

Abstract
Objectives
Dysfunctional connectivity and preexisting structural abnormalities of central autonomic network (CAN) regions have been shown on magnetic resonance imaging (MRI) in sudden unexpected death in epilepsy (SUDEP) and may be mechanistically relevant. In a previous postmortem study we reported increased microglia in CAN regions, including the superior temporal gyrus (STG) in SUDEP. In this current study we investigated mammalian target of rapamycin (mTOR) pathway activation and neuronal c‐Fos activation in CAN regions in SUDEP compared to control groups.
Methods
In a series of 59 postmortem cases (SUDEP, n = 26; epilepsy controls [EPCs], n = 14; and nonepilepsy controls [NECs], n = 19), we quantified pS6‐240/4, pS6‐235/6 (markers of mTOR activation) and c‐Fos neuronal densities and labeling index in the STG, anterior cingulate, insula, frontobasal, and pulvinar regions using immunohistochemistry with whole‐slide automated image analysis.
Results
Significantly more pS6‐positive neurons were present in the STG in cases with a history of recent seizures prior to death and also in SUDEP compared to other cause of death groups. No differences were noted for c‐Fos neuronal labeling in any region between cause of death groups. Cortical neuronal hypertrophy in the STG was observed in some SUDEP cases and associated with pS6‐240/4 expression. pS6‐235/6 highlighted neuronal intranuclear inclusions, mainly in SUDEP cases and in the STG region.
Significance
Neuronal labeling for pS6 in the STG correlated with both seizure activity in the period prior to death and SUDEP. Further investigations are required to explore the significance of this region in terms of autonomic network dysfunction that may increase the vulnerability for SUDEP.
Keywords: C‐Fos, mTOR, SUDEP, superior temporal gyrus
Key Points.
The superior temporal gyrus showed significantly more mammalian target of rapamycin (mTOR) pathway activation in sudden unexpected death in epilepsy (SUDEP) postmortems compared to other groups.
c‐Fos and mTOR expression in other autonomic regions, including the anterior cingulate, pulvinar, and insular cortex, did not distinguish SUDEP from other causes of death.
The superior temporal gyrus showed increased microglial activation in a previous study, highlighting the potential relevance of this region to autonomic dysregulation in SUDEP.
1. INTRODUCTION
Current theories propose that sudden unexpected death in epilepsy (SUDEP) may be precipitated by a suppression or dysfunction of autonomic systems that regulate respiratory and cardiac functions occurring at around the time of a seizure. Recent attention has focused on the central autonomic networks (CANs), which modulate vital brainstem centers. CANs are interconnected cortical and subcortical networks that enable adaptation to physiological challenges, and knowledge of specific functions associated with brain regions continues to evolve through functional imaging, tractography, as well as stimulation studies. 1 , 2 , 3 , 4 , 5
There is growing evidence from magnetic resonance imaging (MRI) studies in SUDEP for specific patterns of alteration in CAN regions, both representing potential risk biomarkers and of mechanistic relevance to understanding causation. Altered gray matter volumes have been reported in SUDEP, including in temporal, cingulate cortex, and thalamus, 6 , 7 , 8 , 9 and altered connectivity between CAN regions on functional MRI (fMRI), suggesting disorganization of interconnections. 6 , 10 , 11 , 12 Furthermore, electrode stimulation studies in CAN regions in patients with epilepsy have provided important insights on regional cortical autonomic functions of relevance in SUDEP. 13 , 14 , 15
In a previous postmortem study of CAN in SUDEP, we noted higher microglial densities in the superior temporal gyrus (STG) and thalamic regions compared to control groups. 16 In addition, we noted increased microglia in patients with reported recent seizures occurring in the 10 days prior to death. Recent seizures, including seizure clusters,17 have been considered as a risk‐factor for SUDEP. In the landmark mortality in epilepsy monitoring unit study reviewing SUDEP on monitoring units, one or more generalized seizure (GS) occurred in the 12 h prior to death in 12 of 16 cases. 18 The mechanisms and relationships between recent seizures and SUDEP are unclear but transient neuronal dysfunction, including reversible alterations of modulatory neuropeptides, 19 may represent a period of vulnerability, particularly if involving the critical CAN regions.
Our aim was to investigate c‐Fos, an early/immediate proto‐oncogene and inducible transcription factor used to monitor temporospatial cellular injury including following epileptiform activity 20 in SUDEP postmortem cases. In parallel we investigated mammalian target of rapamycin (mTOR) pathway activation, which regulates diverse and critical cellular functions and is upregulated following seizures, 21 using phosphorylation of the downstream ribosomal protein S6 as a surrogate marker of pathway activity. 22 This would be investigated in selected CAN regions to explore any relationship to recent seizure histories and other clinicopathological factors.
2. METHOD
2.1. Case groups
SUDEP cases (n = 26), primarily from adults, were obtained through the Epilepsy Society Brain and Tissue Bank (ESBTB) at University College London (UCL). The study has been ethically approved and cases consented for use in research. All of the SUDEP cases were initially coroner's autopsies conducted for investigations into the cause of death and subsequently donated for research to the tissue bank. These included definite SUDEP cases, with four cases of possible/probable SUDEP according to criteria of Nashef. 23 We included seven SUDEP cases, in which a degree of cortical neuronal hypertrophy had been noted during diagnostic evaluation on routine examination using hematoxylin and eosin (H&E), Cresyl violet/Luxol fast blue and neurofilament stains. In none of these seven cases was there a clinical, neuroimaging or genetic diagnosis of focal cortical dysplasia during life. Neuronal hypertrophy was evident mainly in the STG region, with scattered enlarged neurons but without the hallmark pathology criteria of cortical dyslamination, balloon cells, and white matter changes for a definitive diagnosis of focal cortical dysplasia. 24 (Further detail in Table S1).Non‐SUDEP epilepsy cases (n = 14) were used as a control groups, from the ESBTB and the ERUK Corsellis epilepsy brain collection, including a subset with documented recent status epilepticus or seizures prior to death (epilepsy controls, EPCs). Nonepilepsy control (NEC) cases were obtained from the ESBTB and Medical Research Council (MRC) Edinburgh brain bank (n = 19). All postmortem cases were collected between 1971 and 2019 (summary group details are shown in Table 1 and further case details regarding epilepsy history, circumstances of death, and neuropathology findings are shown in Table S1).
TABLE 1.
Summary details of cases in three cause of death groups (case detail shown in Table S1)
| Groups | Age at death, mean (years); range | Gender M: F | Age at onset of epilepsy, mean (years); range | Cases with recent seizures reported recent: last 10 days | Focal brain pathology (relevant to epilepsy) Yes: No | Brain weight, mean (g) range | Fixation time, a mean (days) (range) | Postmortem interval, mean (days) (range) |
|---|---|---|---|---|---|---|---|---|
|
SUDEP N = 26 |
36.38 (15–68) | 15: 11 | 20.13 (5–49) | 2: 7 | 11: 15 | 1354 g (812–1664) | 45 (7–210) | 3.4 (1–7) |
|
Epilepsy controls (EPCs) N = 14 |
48.9 (1–88) | 8: 6 | 31.2 (0–82) | 8: 3 | 5: 9 | 1220 g (850–1421) | 52 (9–83) | 2.55 (1–6) |
|
Nonepilepsy controls N = 19 |
48.9 (23–90) | 10: 9 | NA | NA | NA | 1358 g (1060–1580) | 29 (12–140) | 4.4 (2–12) |
| Significance between cause of death groups | p = .05 (all three groups) | N/S (all three groups) | N/S (SUDEP and EPC groups) | p = .01 (SUDEP and EPC groups) | N/S (SUDEP and EPC groups) | N/S | p = .013 (all three groups) | N/S (all three groups) |
Note: Recent = Seizure reported at the time of, or just prior to death, including status epilepticus.
Fixation times shown for macroscopic brain examination and diagnostic sampling; detailed fixation times for each region shown in Table S1, as for some cases certain brain regions were sampled at a later time. Brain weight is recorded as the fixed weight prior to macroscopic dissection for the majority of cases where available data (see also Table S1). NA = not applicable. Differences between cause of death groups in the bottom row shown with nonparametric tests.
2.2. Rationale for the CAN brain regions selected
We evaluated four brain regions based on the existing evidence of their involvement in SUDEP: (1) The subgenual anterior cingulate cortex (ACC), includes Brodmann area 25 (BA25), BA24, and BA33; stimulation of BA25 was shown to induce hypotension in monitored patients with epilepsy, 14 and this region has known roles in cardiovascular/vagal responses 2 , 3 as well as in sympathetic regulation. 1 Furthermore, increased gray matter volumes have been reported in the subcallosal cortex in SUDEP 8 , 25 and altered functional connectivity in MRI studies. 6 , 11 (2) Superior temporal gyrus (or STG) (BA 21/22/41/42): cortical thinning is reported in this region in epilepsy series 26 and tractography studies confirm the integration of this hub in CAN networks 4 with recognized roles in parasympathetic regulation. 1 In a recent study of patients with severe peri‐ictal hypoxia, decreased gray‐matter volumes were reported in the right STG, 7 evidence that this region is modulated by seizures, and in our previous postmortem study, higher STG microglial densities were observed in SUDEP compared to control groups. 16 (3) Insular cortex (BA13/14): stimulation of both the left and right insular cortex during the course of implantations for the investigation of epilepsy induced a decrease in heart rate and cardiac output, 5 and direct autonomic dysfunction of this region has been implicated in SUDEP. 27 , 28 Furthermore the insula forms a central hub of the CAN 4 ; cortical thickening of this region has been reported in patients with generalized seizures, 8 increased functional connectivity was associated with SUDEP risk, 11 and recently, hypometabolism of the right posterior insula was shown on positron emission tomography (PET) in patients with epilepsy with ictal asystole. 29 (4) The pulvinar region of the thalamus is recognized as a respiratory and sympathetic regulatory brain region. 1 , 30 Decreased pulvinar volumes bilaterally have been reported in SUDEP on MRI, 9 and in a previous postmortem study we reported increased microglia in this region in definite SUDEP cases. 16
2.3. Tissue preparations
Sections were cut at a 5 μm thickness from the ACC, STG, insular, and pulvinar regions from both the left and right sides, where available. Sections were stained with Cresyl violet for confirmation of the cytoarchitectural regions, which is particularly relevant for ACC to distinguish BA25 from BA33 and BA24. Sections from all regions were immunolabeled for c‐Fos (Santa Cruz, Biotechnology SC‐166940, 1: 50), from all regions (except ACC) for pS6‐240/4 (Cell signaling technology, 5364, 1: 500) and pS6‐235/6 (Cell signaling technology, 4857S, 1: 50), and in the pulvinar region for glial fibrillary acidic protein (GFAP) (Dako, Z0334. 1: 2000) using a Leica BondMax (see Appendix S1 for details of immunohistochemistry). For the demonstration of pS6, we utilized two antibodies that recognized different phosphorylation sites of pS6: pS6‐240/4 is specific for mammalian target of rapamycin complex 1 (mTORC1) pathway activation, whereas pS6‐235/236 may also occur through mTOR independent pathways including Ras‐mitogen activated protein kinase pathway activation and both antibodies in conjunction have been widely used in studies. 22 Because many cases were archival coronial postmortems, not all regions were available from all cases and some regions were sampled at a later interval. The cases used and fixation times for each region are detailed in Tables 2 and S1. Double immunofluorescence labeling was carried out for pS6‐240/4 and pS6‐235/6 and with neuronal markers (MAP2 [microtubule associated protein] and synaptophysin), GFAP, and microglial marker Iba1 in the cortical and pulvinar regions in selected SUDEP and NEC cases (see Appendix S1 for details).
TABLE 2.
Results of quantitative analysis of c‐Fos and pS6 labeling in STG, pulvinar, and insular regions
| Region | SUDEP | Definite SUDEP | Epilepsy controls | Nonepilepsy controls | |
|---|---|---|---|---|---|
|
c‐Fos Neuronal density/mm2, mean (SD) (N = cases a ) Percentage positive neurons (SD) |
STG |
348.43 (260) N = 19 52.3% (26) |
279.9 (172.1) N = 15 48.9% (25) |
408.15 (285.5) N = 7 48.9% (26) |
298.49 (193.3) N = 18 48.2% (25) |
| Insular |
277.9 (245) N = 19 37.7% (24) |
287.18 (249.4) N = 18 38.6% (25) |
325.0 (137.8) N = 7 41.5% (10) |
174.57 (164.05) N = 8 26.8% (19.7) |
|
| Pulvinar |
49.05 (61.8) N = 19 7.6% (7) |
40.56 (42.7) N = 17 6.9% (6.2) |
24.2 (2.9) N = 3 6.5% (1.5) |
47.49 (54.87) N = 8 8.8% (10.5) |
|
|
pS6‐240/4 (N = cases a ) Neuronal density/mm2, mean (SD) Percentage positive neurons (SD) |
STG |
28 (40)/mm2 N = 15 6.6% (8) |
18 (32)/mm2 N = 13 7% (9.2) |
1 (2)/mm2 N = 6 6.6% (2.9) |
6 (10)/mm2 N = 18 1.2% (.53) |
| Insular |
5 (14)/mm2 N = 18 3.9% (4.7) |
6 (15)/mm2 N = 15 4.1% (5.1) |
0.8 (0.5)/mm2 N = 8 3.5% (2) |
0.7 (0.7)/mm2 N = 7 2.1% (1.6) |
|
| Pulvinar |
5 (10)/mm2 N = 17 6.9% (8.7) |
6 (11)/mm2 N = 15 7.2% (9.1) |
0.1 (0.05)/mm2 N = 3 3% (1.8) |
0.2 (0.1)/mm2 N = 8 2.6% (3.5) |
|
|
pS6‐235/6 (N = cases a ) Labeling index (%), mean (SD) |
STG |
0.26 (0.35) N = 19 |
0.31 (0.37) N = 15 |
0.075 (0.08) N = 6 |
0.071 (0.06) N = 18 |
| Insular |
0.13 (0.15) N = 11 |
0.13 (0.16) N = 10 |
0.086 (0.1) N = 3 |
0.084 (0.1) N = 5 |
|
| Pulvinar |
0.76 (0.88) N = 16 |
0.65 (0.8) N = 14 |
0.67 (0.3) N = 5 |
0.98 (0.59) N = 7 |
Refers to the number of cases evaluated in each group for each region (not all regions were available in all cases).
2.4. Quantitative analysis and regions of interest
A digital slide scanner (Leica SCN400F, Leica Microsystems or Hamamatsu 360, Hamamatsu Photonics) was used to scan all slides at ×40 magnification. The scanned whole slide images (WSIs) were stored in a computer server and were uploaded to either Definiens (Definiens AG) for c‐Fos or QuPath platform 31 for pS6 and GFAP image analysis. The following regions of interest (ROIs) were outlined. ACC: Evaluation of the cytoarchitectural of the subcallosal coronal blocks on CV was designated as either BA25 or BA33/24/32 based on previous studies 32 ; in 10 cases both of these BA regions were available in separate blocks. The entire extent and full thickness (pia to white matter border) of these BA regions were outlined, to include both superficial and deep cortical layers but excluding white matter. A further cortical ROI was drawn on frontobasal cortex (BA11), representing a nonautonomic control brain region. STG: ROIs were placed on either side of the gyrus at the mid‐point between the crown and sulcus, representing the full thickness of cortex but not including white matter. Insular cortex: The entire extent and full cortical thickness representing BA 13/4 were outlined on the sections, overlying the claustrum, with care not to include the white matter or adjacent gyri. Pulvinar: A rectangular ROI of ~45 mm2 was placed within the nucleus deep to the subependymal surface in all cases. (Further details of ROIs are detailed in Figure S1A–D.)
For c‐Fos sections using Definiens Tissue Studio Software (Definiens AG) thresholds were set following analysis of a pilot series of a range of labeling intensities and densities to delineate single neuronal cells, and the neuronal density (ND) (positive cells/area) was calculated (Figure S1C). Different thresholds were set for the ACC regions from the STG, insular, and pulvinar regions, but the thresholds were kept constant for each region across all cases. Scanned images of pS6‐240/4 and pS6‐235/6 were uploaded onto the Qupath platform, thresholds similarly optimized and neuronal density/mm2 (or ND) and overall labeling index (LI) measured. The LI represents the field fraction of immunolabeling for the whole region and, therefore, provides a measure of the overall labeling of cells and processes. In pulvinar GFAP sections, using similar ROIs, the GFAP LI was evaluated. For c‐Fos and pS6‐240/4 in the insular, pulvinar, and STG regions, positively labeled neurons as a percentage of all neuronal cells in each region were also calculated (see Appendix S1 and Figure S1E).
Statistical analysis between the cause of death groups was carried out using SPSS (version 25, IBM incorporation) using nonparametric tests (Mann‐Whitney, Kruskal‐Wallis tests), Spearman's correlation, and linear regression analysis. Graph pad (Prism, version 9) was used for the graphical representation of the data.
3. RESULTS
3.1. Qualitative patterns
3.1.1. c‐Fos
Neuronal labeling (nuclear and cytoplasmic) was present in the cortical regions in epilepsy and control groups but variable between cases. In the ACC (BA33, 24, 32, and 25) common patterns were labeling of layer II neurons alone, a “tram‐track: pattern of layer II and layer V/VI neurons, or pan‐cortical labeling, the latter pattern particularly noted in BA25 (Figure 1A). In the adjacent control BA11 region, variable laminar neuronal‐staining patterns (II, V/VI or pan‐cortical) was also observed. In insular cortex and STG, scattered positive neuron, laminar patterns of labeling (layers II, IV, V/VI), or pan‐cortical labeling was variably observed but with no pattern dominating in any cause of death group (Figure 1B–D). There was no evidence of glial labeling with c‐Fos. In the pulvinar, the impression was of overall fewer c‐Fos‐positive neurons compared to the cortical regions (Figure 1E).
FIGURE 1.
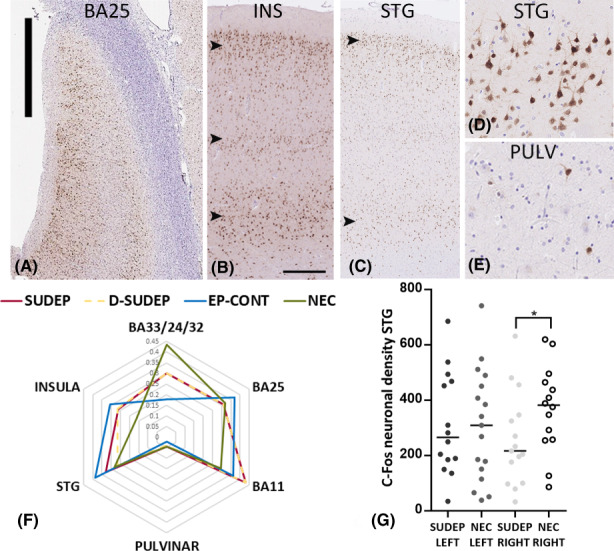
c‐Fos immunostaining patterns and quantitative analysis in sudden unexpected death in epilepsy (SUDEP). (A) Low‐power image of Brodmann area 25 (BA25) and c‐Fos labeling showing a pan‐cortical to laminar labeling pattern of neurons. (B) Insular cortex (INS) with neuronal labeling in layers II, IV, and V/VI (arrowheads) showing a laminar pattern. (C) Superior temporal gyrus (STG) with neuronal labeling predominantly in layers II and VI (arrowheads). (D) Distinct neuronal labeling, including pyramidal cells with a lack of glial labeling was observed with c‐Fos immunohistochemistry. (E) In comparison, the pulvinar region (PULV) showed only scattered positive neuronal cells. (F) Radar plots of c‐Fos mean neuronal densities were generated to illustrated relative differences between regions in cause of death groups (SUDEP, D‐SUDEP (definite), EP‐Cont (Epilepsy controls) and NECs (nonepilepsy controls). Because different thresholds for detection were used in the anterior cingulate regions to other areas, the mean neuronal density is expressed as a ratio of the maximum value for each region. Lower levels of c‐Fos were noted in the pulvinar region in all cause of death groups. (G) Scatter plots of c‐Fos neuronal densities in the STG region were significantly lower in SUDEP cases on the right side compared to NECs. Bar in (A) = 2 mm, Bar shown in (B) approximately equivalent in (B and C) to 300 μm and in (D and E) to 100 μm.
3.1.2. pS6‐240/4
In the STG region, neuronal cytoplasmic labeling primarily of pyramidal cortical cells was observed both in epilepsy cases and controls, including the hypertrophic neurons described in SUDEP cases (Figure 2A). The extent of labeling in cases varied from scattered neurons to laminar patterns involving layers II, III, V, and VI, with relative sparing of layer IV (Figure 2B) and rare astroglial labeling. pS6‐240/4 neuronal labeling was present in the insular region but appeared less extensive than STG (Figure 2C), whereas the pulvinar region showed predominantly infrequent scattered positive neurons with rare astroglial labeling (Figure 2D).
FIGURE 2.
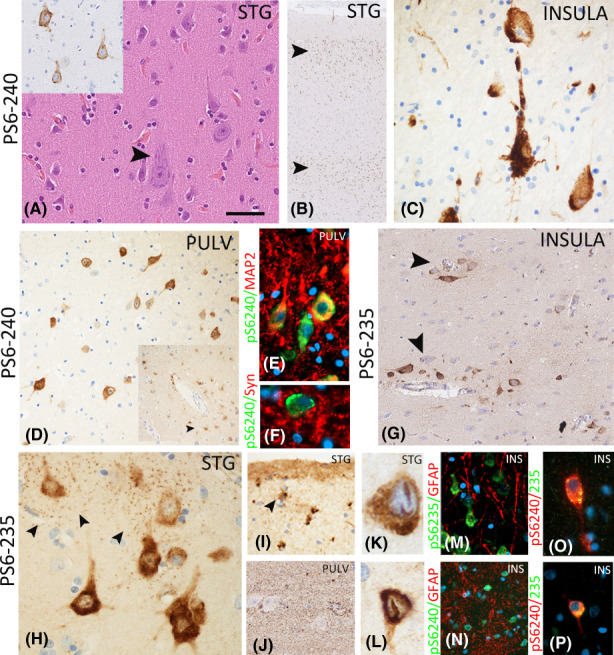
Phospho‐S6 immunohistochemistry patterns in the central autonomic regions. (A–D), pS6 240–44: (A) Scattered hypertrophic neurons were noted, particularly in the superior temporal gyrus region (STG) in a proportion of sudden unexpected death in epilepsy (SUDEP) cases, but lacking the balloon cells of other specific pathological features of a focal cortical dysplasia. These neurons were often pS6 positive (inset). (B) Low power of STG in a SUDEP case showing a laminar pattern of neuronal labeling with pS6‐240. (C) In the insular region, frequent neuronal pS6‐240 expression was also noted. (D) Scattered but variable numbers of pS6‐positive neurons were observed in the pulvinar region (PULV), and in some cases astroglial labeling was noted (inset, arrowed); there was focal co‐localization of labeling of pS6‐240 with neuronal markers MAP2 (E) and synaptophysin (F). (G–L), pS6 235–6: (G) A prominent perivascular distribution of pS6 labeling was noted in the insular cortex, rather than a laminar pattern. (H) In addition to neuronal perikaryal labeling, a striking perineuronal synaptic granular pattern of labeling was noted, shown here in the STG. (I) Labeling of glial cells in layer I was noted (arrowed) in some cases. (J) In the pulvinar region, a more striking granular labeling of the parenchyma was appreciated. (K and L) Intranuclear linear inclusions in neurons in the STG were more frequently seen in SUDEP cases. (M and N) Double labeling of pS6 markers with GFAP showed an overall prominent neuronal pattern with minimal localization with glial processes. (O and P) Focal co‐labeling of pS6‐240 and pS6‐235 was observed in neuronal cells. The bar shown in (A) for (A, C, E, F, M, O, P, and H) is equivalent to ~50 microns; in (D, G, I and H) to ~150 microns, and in (B) to ~300 microns.
3.1.3. pS6‐235/6
Neuronal cytoplasmic labeling with pS6‐235/6 was observed in the STG and to a lesser extent in the insular cortex. Scattered small and large neurons were variably present, randomly scattered through laminae and often in perivascular locations rather than a laminar distribution (Figure 2G). Peri‐neuronal granular, synaptic‐like labeling was also evident (Figure 2H). In the pulvinar, there was less frequent neuronal labeling and a more prominent synaptic‐like labeling (Figure 2J). Cortical astroglial labeling was noted, particularly in layer I astrocytes (Figure 2I). pS6‐235/6‐positive, linear to round intranuclear neuronal inclusions were noted mainly in the STG region (Figure 2K,L) and present in 57% of SUDEP cases compared to 16% of NECs but not in EPCs.
Double labeling confirmed neuronal co‐localization of pS6‐240/4 and pS6‐235/6 in a proportion of neurons (Figure 2O,P), and co‐expression with mainly neuronal markers MAP2 and synaptophysin (Figure 2E,F) over glial markers (Figure 2M,N).
4. QUANTITATIVE ANALYSIS
4.1. Cause of death groups
4.1.1. c‐Fos
There was no significant difference between the mean c‐Fos neuronal densities for any region (anterior cingulate regions [BA25, BA33/24/32], BA11, STG, insular, and pulvinar) between SUDEP, EPCs, and NECs (Tables 2 and S2, Figure 1F). This was also the case when evaluating definite SUDEP cases compared to control groups. Significantly lower c‐Fos neuronal densities and also the percentage of c‐Fos‐positive neurons were confirmed in the pulvinar compared to the STG and insular regions in both SUDEP and NECs, reaching the greatest significance in the SUDEP group (p < .001 and p < .0001) (Figure 1F). Considering left or right sides separately between the cause of death groups, the mean c‐Fos ND (and percentage positive neurons) was found to be significantly lower in the right STG in SUDEP (and for definite SUDEP) compared to NECs (p ≤ .05) (Figure 1G). Comparing left and right sides within cause of death groups, significantly higher c‐Fos ND was noted on the left compared to the right insular cortex in the SUDEP group only (p = .02).
4.1.2. pS6‐240/4 and pS6‐235/6
Higher mean neuronal densities were noted with pS6‐240/4 in SUDEP compared to other cause of death groups in all regions, with the highest values in the STG region (Table 2, Figure 3A). Significantly higher mean LI for pS6‐235/6 was present in the STG in SUDEP cases (and definite SUDEP cases) compared to NECs (p = .004 and p = .003 [definite SUDEP]) (Table 2, Figure 3B). In contrast, in the pulvinar region the mean pS6‐235/6 LI was lower in the SUDEP group (and definite SUDEP cases) compared to NECs (p = .046, Figure 3B). Considering hemispheres separately between cause of death groups, higher LI for pS6‐235/6 in SUDEP than NECs was noted for both the right and left STG (p < .0001, p < .005). Comparing hemispheres within cause of death groups, a lower percentage of positive pS6‐240/4 neurons in the left than the right insula was noted in SUDEP cases (p = .03). There was a significant correlation between pS6‐235 and pS6‐240 labeling in the STG and insular regions (p < .05 and p = .003, Spearman's correlation) but not in the pulvinar region. There was a correlation between the percentage of positive neuron labeling with c‐Fos and pS6‐240/4 in epilepsy groups in the pulvinar region (p < .05) but not in controls or other regions.
FIGURE 3.
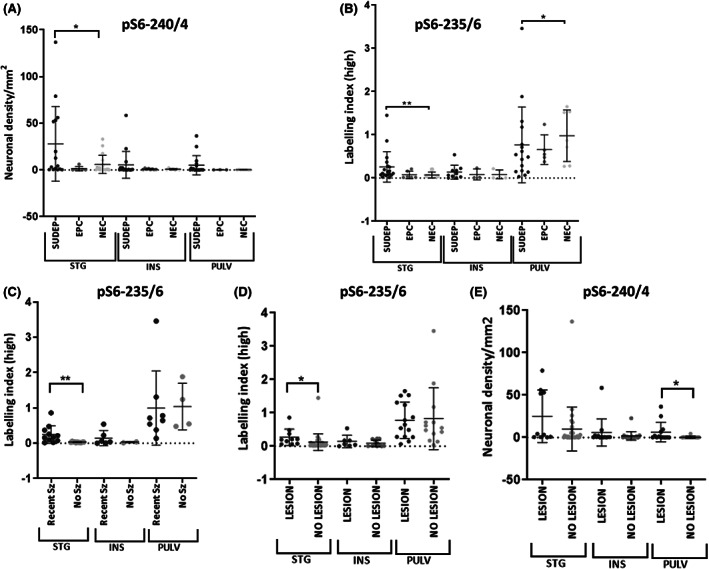
Scatter graphs of phospho‐S6 quantitative labeling in brain regions. (A) pS6‐240/4 neuronal densities in cause of death groups (sudden unexpected death in epilepsy [SUDEP], epilepsy controls [EPCs], and nonepilepsy controls [NECs]) in superior temporal gyrus (STG), insular (INS), and pulvinar (PULV) regions showing higher densities in the SUDEP group in all regions, reaching significance in the STG, when fixation time factored with multivariate analysis. (B) pS6‐235/6 showed significantly higher labeling indices in SUDEP in the STG but lower labeling in the pulvinar region compared to the control groups. (C) In cases with recent generalized seizures in the last 10 days prior to death (Recent Sz group) compared to cases without this history (No Sz) significantly greater pS6‐235/6 labeling was observed in the STG in the group with reported recent seizures. (D) In cases with a focal epilepsy‐related lesional pathology identified at postmortem, significantly greater labeling was noted with pS6 235/6 in the STG and (E). pS6 240/4 in the pulvinar region.
4.1.3. GFAP
In view of a lower level of neuronal labeling in the pulvinar compared to cortical regions, quantitative evaluation of GFAP was also carried out. There was no correlation between GFAP labeling index, pS6‐235/6, or pS6‐240/4 in any cause of death group. No difference in the mean pulvinar GFAP labeling index between cause of death groups was noted, with lowest mean values in the SUDEP group (Figure S3).
4.2. Clinical and pathological correlations
Regarding seizure history, there was no significant difference between in regional pS6‐235/6 and pS6‐240/4 measurements in epilepsy cases with or without a history of status epilepticus prior to death. However, in cases with a history of generalized seizures reported in the 10 days prior to death, significantly higher pS6‐235/6 was observed in the STG than cases with no seizures (p = .004, Mann‐Whitney test, Figure 3C). Higher pS6‐240/4 neuronal densities were also noted in all regions in cases with recent seizures, but without reaching significance. There was no significant association of c‐Fos neuronal densities in any region in relation to status epilepticus or seizures reported 10 days prior to death.
A significant positive correlation between pulvinar GFAP labeling with age at death was observed in the SUDEP group only (p = .006) (Figure S3) but was not noted with other markers. In patients with epilepsy with any focal brain pathologies relevant to seizures in any region (including old surgical scars, hippocampal sclerosis [see Table S1 for detail]), a significantly higher LI with pS6‐235/6 was noted in the STG (p = .02, Figure 3D) and pS6‐240/4 neuronal density in the pulvinar (p = .016, Figure 3E) compared to neuropathology‐negative epilepsy cases. In SUDEP cases with neuronal hypertrophy, although higher pS6‐ pS6‐240/4 and 235/6 labeling was noted in the STG and insular cortex, there were no statistically significant difference from epilepsy cases without this feature (Figure S2A,B).
There was no statistical association between c‐Fos ND and the presence of an underlying brain pathology or neuronal hypertrophy.
4.3. Effects of fixation times and postmortem intervals
There were no significant differences in mean postmortem intervals between the cause of death groups (Table 1) and there were no significant associations between postmortem interval and c‐Fos, pS6 240/4, or 235/6 measurements. There was a significant variation in mean brain fixation times between the cause of death groups (Table 1, p < .05), with the shortest fixation times in NECs. There was no correlation between regional tissue fixations times and pS6 labeling in the pulvinar or insular sections. There was an inverse correlation between pS6‐240/4 neuronal densities in the STG and fixation time; using multivariate analysis to factor the variable of fixation time, linear regression analysis confirmed significantly higher pS6‐240/4 labeling in the STG in SUDEP compared to other cause of death groups (p = .02, Figure 3A). There were no significant associations in c‐Fos measures and fixation time for any brain region (Figure S1F).
5. DISCUSSION
There is evidence implicating CAN in the peri‐ictal autonomic dysfunction that precipitates SUDEP. Using a postmortem series to evaluate four autonomic brain regions, we have shown increased mTOR pathway activation in the STG region in SUDEP that also correlated with recent seizures prior to death. In contrast, there was less evidence that c‐Fos neuronal expression patterns in CAN distinguished SUDEP from other causes of death. In a previous study on a different postmortem cohort, increased microglial densities were identified in the STG in SUDEP, which also associated with recent seizures. 16 These current observations reinforce that the STG is of potential relevance to the autonomic pathways linked to SUDEP.
Identification of specific tissue biomarkers for acute neuronal dysfunction and their application in the investigation of epilepsy‐related and sudden deaths has been an area of recent research, as both a much‐needed diagnostic tool as well as furthering our understanding of brain regions that may be critically involved in SUDEP. 33 , 34 mTOR is a major kinase that forms two complexes—mTORC1 and mTORC2—with different activation pathways, regulating many fundamental cellular functions 35 , 36 and influencing neuronal size and synaptic neurotransmission. Activation of this pathway is observed in broad spectrum of focal epilepsy pathologies and nonlesional tissue in both neuronal and glial cell types. 37 , 38 , 39 Experimental epilepsy models confirm mTOR activation following seizures, 40 occurring within 2 h following status epilepticus in the kainic acid model, 41 and mTOR activation may play a critical role in cellular responses and epileptic networks following status epilepticus, although little explored in human tissues to date. 42
We utilized quantitative immunohistochemistry for phosphorylation of the downstream ribosomal protein S6 at Ser235/6 and Ser240/4, as mTOR‐dependent and mTOR‐independent phosphorylation sites, as used widely in previous studies to assess pathway activation. 22 Differences in pS6 patterns were noted across the SUDEP cases, which may reflect the clinical and pathological heterogeneity of this group as well as dysfunctional cellular mechanism prior to death. For example, we noted greater pS6 labeling in relation to any underling focal neuropathology identified postmortem. However, we also observed a relationship with reported seizures occurring in the last 10 days before death, which may suggest a more sustained mTOR activation in these cases. In experimental models, mTOR activation evaluated by pS6 immunohistochemistry occurs from 6 h to 5 weeks following seizure onset. 21 Furthermore, mTOR activation following experimental seizures induces inflammatory pathways and activates microglia 42 ; of interest, our previous observations of increased regional microglia in SUDEP, particularly with recent seizures prior to death, 16 could be linked mechanistically to the observed mTOR activation. In seven SUDEP cases we also noted hypertrophic neurons in the cortical regions, particularly the STG, which did not amount to focal cortical dysplasia based on pathology criteria, although genetic testing had not been conducted. We also observed pS6 expression in these hypertrophic neurons similar to the ubiquitous expression in hypertrophic and dysmorphic neurons reported in focal cortical dysplasias associated with mTOR pathway mutations. 22 Of interest, increased rates of SUDEP are reported in some focal epilepsies with DEPDC5 mutations, an mTOR pathway inhibitor, 43 and associated with focal dysplasia, 44 although the precise cellular mechanisms leading to the sudden death and any autonomic dysfunction is as yet unclear. 45 Hypertrophic neurons have long been recognized in acquired epilepsy pathologies, 46 , 47 considered to reflect altered neuronal function, metabolism, cellular stress, or connectivity as a result of seizures. The identification of isolated hypertrophic neurons and more widespread mTOR activation in our SUDEP series is unlikely to represent mTOR pathway mutations but raises potential mechanistic links with DEPDC5‐related epilepsies as a future line of investigation.
This study highlights the STG as a brain region of relevance in SUDEP (Figure S4). Cortical thinning, including progressive atrophy, 48 has been demonstrated in the STG in large epilepsy cohorts on MRI. 26 There are few data on specific autonomic regulatory functions associated with the STG, but parasympathetic autonomic regulation is recognized 1 to be of relevance to SUDEP. In addition, tractography studies of the CAN place the temporal lobe, particularly the STG as an integral part of autonomic networks including the parieto‐anterior‐temporal pathway, which connects STG to amygdala and insular cortex. 4 In this current study only a minority (eight of the SUDEP cases) were also included in our previous study reporting increased microglia in the STG in SUDEP; this reinforces that pathological alteration in this region may be relevant to SUDEP across cohorts. Concerning lateralization, there was some evidence in this series for greater mTOR activation on the right than left STG and lower c‐Fos activation in the left STG in SUDEP compared to controls. In a recent study of patients with severe ictal‐hypoxia during generalized seizures, considered a risk‐factor for SUDEP, lower gray matter volumes were observed on the right but not on the left STG. 7 Although not all SUDEP cases showed increased pS6 labeling, likely reflecting the heterogeneity of underlying causes, these collective studies suggest that further study of specific autonomic functional networks associated with the right STG would be relevant for some cases.
The subcallosal (or subgenual) cingulate gyrus comprises BA25 as well as parts of BA33, 24, and 32 as cytoarchitectonically distinct regions 32 with different functional connectivity profiles; BA25 is activated with autonomic processing and BA33/24 is co‐activated with the amygdala and anterior cingulate, among other regions. 49 In tractography studies, the ACC forms part of the rostral limbic pathway, which is connected to the insular region. 4 BA25 has been regarded as the principal site of autonomic regulation in the frontal lobe, 50 including cardiovagal 2 , 3 as well as sympathetic regulation. 1 Stimulation of Brodmann area 25 (subcallosal neocortex) in patients with epilepsy produced striking systolic hypotensive changes. 14 Furthermore, reduced functional connectivity of the ACC in fMRI has been associated with SUDEP risk 11 and network alterations in SUDEP. 10 In structural MRI studies, increased thickness of subgenual cingulate cortex was shown in patients with GS 8 and increased gray matter volumes reported in the subcallosal cortex in SUDEP vs Control groups. 25 This is the first pathological study of BA25 in epilepsy but despite striking laminar neuronal labeling with c‐Fos in SUDEP cases, we failed to identify distinct difference from controls, supported by quantitative analysis.
The insula is also regarded as a central hub in connectome studies of CAN using tractography. 4 Cortical thickening in the insular region has been reported in patients with generalized seizures on MRI, 8 increased functional connectivity recognized in SUDEP,11 and surgical resection associated with autonomic dysfunction. 27 There is robust evidence for a role of the insula in the regulation of heart rate, 51 with inconsistent evidence for lateralization 52 ; PET hypometabolism in the right posterior insula was observed in patients with epilepsy with ictal asystole 29 but electrode implantation and stimulation of either left or right insula was shown to induce changes in heart rate and cardiac output but without effects on blood pressure or respiration. 5 We noted some lateralization of pathology with greater c‐Fos labeling and reduced pS6‐240 labeling in the left compared to the right insula in the SUDEP group, but no overall significant differences from controls. The pulvinar region showed less mTOR and c‐Fos expression than cortical regions but again no specific patterns emerged in SUDEP cases. In addition, despite evidence for reduced posterior thalamic volumes in SUDEP in structural MRI, 9 , 25 we also did not identify increased gliosis in the pulvinar region in SUDEP, only a significant association with age, and the pathological basis for these imaging alterations remains unclear.
There are several limitations to this postmortem study, in that not all regions were available in all cases and only limited CAN regions were selected for this study. We included cases in the study with reports of a seizure occurring in the last days prior to death from the clinical records; we cannot exclude that in cases without this history, a recent seizure occurred but was not documented. Furthermore, in many cases, details of seizure types (including for status epilepticus), duration, and localization through recent EEG and MRI investigations were not always available to further stratify the cases. In addition, different fixation times for brain regions were factored into our analysis. This is a coroner's postmortem SUDEP cohort, and genetic testing is not available to correlate with pathology findings.
In summary, neuronal labeling for pS6 in the STG correlated with seizure activity in the period prior to death and was enhanced in SUDEP, highlighting this as a potential critical region. Further clinical and functional studies, including the role of this region within the CAN, is therefore warranted in SUDEP.
AUTHOR CONTRIBUTION
SP and HEH were involved in tissue sampling protocols and preparation. SP, FC, and IS carried out immunostaining and quantitation. YL developed image analysis protocols. SP and MT were involved in data analysis, and all the authors were involved in manuscript preparation.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Appendix S1
Table S1
Table S2
ACKNOWLEDGMENTS
University College London (UCL) is part of the Center for SUDEP Research (CSR) supported through a National Institute of Neurological Disorders and Stroke of the National Institutes of Health (NIH; Award Numbers neuropathology of SUDEP: 5U01NS090415 and SUDEP admin core grant: U01‐NS090405). All tissues are provided by the Epilepsy Society Brain and Tissue Bank at UCL and MRC tissue bank in Edinburgh, UK. The Epilepsy Society through the Katy Baggott Foundation supports the UCL Epilepsy Society Brain and Tissue Bank. Epilepsy Research UK supports the Corsellis Epilpesy collection and HEH. This work was undertaken at University College Hospital London/UCL, which received a proportion of funding from the Department of Health's National Institute of Health Research Biomedical Research Centres' funding scheme.
Patodia S, Lim YM, Chung F, Stylianou I, El Hachami H, Thom M. Cortical neuronal hypertrophy and mTOR pathway activation in CAN regions in SUDEP . Epilepsia. 2022;63:2427–2438. 10.1111/epi.17335
REFERENCES
- 1. Beissner F, Meissner K, Bar KJ, Napadow V. The autonomic brain: an activation likelihood estimation meta‐analysis for central processing of autonomic function. J Neurosci. 2013;33(25):10503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Valenza G, Sclocco R, Duggento A, Passamonti L, Napadow V, Barbieri R, et al. The central autonomic network at rest: uncovering functional MRI correlates of time‐varying autonomic outflow. Neuroimage. 2019;197:383–90. [DOI] [PubMed] [Google Scholar]
- 3. Valenza G, Passamonti L, Duggento A, Toschi N, Barbieri R. Uncovering complex central autonomic networks at rest: a functional magnetic resonance imaging study on complex cardiovascular oscillations. J R Soc Interface. 2020;17(164):20190878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reisert M, Weiller C, Hosp JA. Displaying the autonomic processing network in humans ‐ a global tractography approach. Neuroimage. 2021;231:117852. [DOI] [PubMed] [Google Scholar]
- 5. Sanchez‐Larsen A, Principe A, Ley M, Navarro‐Cuartero J, Rocamora R. Characterization of the insular role in cardiac function through intracranial electrical stimulation of the human insula. Ann Neurol. 2021;89(6):1172–80. [DOI] [PubMed] [Google Scholar]
- 6. Allen LA, Harper RM, Lhatoo S, Lemieux L, Diehl B. Neuroimaging of sudden unexpected death in epilepsy (SUDEP): insights from structural and resting‐state functional MRI studies. Front Neurol. 2019;10:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Allen LA, Harper RM, Vos SB, Scott CA, Lacuey N, Vilella L, et al. Peri‐ictal hypoxia is related to extent of regional brain volume loss accompanying generalized tonic‐clonic seizures. Epilepsia. 2020;61:1570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ogren JA, Tripathi R, Macey PM, Kumar R, Stern JM, Eliashiv DS, et al. Regional cortical thickness changes accompanying generalized tonic‐clonic seizures. Neuroimage Clin. 2018;20:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wandschneider B, Koepp M, Scott C, Micallef C, Balestrini S, Sisodiya SM, et al. Structural imaging biomarkers of sudden unexpected death in epilepsy. Brain. 2015;138(Pt 10):2907–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. La A, Rm H, M G, R K, Ja O, Sb V, et al. Altered brain connectivity in sudden unexpected death in epilepsy (SUDEP) revealed using resting‐state fMRI. Neuroimage Clin. 2019;24:102060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allen LA, Harper RM, Kumar R, Guye M, Ogren JA, Lhatoo SD, et al. Dysfunctional brain networking among autonomic regulatory structures in temporal lobe epilepsy patients at high risk of sudden unexpected death in epilepsy. Front Neurol. 2017;8:544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang Y, Chen Q, Yu X, Xia W, Luo C, Huang XQ, et al. A resting‐state functional connectivity study in patients at high risk for sudden unexpected death in epilepsy. Epilepsy Behav. 2014;41:33–8. [DOI] [PubMed] [Google Scholar]
- 13. Lacuey N, Zonjy B, Londono L, Lhatoo SD. Amygdala and hippocampus are symptomatogenic zones for central apneic seizures. Neurology. 2017;88(7):701–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lacuey N, Hampson JP, Theeranaew W, Zonjy B, Vithala A, Hupp NJ, et al. Cortical structures associated with human blood pressure control. JAMA Neurol. 2018;75(2):194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rhone AE, Kovach CK, Harmata GIS, Sullivan AW, Tranel D, Ciliberto MA, et al. A human amygdala site that inhibits respiration and elicits apnea in pediatric epilepsy. JCI Insight. 2020;5(6):e134852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Somani A, el‐Hachami H, Patodia S, Sisodiya S, Thom M. Regional microglial populations in central autonomic brain regions in SUDEP. Epilepsia. 2021;62(6):1318–28. [DOI] [PubMed] [Google Scholar]
- 17. Ochoa‐Urrea M, Lacuey N, Vilella L, Zhu L, Jamal‐Omidi S, Rani MRS, et al. Seizure clusters, seizure severity markers, and SUDEP risk. Front Neurol. 2021;12:643916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12(10):966–77. [DOI] [PubMed] [Google Scholar]
- 19. Somani A, Perry C, Patodia S, Michalak Z, Ellis M, Sisodiya SM, et al. Neuropeptide depletion in the amygdala in sudden unexpected death in epilepsy: a postmortem study. Epilepsia. 2020;61(2):310–8. [DOI] [PubMed] [Google Scholar]
- 20. Schlabitz S, Monni L, Ragot A, Dipper‐Wawra M, Onken J, Holtkamp M, et al. Spatiotemporal correlation of epileptiform activity and gene expression in vitro. Front Mol Neurosci. 2021;14:643763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sha LZ, Xing XL, Zhang D, Yao Y, Dou WC, Jin LR, et al. Mapping the spatio‐temporal pattern of the mammalian target of rapamycin (mTOR) activation in temporal lobe epilepsy. PLoS One. 2012;7(6):e39152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marsan E, Baulac S. Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol Appl Neurobiol. 2018;44(1):6–17. [DOI] [PubMed] [Google Scholar]
- 23. Nashef L, So EL, Ryvlin P, Tomson T. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. 2012;53(2):227–33. [DOI] [PubMed] [Google Scholar]
- 24. Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia. 2011;52(1):158–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Allen LA, Vos SB, Kumar R, Ogren JA, Harper RK, Winston GP, et al. Cerebellar, limbic, and midbrain volume alterations in sudden unexpected death in epilepsy. Epilepsia. 2019;60(4):718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whelan CD, Altmann A, Botía JA, Jahanshad N, Hibar DP, Absil J, et al. Structural brain abnormalities in the common epilepsies assessed in a worldwide ENIGMA study. Brain. 2018;141(2):391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lacuey N, Garg V, Bangert B, Hampson JP, Miller J, Lhatoo S. Insular resection may lead to autonomic function changes. Epilepsy Behav. 2019;97:260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lacuey N, Zonjy B, Theerannaew W, Loparo KA, Tatsuoka C, Sahadevan J, et al. Left‐insular damage, autonomic instability, and sudden unexpected death in epilepsy. Epilepsy Behav. 2016;55:170–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lagarde S, Singh R, Bartolomei F, Guedj E. Insular interictal positron emission tomography hypometabolism in patients with ictal asystole. Epilepsia. 2021;62(8):e117–22. [DOI] [PubMed] [Google Scholar]
- 30. Macey PM, Woo MA, Macey KE, Keens TG, Saeed MM, Alger JR, et al. Hypoxia reveals posterior thalamic, cerebellar, midbrain, and limbic deficits in congenital central hypoventilation syndrome. J Appl Physiol (1985). 2005;98(3):958–69. [DOI] [PubMed] [Google Scholar]
- 31. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Palomero‐Gallagher N, Mohlberg H, Zilles K, Vogt B. Cytology and receptor architecture of human anterior cingulate cortex. J Comp Neurol. 2008;508(6):906–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michalak Z, Obari D, Ellis M, Thom M, Sisodiya SM. Neuropathology of SUDEP: role of inflammation, blood‐brain barrier impairment, and hypoxia. Neurology. 2017;88(6):551–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patodia S, Somani A, Thom M. Review: neuropathology findings in autonomic brain regions in SUDEP and future research directions. Auton Neurosci. 2021;235:102862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim JK, Lee JH. Mechanistic target of rapamycin pathway in epileptic disorders. J Korean Neurosurg Soc. 2019;62(3):272–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361–71. [DOI] [PubMed] [Google Scholar]
- 37. Talos DM, Jacobs LM, Gourmaud S, Coto CA, Sun H, Lim KC, et al. Mechanistic target of rapamycin complex 1 and 2 in human temporal lobe epilepsy. Ann Neurol. 2018;83(2):311–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu J, Reeves C, Michalak Z, Coppola A, Diehl B, Sisodiya SM, et al. Evidence for mTOR pathway activation in a spectrum of epilepsy‐associated pathologies. Acta Neuropathol Commun. 2014;2:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rossini L, Villani F, Granata T, Tassi L, Tringali G, Cardinale F, et al. FCD Type II and mTOR pathway: evidence for different mechanisms involved in the pathogenesis of dysmorphic neurons. Epilepsy Res. 2017;129:146–56. [DOI] [PubMed] [Google Scholar]
- 40. Switon K, Kotulska K, Janusz‐Kaminska A, Zmorzynska J, Jaworski J. Molecular neurobiology of mTOR. Neuroscience. 2017;341:112–53. [DOI] [PubMed] [Google Scholar]
- 41. Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414(1):57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crino PB. Mechanistic target of rapamycin (mTOR) signaling in status epilepticus. Epilepsy Behav. 2019;101(Pt B):106550. [DOI] [PubMed] [Google Scholar]
- 43. Baldassari S, Picard F, Verbeek NE, van Kempen M, Brilstra EH, Lesca G, et al. The landscape of epilepsy‐related GATOR1 variants. Genet Med. 2019;21(2):398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sahly AN, Shevell M, Sadleir LG, Myers KA. SUDEP risk and autonomic dysfunction in genetic epilepsies. Auton Neurosci. 2021;237:102907. [DOI] [PubMed] [Google Scholar]
- 45. Bacq A, Roussel D, Bonduelle T, Zagaglia S, Maletic M, Ribierre T, et al. Cardiac investigations in sudden unexpected death in DEPDC5‐related epilepsy. Ann Neurol. 2021;91(1):101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ryufuku M, Toyoshima Y, Kitaura H, Zheng Y, Fu YJ, Miyahara H, et al. Hypertrophy of hippocampal end folium neurons in patients with mesial temporal lobe epilepsy. Neuropathology. 2011;31(5):476–85. [DOI] [PubMed] [Google Scholar]
- 47. Bothwell S, Meredith GE, Phillips J, Staunton H, Doherty C, Grigorenko E, et al. Neuronal hypertrophy in the neocortex of patients with temporal lobe epilepsy. J Neurosci. 2001;21(13):4789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Galovic M, van Dooren VQH, Postma TS, Vos SB, Caciagli L, Borzì G, et al. Progressive cortical thinning in patients with focal epilepsy. JAMA Neurol. 2019;76:1230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Palomero‐Gallagher N, Eickhoff SB, Hoffstaedter F, Schleicher A, Mohlberg H, Vogt BA, et al. Functional organization of human subgenual cortical areas: relationship between architectonical segregation and connectional heterogeneity. Neuroimage. 2015;115:177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lane RD, Weidenbacher H, Smith R, Fort C, Thayer JF, Allen JJB. Subgenual anterior cingulate cortex activity covariation with cardiac vagal control is altered in depression. J Affect Disord. 2013;150(2):565–70. [DOI] [PubMed] [Google Scholar]
- 51. Oppenheimer S, Cechetto D. The insular cortex and the regulation of cardiac function. Compr Physiol. 2016;6(2):1081–133. [DOI] [PubMed] [Google Scholar]
- 52. Kimmerly DS. A review of human neuroimaging investigations involved with central autonomic regulation of baroreflex‐mediated cardiovascular control. Auton Neurosci. 2017;207:10–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Appendix S1
Table S1
Table S2