

Abstract
The production of L‐lactide from L‐lactic acid involves a substantial formation of meso‐lactide as an impurity, and, upon polymerization with the industrial catalyst tin octanoate, results in poly(L‐lactic acid) of reduced crystallinity due to stereoerrors randomly distributed along the polymer chains. We describe a new approach wherein, instead of avoiding stereoerrors by removing the meso‐lactide prior to polymerization, the stereoerrors in the polymer are tolerated, by crowding them in a stereogradient copolymer. A zirconium complex of an amine tris(phenolate) ligand is found to exhibit very high syndioselectivity in the ring opening polymerization catalysis of meso‐lactide at room temperature, and gives rise to stereogradient copolymers in the polymerization of mixtures of meso‐lactide/L‐lactide in the melt at 180 °C. Relative to the stereo‐random copolymers obtained with tin octanoate, the stereogradient copolymers exhibit enhanced crystallinities manifested in lower solubilities and higher melting temperatures and enthalpies.
Keywords: Copolymerization, Lactide, Ring-Opening Polymerization, Stereogradient Copolymers, Tripodal Ligands
Polymerization of meso‐lactide/L‐lactide mixtures was carried out at 180 °C by employing a highly syndioselective catalyst based on a zirconium complex of an amine tris(phenolate) ligand. Stereogradient copolymers that exhibit enhanced crystallinities relative to the stereo‐random copolymers were obtained with the industrial catalyst.

Introduction
Poly(lactic acid) (PLA) is derived from annually renewable resources, and may be composted or recycled post‐consumption. It has found both commodity and biomedical applications, and has the potential to replace traditional plastics like polyethylene, polystyrene and polyethylene terephthalate.[ 1 , 2 , 3 , 4 ] The industrial production of PLA relies on the ring opening polymerization (ROP) of its dilactone—lactide, in the presence of a catalyst—tin octanoate. The chirality of lactic acid gives rise to three lactide stereoisomers: L‐(S,S), D‐(R,R), or meso‐(S,R) lactide. The physical and chemical properties of the PLA depend on its stereoregularity, which is an outcome of the monomer constitution and the stereoselectivity of the ROP catalyst employed.[ 5 , 6 ] Of all forms of PLA, the most valuable one is poly(L‐lactic acid) (PLLA) formed by the ring opening polymerization of L‐lactide. Pure L‐lactide leads to a semicrystalline PLLA having a melting temperature of 180 °C. L‐lactide is industrially produced by the high‐temperature “two‐step” process wherein L‐lactic acid is first condensed to an oligo(L‐lactic acid) and then undergoes a catalytic depolymerization. [7] This process is accompanied by epimerization which leads to substantial formation of stereoisomers and in particular meso‐lactide, estimated to form in as much as 5–10 % depending on the specific process. [8] Tin octanoate shows very little discrimination between the different lactide stereoisomers, and, therefore, the PLLA formed from polymerization of stereo‐impure L‐lactide is essentially a stereo‐random copolymer in which the stereoerrors are statistically distributed.[ 1 , 9 , 10 , 11 ] An approximate 5 °C decrease in the melting temperature of the polymer for every 1 % of meso‐lactide in the L‐lactide stream, and complete loss of crystallinity at ca. 10–12 % of meso‐lactide were reported.[ 12 , 13 ] Therefore, to manufacture crystalline PLLA, the current production processes require an additional step of meso‐lactide removal, achieved e.g., by vacuum distillation. The recovered meso‐lactide is either discarded, returned to the L‐lactide stream, or transformed into racemic lactide and reacted further.[ 13 , 14 ] In recent years, vast efforts have been invested in developing of alternative methods for production of L‐lactide wherein the meso‐lactide contamination would be minimized, including “one‐step” processes.[ 8 , 15 , 16 , 17 ] To our knowledge, though, these processes are yet to be implemented on an industrial scale. [18] Herein we describe a new approach for attaining highly crystalline PLLA which relies on accommodating rather than removing lactide stereoisomers. We show that by employing a catalyst which is based on a zirconium complex of an amine tris(phenolate) ligand which features a strong preference for polymerization of meso‐lactide in meso‐lactide/L‐lactide monomer mixtures, stereogradient rather than stereo‐random PLA‐copolymers are formed. These novel stereogradient copolymers exhibit enhanced crystallinities relative to the stereo‐random copolymers, which are manifested in their reduced solubilities and increased melting temperatures and enthalpies. Most importantly, these polymers are produced under industrially relevant conditions, namely, in the melt at 180 °C.
Results and Discussion
We hypothesized that catalysts that exhibit high syndioselectivities in polymerization of meso‐lactide following the heteroselective chain‐end stereocontrol mechanism, [19] should also consume meso‐lactide preferentially in polymerizations of meso‐lactide/L‐lactide mixtures. As outlined in Figure 1, such catalysts prefer to open a meso‐lactide ring next to a stereogenic center which has the opposite configuration of the last inserted one to form a racemo‐dyad (r‐dyad—a pair of adjacent stereogenic centers of opposite configurations), [19] and consequently regenerate the stereogenicity at the metal center, favoring another insertion of meso‐lactide (Path a). [20] An unfavored insertion of L‐lactide would retain the stereogenicity at the metal center which would still favor the insertion of meso‐lactide (Path b). An opposite insertion of meso‐lactide may correct itself by insertion of L‐lactide (Path c), and altogether this would generate a self‐repairing mechanism for enhanced meso‐lactide consumption.
Figure 1.
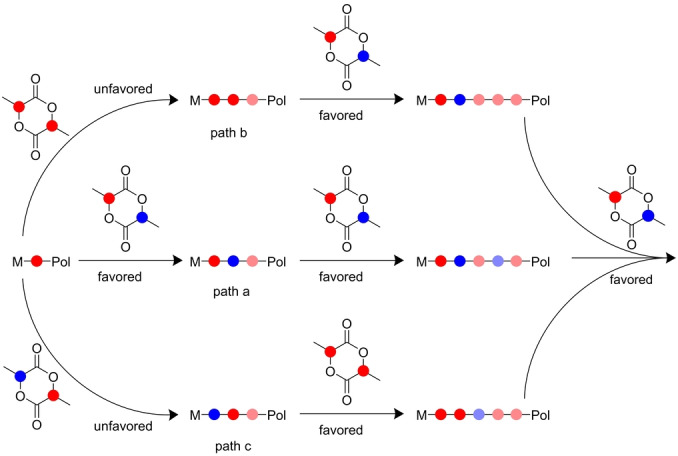
Self‐repairing mechanism for favored meso‐lactide consumption. Stereogenic centers are marked in blue and red. Stereogenic centers of previous insertions are faded for clarity. Path a: A sequence of favored insertions of meso‐lactide. Path b: An unfavored insertion of L‐lactide retains the stereogenicity of the last inserted unit favoring the insertion of meso‐lactide. Path c: An unfavored opposite insertion of meso‐lactide favors the insertion of the dominant L‐lactide stereoisomer favoring the insertion of meso‐lactide.
The resulting polymers would best be defined as stereogradient copolymers, [21] wherein the segment of the chains proximal to the initiation point would be enriched in stereoerrors, while the segment(s) remote from the initiation point would be depleted of stereoerrors, as schematically shown in Figure 2.
Figure 2.

Schematic representation of polymerization of a meso‐lactide/L‐lactide mixture employing different catalysts and initiators. a) Employing a nonselective catalyst and a single‐headed initiator giving a stereo‐random copolymer. b) Employing a syndioselective catalyst and a single‐headed initiator giving a stereogradient copolymer featuring a single segment depleted of stereo‐errors. c) Employing a syndioselective catalyst and a double‐headed initiator giving a stereogradient copolymer featuring two segments depleted of stereo‐errors.
The crystallinity of stereoregular polymers is mainly determined by the length of stereoerror‐free segments.[ 13 , 22 ] We hypothesized that, for a given proportion of stereoerrors in either stereogradient copolymers or stereo‐random copolymers, the extended lengths of stereoerror depleted segments in the stereogradient copolymers would lead to their enhanced crystallinities relative to the stereo‐random copolymers. For such a process to be feasible, a catalyst that exhibits an appreciable degree of syndioselectivity at the high temperatures characteristic of the industrial processes should be devised.
Group 4 metal complexes of amine tris(phenolate) ligands were previously reported to catalyze the ring opening polymerization of lactide to give PLA. [23] These complexes were also found to be stereoselective.[ 24 , 25 ] In particular, complex 1 (Figure 3), including tert‐butyl substituents on the phenolate rings was reported to show moderate syndioselectivity in the ROP of meso‐lactide. [26] Very recently, we described a zirconium complex of an amine tris(phenolate) ligand that included ortho‐mesityl phenolate substituents (complex 2). [27] Complex 2 exhibited remarkable reactivity in the ROP of L‐lactide at 180 °C when employed at the low ppm range, and gave rise to thermally stable poly(L‐lactic acid).
Figure 3.
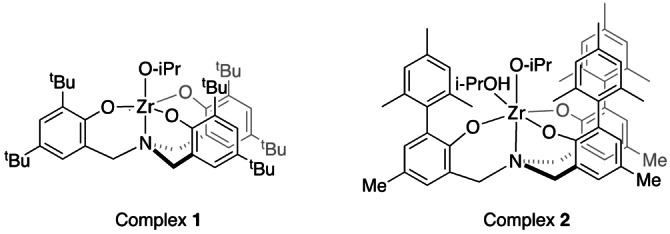
Zirconium complexes of amine tris(phenolate) ligands described in this work.
To estimate whether this catalyst would be suitable for the production of stereogradient copolymers from meso‐lactide/L‐lactide monomer mixtures we conducted several preliminary experiments at room temperature. We first tested the stereoselectivity of this catalyst by polymerization of meso‐lactide in dichloromethane solution.[ 26 , 28 , 29 ] We found that, in the presence of 10 equiv of 2‐propanol as initiator, 1000 equiv of meso‐lactide were fully converted to PLA within 10 min, signifying a very high catalytic activity towards meso‐lactide. The obtained PLA had a number average molecular weight of M n=12 100 consistent with the theoretical value of 12 000, and a narrow molecular weight distribution of Ð=1.11 supporting a living/immortal polymerization (Table S‐1). [30] Most importantly, the polymer featured a very high degree of syndiotacticity of P s=0.93 (see the Supporting Information), according to the ratio of tetrads in the homodecoupled 1H‐NMR spectrum, matching the highest degree of syndiotacticity attained with catalysts which obey the chain‐end stereocontrol mechanism (Figure 4, left, Figure S‐1).[ 26 , 29 ] The distribution of tetrads in the 13C‐NMR spectrum, and in particular the very low abundance of the mrm tetrad support the chain‐end stereocontrol mechanism as being responsible for this syndiotacticity (Figure S‐2, left). [26] There are two precedents for slightly higher syndiotacticities in polymerization of meso‐lactide, however those were attained with very sluggish aluminum‐based catalysts that obeyed different stereocontrol mechanisms.[ 20 , 31 ] Differential scanning calorimetry revealed multiple first heating melting transitions, the highest of which at 132.5 °C, with ΔH melting of 28.2 J g−1 (Figure S‐3), consistent with the high syndiotacticity. [20]
Figure 4.
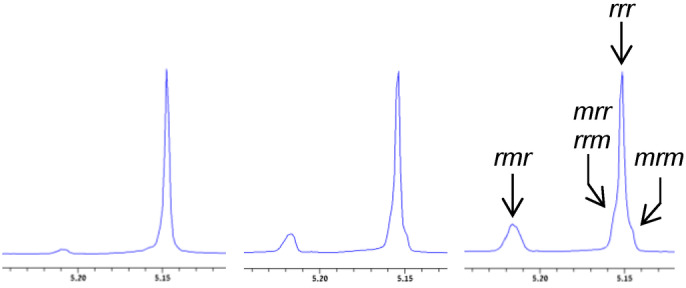
The methine region of the HD 1H‐NMR spectra of PLA samples prepared from meso‐lactide under different conditions and showing different degrees of syndiotacticity. Tetrad assignments appear on the spectrum of the least stereoregular polymer. Left: With complex 2 and 2‐propanol at room temperature, P s=0.93. Centre: With complex 2 and benzyl alcohol at 180 °C, P s=0.76. Right: With tin octanoate and benzyl alcohol at 180 °C, P s=0.58.
In comparison, complex 1 was reported to fully polymerize 100 equiv of meso‐lactide within 48 h, to give syndiotactic PLA with P s=0.85. [26] Namely, complex 2 is estimated to be at least three orders of magnitude more active than complex 1 towards meso‐lactide while exhibiting significantly higher syndioselectivity. Next, we followed the consumption of the two monomers in d8‐toluene solutions by 1H‐NMR spectrometry (Figure 5) and found first order kinetics with k app=4.8 h−1 for meso‐lactide and k app=0.49 h−1 for L‐lactide (0.32 mM catalyst; 400 : 1 lactide/catalyst ratio), namely, a ratio of rate constants of approximately 10 : 1 signifying a very strong bias towards meso‐lactide.
Figure 5.
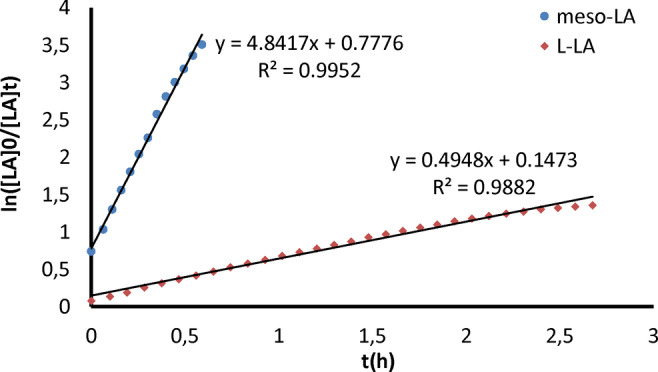
Kinetic plots for the polymerization of meso‐lactide and L‐lactide by complex 2 attained from 1H‐NMR measurements. 0.32 mM of complex 2 and 125 mM of lactide in toluene‐d8 at 25 °C relatively to 1,4‐bis(trimethylsilyl)benzene internal standard (4.5 mM).
Finally, we polymerized a mixture of meso‐lactide/L‐lactide of 10 : 90 ratio (complex 2/2‐propanol/total lactide mixture of 1 : 20 : 2000 molar ratios in dichloromethane with aliquots taken every 5 min, Table S‐2, Figures S4–S10). We found that the meso‐lactide was consumed preferentially, and that at 24 % monomer conversion (attained after 20 min), the unreacted monomer consisted of 1 % of meso‐lactide and 99 % of L‐lactide. Namely, while more than 90 % of the meso‐lactide have reacted, only 16 % of the L‐lactide have reacted. After 24 h, 66 % of the monomer mixture was converted to PLA, and no meso‐lactide could be detected. Thus, the initial segment (0‐to‐24 %) of the polymer chains is composed of ca. 38 % of meso‐lactide and 62 % of L‐lactide, whereas the next segment (24 %‐to‐66 %) is composed of ca. 2 % of meso‐lactide and 98 % of L‐lactide, strongly supporting the proposed stereogradient structure of the formed PLA copolymer.
Of course, the selectivity is expected to decrease as the temperature is raised. Polymerization of neat meso‐lactide at 180 °C in a ratio of 2/benzyl alcohol (BnOH)/meso‐lactide of 1 : 10 : 10000 resulted in 95 % monomer conversion after 5 min to give a polymer of M n=71800 and Ð=1.31. Notably, a syndiotacticity degree of P s=0.76 was found, which, albeit lower than that obtained at room temperature, indicates that the catalyst retains some of its syndioselectivity even under these harsh conditions. A parallel polymerization with tin octanoate resulted in 70 % monomer conversion to give a polymer of M n=51200 and Ð=1.41 having a substantially lower degree of syndiotacticity of P s=0.58—signifying an almost atactic polymer (Table S‐3, Figures 4, S11–S14). The following polymerization runs were conducted on a 10 gram scale in a pressure glass vessel with magnetic stirring employing a ratio of catalyst/initiator/monomer of 1 : 59 : 59000 (Table S‐4). We first conducted a short polymerization run of 10 min of a 10 : 90 mixture of meso‐lactide/L‐lactide with complex 2 as catalyst and benzyl alcohol as initiator. 1H‐NMR analysis of the polymerization mixture revealed that 76 % of the total monomer was converted to polymer. The remaining monomer was found to consist of >99 % L‐lactide, signifying that the meso‐lactide was essentially completely consumed. Namely, this catalyst is selective towards meso‐lactide even under the harsh melt conditions employed. Performing a similar polymerization run with tin‐octanoate as catalyst resulted in a very similar conversion of 74 %. However, the remaining monomer comprised both meso‐lactide and L‐lactide, in a proportion similar to that of the original polymerization mixture, signifying that tin octanoate does not exhibit noticeable preference for meso‐lactide over L‐lactide (compare Figures S‐15 and S‐25). These polymerization runs are consistent with the formation of a stereogradient copolymer and an essentially stereo‐random copolymer, respectively.
To gain insight on the properties of the stereogradient copolymers as a function of the relative amounts of stereoerrors, we ran a series of melt polymerizations under the conditions described above (see the Supporting Information). The meso‐lactide/L‐lactide monomer compositions included 10 : 90, 5 : 95, 2.5 : 97.5, 1 : 99 ratios, as well as pure L‐lactide. These ratios are proposed to represent typical compositions of lactide stereoisomers generated by various two‐step as well as one‐step lactide production processes. Three different catalyst/initiator combinations were tested (Figure 2): 1) Complex 2 and benzyl alcohol (a single‐headed initiator) expected to produce stereogradient copolymers wherein the stereoerrors would concentrate in the terminus of the chain proximal to the initiator; 2) Complex 2 and 1,4‐benzenedimethanol (a double‐headed initiator) expected to produce stereogradient copolymers wherein the stereoerrors would concentrate in the central part of the chains proximal to the initiator; and 3) Tin octanoate and benzyl alcohol expected to produce essentially stereo‐random copolymers. The polymerizations are summarized in Table S‐4. We observed that the viscosity of the polymerization mixtures increased with time, and after several minutes (2–3 minutes for complex 2, and 6–7 minutes for tin octanoate) the stir‐bar ceased to rotate. The polymerizations were quenched after either 20 or 30 minutes, attempting to attain high conversions while minimizing side‐reactions. According to 1H‐NMR analysis of the polymerization mixtures, high conversions of 90–94 % were obtained with catalyst 2 and slightly lower conversions of 81–88 % were obtained with tin octanoate, signifying a higher activity of catalyst 2. 1H‐NMR analysis of the crude polymerization mixtures revealed that, as expected, for all the polymerizations conducted with catalyst 2, no meso‐lactide could be detected in the remaining monomer (Figures S‐17–S‐24). In contrast, for the 10 : 90 and 5 : 95 monomer compositions polymerized with tin octanoate, appreciable amounts of meso‐lactide were detected in the remaining monomer albeit the relatively high conversions (Figures S‐27 and S‐29). When taking in consideration that, in the 10 minute polymerization run of the 10 : 90 meso‐lactide/L‐lactide monomer mixture with complex 2, essentially all meso‐lactide has been embedded in the polymer chains after 76 % conversion, then in the 30 minute polymerization run the 76 %‐to‐90 % segment of the polymer chains is expected to be stereoerror‐free (assuming that trans‐esterification does not play a significant role). This is consistent with a stereoblock microstructure, and is proposed to account for the differences in the thermal properties between these two samples (see below). For the monomer compositions lower in meso‐lactide, the lengths of the stereoerror free segments attained with complex 2 are expected to be even longer. A similar preference for meso‐lactide was found when benzenedimethanol was employed as initiator together with complex 2 (Figures S‐35–S‐42), supporting the formation of stereogradient copolymers.
Removal of the remaining monomer gave the polymers as white powders which appeared similar to each other. All polymers were found to be of high molecular weights albeit somewhat lower than the theoretical values (Table S‐4). Narrower molecular weight distributions were found for the polymers derived from complex 2 relative to those derived from tin octanoate consistent with a better behaved polymerization by the former. The 1H‐NMR and 13C‐NMR spectra were found to reflect the total number of stereoerrors in the obtained polymers, but could not differentiate between the polymer microstructures. Yet, the difference between the PLA samples became apparent when their properties were assessed. A simple measure of the crystallinity of PLA is its solubility in THF.[ 4 , 32 ] As expected, for all catalyst/initiator combinations, a decrease in solubility was found as the relative amount of meso‐lactide in the monomer mixture decreased (Table S‐4). With the exception of the PLLA samples prepared from pure L‐lactide, the samples prepared with the stereoselective catalyst 2 and either benzyl alcohol or 1,4‐benzenedimethanol were consistently less soluble than the samples prepared with tin octanoate, supporting the higher crystallinity of the stereogradient copolymers relative to the stereo‐random copolymers. The differences in solubility are particularly notable for the high meso‐lactide compositions. For example, the polymers prepared with tin octanoate and either 10 % or 5 % of meso‐lactide showed a solubility of ≥100 mg mL−1, whereas those prepared with complex 2 and benzyl alcohol showed a solubility of 70 and 44 mg mL−1, respectively. Since the conversions are lower than 100 % for all monomer compositions, the total number of stereoerrors in the stereogradient copolymers prepared with complex 2, for which all meso‐lactide has been embedded, are higher than those found in the stereo‐random copolymers prepared with tin octanoate, for which not all meso‐lactide has been embedded. The higher crystallinity of the stereogradient copolymers relative to the stereo‐random copolymers prepared from the same monomer mixtures, is therefore even more impressive. As Table S‐4 shows, for all monomer compositions polymerized with complex 2, slightly lower solubilities were found for the polymers prepared with benzyl alcohol relative to 1,4‐benzenedimethanol. This may imply higher crystallinities for stereogradient copolymers which include a single long stereoerror‐free segment rather than two shorter ones (Figure 2).
Differential scanning calorimetry (DSC) revealed several trends that were distinctly different for the polymers synthesized using complex 2 and tin octanoate (Table S‐5, Figures S‐61–S‐77). For the analysis, the melting‐crystallization behavior of the polymer samples with identical thermal history was comparatively examined. DSC thermograms, recorded during the second heating at 10 °C per minute temperature ramps after erasing the preceding thermal history and controlled cooling, were characterized in terms of temperatures and energies of thermal effects associated with melting and reorganization of the crystalline structures. As expected, all polymer samples prepared from monomer mixtures richer in L‐lactide showed higher melting temperatures and enthalpies. The polymers synthesized using complex 2 and benzyl alcohol showed slightly higher melting temperatures in comparison to the polymers synthesized using complex 2 and 1,4‐benzenedimethanol indicating a better organized crystalline structure of the stereogradient copolymers having a single stereoerror‐free segment. Most significantly, for all monomer compositions, the polymers synthesized using complex 2 showed systematically higher melting temperatures and enthalpies in comparison to their tin octanoate synthesized counterparts. These differences were more pronounced for the polymers synthesized from the monomer mixtures richer in meso‐lactide. For example, for the polymers synthesized from the monomer mixture containing 10 % meso‐lactide, the sample synthesized using tin octanoate showed no melting endotherm, whereas the sample prepared using complex 2 and benzyl alcohol showed a melting peak temperature of 164.3 °C and a melting enthalpy of 5.9 J g−1 (Figures S‐67 and S‐62). Similarly, for the monomer mixture containing 5 % meso‐lactide, the polymer synthesized using tin octanoate showed a broad low intensity melting endotherm with peak temperatures and melting enthalpies of 161.3 °C/1.6 J g−1, respectively, while the polymer prepared with complex 2 demonstrated a prominent melting endotherm with two peaks (167.1 and 172.4 °C) and a melting enthalpy of 28.7 J g−1 (Figure 6, Figures S‐68 and S‐63).
Figure 6.
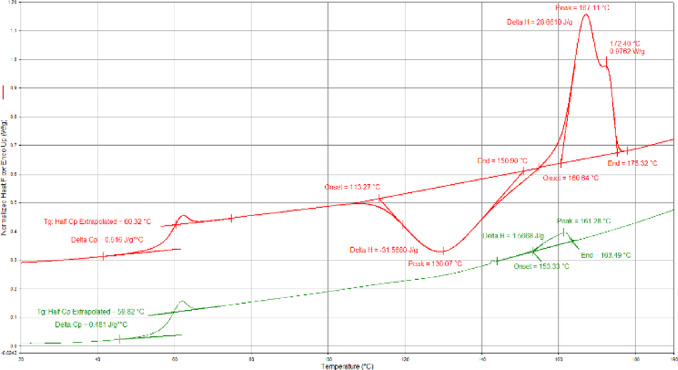
Differential scanning calorimetry second heating run of PLA samples obtained from polymerization of 5 % meso‐lactide/95 % L‐lactide at 180 °C. Top: Prepared with complex 2 and benzyl alcohol. Bottom: Prepared with tin octanoate and benzyl alcohol.
It is also noteworthy that for the two polymer samples attained from the monomer composition containing 10 % meso‐lactide employing complex 2 and benzyl alcohol, the sample wherein higher conversion was reached also showed a higher melting temperature and enthalpy (compare entries 11 and 12 in Table S‐5 and Figures S‐61 and S‐62). We attribute this difference to elongation of the stereoerror‐free segment as the polymerization proceeds in these stereogradient copolymers.
The differences in melting‐crystallization behavior of the two types of the polymers, reflecting their important structural difference, are clearly seen in Figure 7 presenting the melting temperatures as a function of the meso‐lactide comonomer content in the monomer mixture. A monotonous decrease of the melting temperatures observed for the polymers derived from tin octanoate is characteristic of copolymers with random insertion of the comonomer units. In contrast, the essential independence of the melting temperatures at low comonomer contents followed by a gentle decrease at the higher contents, observed for the polymers produced using complex 2, are more in line with a block‐type copolymers structure.[ 33 , 34 , 35 ] Comparative analysis of double‐peak endotherms observed in some thermograms and associated with melting‐reorganization of α′ and α PLA crystalline phases (Figure S‐80) further implies that the polymers derived from complex 2 have significantly higher tendency for forming stable and better ordered α‐crystallites than their tin octanoate derived counterparts. [12]
Figure 7.
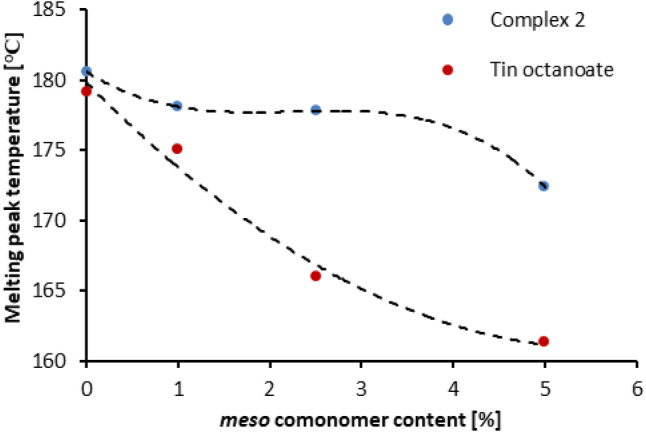
Melting temperatures of PLA as a function of meso‐lactide content in the monomer stream. Blue dots: PLA samples prepared with complex 2 and benzyl alcohol. Red dots: PLA samples prepared with tin octanoate and benzyl alcohol. Dashed lines represent fitting of the result trends using second (blue) and third (red) order polynomials.
Finally, we attempted to produce stereogradient PLA under more demanding conditions. First, we employed a lower loading of catalyst 2 for a prolonged period of time. Thus, melt polymerization of a meso‐lactide/L‐lactide mixture (5 : 95) employing complex 2/benzyl alcohol/monomer ratios of 1 : 214 : 214000 (4.7 ppm of catalyst) for 60 min led to 94 % monomer conversion in which all meso‐lactide was consumed, and the PLA obtained was of high molecular weight and narrow dispersity of 95700/1.18 (Figures S‐47, S‐48). Notably, the THF solubility (36 mg mL−1) and thermal characteristics (T m=166.6/173.6 °C, ΔH m=31.7 J g−1, Figure S‐77) are consistent with those of the stereogradient copolymers described above and attained with higher catalyst loading. Then, we attempted to produce stereogradient PLA from simulated “industrial‐grade” lactide. Monomer mixtures composed of meso‐lactide/technical L‐lactide (see the Supporting Information) in the ratios of 10 : 90 and 5 : 95 were polymerized at 180 °C for 60 min to promote high conversions for this slower‐reacting technical monomer, [27] employing complex 2/benzyl alcohol/monomer ratios of 1 : 59 : 59000. In both cases high conversions of 94 % were found, and the remaining monomer contained no meso‐lactide, consistent with formation of stereogradient PLA copolymers (Figures S‐49–S‐52). In comparison to the samples prepared from the purified monomers, the THF‐solubilities of the current samples (84 and 55 mg mL−1, respectively) were slightly higher (Figures S‐53–S‐60), possibly because of their lower molecular weights, while their heats of melting (6.4 and 31.0 J g−1, respectively) were slightly higher consistent with the higher conversions (Figures S‐78 and S‐79). Diffusion NMR experiments (Figures S‐81 and S‐82), albeit being of relatively high errors (in particular for the 5 : 95 polymer sample), pointed to identical diffusion coefficients for the stereo‐error containing and the stereo‐error depleted portions of the polymer samples, giving further support for a stereogradient copolymer microstructure rather than a mixture of polymers of different degrees of stereoregularities. Namely, stereogradient copolymers are attained even when employing very low catalyst loadings or technical‐grade lactide and are not perturbed markedly by side reactions even after extended reaction times.
Conclusion
We describe stereogradient copolymers derived from mixtures of meso‐lactide and L‐lactide. These stereogradient copolymers were found to exhibit enhanced crystallinities relative to the corresponding stereo‐random copolymers derived from the same monomer mixtures and employing the non‐stereoselective tin octanoate. The formation of such stereogradient copolymers requires catalysts that exhibit appreciable stereoselectivities even under the harsh industrial melt conditions. Currently, complex 2 which features very high syndioselectivity in meso‐lactide polymerization combined with very high activity and turnover numbers in the melt, stands out as a unique catalyst suitable for attaining such copolymers. However, we expect that further research would yield additional effective catalysts. By employing such catalysts, polymers of desired crystallinity would be obtained from monomer mixtures of lower stereochemical purity. Tolerating the stereoerrors in the polymer backbone rather than having to remove the impurities in the monomer stream that caused them, may help in lowering the cost of PLLA production, facilitating its widespread replacement of non‐degradable plastics. [36] We are currently investigating the properties of these polymers, and developing highly stereoselective catalysts and their applications.
Conflict of interest
M. K. and R. H. are inventors on U.S. provisional patent application 63/283253, and M. K., R. H. and M. S. are inventors on U.S. provisional patent application 63/283254 submitted by Ramot, Tel Aviv University Tech Transfer Company which cover the catalyst and the stereogradient copolymers described herein. V. V. declares no competing interests.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors acknowledge financial support by grant No. 553/18 of the Israel Science Foundation, grant No. 2711/17 of the Israel Science Foundation and the University Grant Commission of India, and grant No. 86599 of the Ministry of Science Technology and Space. We thank Ramona Orlacchio (University of Salerno) for help with some of the DSC measurements.
R. Hador, M. Shuster, V. Venditto, M. Kol, Angew. Chem. Int. Ed. 2022, 61, e202207652; Angew. Chem. 2022, 134, e202207652.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Drumright R. E., Gruber P. R., Henton D. E., Adv. Mater. 2000, 12, 1841–1846. [Google Scholar]
- 2. Inkinen S., Hakkarainen M., Albertsson A.-C., Södergård A., Biomacromolecules 2011, 12, 523–532. [DOI] [PubMed] [Google Scholar]
- 3. Castro-Aguirre E., Iñiguez-Franco F., Samsudin H., Fang X., Auras R., Adv. Drug Delivery Rev. 2016, 107, 333–366. [DOI] [PubMed] [Google Scholar]
- 4. Farah S., Anderson D. G., Langer R., Adv. Drug Delivery Rev. 2016, 107, 367–392. [DOI] [PubMed] [Google Scholar]
- 5. Thomas C. M., Chem. Soc. Rev. 2010, 39, 165–173. [DOI] [PubMed] [Google Scholar]
- 6. Stanford M. J., Dove A. P., Chem. Soc. Rev. 2010, 39, 486–494. [DOI] [PubMed] [Google Scholar]
- 7. Van Wouwe P., Dusselier M., Vanleeuw E., Sels B., ChemSusChem 2016, 9, 907–921. [DOI] [PubMed] [Google Scholar]
- 8. Dusselier M., Van Wouwe P., Dewaele A., Jacobs P. A., Sels B. F., Science 2015, 349, 78–80. [DOI] [PubMed] [Google Scholar]
- 9. Thakur K. A. M., Kean R. T., Hall E. S., Kolstad J. J., Munson E. J., Macromolecules 1998, 31, 1487–1494. [Google Scholar]
- 10. Huang J., Lisowski M. S., Runt J., Hall E. S., Kean R. T., Buehler N., Lin J. S., Macromolecules 1998, 31, 2593–2599. [Google Scholar]
- 11. Auras R., Harte B., Selke S., Macromol. Biosci. 2004, 4, 835–864. [DOI] [PubMed] [Google Scholar]
- 12. Saeidlou S., Huneault M. A., Li H., Park C. B., Prog. Polym. Sci. 2012, 37, 1657–1677. [Google Scholar]
- 13. Benson R. D., Sumner E. S., Schroeder J. D., WO 105143A2, 2010.
- 14. Zhu J.-B., Chen E. Y.-X., J. Am. Chem. Soc. 2015, 137, 12506–12509. [DOI] [PubMed] [Google Scholar]
- 15. De Clercq R., Dusselier M., Sels B. F., Green Chem. 2017, 19, 5012–5040. [Google Scholar]
- 16. De Clercq R., Dusselier M., Makshina E., Sels B. F., Angew. Chem. Int. Ed. 2018, 57, 3074–3078; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3128–3132. [Google Scholar]
- 17. Zhang Y., Qi Y., Yin Y., Sun P., Li A., Zhang Q., Jiang W., ACS Sustainable Chem. Eng. 2020, 8, 2865–2873. [Google Scholar]
- 18. Botvin V., Karaseva S., Salikova D., Dusselier M., Polym. Degrad. Stab. 2021, 183, 109427. [Google Scholar]
- 19. Chamberlain B. M., Cheng M., Moore D. R., Ovitt T. M., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 2001, 123, 3229–3238. [DOI] [PubMed] [Google Scholar]
- 20. Hador R., Botta A., Venditto V., Lipstman S., Goldberg I., Kol M., Angew. Chem. Int. Ed. 2019, 58, 14679–14685; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14821–14827. [Google Scholar]
- 21. Pilone A., Press K., Goldberg I., Kol M., Mazzeo M., Lamberti M., J. Am. Chem. Soc. 2014, 136, 2940–2943. [DOI] [PubMed] [Google Scholar]
- 22. Baratian J. S., Hall E. S., Lin J. S., Xu R., Runt J., Macromolecules 2001, 34, 4857–4864. [Google Scholar]
- 23. Gendler S., Segal S., Goldberg I., Goldschmidt Z., Kol M., Inorg. Chem. 2006, 45, 4783–4790. [DOI] [PubMed] [Google Scholar]
- 24. Chmura A. J., Davidson M. G., Frankis C. J., Jones M. D., Lunn M. D., Chem. Commun. 2008, 1293–1295. [DOI] [PubMed] [Google Scholar]
- 25. Sauer A., Kapelski A., Fliedel C., Dagorne S., Kol M., Okuda J., Dalton Trans. 2013, 42, 9007–9023. [DOI] [PubMed] [Google Scholar]
- 26. Buffet J. C., Kapelski A., Okuda J., Macromolecules 2010, 43, 10201–10203. [Google Scholar]
- 27. Hador R., Shuster M., Lipstman S., Kol M., ACS Catal. 2022, 12, 4872–4879. [Google Scholar]
- 28. Buffet J.-C., Okuda J., Polym. Chem. 2011, 2, 2758–2763. [Google Scholar]
- 29. Kapelski A., Okuda J., J. Polym. Sci. Part A 2013, 51, 4983–4991. [Google Scholar]
- 30. Aida T., Inoue S., Acc. Chem. Res. 1996, 29, 39–48. [Google Scholar]
- 31. Ovitt T. M., Coates G. W., J. Am. Chem. Soc. 2002, 124, 1316–1326. [DOI] [PubMed] [Google Scholar]
- 32. Li T., Strunz S., Radke W., Klein R., Hofe T., Polymer 2011, 52, 40–45. [Google Scholar]
- 33. Alamo R. G., Mandelkern L., Thermochim. Acta 1994, 238, 155–201. [Google Scholar]
- 34. Shan C. L. P., Hazlitt L. G., Macromol. Symp. 2007, 257, 80–93. [Google Scholar]
- 35. Fijten M. W. M., Johannes M., Kranenburg J. M., Thijs H. M. L., Paulus R. M., van Lankvelt B. M., de Hullu J., Springintveld M., Thielen D. J. G., Tweedie C. A., Hoogenboom R., Van Vliet K. J., Schubert U. S., Macromolecules 2007, 40, 5879–5886. [Google Scholar]
- 36. Zhu Y., Romain C., Williams C. K., Nature 2016, 540, 354–362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.