 13 | B |
| PF16094 | Proteasome assembly chaperone 4 | PF09754 Pac2 family (proteasome assembly chaperone) | 3wz2A | 17.5 | 8 | B |
| PF16887 | Domain of unknown function (DUF5081). Believed to be involved in type VII secretion system. | PF14011 EspG family (secretion‐associated proteins) | 4w4lC | 11.3 | 8 | T |
| PF17184 | Rit1 N‐terminal domain of tRNA modifying enzyme | PF00156 Phosphoribosyltransferase domain (CL0533) | 7kl7A | 4.4 | 9 | E |
| PF18143 | HAD domain phosphoesterases in Swiss Army Knife RNA repair proteins | Inorganic pyrophosphatase, member of the haloacid dehalogenase superfamily (CL0137) | 3qu2D | 8.2 | 10 | E |
| PF19043 | Nuclear cap binding complex subunit CBP66 | PF07065 Cell division cycle protein 123 | 4zgoA | 16.4 | 14 | B |
| PF19306 | Helicase Lhr winged helix domain | Winged helix domain of ATP‐dependent DNA helicase (Jones et al., 2018). | 5v9xA | 14.1 | 25 | B |
Note: Query family has no structural information in Pfam 35.0. Type: B, binding; E, enzyme; S, structural; T, transport.
TABLE 4.
Putative function transfer
| pfamid | pfam_description | PDB hit's family | PDB‐id | Z | %ide | Shared motif |
|---|---|---|---|---|---|---|
| PF00674 | DUP family | Myb‐like DNA‐binding domain | 2m3aA | 5.3 | 18 | Conserved Pro, Trp |
| PF04755 | Plastid lipid associated protein and fibrillins | PF00061 Lipocalin/cytosolic fatty‐acid binding protein | 2wq9A | 9.5 | 11 | Conserved Trp |
| PF04937 | DUF659 Protein of unknown function | Transposase | 2bw3A | 7.9 | 13 | Conserved Trp, Asp, Cys |
| PF05212 | Protein of unknown function (DUF707) | PF00535 glycosyltransferase family 2 | 6h2nB | 12.7 | 7 | Conserved DxD contact substrate |
| PF05444 | Protein of unknown function (DUF753) | LY6/PLAUR domain | 6ionA | 5.4 | 23 | Five conserved disulfide bridges |
| PF05912 | Caenorhabditis elegans protein of unknown function (DUF870) | PF01060 Transthyretin‐like family (CL0287) | 3uafA | 9.5 | 17 | C, HxC form conserved disulfide bridge |
| PF07505 | Protein of unknown function (DUF5131) |
Spore photoproduct lyase |
4rh1A | 14.1 | 11 |
Three conserved Cys and Asp |
| PF07712 | Stress up‐regulated Nod 19 | Pf01082 & PF03712 Copper type II ascorbate‐dependent monooxygenase, N‐terminal & C‐terminal domains (CL0612) | 6alaA | 17.1 | 11 | Conserved HH, HxH, M contact cofactor |
| PF08795 | Putative papain‐like cysteine peptidase (DUF1796) | PF08942 Domain of unknown function (DUF1919) | 2g6tA | 7.3 | 11 | Conserved catalytic dyad Cys30/Cys119, His90/His192 |
| PF09725 | Folate‐sensitive fragile site protein Fra10Ac1 | N‐terminal zinc finger domain of transcription repressor Val1 | 5yugE | 2.7 | 16 | Conserved CCCC zinc finger |
| PF09887 | Uncharacterized protein conserved in archaea (DUF2114) | PF03702 Anhydro‐N‐acetylmuramic acid kinase (CL0108) | 4bgbB | 18.5 | 13 | GN, G, D ‐ Asp233/Asp165 contact substrate |
| PF09892 | Uncharacterized protein conserved in archaea (DUF2119) | PF04952 Succinylglutamate desuccinylase/Aspartoacylase family | 3cdxF | 10.6 | 13 | Conserved calcium binding residues GxHGxE, H, E |
| PF09909 | Uncharacterized protein conserved in bacteria (DUF2138) | Fusion of (i) the treponema porin (t‐por) family, and (ii) PF14032 PknH‐like extracellular domain (CL0619) |
3k8iA 4esqA |
7.9 6.2 |
9 7 |
Structural modeling suggests it is a fusion of porin and extracellular sensor domain |
| PF10118 | Predicted metal‐dependent hydrolase | PF00268 ribonucleotide reductase, small chain | 1w69A | 14.0 | 13 | Conserved E, H, E, H in active site |
| PF10170 | Cysteine‐rich domain | PF02318 FYVE‐like zinc finger (CL0390) | 2cjsC | 4.4 | 23 | Two CCCC zinc fingers |
| PF10223 | Uncharacterized conserved protein (DUF2181) | PF03009 phosphodiesterase (CL0384) | 4oecB | 16.3 | 19 | Conserved H, ExD, H bind Mg ion |
| PF10561 | C2orf69 | PF00756 Putative esterase | 6gi0A | 6.0 | 10 | Conserved GxSxGG motif in catalytic site (Perraud et al., 2018) |
| PF10936 | Protein of unknown function (DUF2617) | PF01536 Adenosylmethionine decarboxylase | 1jl0A | 8.1 | 12 | Conserved catalytic His (Ekstrom et al., 2001) |
| PF10974 | Protein of unknown function (DUF2804) | PF07143 CrtC N‐terminal lipcalin domain; PF17186 Lipocalin‐like domain | 2ichA | 20.2 | 10 | Conserved Trp |
| PF11296 | Protein of unknown function (DUF3097) | Class 2 old family nuclease | 6nk8A | 9.7 | 10 | Conserved Asp, Glu bind Mg |
| PF11443 | Domain of unknown function (DUF2828) | PF05731 TROVE domain – RNA binding protein | 1yvrA | 16.9 | 11 | Large common core (403 residues) |
| PF12038 | Domain of unknown function (DUF3524) | PF13579 Glycosyl transferase 4‐like domain. | 6kihD | 13.5 | 15 | Essential His near UDP at domain interface |
| PF14001 | YdfZ protein. | KOW domain, involved in RNA binding | 5oikZ | 7.7 | 18 | DxxxN, GxxG |
| PF15016 | Domain of unknown function (DUF4520) | PF00659 POLO box duplicated region | 4x9vA | 11.8 | 13 | Structural resemblance |
| PF15025 | Domain of unknown function (DUF4524) | PF00659 POLO box duplicated region | 4xb0A | 9.3 | 11 | Structural resemblance |
| PF15094 | Domain of unknown function (DUF4556). | Zona pellucida sperm‐binding protein | 6gf6B | 5.6 | 12 | Conserved disulfide bridge |
| PF15474 | Meiotically up‐regulated gene family | PF09044 KP4 killer toxin. | 1kptA | 10.2 | 19 | Two conserved disulfide bridges |
| PF15479 | MRN‐interacting protein | PF09082 Domain of unknown function (DUF1922) | 1gh9A | 2.9 | 17 | Conserved CCCC zinc finger |
| PF15866 | Domain of unknown function (DUF4729) | PF03145 Seven in absentia protein family (CL0389( | 4ca1A | 11.5 | 9 | Conserved CCHH zinc finger |
| PF16044 | Domain of unknown function (DUF4796) | PF05903 PPPDE peptidase domain (CL0125) | 3ebqA | 7.6 | 12 | Conserved H, C bind metal ion |
| PF17249 | Family of unknown function (DUF5318) | PF03367 ZPR1 zinc‐finger domain (CL0167) | 2qkdA | 3.6 | 17 | Conserved CCCC zinc finger |
Note: Query family has no structural information in Pfam 35.0; pfamid is hyperlinked to web page visualizing the structural comparison to PDB hit. Protein families of unknown function are highlighted in red.
All analyses can be re‐run by launching a PDB search, PDB25 search, or pairwise structure comparison on the DALI server; note that database search results may change due to weekly PDB updates. The representative domain models for Pfam families can be downloaded as http://ekhidna2.biocenter.helsinki.fi/barcosel/pfamtest/PFnnnnn/s001.brk where PFnnnnn is the Pfam identifier. Note that there may be small differences in the structural alignment statistics (e.g., DALI Z‐score) depending on whether the query is a cropped domain model, a domain model, or a full protein model.
The visual screening focused on cases that showed identical highly conserved residues in the stacked sequence logos (e.g., Figure 2d). In addition, large structural matches involving complex folds were retained for further analysis. Note that we did not pursue partially matched domains or very common folds, such as four‐helical bundles that did not exhibit sequence conservation. The sequence conservation was quantified based on family‐wise error rate (FWER, discussed below), which showed to be highly selective yet conducive to many false negatives when compared with expert classification.
FIGURE 2.
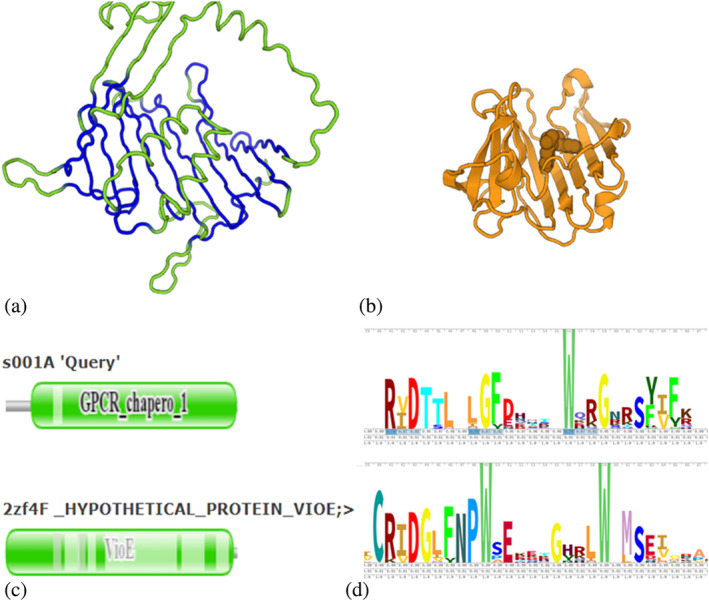
PF11904 joins the LolA/B superfamily (CL0048). (a) PF11904 representative AF‐Q7K4M9‐F1‐model_v1/118‐496, structurally aligned segments are shown in blue. Shown in green are large insertions in the GPCR‐chaperone domain, which are modeled with lower confidence. (b) VioE family representative 2zf4F in the same orientation, showing a bound lipid molecule above the flat beta sheet of the common core. (c) Pfam domains mapped to structural alignment. Note that structural equivalence extends to the left of Pfam domain boundary on the query. (d) Stacked sequence logos. The RxD motif contacts the bound lipid in the VioE family. The RxD motif appears specific to the VioE family and PF11904.
Expert classification assesses evidence for structural similarity, functional similarity, and sequence conservation. Evidence was collected using the interactive visualization tools on the DALI website using the protocol outlined below.
2.2.1. Functional descriptions
Although the summary from DALI echoes information from the COMPND record of the PDB entry, this information may be misleading insofar as it may refer to another subunit or another domain. So, we primarily checked the functional annotations in Pfam based on the Pfam cartoon view (e.g., Figure 2c). And if these were uninformative, we would additionally check: (i) Pannzer predictions (Törönen et al., 2018); (ii) the crystallographic literature at the RCSB site (https://www.rcsb.org/; Berman et al., 2000) or (iii) crosslinks to InterPro (Blum et al., 2021) in the UniProt entry via Pannzer/SANS links (Somervuo & Holm, 2015) on the web page. From 2023 onwards, the Pfam documentation can be accessed at https://www.ebi.ac.uk/interpro/entry/pfam/PFnnnnn/, substituting the relevant Pfam family identifier “PFnnnnn.”
2.2.2. Domain boundaries
Pfam domain boundaries are based on conserved sequence blocks (Eddy, 2008). Since domain boundaries did not always coincide with globular domains in the AlphaFold models, they had to be manually corrected throughout the analyses. In cases where there were conspicuous gaps in the 3‐D superposition, the pairwise alignment with the complete protein model was repeated using the DALI server. (The DALI identifier for the AFDB1 entry can be recovered using the SANS link on the web page.) In the 3‐D superposition, one could then see the gaps filled by N‐ or C‐terminal extensions of the Pfam domain. The revised domain boundaries were validated through a parallel examination of the respective Pfam cartoon view.
2.2.3. Multidomain query families
Until now, the analysis was restricted to the best PDB hit retrieved by screening, however some Pfam families are composed of multiple domains. For cases where the domains comprised repeats of the same fold, substructures of the model based on its fold were manually extracted into PDB‐formatted coordinate files. From here, the domains were aligned individually against the PDB structure (c.f., OB‐fold and POLO domain examples). If the domains are different, a systematic PDB25 search reports matches to every part of the query structure (c.f., receptor‐sensor fusion example, Figure 4).
FIGURE 4.
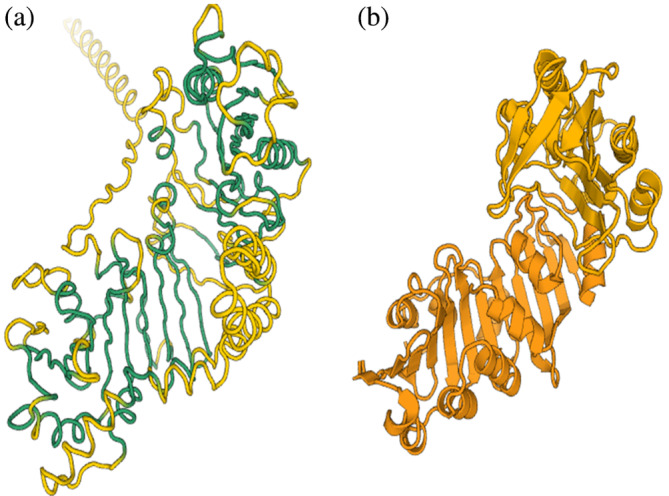
Fusion of receptor and sensor domains in PF09909. (a) PF09909 model. Structurally aligned regions are shown in green. (b) Receptor and sensor domain matches in the same orientation and colored darker or lighter orange, respectively. Reproduce the superposition by launching a DALI pairwise alignment of eszA as first structure, 3k8hA and 4esqA as second structures.
2.2.4. 3‐D superimposition
DALI optimizes the similarity of 2‐D distance matrices, which sometimes yields large RMSD values on rigid‐body superimpositions. For such cases, the coloring of the structure conservation helps to unveil the common core (c.f., Figure 2a).
2.2.5. Functional residues
Conserved functional residues tend to cluster around bound ligands. By default, the 3‐D structure viewer displays the superimposed CA traces along with an option that enables side chains or ligands to be displayed. When combined, the sliding ruler accompanied by a sequence conservation threshold is useful for localizing the most conserved residues within the 3‐D structure (c.f., Figure 3).
FIGURE 3.
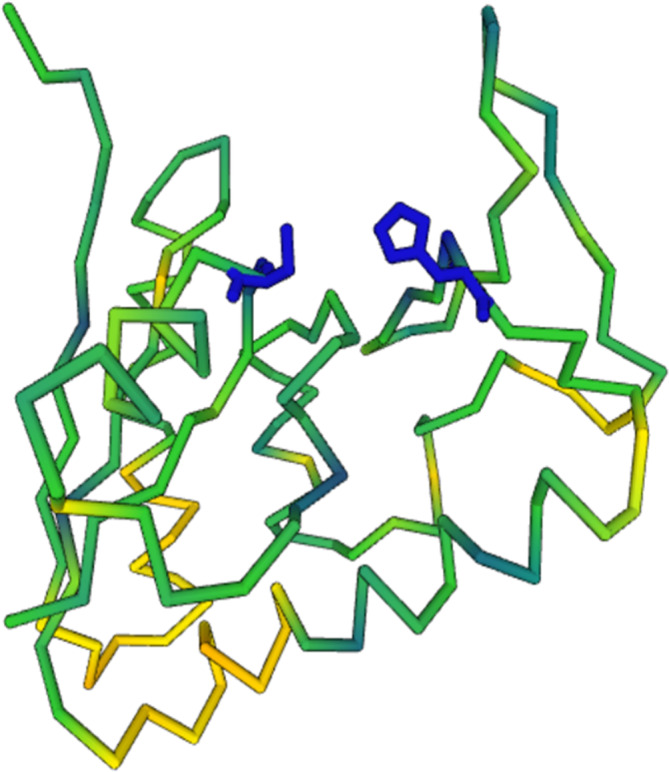
Papain‐like conserved active site. The AlphaFold model for a representative of PF08795 is colored by sequence conservation versus 2g6tA. His and Cys of the papain‐like catalytic dyad (shown in dark blue) are invariantly conserved in both protein families.
2.3. Sequence Logo Signatures
Structural alignment can reveal distant homologies even when only a few conserved residues in structurally equivalent positions are available—we call such coincidences Sequence Logo Signatures. We generated sequence profiles based on a pile‐up alignment of 1000 sequence neighbors from the UniProt database onto the query sequence (Somervuo & Holm, 2015). Separate alignments were generated for either protein of a structurally matched pair. Gap columns were inserted to the alignments according to the structural alignment, so that Sequence Logos generated from the alignments, using SkyLign (Wheeler et al., 2014), have the same length and show structurally equivalent positions in the same columns. The sequence profiles are stored as data1.json and data2.json at the same website as the results (c.f., Tables 1, 2, 3, 4). We defined as conserved residues those that have an amino acid frequency >80% in a column of a sequence profile. By comparing two structurally aligned sequence profiles, we were able to test whether there were more identical conserved residues than expected by chance.
We estimated the statistical significance of matches in sequence profiles as follows: Let N obs be the observed number of positions with identical conserved residues, and let N exp be the expected number of positions with identical conserved residues. We are comparing two sequence profiles of equal length. Keeping one sequence profile fixed, N exp is estimated by randomly permuting the columns of the other sequence profile 1000 times and calculating the number of columns that have an identical conserved residue between the fixed and permuted sequence profiles. Next, a p‐value is calculated using the binomial distribution,
where is the total number of columns in the structural alignment, x is the observed number of matches, and p is the probability of a random match, p = N exp/L. The binomial distribution f(x,L,p) was used to estimate a p‐value, This test was repeated for each pair of sequence profiles and the resulting p‐values were adjusted using the Bonferroni‐correction method (Abdi, 2007) of controlling the FWER (the probability of at least one false positive when multiple comparisons are being tested).
The Bonferroni method of adjusting p‐values is very conservative. Consequently, if on one hand the Bonferroni method is conducive to a considerable amount of false negatives when compared with expert curation, on the other, it results in very few false discoveries. Altogether, this is a simple test that allows us to pick, so to speak, low‐hanging fruits with quantitative support. It is important to mention that the test neither monitors similarities in less conserved columns (similar mixture in both profiles) nor does it consider dependencies between spatial neighbors columns in the 3‐D structure. False negatives were rescued in the qualitative assessment steps of expert classification.
2.4. Alternative sequence similarity tests
We argue that the above simple binomial test has a straightforward interpretation by identifying residues that are critical for molecular function, and that we are looking at cases with marginal sequence similarity where structural conservation as well as more qualitative attributes, like function, need to be considered to assert homology. The pros and cons of a multinomial test are elaborated in the Supplementary Methods.
Given that AlphaFold (AF) uses multiple sequence alignments as one of its inputs, it is important to understand how reliant AF eventually is on sequence‐level connections. To address this question, we performed hhblits (Steinegger et al., 2019) comparisons of Pfam.35 profiles against the subset PDB70, whose data were downloaded from the hhsuite in March 2022. For each query family in the nopdb set, we extracted the top match in PDB70 as given by hhblits. hhblits matches had to fulfil two criteria to be considered positive: first, we required that the profile‐profile alignment by hhblits had an e‐value lower than 1; second, because hhblits is based on profile‐profile comparison, it can yield a different top match than structural comparison by DALI. For this reason, it was necessary to compare DALI Z‐score between (i) the query and the top match from a PDB search by DALI, yielding Z DALI, and (ii) the query and the top match suggested by hhblits, yielding Z hhblits. We considered a hhblits match structurally valid if the ratio Z hhblits/Z DALI was greater than 80%. The data are shown in Table S1.
3. RESULTS
The most up‐to‐date release of Pfam, Pfam 35.0 released in November 2021, was used in this study as a state‐of‐the‐art reference in protein family classification, ascribed to the yearly update cycle underpinned by an extensive manual curation carried out by domain experts (Finn et al., 2016; Mistry et al., 2020). The release of AFDB1 in July 2021 created a window of opportunity, which we explored in this study to find remote homologies that are not yet recorded in Pfam 35.0. A trickle of close homologs of recently released PDB structures was purged from the nopdb set as of March 2022.
Screening the nopdb set against the PDB yielded 100 novel homologous relationships. Below, results are divided into four subsets. First, though the nopdb set was restricted to entries for which no solved PDB structure still existed in Pfam 35.0, some of the entries have now a recently released PDB structure (Table 1). In the second subset, homology is supported by both structural resemblance and statistically significant Sequence Logo Signatures (Table 2). In the third subset, homology is supported by both structural resemblance and functional similarity (Table 3). In the fourth subset, whereas one of the homologs is functionally uncharacterized, structural resemblance as well as conservation of functional residues support the possibility of a similar function (Table 4). For every table, the provided web links redirect the user to examine the evidence for homology using DALI's interactive visualization tools. The user interface has been described previously (Holm, 2020; Holm & Laakso, 2016) and tutorials are available on the website (http://ekhidna2.biocenter.helsinki.fi/dali).
3.1. Experimentally validated AlphaFold predictions
The nopdb set was checked for Pfam families that have a structural alignment to PDB with >25% sequence identity. We found matches both to structures added to the PDB after the release of Pfam.35 and a few older structures, which were missing from Pfam's pdbmap table. In total, 23 Pfam families were removed (see Table S1). Fifteen Pfam families matched recently released PDB structures with >25% sequence identity and >80% query coverage (Table 1). These 15 proteins render a valuable internal control since they provide experimental validation of AlphaFold predictions. The predicted AlphaFold models are remarkably accurate, displaying low RMSD values and high Z‐scores, and add to the record of successful predictions by AlphaFold (Jumper et al., 2021; Pereira et al., 2021) (Table 1).
3.2. Sequence Logo Signatures
In a second approach, potential homologs were paired based on structural resemblance and Sequence Logo Signatures at p < 0.05 (Table 2). For these cases, the sequence logos showed several residues that were (almost) invariant and occurred in structurally equivalent positions. The test emulates a human expert eyeing stacked sequence logos.
For half of the pairs in this subset, a function has been previously assigned on both sides, indicating that these proteins are potentially involved in similar biological processes.
Obvious cases share the same or synonymous terms in their descriptions as experimentally determined structures (PF12062 deacetylases, PF13898 deubiquitinases, PF14130 endonucleases, PF18168 DNA primase subunits, PF10307 RNA repair proteins, and PF12222 peptide‐N‐glycosidase).
PF08889 are putative glycine transferases with shared conserved residues and structural similarity to cysteine‐adding enzymes in the family PF10079 (CL0039).
The exostosin glucosyltransferases (PF03016) join the GT‐B clan (CL0113), which contains diverse glucosyltransferases with the Rossmann fold. Function loss has been implicated in some exostosin subfamilies (Wilson et al., 2022).
Cell wall proteins pertain to the family PF11765, whose members are involved in sialidase activity and are structurally similar to pectate lyases.
The spin‐doc zinc‐finger (PF18658) matches the zinc finger domain of PF19088, a member of a diverse nucleotidyltransferase superfamily (CL0260).
Carbonic anhydrase is involved in CO2 transport, a common denominator of probable inorganic carbon transporters (PF10070) and carboxysome shell carbonic anhydrase (PF08936).
There are no structures associated to clan CL0370 of membrane transporters in Pfam 35.0. Despite this, our more recent PDB search found a match between two members of the bacterial PF04632 and plant PF11744 families. Results indicated that the bacterial form underwent gene duplication as well as fusion of interacting subunits. The internal DALI identifier for the representative of PF04632 is efjA. Pairwise DALI alignment of eufjA versus 7vq3A shows that the resistance protein family (PF04632) structurally aligns beyond the domain boundaries defined in Pfam, over the whole length of the protein. There is an internal duplication in the model structure, while the PDB match is a dimer. The PDB structure was solved recently and the region covered by the Pfam domain has a chain of conserved Arg, Arg, Tyr, and Lys residues inside a channel formed by 12 transmembrane helices (Wang et al., 2022). This sequence motif is conserved in PF04632.
The C‐terminal domain of DnaJ‐like protein C11 (PF11875) is critical for protein–protein interactions. Intriguingly, it has a Sequence Logo Signature FxDPCxxxxxK in common with the carbohydrate‐binding loop of a lectin domain of pattern recognition proteins (PF02140) (Shirai et al., 2009). The structural alignment shows a cyclical permutation in that the two C‐terminal beta strands of PF11875 align to two N‐terminal beta strands of PF02140. The sequence signature suggests homology, though the functions are different.
An immunoglobulin‐like domain is found in eukaryotic MYCBP‐associated proteins (PF14646), which may be involved in synaptic processes and spermatogenesis. The nearest PDB match was a member of the PapD‐like superfamily of periplasmic chaperones (IPR008962) that mediate the attachment of many pathogenic bacteria to host tissues. Conserved residues are aromatic and have a structural role.
The GPCR‐chaperone family (PF11904) joins the LolA/B superfamily of lipoprotein sorting proteins (Figure 2). The structural alignment of the Pfam domain to the best PDB match had a gap in the middle of a beta sheet. The gap closed when the domain boundary was adjusted toward the N‐terminus. The best PDB match belongs to the VioE family. Other LolA/B superfamily members occupy the top ranks in a PDB search. A conserved RxD motif is specific to members of the GPCR‐chaperone and VioE families.
The other half of the cases have in common that one member of the homologous pair is a domain of unknown function (red‐highlighted in Table 2). For most of these families, a function can be suggested based on an evolutionary relationship.
Structural similarity and sequence conservation link 11 families of unknown function (PF03690/UPF0160, PF04250/DUF429, PF06356/DUF3064, PF07759/DUF1615, PF11001/DUF2841, PF13258/DUF4049, PF14033/DUF4246, PF14976, PF15851/DUF4723, PF16062/DUF4804, and PF17720/DUF5565) to proteins that were experimentally characterized, thus enabling homology transfer of function.
Conversely, a bacterial family involved in spore complex formation (PF04293) suggests a biological process to a structural genomic target, the bacterial family PF05960/DUF885 of putative lipoproteins (CL0126).
An invariantly conserved DxTW motif defines the eukaryotic‐bacterial Pfam family PF03942. Structural comparison unifies this family with archaeal and eukaryotic ribosomal RNA biogenesis proteins (PF04034), whose corresponding signature is DCSW.
PF17659 (DUF5521) contains four oligonucleotide/oligosaccharide binding OB‐fold domains, whose closest match is replication protein A.
PF14923 is a helical domain that has a large structural overlap (329 residues) with Sec10, a component of the exocyst complex.
PF11070/DUF2871 are short, uncharacterized, mostly bacterial proteins that match four of the 12 transmembrane helices of cytochrome c and quinol oxidase subunit I (PF00115). A HxH motif that binds two haem cofactors in PF00115 is conserved in both. A DALI search of the AFDB showed that both Escherichia coli and Staphylococcus aureus encode full‐length quinol oxidase subunits 1 that are orthologs of the eukaryotic cytochrome oxidase subunit 1 (Cox1). The high Z‐score shows that AlphaFold has clearly modeled PF11070 using Cox1 as a structural template, but there is no evidence for the involvement of PF11070 in any Cox1‐like assembly.
In one case, we got a fortuitous signal of sequence conservation due to incorrect domain boundaries.
-
7
A family of iron–sulfur binding proteins (PF12617) has a (βα)8 fold, a so‐called TIM barrel. In contrast to enzyme matches, the shared conserved residues comprise seven structural glycines. The Pfam domain covers a part of the barrel. The TIM barrel fold has rotational symmetry, and the alignment is shifted by two βα units. A DALI search with the whole protein model against PDB matches many TIM barrel families with similar high Z‐scores, ergo there is no conserved sequence signature. Moreover, inaccurate domain boundaries were cause for fortuitous significant sequence conservation signals and as a consequence, no functional inference could be made for these cases from the structural resemblance.
3.3. Related known functions
In a third subset, the criteria supporting homology were structural resemblance and functional similarity despite the absence of a clear Sequence Logo Signature. The analysis showed that 17 pairs are enzymes, 14 pairs are proteins involved in either protein‐ or DNA‐binding functions, 4 pairs function as transporters, and 2 pairs are structural proteins. Overall, the Z‐scores were high, which was indicative of a strong structural similarity (Table 3). On the other hand, for eight cases the Z‐scores were below 8, owing to the small size of their domains (see Table S1).
Fifteen enzymes share the same or synonymous terms in their descriptions as experimentally determined structures: PF01548 transposases/ribonuclease, PF01955 amidohydrolase/peptidase, PF04666 acetylglucosaminyltransferase, PF04724 and PF04765 glycosyltransferases (CL0110), PF06189 nucleotidase/polynucleotide kinase, PF07906 protease, PF09317 acyl‐CoA dehydrogenase, PF10343 DNA glycosidase/DNA lyase, PF14249 cyclase, PF15051 kinase (CL0016), PF18143 phosphoesterase/pyrophosphatase (CL0137), PF16094 proteasome assembly protein. In addition, PF15704 is associated with mitochondrial ATP synthase, and colipase‐like PF15083 matches colipases (CL0621).
Inovirus Gp2 (PF11726) is involved in viral DNA replication via a phospho‐Tyr mechanism. The active site is conserved in a hypothetical protein ORF119 from Sulfolobus islandicus rod‐shaped virus 1, a structural genomics target that has been functionally characterized as a member of the HUH‐endonuclease superfamily (Oke et al., 2011).
The COBRA domain PF04833 is implicated in cellulose deposition and has structural similarity with the cellulose‐binding domain PF03071.
The N‐terminal domain of hormone PF04709 has a similar fold to a secreted component of a TGF‐beta complex.
Similar substrates are recognized by PF12141 beta‐mannosyltransferases and beta‐1,4‐mannooligosaccharide phosphorylase (PF04041), by the tRNA modifying enzyme PF17184 and phosphoribosyltransferases (PF00156), and by proteins of unknown function involved in uracil catabolism (PF07958/DUF1688) and N‐glycosylase/DNA lyases (PF09171).
Eight families were involved in DNA binding. The CG‐1 domain (PF03859) joins the WRKY/GCM1 clan (Holm, 2022). PF06524, PF05458, and PF14616 are zinc fingers. PF10238 is associated with a transcription factor. PF19306 is a winged helix DNA binding domain. PF06420 joins the DNA repair protein Rad52/59/22 superfamily. The fold of cell division cycle protein 123 (PF07065) is reused in the C‐terminal part of PF19043, a subunit of the nuclear cap binding complex.
Three families resemble helical transmembrane proteins. PF06081 are putative exporters, and the model matches the recently released PDB structure of a remote homolog in the same family. PF04479 are fungal proteins thought to bind toxic substances, with six TM helices modeled in similar arrangement to bacteriorhodopsin. PF10337 matches a recently solved structure within the transmembrane exporter clan CL0307, which also includes PF04632 from Table 2.
PF16887 and PF14011 are involved in secretion.
S‐layer proteins (PF05123, PF05124) match a recently solved structure from the same family, which was not yet recorded in Pfam.35.
Two cases share unspecific functions, increasing the challenge to prove evidence for homology.
-
10
Biosynthetic protein PF10113 shares a TIM barrel fold with several enzymes, but there is no conspicuous shared sequence motif.
-
11
Protein binding is common to PF05428 and its closest structural match, a CUB domain (CL0164).
3.4. Putative function transfer
Results transpiring from the fourth subset showed that this group had generally high Z‐scores (Z > 8) as an indication of strong structural resemblance. The exceptions included cases covered by small domains, such as, for example, zinc fingers and the KOW domains. Furthermore, folds containing an open beta sheet or an array of decorations around the common core were also reason for lower Z‐scores, and that is because Z‐scores favor complete domain matches. In particular, PF10561 matched only half a domain due to low confidence modeling. As given in Table 4, most cases shared conserved motifs as evidence of homology, while in others the resemblance involves special structural features.
Zinc fingers were identified in five families: PF09725, PF15866 (DUF4729), PF17249 (DUF5318), PF15794 similar to PF090082 (DUF1922), and two zinc fingers in cysteine‐rich domain PF10070. Zinc fingers typically function as interaction modules that bind DNA, RNA, proteins, or small molecules. Zinc finger domains have relatively low Z‐scores because of their small size.
Conserved catalytic residues suggest an enzymatic function to 11 families: transposase activity to PF04937/DUF659, glycosyltransferase to PF05212 (DUF707), monooxygenase to stress up‐regulated Nod 19 (PF07712), kinase to PF09887 (DUF2114), desuccinylase/aspartoacylase (CL0035) to PF09892, phosphodiesterase to PF10223 (DUF2181), esterase (CL0028) to PF10561, adenosylmethionine decarboxylase to PF10936 (DUF2617), nuclease to PF11296 (DUF3097), glucosyltransferase (CL0113) to PF12308, peptidase to PF16044, and papain‐like cysteine protease to PF08795 (DUF1976) and the uncharacterized PF08942 (DUF1919) (Figure 3).
PF07505/DUF5131 shares a similar fold with that of the spore photoproduct lyase by displaying the conservation of three Cys residues responsible for binding an iron–sulfur cluster.
PF10118 is an uncharacterized bacterial family of predicted metal‐dependent hydrolases. The predicted fold matches that of ribonucleotide reductase (PF00268/CL0044), comprising seven alpha helices and over 200 residues. There are two glutamic acids and two conspicuously conserved Fe binding histidines in the active site, suggesting metal‐assisted catalysis as a common functional feature.
A conserved Trp is the hallmark of the lipocalin fold (CL0116), which was found in two families (PF04755, PF10974/DUF2804). Lipocalins act as transporters of small hydrophobic molecules.
Folds with conserved disulfide bridges were observed between PF05444 (DUF753) and a LY6/uPAR/alpha‐neurotoxin domain (Jiang et al., 2020), between PF05912 (DUF870) and the transthyretin clan (CL0287), between PF15094 (DUF4556) and Zona pellucida sperm‐binding protein, and between the meiotically up‐regulated gene family PF15474 and KP4 killer toxin (PF09044). The killer toxin impairs calcium uptake, thereby compromising cell cycle regulation.
PF14001 shares conserved motifs and is structurally similar to the RNA‐binding KOW domain.
PF00674 is a small domain with similarity to a Myb‐like DNA binding domain. Function transfer is supported by several positively charged residues and common conserved structural residues (Pro, Trp).
PF11443 (DUF2828) shares a large common core with the TROVE domain (PF05731) of RNA binding proteins, suggesting its involvement in a similar function.
PF15016 and PF15025 are domains of unknown function that appear in tandem. Our representative sequences were the uncharacterized Q96MH7 and Q3UJC8, the human and mouse homologs, respectively. Structural comparison showed that the protein is composed of four POLO box domains (CL0708). DALI alignment of the individual POLO box domains onto 4x9vA yielded Z‐scores ranging from 7.0 to 11.4. The crystal structure has duplicated POLO box domains joined by a flexible linker, so it is not surprising that the AlphaFold models of the full proteins do not reproduce the relative orientation of the duplicated POLO domains. POLO box domains owe their name to POLO‐like kinases and act as mediators for protein–protein interactions (IPR000959).
Uncharacterized bacterial protein family PF09909 was found to be a fusion of membrane permeability and extracellular sensor domains (Figure 4). Despite a lack of sequence conservation, the combination of the functions is plausible and the protein could thus act in signaling.
3.5. Profile‐profile comparison
We selected 100 examples of putative remote homologs based on structural comparison, Sequence Logos, known functions and clusters of conserved residues (Tables 2, 3, 4). Given that Sequence Logo Signatures were only detected in 32% of the cases, we tested more sophisticated profile‐profile comparisons in an attempt to broaden the analysis to large datasets. Following this approach, 53 of the 100 examples passed our dual filtering criteria of hhblits e‐value <1 and a near‐optimal structural match as compared to a DALI search of the PDB. Thus, structural comparison is required to uncover interesting relationships not picked up by profile‐profile comparison (Figure 5). The dual filtering criteria yielded 9% of positives in the rest of the nopdb set (see Table S1). This latter set of potential candidates is enriched in helical models with an armadillo repeat‐like structure or partial overlap with helical transmembrane proteins.
FIGURE 5.
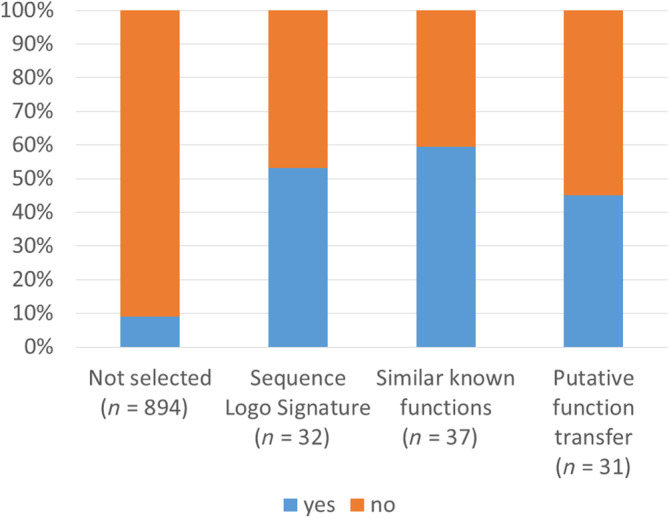
Fraction of nopdb families with hhblits e‐value <1 and DALI Z‐score ratio >0.8 (blue) or not meeting the criteria (orange).
4. DISCUSSION
We mined nopdb, a dataset that contains protein families with no prior structural information in Pfam 35.0 but to which AlphaFold Database version 1 (AFDB1) provided a confident structural model. Analyses were carried out using the DALI web server. It includes features for visualization that provide elementary yet useful functionalities, among them: visualization in 1‐D of stacked structural alignments annotated with secondary structure, protein families, sequence logos, and 3D structure superimposition highlighted in color‐coded sequence and structure conservation. The user can interactively switch between views through clickable menu options. Hyperlinks and cross‐linked identifiers ease the navigation between protein sequence, family, and structure databases. In this work, we found 100 novel remote homology relationships with functional implications. Notably, all the reported findings involve pairs of proteins that share less than 25% sequence identity. For about half of the cases to which remote homology could be assigned, there was the involvement of an uncharacterized protein family; for these cases, homology transfer suggests either a molecular mechanism or a putative involvement in a biological process, accounting for a valuable in silico prediction that can open or expand a working hypothesis.
We inferred homology based on expert assessment, considering structural and functional features in addition to sequence conservation. DALI Z‐scores for structural similarity and a statistical test for sequence conservation providing quantitative support for homology. The structural alignment of the best PDB match usually covered complete domain folds with high Z‐scores. The test for Sequence Logo Signatures was conditioned to the structural alignment, and it retrieved a positive signal (p < 0.05) even when only a small number of conserved residues were occupying structurally equivalent positions. It should be pointed out that the test rejects the null hypothesis that two proteins are unrelated; a negative result neither proves nor disproves homology. Indeed, many pairs of protein families deemed homologous did not pass the threshold of statistical significance. Sequence comparison by profile‐profile alignment is more sensitive than the binomial test for Sequence Logo Signatures and applicable to quickly screen large data sets. Nevertheless, structural comparison is required to validate the inferred homologies. About half of our manually selected examples were supported by statistically significant profile‐profile similarity, emphasizing that structural comparison is paramount to detect homologies that would otherwise be missed by sequence comparisons. As the classification of folds as homologous or as analogous relies on careful manual examination of alignment‐derived features, the automation of this classification process seems currently challenging to unfeasible.
Here, we searched for remote homology using predicted structural models. AlphaFold models have been shown to be remarkably accurate in comparison to crystallographic structures (Pereira et al., 2021; Table 1). However, not all Pfam families had credible models. We discarded about one‐third of Pfam family models as un‐folded (low confidence), ill‐folded (noncompact) or too short (nonglobular) (Figure 1). The possibility of inaccurate modeling does not affect our conclusions, and that is because we restricted our analysis to cases that showed strong structural resemblance to a PDB structure. Some AlphaFold models are extremely close to a PDB structure, indicating that the prediction has been guided by a template structure. There is an important difference to traditional template‐based homology modeling (e.g., SwissModel; Waterhouse et al., 2018), which forces a given alignment—possibly incorrect—between the prediction target and template structure. In contrast, AlphaFold starts from a gas‐like configuration of atoms from which interatomic distances are iteratively refined using information from evolutionary profiles, empirical potentials, and geometrical constraints (Jumper et al., 2021). This approach enables AlphaFold to effectively solve difficult sequence‐structure alignment problems via modeling. The predicted lysozyme family PF07759 provides a good example since it has long insertions between blocks of conserved residues (Table 2). Remarkably, AlphaFold has also successfully predicted folds not represented in the PDB at the time of prediction, for example, meckelin (PF09773, Table 1; Liu et al., 2021).
Upon the conclusion of the analyses reported here, DALI was upgraded with the AlphaFold Database version 2. We foresee that the enlarged coverage, including all the Swiss‐Prot entries, will open the possibility for more Pfam families to be explored. The findings reported here will be incorporated into the Pfam database.
5. CONCLUSION
Although it is well‐known that structural alignments are more accurate and cover a wider evolutionary divergence than sequence alignments, current methods for meta‐analysis are still largely based, although not dependent on, pairwise alignments that rely on the primary structure of proteins. In this study, we showcased—by 100 examples—how the DALI web server can be used to investigate ancient homologous relationships in an interactive and freely accessible way. This contribution, along with the recent upgrade of AlphaFold Database version 2 in DALI, will widen the horizons of structural and evolutionary biologists alike, who will have now another tool at their disposal for generating accurate and fast function hypotheses.
AUTHOR CONTRIBUTIONS
Liisa Holm: Conceptualization (lead); investigation (lead); writing – original draft (lead). Aleksi Laiho: Formal analysis (equal). Petri Törönen: Formal analysis (equal). Marco Salgado: Writing – original draft (supporting).
Supporting information
Appendix S1: Supporting Information.
Table S1: Supporting Information.
ACKNOWLEDGMENTS
This work was funded by NNF20OC0065157 of the Novo Nordisk Foundation. Computations were done using resources in Biocenter Finland's Bioinformatics platform. The authors have no conflict of interest.
Holm L, Laiho A, Törönen P, Salgado M. DALI shines a light on remote homologs: One hundred discoveries. Protein Science. 2023;32(1):e4519. 10.1002/pro.4519
Review Editor: Nir Ben‐Tal
Funding information Novo Nordisk Foundation, Grant/Award Number: NNF200C0065157
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available at http://ekhidna2.biocenter.helsinki.fi/dali/Tables_examples.html.
REFERENCES
- Abdi H. The Bonferroni and Sidak corrections for multiple comparisons. In: Salkind NJ, editor. Encyclopedia of measurement and statistics. Thousand Oaks: Sage; 2007. [Google Scholar]
- Aderinwale T, Bharadwaj V, Christoffer C, Terashi G, Zhang Z, Jahandideh R, et al. Real‐time structure search and structure classification for AlphaFold protein models. Commun Biol. 2022;5:316. 10.1038/s42003-022-03261-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenhoff AM, Train CM, Gilbert KJ, Mediratta I, Mendes de Farias T, Moi D, et al. OMA orthology in 2021: Website overhaul, conserved isoforms, ancestral gene order and more. Nucleic Acids Res. 2021;49:D373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum M, Chang HY, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021;49:D344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordin, N. , Sillitoe, I. , Nallapareddy, V. , Rauer, C. , Lam, S.D. , Waman, V.P. , Sen, N. , Heinzinger, M. , Littmann, M. , Kim, S. , Velankar, S. , Steinegger, M. , Rost, B. , Orengo, C. (2022) AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms. BioRxiv preprint, https://www.biorxiv.org/content/early/2022/06/03/2022.06.02.494367.full.pdf. [DOI] [PMC free article] [PubMed]
- de Crécy‐Lagard V, Amorin de Hegedus R, Arighi C, Babor J, Bateman A, Blaby I, et al. A roadmap for the functional annotation of protein families: A community perspective. Database (Oxford). 2022;2022:baac062. 10.1093/database/baac062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput Biol. 2008;4:e1000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrom JL, Tolbert WD, Xiong H, Pegg AE, Ealick SE. Structure of a human S‐adenosylmethionine decarboxylase self‐processing ester intermediate and mechanism of putrescine stimulation of processing as revealed by the H243A mutant. Biochemistry. 2001;40:9495–504. [DOI] [PubMed] [Google Scholar]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes WA, Tomczak A, Khatri P. Gene annotation bias impedes biomedical research. Sci Rep. 2018;8:1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Laakso LM. Dali server update. Nucleic Acids Res. 2016;44:W351–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. Benchmarking fold detection by DaliLite v.5. Bioinformatics. 2019;35:5326–7. [DOI] [PubMed] [Google Scholar]
- Holm L. DALI and the persistence of protein shape. Protein Sci. 2020;29:128–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022;50:W210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta‐Cepas J, Szklarczyk D, Heller D, Hernández‐Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lin L, Chen S, Jiang L, Kriegbaum MC, Gårdsvoll H, et al. Crystal structures of human C4.4A reveal the unique association of Ly6/uPAR/α‐neurotoxin domain. Int J Biol Sci. 2020;16(6):981–93. 10.7150/ijbs.39919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JC, Kumar G, Barman S, Najera I, White SW, Webby RJ, et al. Identification of the I38T PA substitution as a resistance marker for next‐generation influenza virus endonuclease inhibitors. MBio. 2018;9:e00430–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22:2577–637. [DOI] [PubMed] [Google Scholar]
- Liu D, Qian D, Shen H, Gong D. Structure of the human Meckel‐Gruber protein Meckelin. Sci Adv. 2021;7:eabj9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020;49:D412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–40. [DOI] [PubMed] [Google Scholar]
- Oke M, Kerou M, Liu H, Peng X, Garrett RA, Prangishvili D, et al. A dimeric rep protein initiates replication of a linear archaeal virus genome: Implications for the rep mechanism and viral replication. J Virol. 2011;85:925–31. 10.1128/JVI.01467-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CL, Zhang Y, et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol. 2017;2:16258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira J, Simpkin AJ, Hartmann MD, Rigden DJ, Keegan RM, Lupas AN. High‐accuracy protein structure prediction in CASP14. Proteins. 2021;89:1687–99. 10.1002/prot.26171 [DOI] [PubMed] [Google Scholar]
- Perraud Q, Moynié L, Gasser V, Munier M, Godet J, Hoegy F, et al. A key role for the periplasmic PfeE esterase in iron acquisition via the siderophore enterobactin in Pseudomonas aeruginosa . ACS Chem Biol. 2018;13:2603–14. [DOI] [PubMed] [Google Scholar]
- Plaxco KW, Simons KT, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J Mol Biol. 1998;277:985–94. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Russell RR. The natural history of protein domains. Ann Rev Biophys Biomed. 2002;31:45–71. [DOI] [PubMed] [Google Scholar]
- Schlieker C, Weihofen WA, Frijns E, Kattenhorn LM, Gaudet R, Ploegh HL. Structure of a herpesvirus‐encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol Cell. 2007;25:677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai T, Watanabe Y, Lee MS, Ogawa T, Muramoto K. Structure of rhamnose‐binding lectin CSL3: Unique pseudo‐tetrameric architecture of a pattern recognition protein. J Mol Biol. 2009;391:390–403. [DOI] [PubMed] [Google Scholar]
- Somervuo P, Holm L. SANSparallel: Interactive homology search against Uniprot. Nucl Acids Res. 2015;43:W24–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M, Meier M, Mirdita M, Vöhringer H, Haunsberger SJ, Söding J. HH‐suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019;20:473. 10.1186/s12859-019-3019-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeger T, Gerlach M, Morimoto RI, Nunes Amaral LA. Large‐scale investigation of the reasons why potentially important genes are ignored. PLoS Biol. 2018;16:e2006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törönen P, Medlar A, Holm L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018;46:W84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50:D439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kempen, M. , Kim, S.S. , Tumescheit, C. , Mirdita, M. , Gilchrist, C.L.M. , Soding, J. , Steinegger, M. (2002) Foldseek: Fast and accurate protein structure search. BioRxiv preprint, 10.1101/2022.02.07.479398. [DOI] [PMC free article] [PubMed]
- Wang J, Yu X, Ding ZJ, Zhang X, Luo Y, Xu X, et al. Structural basis of ALMT1‐mediated aluminum resistance in Arabidopsis. Cell Res. 2022;32:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS‐MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TJ, Clements J, Finn RD. Skylign: A tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinform. 2014;15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson LFL, Dendooven T, Hardwick SW, Echevarría‐Poza A, Tryfona T, Krogh KBRM, et al. The structure of EXTL3 helps to explain the different roles of bi‐domain exostosins in heparan sulfate synthesis. Nat Commun. 2022;13:3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information.
Table S1: Supporting Information.
Data Availability Statement
The data that support the findings of this study are openly available at http://ekhidna2.biocenter.helsinki.fi/dali/Tables_examples.html.