
Figure 6.
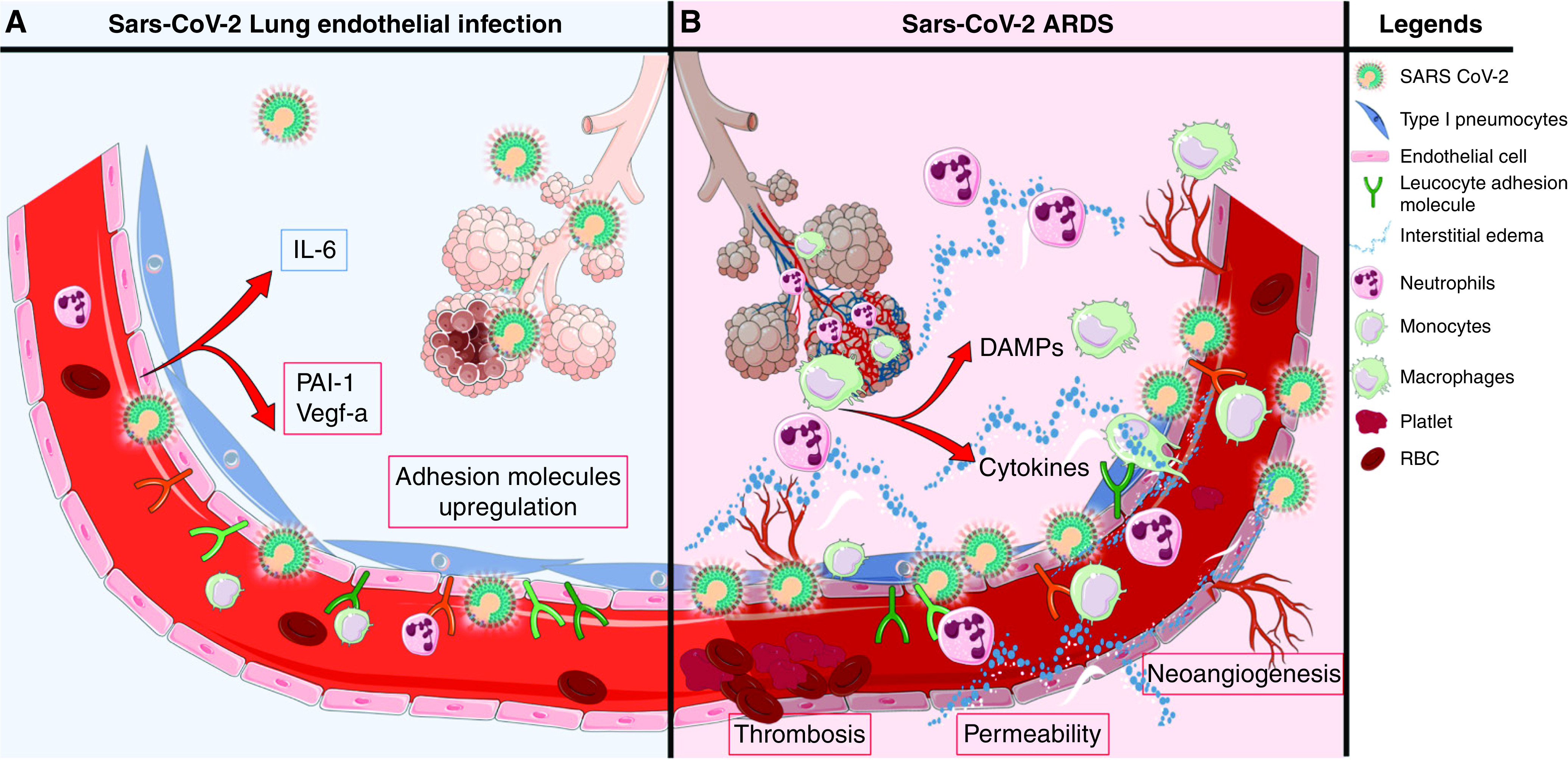
Hypothetical model of coronavirus disease (COVID-19)-associated lung microvascular endotheliopathy. On the basis of our experimental results, we speculate that viral infection and secondary injury of the endothelium by inflammatory mediators and immune cells contribute to the lung endotheliopathy of COVID-19 and, in turn, COVID-19–associated acute respiratory distress syndrome (ARDS). (A) Endothelial cell exposure to live severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) could occur early or later in the course of the disease if there is persistence of virus in the host, including in alveolar epithelial cells, which are in proximity to microvascular endothelial cells. In susceptible individuals, immune dysfunction could lead to increased viral load and release by alveolar epithelial cells. In this speculative model, SARS-CoV-2 infection of lung microvascular endothelial cells induces permeability, upregulates surface expression of leukocyte adhesion molecules and secretion of plasminogen activator inhibitor 1 (PAI-1) and vascular endothelial growth factor (VEGF) secretion, and induces low levels of secretion of cytokines and chemokines. In this context, infection of endothelial cells may promote and/or amplify endotheliopathy and ARDS. (B) Secondary injury to the lung endothelium is likely to be a principal determinant of COVID-19–induced endotheliopathy. Secondary injury may result from the actions of circulating inflammatory mediators and toxins and of activated pulmonary leukocytes and damaged alveolar epithelial cells. In susceptible individuals, impaired viral clearance and persistent viral replication in epithelial or endothelial cells may promote leukocyte recruitment to the lung and cell death and the release of damage-associated molecular patterns (DAMPs). These DAMPs could, analogously to their roles in sepsis and injury, further exacerbate the endotheliopathy, as well as acute lung injury and ARDS. RBC = red blood cells.