

Abstract
Skin cancers are by far the most frequently diagnosed human cancers. The closely related transcriptional co‐regulator proteins YAP and TAZ (WWTR1) have emerged as important drivers of tumour initiation, progression and metastasis in melanoma and non‐melanoma skin cancers. YAP/TAZ serve as an essential signalling hub by integrating signals from multiple upstream pathways. In this review, we summarize the roles of YAP/TAZ in skin physiology and tumorigenesis and discuss recent efforts of therapeutic interventions that target YAP/TAZ in in both preclinical and clinical settings, as well as their prospects for use as skin cancer treatments.
Keywords: basal cell carcinoma, epidermis, Hippo signalling, melanoma, skin cancer, squamous cell carcinoma
1. INTRODUCTION
Skin is the largest organ of the human body and has important barrier, sensory and immune functions. 1 , 2 In the skin, various cell populations cooperate to provide protection from daily wear and tear, harmful microorganisms and other attacks from the external environment. In addition, skin enables thermoregulation and tactile sensations. 1 , 2
Skin cancers are by far the most frequently diagnosed human cancers. 3 , 4 , 5 , 6 While non‐melanoma skin cancers are more common, melanoma is the most dangerous type due to its ability to metastasize. 5 , 7
In the skin, the Hippo signalling pathway and its downstream effectors, the transcriptional co‐regulator proteins Yes‐associated protein (YAP) and transcriptional co‐activator with PDZ‐binding motif (TAZ, also called WW Domain Containing Transcription Regulator 1 (WWTR1)), regulate diverse tissue‐specific functions during development, homeostasis and regeneration. 8 The Hippo pathway is a tumor suppressor pathway, since its deregulation and the resulting YAP/TAZ hyperactivation promote development and progression of many cancer types, including skin cancers. 8 , 9 , 10 , 11 Importantly, there is strong evidence that YAP/TAZ are essential in both melanoma and non‐melanoma skin cancers, 8 , 10 but they appear to be largely dispensable for normal tissue homeostasis, 12 , 13 , 14 pinpointing YAP/TAZ as interesting novel therapeutic targets.
We begin this review by giving a brief overview of the contributions of different skin cell populations to tissue homeostasis and repair and the roles of these cells in cancer development. In the second part, we summarize the specific roles of Hippo/YAP/TAZ signalling in controlling cell functions in healthy skin, review the roles of Hippo/YAP/TAZ signalling in skin cancer development and progression and discuss potential therapeutic approaches. We close this review with discussing major open questions.
2. EPIDERMAL TISSUE HOMEOSTASIS AND REPAIR
Skin epidermis comprises the interfollicular epidermis (IFE) and associated epidermal appendages (Figure 1). These include pilosebaceous units, consisting of a hair follicle (HF) and sebaceous and sweat glands. 2 , 15 , 16 In addition to keratinocytes, mammalian epidermis contains several other cell types, including melanocytes, Merkel cells, gamma delta (γδ) T‐cells and Langerhans cells 1 (Figure 1). The IFE consists of several layers of suprabasal keratinocytes at various stages of a terminal differentiation programme, and a basal layer of proliferative keratinocytes which express keratins KRT5 and KRT14 and are attached to the underlying basement membrane (BM) via integrin (ITG) extracellular matrix receptors. 1 , 2 , 8
FIGURE 1.
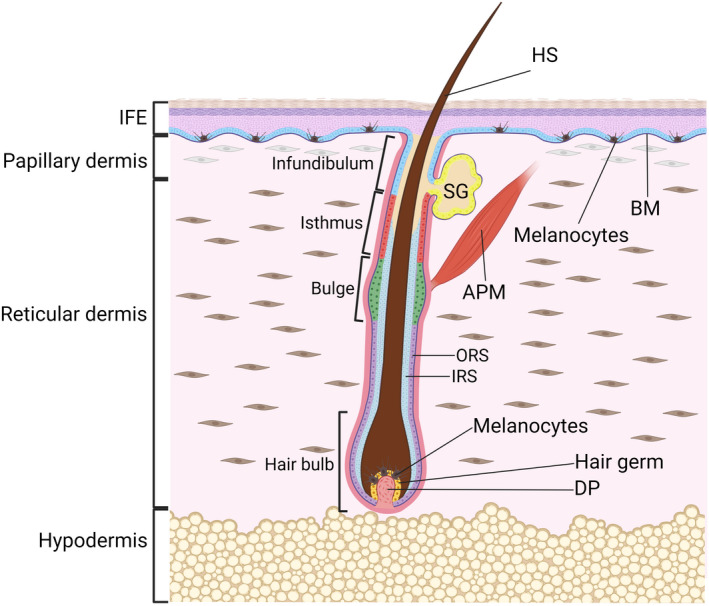
Morphology of the skin. The epidermis and the underlying dermis are separated by a basement membrane (BM). Multiple, spatially distinct stem cell populations have been identified in the interfollicular epidermis (IFE) and the bulge, isthmus, infundibulum and sebaceous gland (SG) parts of the hair follicle and are indicated by different colours. Two populations of fibroblasts populate the dermis: papillary fibroblasts are in proximity to the BM, while reticular fibroblasts are found in the central dermis. The hair follicle is depicted in the growth phase (anagen), when a transient population of stem cells in the hair germ create an inner root sheath (IRS) and hair shaft (HS, protruding out of the skin surface), while stem cells in the permanent bulge region of the hair follicle give rise to the outer root sheath (ORS). The hair germ rests above the dermal papilla (DP), a population of mesenchymal cells that provides inductive signalling for hair growth and modulates hair follicle regeneration. Pigment‐producing melanocytes are present in the hair follicle and the IFE. APM, arrector pili muscle. Figure graphics were created with BioRender.com.
Continuous renewal of the IFE and its appendages throughout life is ensured by stem cells (SCs) and progenitor cells that balance proliferation and differentiation to replace dead and terminally differentiated cells. 1 , 2 , 8 , 15 , 16 During tissue homeostasis, HFs undergo continuous cycles of growth (anagen) and degeneration (catagen), followed by a resting stage (telogen). 1 The SCs responsible for cyclic HF regeneration are located in the permanent non‐cyclic HF portion called the bulge 1 , 15 , 16 (Figure 1). The upper portion of the HF does not cycle but turns over frequently, which is governed by multiple resident SC pools 15 , 16 , 17 (Figure 1). The identity, organization and dynamics of SCs within the IFE are still matters of debate. 18 Current models of mouse IFE homeostasis suggest that each basal cell appears to be equipotent and generates progeny that have equal probability to self‐renew or differentiate. 18 In contrast, the human IFE appears to be maintained by a hierarchy of SCs that generates actively dividing progenitor cells which ultimately commit to terminal differentiation. 18 , 19 , 20
In wounded mouse skin, several epidermal cell populations, including those in HFs distal to the wound site, contribute to the skin wound repair process. 15 , 16 Interestingly, lineage restriction and spatial confinement of HF‐resident SC pools are transiently lost during wound repair, allowing contribution of multiple SC populations. 1 , 15 , 16 This lineage plasticity is not only critical in wound repair but is also functional in skin cancer development. 16 , 21
3. SKIN CANCER
Skin cancers are among the most frequently diagnosed human malignancies world‐wide, with over a million cases detected each year. 4 , 22 Skin cancers can be divided into cutaneous melanomas (CM, also referred to as malignant melanoma of the skin, or melanoma skin cancer) and non‐melanoma skin cancers (NMSC). 4 , 6 , 22 NMSCs comprise several different types of carcinomas, such as basal cell carcinomas (BCC), cutaneous squamous cell carcinomas (cSCC), keratoacanthomas, Merkel cell carcinomas and various rare adnexal tumors, as well as angiosarcomas and cutaneous lymphomas. 3 , 4 , 6 , 8 Approximately 80% of NMSCs are BCC, and 20% are cSCC. 3 , 4 These two most common types of NMSC, originating from epidermal keratinocytes, are nowadays also referred to as keratinocyte cancers. 3 , 23
3.1. Non‐melanoma skin cancer
Non‐melanoma skin cancers incidence greatly outnumbers CM, but most NSMCs are much easier to treat and have much better long‐term prognosis, especially when detected at their initial stages. 6 NMSC most frequently occur on commonly sun‐exposed parts of the body, and affect mostly persons with fair skin, who tend to burn easily rather than suntan when exposed to sunlight. 3 , 4 The long‐term, repeated exposure to ultraviolet (UV) radiation, both solar and artificial, is a causative factor for nearly 90% of NMSCs. 3 , 4 , 6 Yet, despite increasing public awareness of the harmful effects of sun exposure and health costs, NMSC incidence has been increasing by 4% each year. 3 , 22
Basal cell carcinoma is a malignant cancer that arises from the basal epidermal cell layer. 4 , 24 BCC is generally only locally invasive and rarely metastasizes. 3 , 4 BCCs do not proliferate rapidly, but, if ignored and left untreated, they are prone to destroy local underlying tissues. 3 Chronic exposure to UV radiation plays the most important role in BCC pathogenesis. 3 , 4 However, individual risk factors for BCC also include gender, age and genetic diseases (e.g. Gorlin–Goltz syndrome), 3 , 6 , 24 exposure to ionizing radiation, carcinogenic chemicals (especially arsenic) and immune suppressive drugs. 4 Deregulation of the hedgehog(HH)/PTCH1/SMO signalling pathway is central to BCC development. 24 Hyperactivation of the HH pathway, either through deletion of PTCH1, mutational activation of SMO or overexpression of GLI1 or GLI2, has been reported in human and mouse BCC. 3 , 4 , 24 BCCs display different growth patterns and can be classified into various subtypes based on tumor location, gender, age and skin type. 3 , 24 A number of lineage tracing studies (summarized in 24 ) that used Cre‐mediated cell‐specific targeting in mice, either by lineage tracing or by the activation of oncogenic HH signalling in distinct cell populations, have provided strong support for a stem/progenitor cell origin of BCC. The results from these studies also suggest that oncogenic HH signalling can drive BCC initiation in several different epithelial stem‐ and progenitor cell populations in mouse skin, including SCs in the bulge and isthmus regions of the HF and GLI1‐positive cells in touch dome epithelia, although the tumor morphology and the final outcome of BCC development are also influenced by the mutated HH signalling pathway member, and the strength of oncogenic HH signalling. 24 , 25 , 26 In the context of human skin, experimental evidence suggests that BCCs originate from CD200‐positive SCs in the HF bulge. 27
While BCC contributes minimally to the keratinocyte cancer mortality rate, cSCC accounts for about 75% of keratinocyte cancer‐related deaths. 3 , 4 , 23 Like BCC, cSCC also arises from basal epidermal cells. It is characterized by infiltrative and metastatic behaviour as well as destructive growth. 3 , 28 Once cSCC has progressed to an invasive stage, it has the potential to recur after surgical removal and to metastasize, with a variable metastatic rate of 0.1%–9.9%, and with transplant recipients and immunocompromised patients being at greater risk of developing metastatic disease. 3 , 4 , 29 The mortality rate of patients with distant metastatic cSCC is very high (70%–89%), and a curative therapeutic approach is still lacking. 3 , 29 Similar to BCC, solar UV radiation is an important risk factor for the development of cSCC and leads to genetic and epigenetic changes both in basal epidermal cells and cells of the underlying dermal stroma. 23 , 28 , 30 Consequently, cSCC most commonly arises in sun‐damaged skin. One of the strongest predictors of cSCC development in previously unaffected people is the presence of actinic keratoses (AKs, also known as solar keratoses or cSCC in situ), which are benign scaly dysplastic keratinocyte‐derived tumors caused by cumulative sun exposure. 23 , 28 Other risk factors include infection with human papillomaviruses and genetically inherited cutaneous diseases, such as albinism, xeroderma pigmentosum and epidermodysplasia verruciformis. 4 , 23 However, there are also clinical situations in which increased cSCC formation in patients is associated with therapeutic treatments. 28 A noticeable example is cSCCs induced by long‐term treatment with immunosuppressive drugs. 31 cSCC development is a complex process, but it is frequently associated with mutations in RAS GTPases (HRAS and KRAS), cell cycle regulators such as TP53 and CDKN2A, regulators of squamous cell differentiation such as Notch signalling receptors (NOTCH1, NOTCH2 and NOTCH3), chromatin remodelling factors such KMTC2 and KMTD2, and FAT1 cadherin. 4 , 28 , 30 Dysregulated RAS/receptor tyrosine kinase/PI3K and cell cycle pathways appear to be particularly involved in aggressive cSCC. 32 In mice, cSCC can be induced through multistage carcinogenesis models that use chemical carcinogens, UV irradiation or forced expression of oncogenes targeted to epidermal SC populations. 28 , 30 , 33 , 34 Using transgenic mice where the expression of oncogenic KRAS was targeted to different compartments of adult epidermis, studies found that both IFE and HF SCs are cells of origin of mouse cSCC. 30 However, targeting of oncogenic KRAS to HF SCs led to formation of more aggressive cSCCs with features of epithelial to mesenchymal transition (EMT). 30 Of note, a hybrid EMT state seems to be associated with cSCC metastasis. 35
3.2. Cutaneous melanoma
Melanoma skin cancer is a malignancy of melanocytes. The cutaneous form of melanoma (CM) causes the majority (75%) of deaths related to skin cancers. 22 , 36 , 37 CM is characterized by an extensive degree of heterogeneity in terms of clinical, dermatological and histopathological presentation, 36 , 37 , 38 genomic profile, 37 , 38 , 39 and risk factors. 36 , 37 , 38 , 39 The development of fully evolved CM from pre‐neoplastic lesions is complex and not represented by a single evolutionary pattern. 37 , 38 , 39 The spectrum of melanocytic neoplasms ranges from benign nevi (circumscribed proliferations of melanocytes), which are common and have only a very marginal risk of progressing, to invasive melanomas, which have the potential to metastasize. 37 , 38 In between are several intermediate stages that include dysplastic naevi and non‐invasive (in situ) CM. 37 , 38 Sun (UV) exposure is the main environmental risk factor for CM development, and CM occurs mainly in white populations with fair skin. 36 , 39 According to the 2018 “WHO Classification of Skin Tumors,” melanomas can be divided into thosethat are causally related to sun exposure and those that are not, as determined by their mutational signatures, anatomic site and epidemiology. 38 Based on their origins from skin that is or is not chronically sun damaged (CSD), CMs can be broadly categorized into high‐CSD, low‐CSD and non‐CSD. 37 , 38 High‐CSD CMs encompass lentigo maligna and desmoplastic melanomas, and low‐CSD CMs include superficial spreading melanomas. 38 The non‐CSD (or variable/incidental UV radiation exposure) category includes acral melanomas, some melanomas in congenital nevi, melanomas in blue nevi, Spitz melanomas, but also non‐cutaneous melanomas, such as mucosal melanomas and uveal melanomas. 38 High‐CSD CMs commonly arise from in situ CM on skin of older (>55 years of age) individuals with a history of long‐term exposure to UV radiation, and have a high mutational burden associated with NF1, NRAS, BRAF nonV600E or KIT driver mutations, leading to aberrant activation of the MAPK pathway. 37 , 39 In contrast, low‐CSD CMs most commonly arise from benign or dysplastic naevi, affect the more sporadically sun‐exposed areas of younger individuals (<55 years of age), and are often associated with a moderate mutational burden and predominance of BRAF V600E driver mutations. 37 , 38 In the absence of other driver mutations, the BRAF V600E mutation causes limited melanocyte proliferation which is kept in check by oncogene‐induced cell senescence. 37 The resulting nevus remains stable for decades, also due to immune surveillance. 37 In fact, melanomas are among the most immunogenic tumors. 40 Progression of low‐ and high‐CSD CMs to invasive CM is usually associated with secondary and tertiary mutations, such as TP53 and PTEN mutations, telomerase reverse transcriptase (TERT) promoter mutations and bi‐allelic loss of CDKN2A. 37 , 39 No conclusive hierarchic mutation pattern associated with metastasis has been identified, suggesting that metastatic progression involves distinct transcriptional programmes. 37 , 41 In the non‐CSD category of CM, Spitz melanomas are characterized by driver fusion genes including the kinase domains of ALK, ROS1, NTRK1, NTRK3, MET, RET, BRAF and MAP3K8, while Acral melanomas display a high incidence of copynumber variation with gene amplifications of CCND1 and KIT. 38
4. THE HIPPO SIGNALLING PATHWAY
The Hippo pathway is a highly conserved signalling pathway that was first characterized in Drosophila melanogaster for its role in larval growth and was later implicated in human cancers as a major tumour suppressor pathway. 42 , 43 , 44 , 45 The pathway (Figure 2) consists of a core kinase cascade beginning with MST1 (Ste20‐like kinase 1; also known as STK4) and MST2 (also known as STK3), which phosphorylate and activate large tumour suppressor kinases LATS1 and LATS2. 42 , 44 , 45 MST1/2 are activated either by TAO1/2/3 kinase‐mediated phosphorylation, or by trans‐autophosphorylation, of their activation loop. 42 , 44 Active MST1/2 phosphorylate Salvador homologue 1 (SAV1) and MOB (monopolar spindle‐one‐binder proteins) kinase activator 1A and 1B (MOB1A/MOB1B), two scaffold proteins that assist MST1/2 in the recruitment and phosphorylation of LATS1/2. 45 , 46 , 47 , 48 , 49 , 50 Two groups of MAP4Ks (mitogen‐activated protein kinase kinase kinase kinase), MAP4K1/2/3/5 and MAP4K4/6/7, work in parallel to MST1/2 and can also directly phosphorylate and activate LATS1/2. 51 , 52 Another important player in the Hippo pathway is neurofibromatosis type 2 (NF2)/Merlin, which directly interacts with LATS1/2 and facilitates LATS1/2 phosphorylation by the MST1/2–SAV1 complex. 53 Upon phosphorylation at their hydrophobic motif by upstream kinases, LATS1/2 subsequently undergo autophosphorylation and are activated. 44 , 49 , 50 In addition to LATS1/LATS2, nuclear Dbf2‐related kinases NDR1 (STK38) and NDR2 (STK38L) also function as YAP/TAZ kinases. 54 NDR1/2 kinases phosphorylate the paralogous transcriptional co‐regulators YAP and TAZ at consensus HXRXXS motifs, of which YAP has five and TAZ has four. 55 , 56 , 57 The most relevant phosphorylation sites that keep YAP/TAZ inhibited are S127 and S381 in human YAP, and S89 and S311 in human TAZ. 42 , 43 , 44 Phosphorylation at different residues can regulate independent fates of YAP/TAZ. LATS1/LATS2‐mediated YAP‐S127 and TAZ‐S89 phosphorylation creates a binding site for 14‐3‐3 proteins which contribute to keeping YAP/TAZ in the cytoplasm and therefore transcriptionally inactive. 55 , 56 , 58 However, in many cellular contexts this signalling input alone does not appear to be sufficient to keep YAP/TAZ in the cytoplasm, as S127/S89‐phosphorylated YAP/TAZ have also been detected in the nucleus. 59 , 60 , 61 , 62 Consequently, nuclear localization alone may not always be a reliable surrogate of YAP/TAZ activity, since mice carrying a YapS112A (similar to S127 in human YAP) knock‐in mutation are surprisingly without phenotype despite nuclear localization of the mutant YAP protein. 63 Indeed, feedback activation of Hippo signalling was found to ensure physiological levels of YAP/TAZ activity in mammalian cells. 63 , 64 , 65 S381/S311 phosphorylation of YAP/TAZ primes a phosphodegron that can be further phosphorylated by CK1δ/ε to recruit the SCFβ‐TRCP E3 ubiquitin ligase complex to tag YAP/TAZ for proteasomal degradation. 55 , 56 , 58 It is worth noting here that YAP/TAZ regulation is not static, but rather dynamic. Indeed, YAP/TAZ undergo constant phosphorylation and dephosphorylation, and are rapidly shuttled between the cytoplasm and the nucleus. 66 , 67 , 68
FIGURE 2.
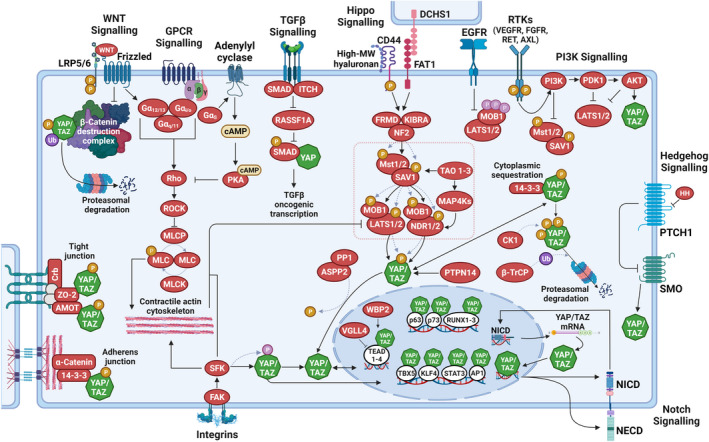
Key signals regulating YAP/TAZ activity. The transcription co‐regulators Yes‐associated protein (YAP) and transcriptional co‐activator with PDZ‐binding motif (TAZ), are predominantly regulated by phosphorylation (serine phosphorylation, orange; tyrosine phosphorylation, pink). Serine‐phosphorylated YAP/TAZ are exported from the nucleus and are either degraded in the cytoplasm via the proteasome or sequestered in the cytoplasm via 14‐3‐3 proteins or at tight‐ and adherens junctions. In their non‐serine‐phosphorylated and tyrosine‐phosphorylated states, YAP/TAZ accumulate in the nucleus, where they bind to various transcription factors, most notably those of the TEA domain (TEAD) family, to control target gene expression. In the nucleus, vestigial‐like family member 4 (VGLL4) competes with YAP/TAZ in binding to TEADs, while WW domain binding protein‐2 (WBP2) enhances the co‐activator functions of YAP/TAZ. The core of the Hippo pathway (dotted pink box) is defined by a kinase cascade composed of MST1 and MST2 kinases, large tumour suppressor (LATS)1 and LATS2 kinases and their co‐factors SAV1 and MOB1A and MOB1B. Membrane‐associated signalling events causing Hippo pathway activation include high‐molecular‐weight hyaluronan‐mediated clustering of CD44 and cell–cell signalling via Dachsous cadherin‐related 1 (DCHS1)/FAT1. Hippo pathway activation involves the phosphorylation of the core Hippo kinases, MST1/2 and LATS1/2: MST1/2 are autophosphorylated and subsequently phosphorylate LATS1/2. MST1/2 are also activated by TAO kinases. Activation of LATS1/2 causes the serine‐phosphorylation of YAP and TAZ and inhibits their transcription co‐regulator functions. PP1, together with apoptosis‐stimulating protein of p53 2 (ASPP2), antagonizes Hippo pathway activity by de‐phosphorylating YAP/TAZ. In addition to MST1/2, various upstream effectors of the LATS1/2 have been identified, including the MAP4K and TAOK families of kinases, which phosphorylate and activate LATS1/2. Nuclear Dbf2‐related (NDR)1/2 kinases act in parallel to LATS1/2 in the Hippo pathway to inactivate YAP/TAZ. The activities of the core Hippo pathway components are regulated by several upstream mechanisms. These involve various scaffolding proteins such as angiomotin (AMOT), neurofibromin 2 (NF2; also known as Merlin), kidney and brain protein (KIBRA; also known as WWC1), the protocadherin FAT1 and zonula occludens (ZO) proteins at tight junctions. Cell polarity and adhesion regulators promote LATS1/2‐mediated regulation of YAP/TAZ by altering actin dynamics and by facilitating Hippo pathway effector association. G protein‐coupled receptors (GPCR) signalling, mechanical cues and signals transduced by the extracellular matrix and matrix‐binding integrins (through FAK and SRC family kinases (SFKs)) can inactivate LATS1/2 by promoting a contractile F‐actin‐myosin cytoskeleton. SFKs also directly regulate YAP/TAZ nuclear abundance, predominantly by controlling their nuclear export rate. Soluble growth factors bind to and activate receptor tyrosine kinases (RTKs) and inactivate the Hippo pathway by stimulating PI3K–PDK1 signalling. EGFR activation causes inhibitory tyrosine phosphorylation of MOB1A/B. RASSF1A is recruited to the activated TGF‐b receptor I and subsequently targeted for degradation by the co‐recruited E3ubiquitin ligase ITCH. RASSF1A degradation then permits YAP association with SMADs and subsequent nuclear translocation of receptor‐activated SMAD2. YAP/TAZ are also regulated by WNT signalling: such as β‐catenin, YAP/TAZ also incorporates into the destruction complex and are targeted for proteasomal degradation. Upon WNT stimulation, inactivation of the destruction complex then drives β‐catenin as well as YAP/TAZ nuclear translocation. YAP/TAZ also interact with the Notch pathway: in the nucleus, YAP/TAZ can induce the gene expression of Notch receptors and/or Notch ligands to regulate Notch signalling, while the transcriptionally active Notch intracellular domain (NICD) can activate YAP1 gene transcription. Activated HH signalling leads to increased nuclear YAP abundance. AKT, Ak strain transforming; AP, activator protein; APC, adenomatous polyposis coli; cAMP, 3′ 5′‐cyclic adenosine monophosphate; CK1, casein kinase 1δ/1ε; Crb, crumbs; DVL, dishevelled segment polarity protein; EGFR, epidermal growth factor receptor; FAK, focal adhesion kinase; FGFR, fibroblast growth factor receptor; FRMD, FERM and PDZ domain containing; GSK, glycogen synthase kinase; HH, hedgehog; KLF, Krüppel‐like factor; LRP, LDL receptor‐related protein; MAP4K, mitogen‐activated protein kinase kinase kinase kinase; MLC, myosin light; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; NECD, Notch extracellular domain; P, phosphorylation; PDK, pyruvate dehydrogenase kinase; PI3K, phosphoinositide 3‐kinase; PKA, protein kinase A; PP1, protein phosphatase 1; PTPN, protein tyrosine phosphatase non‐receptor type; RASSF, RAS association domain family; ROCK, Rho‐associated kinase; RUNX, Runt‐related transcription factor; SFK, SRC‐family kinase; SMAD, suppressor of mothers against decapentaplegic; SMO, smoothened; STAT, signal transducer and activators of transcription; TAO, thousand and one; TBX, T‐box transcription factor; TGF, transforming growth factor; Ub, ubiquitylation; VEGFR, vascular endothelial growth factor receptor. Dotted lines indicate post‐translational modification events. Figure graphics were created with BioRender.com.
Importantly, YAP and TAZ are transcriptional co‐regulators that do not contain DNA‐binding domains. 69 The primary transcriptional binding partners are transcription factors (TF) of the TEA domain family (TEAD1‐4). 43 , 70 , 71 In complex with a TEAD TF, YAP/TAZ bind to gene enhancer elements, and interact with chromatin remodelling factors and modulate RNA polymerase II activity to drive or repress the expression of target genes, which prominently include cell cycle, cell migration and cell fate regulators. 12 , 72 , 73 , 74 , 75 , 76 , 77 , 78 YAP/TAZ transcriptional activity is negatively modulated by VGLL4. 79 , 80 While TEAD TFs are the predominant transcriptional interaction partners of YAP/TAZ, they have also been shown to physically interact with other TFs such as p63, p73, TBX5, RUNX1/2/3, KLF4 and STAT3. 60 , 77 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 YAP‐TEAD complexes appear to cooperate closely with other TFs, most notably those of the AP1 family. 12 , 76 , 89 , 90 YAP and TAZ appear to be have overlapping and non‐redundant roles, 69 , 91 since evidence has accumulated that both paralogues might drive distinct transcriptomes. 87 , 92 , 93 , 94
The Hippo signalling pathway receives myriads of inputs from several intracellular and extracellular cues, which form a complex network to regulate YAP/TAZ localization, abundance and activity (Figure 2). 42 , 43 , 44 , 70 In most scenarios, these signalling cues modulate the core kinase cascade by relaying signals from the plasma membrane. However, only few dedicated transmembrane receptors and extracellular ligands of the Hippo pathway have been identified (Figure 2). Instead, most upstream signalling components have roles in other processes such as the establishment of cell morphology, 61 , 95 , 96 , 97 , 98 , 99 cell–cell and cell‐matrix adhesion, 13 , 62 , 100 , 101 , 102 , 103 , 104 , 105 , 106 , 107 , 108 and cell polarity. 109 , 110 , 111 , 112 , 113 , 114 , 115 , 116 , 117 In addition, there are several proteins that directly regulate YAP/TAZ localization and activation without affecting LATS or NDR kinase activities, such as for example ASPP2/PP1 and PTPN14. 42 , 43 , 70 , 118 Hippo signalling is highly sensitive to mechanical cues including cell density, mechanical stress, ECM stiffness and ECM composition, which regulate YAP/TAZ through changes in cell geometry and cytoskeleton confirmation and tension. 42 , 70 , 119
The Hippo signalling pathway is also modulated by extensive crosstalk with other signalling pathways. 42 , 43 , 44 , 45 These include signalling through G protein‐coupled receptors (GPCRs), activated by either lipids (lysophosphatidic acid and sphingosine‐1‐phosphophate) or hormones (glucagon or adrenaline) 120 , 121 , 122 , 123 ; the WNT pathway, which can regulate YAP/TAZ either through incorporation into the β‐catenin destruction complex or through destruction complex‐independent mechanisms 124 , 125 , 126 , 127 , 128 , 129 ; SRC family kinases that promote YAP/TAZ nuclear localization and transcriptional activity either directly by phosphorylating tyrosine residues or indirectly by repressing LATS1/LATS2 13 , 62 , 129 , 130 , 131 , 132 , 133 , 134 ; TGF‐β signalling, which regulates YAP nuclear translocation by targeting the Hippo pathways scaffold RASSF1 135 ; the PI3K pathway, which either modulates the core Hippo cascade via PI3K‐PDK1 or YAP/TAZ localization via AKT‐mediated phosphorylation 13 , 102 , 136 , 137 , 138 ; the NOTCH pathway, which modulates YAP/TAZ levels and activity 139 , 140 , 141 and the HH pathway which controls nuclear abundance of YAP/TAZ. 142 , 143
5. THE ROLES OF HIPPO/YAP/TAZ SIGNALLING IN SKIN HOMEOSTASIS AND REPAIR
There is now substantial evidence for the importance of YAP/TAZ in driving epidermis homeostasis and repair. Consistent with the predominantly nuclear localization of YAP/TAZ in SC‐containing compartments during HF growth, 13 , 123 , 144 , 145 , 146 tamoxifen‐induced depletion of Yap and Taz in Krt5‐expressing epidermal stem/progenitor cells (K5‐CreERT/Yap/Taz) of adult mice led to progressive hair loss beginning 2 weeks after the first tamoxifen injections, while only causing a moderate reduction of basal cell proliferation in the IFE. 13 This appears to be consistent with the reduced nuclear localization of YAP (and TAZ) in the basal epidermal cell layer of adult compared to foetal and neonatal mice. 13 , 123 , 144 However, conditional knockout of Yap/Taz in adult epidermis significantly impaired epidermal tissue repair upon skin wounding, 13 similar to topical treatment of skin wounds with YAP‐interfering RNAs. 147 Likewise, YAP/TAZ were found to be required for promoting stem/progenitor cell cycling in the IFE in response to mechanical stretching of the epidermis. 148 Mice lacking the YAP/TAZ co‐factor WBP2 did not have hair growth abnormalities but displayed reduced proliferation in the regenerating epidermis in response to skin wounding. 59 The roles of YAP/TAZ in murine epidermis remain somewhat ambiguous, since two studies reported no obvious skin phenotypes in epidermis‐specific conditional Y a p/T a z double knockout mice. 12 , 14 This discrepancy can likely be explained by the use of different promoters to drive conditional Cre transgene expression (bovine Krt5 promoter 13 vs. human KRT14 promoter 12 , 14 ), which are known to have different deletion efficiencies and onsets/timings. Studies using a Cre‐inducible LacZ reporter have indeed found that K5‐Cre drives efficient recombination in both IFE and HFs, whereas very little recombination activity could be detected in HFs of K14‐Cre mice. 25 , 149 , 150 The conflicting reports of the consequences of conditional Y ap/T az knockout in the adult epidermis are thus reminiscent of previous studies on the in vivo functions of ITG beta 1(ITGB1) in the epidermis. 150 , 151 Other factors explaining the disparities in Yap/Taz knockout phenotypes could be tamoxifen dosage and treatment regimens and different genetic backgrounds and age of the mice used (e.g. Elbediwy et al. 13 used intraperitoneal (ip) injections of 0.1 mg tamoxifen/g body weight in 8‐ to 16‐week‐old mice over 8 weeks, three times per week; the study from Zanconato et al. 12 used 6‐ to 8‐week‐old mice and three ip injections of 1 mg tamoxifen per week over two weeks; Debaugnies et al. 14 used P28 mice and 2.5 mg tamoxifen/injection for four consecutive days to knock‐out Y ap and T az), and general differences in the animal colonies (food, air and water) leading to different metabolism and microbiota. Of note, in tamoxifen‐induced K5‐CreERT/Yap/Taz epidermis, basal cells that escaped Cre‐mediated recombination were found to be able to repopulate the mutant tissue in a short time frame, 13 which could have further complicated data interpretation in the different Y ap/T az knock‐out studies. That said, human KRT14 or bovine Krt5 promoter‐driven expression of mutant, hyperactive YAP transgenes with enhanced nuclear localization (YAP‐S127A 100 or NLS‐YAP‐5SA 152 hereafter referred to as K14/YAP‐S127A and K5/NLS‐YAP‐5SA, respectively), in stem/progenitor cells of adult murine epidermis caused severe tissue dysplasia as a consequence of increased stem/progenitor cell proliferation and loss of terminally differentiated cell types, ultimately leading to the formation of cSCC‐like tumors. 100 , 144 , 152 In contrast, mice expressing a different YAP transgene (YAP‐5SA‐DC, lacking the C‐terminal transactivation domain) under control of the bovine Krt5 promoter developed only a mild skin phenotype. 145 , 146 In such mice, hyper‐thickening of the IFE resulted from expansion of both the basal and suprabasal cell compartments as well as hyperkeratinization in the most differentiated cell layers. 145 This suggests that the C‐terminus of YAP may control the balance between epidermal stem/progenitor cell proliferation and differentiation in the IFE. Using cultured human keratinocytes and mice expressing a genetically encoded inhibitor of the interaction of YAP and TAZ with TEADs, recent studies identified a regulatory loop whereby YAP/TAZ/TEADs and KLF4, a TF involved in promoting terminal differentiation, 153 limit each other's activities to balance proliferation and differentiation. 77 , 89 However, these studies did not reveal a direct role of YAP/TAZ/TEAD‐mediated transcription in the regulation of terminal differentiation. 77 , 89 RNAi‐mediated silencing of TEAD expression was also found to impair proliferation of primary mouse and human keratinocytes in culture, highlighting that TEADs might be the predominant transcriptional interaction partners of YAP/TAZ in the epidermis. 59 , 144
Human keratinocytes require sophisticated culture conditions to maintain their full regenerative potential. A cell culture method developed in the mid‐1970s 154 is still regarded as “gold standard” in regenerative medicine settings. Under these culture conditions, the capacity of individual keratinocyte colonies to generate secondary cultures can be quantified via morphological and functional clonal analysis. 155 Based on this procedure, founder colonies (clones) can be categorized as holoclones, meroclones and paraclones. 155 Holoclones possess the greatest (long‐term) proliferative potential and self‐renewal ability; paraclones are colonies with short lifespan where most cells have committed to undergo terminal differentiation, and meroclones have intermediate properties. 155 Clonal tracing of human transgenic epidermis revealed that in situ the holoclone‐forming keratinocytes are indeed self‐renewing, long‐lived SCs, which maintain the epidermis long‐term and give rise to pools of short‐lived progenitors (meroclones and paraclones) that ultimately replenish differentiated cells and contribute to wound healing. 19 Ablation of YAP/TAZ was shown to selectively deplete holoclones and impair regeneration of human epidermal tissue in 3D organotypic skin equivalents, 59 , 105 while enforced YAP expression prevented conversion of SCs into progenitors and indefinitely extended the culture lifespan. 105 YAP expression is dramatically decreased in Junctional Epidermolysis Bullosa (JEB) keratinocytes, which contain only cytoplasmic YAP. 105 This could explain the slow but progressive loss of JEB patient's ability to heal their continuously occurring skin blisters. 105
While YAP/TAZ clearly play important roles in promoting stem‐ and progenitor cell self‐renewal during HF cycling and epidermal tissue repair, there is still ambiguity as to what extent the Hippo signalling pathway is involved in controlling the activity of YAP/TAZ in the epidermis. 8 While epidermis‐restricted (K14‐Cre) conditional knockout of Mst1/Mst2 during mouse development was without consequence for tissue homeostasis and Yap activity even in adult animals, 100 conditional tamoxifen‐induced (K14‐CreER) double knockout of Mob1a and Mob1b in postnatal mouse epidermis led to a marked expansion of the stem/progenitor cell populations through increased nuclear Yap abundance 156 (Figure 3), reminiscent of the epidermal phenotypes of K14/YAP‐S127A and K5/YAP‐5SA‐DC transgenic mice. 100 , 144 , 145 The consequences of epidermis‐restricted LATS1/LATS2 knockout have not been studied yet. However, phosphorylation of LATS1/LATS2 in response to activation of upstream kinases 156 and increased Yap transcriptional activity upon RNAi‐mediated Lats1/Lats2 ablation 130 in mouse keratinocytes support a role of LATS1/LATS2 in controlling YAP/TAZ in mouse epidermis (Figure 3). Moreover, deletion of Gnas (the gene coding for the Gαs heterotrimeric G‐protein) or inactivation of protein kinase A (Pka) in mouse epidermis (K14‐CreER) led to aberrant expansion of the stem/progenitor cell compartment through activation of YAP, likely involving LATS1/2 but not MST1/2 123 (Figure 3). Nuclear localization of YAP in basal epidermal cells appears to also be negatively regulated by PTPN14 157 (Figure 3).
FIGURE 3.
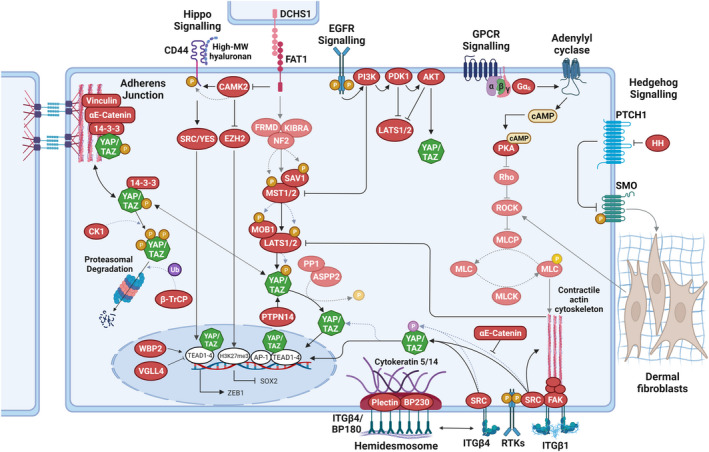
Regulation of YAP/TAZ in normal and neoplastic epidermal cells. Speculative aspects of signalling pathways that are not yet supported by experimental data are indicated by faded graphical elements. Hippo signalling via MOB1A/MOB1B and large tumour suppressor (LATS)1/LATS2 inhibits Yes‐associated protein (YAP) and transcriptional co‐activator with PDZ‐binding motif (TAZ) via serine phosphorylation (orange) to promote cytoplasmic retention and/or proteasomal degradation. Integrin (ITG)–SRC signalling in the basal epidermal cell compartment promotes YAP/TAZ nuclear localization and TEA domain (TEAD) binding. Direct phosphorylation of YAP/TAZ on tyrosine residues (pink) by SRC promotes increased nuclear localization. In the nucleus, vestigial such as family member 4 (VGLL4) competes with YAP/TAZ in binding to TEADs, while WW domain binding protein‐2 (WBP2) enhances the co‐activator functions of YAP/TAZ. The contractile F‐actin‐myosin cytoskeleton stabilizes ITGβ1 adhesions and thus contributes to SRC activation. ITGβ4 adhesions are part of hemidesmosomal complexes that are anchored to keratin 5/14 intermediate filaments. Nuclear localization of YAP in basal epidermal cells is also negatively regulated by PTPN14. At adherens junctions, α‐catenin controls YAP/TAZ activity and phosphorylation by modulating its interaction with 14–3‐3. α‐catenin can also inhibit activation of SRC by ITGβ4. EGFR signalling inactivates the Hippo pathway through the stimulation of PI3K–PDK1. G protein‐coupled receptor (GPCR) signalling, involving Gαs and PKA, suppresses LATS1/2 activation, presumably via decreasing F‐actin‐myosin cytoskeletal contractility downstream of Rho/ROCK. During cSCC progression, loss of function of the protocadherin FAT1 activates a CAMK2‐CD44‐SRC axis that promotes nuclear translocation of YAP and this drives the expression of zinc finger E‐box binding homeobox 1 (ZEB1) that stimulates the mesenchymal state. FAT1 loss of function also inactivates enhancer of zeste homologue 2 (EZH2), promoting SRY‐box transcription factor 2 (SOX2) expression, which sustains the epithelial state. Together, these molecular events promote a hybrid epithelial‐to‐mesenchymal transition (EMT) phenotype. If FAT1 can directly activate the Hippo pathway, is currently not known. In BCC, fibroblast activation and ECM remodelling in papillary dermis as a consequence of increased HH signalling in the epidermis may indirectly activate epidermal ROCK signalling through mechano‐reciprocity. AKT, Ak strain transforming; AP, activator protein; cAMP, 3′ 5′‐cyclic adenosine monophosphate; CAMK, Ca2+/calmodulin‐dependent protein kinase; CK1, casein kinase 1δ/1ε; DCHS, dachsous; EGFR, epidermal growth factor receptor; FAK, focal adhesion kinase; HH, hedgehog; MLC, myosin light chain; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; P, phosphorylation; PDK, pyruvate dehydrogenase kinase; PI3K, phosphoinositide 3‐kinase; PKA, protein kinase A; PP1, protein phosphatase 1; PTPN, protein tyrosine phosphatase non‐receptor type; RASSF, RAS association domain family; ROCK, Rho‐associated kinase; SFK, SRC‐family kinase; SMO, smoothened; Ub, ubiquitylation. Dotted lines indicate post‐translational modification events. Figure graphics were created with BioRender.com.
It is interesting to note that L ats1/L ats2 knockdown in human keratinocytes affected YAP nuclear localization and transcriptional activity only in confluent, fully contact‐inhibited cultures, indicating that efficient control of YAP/TAZ localization and activity likely involves integration of multiple signalling cues, in particular mechanical cues. 59 , 95 Indeed, the adherens junction component αE‐catenin has been identified as a cell density‐dependent YAP regulator in several studies. 100 , 101 , 106 , 130 Genetic deletion of αE‐catenin in murine epidermis (K14‐Cre) or more specifically in the HF bulge (GFAP‐Cre) led to epidermal hyperproliferation, associated with increased nuclear abundance of YAP. 100 , 101 Several mechanisms have been proposed of how αE‐catenin regulates YAP/TAZ. In one mechanism, αE‐catenin promotes YAP S127 phosphorylation and cytoplasmic localization directly by regulating its interaction with 14‐3‐3 proteins 100 (Figure 3). A more recent study found that in adherens junctions, vinculin keeps αE‐catenin in mechanically engaged, stretched/open conformation. The stretched αE‐catenin molecules create binding sites for 14‐3‐3 proteins, which sequester pS127‐YAP at the adherens junctions. 106 This mechanism appears to be conserved also in human keratinocytes, where disruption of actin‐myosin‐mediated cytoskeletal tension at adherens junctions leads to nuclear re‐entry of YAP in contact‐inhibited cultures. 59
In a second mechanism, αE‐catenin suppresses SRC family kinase (SFK)‐mediated tyrosine phosphorylation of YAP, thereby preventing YAP's nuclear localization and TEAD binding 130 (Figure 3). Interestingly, both αE‐catenin‐dependent mechanisms appear to operate independently of LATS1/LATS2. 100 , 130 Integrins not only provide epidermal stem/ progenitor cell markers, but they also regulate stem and progenitor cell fate during homeostasis, tissue repair and cancer progression. 158 , 159 Accordingly, in cultured human keratinocytes, inhibition of ITGB1 or of its downstream effectors SRC and FAK or PI3K impaired YAP/TAZ nuclear localization. 13 , 138 Likewise, epidermis‐restricted deletion of SRC or FAK, or pharmacological inhibition of SFK activity, led to decreased YAP levels and nuclear localization in basal keratinocytes 13 (Figure 3). ITGB1‐mediated activation of YAP appears to depend on the integrity and organization of the F‐actin cytoskeleton, 13 , 70 but less so on actomyosin contractility. 13 , 59 In addition, the hemidesmosome‐associated ITGB4 160 was also shown to control YAP activity via direct SRC‐mediated phosphorylation of YAP, 130 which is negatively regulated by αE‐catenin. 130
6. YAP AND TAZ AS ONCOPROTEINS IN SKIN CANCERS
YAP/TAZ are overexpressed in many different types of murine and human cancers. 9 , 161 , 162 , 163 , 164 Of note, YAP/TAZ appear to be particularly important in squamous cell cancers, which are characterized by frequent amplification of YAP/TAZ. 161 YAP and TAZ are highly expressed and nuclear in different types of BCC in both human and mice. 14 , 59 , 146 , 165 , 166 In cSCC, increased YAP expression was shown to correlate with disease progression. 14 , 152 , 167 In contrast, TAZ expression appears to be more sparse in cSCC, with fewer cells staining positive for nuclear TAZ. 14 Increased nuclear YAP expression was also observed in a subset of kerathoacanthomas with low αE‐catenin expression. 101 YAP overexpression was also documented in pilomatrixoma and trichilemmal carcinoma, rare tumours of HFs. 156 , 168
In accordance with their increased expression in keratinocyte cancers, YAP and TAZ were found to play key roles in the development and progression of cSCC. Several studies found YAP/TAZ to be essential for in vitro proliferation of human cSCC cell lines, by promoting G1/S progression. 35 , 59 , 167 Expression of oncogenic Kras G12D (together with Tp53 knockout) in HF SCs using Lgr5‐CreER/KrasG12D/Tp53KO mice induces cSCCs with varying degrees of squamous differentiation. 14 In this cSCC model, Yap/Taz deletion completely abrogated tumour formation due to rapid cell death of the oncogene‐expressing cells. 14 In a chemical two‐stage skin carcinogenesis mouse model, which involves tumor initiation by the application of a sub‐carcinogenic dose of a carcinogen (e.g. 7,12‐dimethylbenz[a]‐anthracene (DMBA)) and subsequent tumour development by repeated treatment with the tumour‐promoting agent 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA), 169 YAP/TAZ were also shown to be essential for tumor development. 12 Conversely, development of cSCC‐like tumors was observed in K14/YAP‐S127A and K5/NLS‐YAP‐5SA mice, 100 , 152 the latter displaying progression to spindle cell carcinoma at sites of scratch wounding where YAP‐mediated activation of the TF ZEB1 induced an EMT programme. 152 Extending these findings, a recent study found that deletion of the protocadherin Fat1 in a mouse model of cSCC promoted a hybrid EMT phenotype by inducing YAP nuclear translocation and ZEB1 expression that stimulates the mesenchymal state, while increased expression of the cancer SC factor SOX2 170 sustains the epithelial state 35 (Figure 3). A role of YAP in cSCC progression is also supported by in vitro studies demonstrating YAP functions in cell migration and invasion. 167 , 171 Conversely, overexpression of the YAP/TAZ negative regulator VGLL4 was found to reduce growth of human cSCC cells 172 (Figure 3). In human keratoacanthomas and cSCC, strong correlation between low αE‐catenin abundance and nuclear YAP localization has been documented. 100 , 101 , 130 Accordingly, conditional knockout of αE‐catenin in the HF bulge was shown to cause development of keratoacanthomas displaying increased nuclear YAP abundance. 101
Similar to the situation in genetically induced cSCC, epidermis‐restricted deletion of Yap/Taz in a mouse model of BCC (driven by mutant SMO; K14CreER/SmoM2 mice), efficiently prevented tumour initiation. 14 A different study found that conditional deletion of Yap, but not Taz, significantly reduced the tumour burden of K14CreER/SmoM2 mice while not completely abrogating BCC formation. 165 This suggests that in murine BCC YAP is the dominant paralogue. 165 Importantly, clonal tracing of induced BCC tumours demonstrated that Yap‐null clones had a decreased fitness, initially becoming outcompeted by YAP‐positive clones and ultimately becoming depleted as the tumours progressed to an invasive phenotype. 165 Consistent with these findings, inhibition of YAP/TAZ‐Tead binding was found to lead to rapid elimination of tumour cells in BCC lesions. 89 There is also evidence that in BCC, activation of oncogenic HH signalling in the epidermis may be closely linked to activation of Rock‐dependent mechano‐signalling in the dermal stroma, potentially leading to positive feedback activation of YAP/TAZ 143 (Figure 3).
YAP promotes BCC initiation and progression via TEAD TFs to drive JNK‐Jun signalling both at the level of c‐Jun gene transcription but also upstream of c‐Jun by controlling JNK activation. 165 c‐Jun is a component of the functionally diverse AP‐1 TF complex, and in several cell types, YAP/TAZ/TEAD and AP‐1 were shown to cooperate to drive the expression of target genes involved in the cell cycle control of S‐phase entry and mitosis. 8 , 12 , 76 , 77 Indeed, co‐occupation of chromatin regions by TEADs and AP‐1 TFs was observed also in normal keratinocytes and BCC by ChIP sequencing analysis. 77 , 89 , 165 However, recent findings suggest that YAP/TAZ regulate inflammation‐related gene networks in BCC also independently of TEAD. 89 If cSCC initiation and progression also depends on cooperation of TEADs with AP1 factors remains to be firmly established. Of note, impaired tumour formation in the two‐stage skin carcinogenesis model, where TPA treatment activates AP1 TF complexes, does indeed suggest AP1‐YAP/TAZ/TEAD cooperation in driving cSCC development. 12
Several studies found YAP/TAZ expression to be elevated in most benign and dysplastic nevi and in situ CM, however, without significant differences between lesion types. 173 , 174 , 175 Moreover, none of these studies observed striking fluctuations in the YAP/TAZ nuclear/cytoplasmic ratio (which is often used as a proxy for YAP/TAZ activity) in different stages of CM development. 173 , 174 However, using YAP/TAZ target gene expression as a more robust read out of YAP/TAZ activity, a recent study discovered that YAP/TAZ activity was elevated in invasive CM. 176 This is consistent with an unbiased transcriptomics study of human CM tissues, which revealed TEAD TFs as regulators of the invasive cell state. 177 Interestingly, although BRAF inhibitor‐resistant melanoma cells were shown to depend on YAP/TAZ for their proliferation and survival, 178 YAP/TAZ activity is not associated with the mutation status of BRAF and NRAS. 176 This is consistent with findings that YAP/TAZ sensitivity in CM cells does not correlate with either BRAF or NRAS mutation status. 174 Of note, somatic hypermutations of YAP 1 were also detected in CM, but so far only in a single patient. 174 These YAP mutations manifested as seven serine to alanine transpositions. Since four of these serines are the key regulatory residues phosphorylated by the core Hippo pathway kinases LATS1/LATS2 and NDR1/NDR2, 42 , 43 , 44 the mutant YAP allele was consequently shown to code for a hyperactive YAP protein. 174 While the increased expression of YAP/TAZ in non‐invasive CM could suggest a role in tumour development, studies using patient‐derived CM xenografts and established CM cell lines revealed only a variable requirement of YAP/TAZ/TEAD for cell viability. 174 In contrast, important roles for YAP/TAZ in invasive CM and metastasis have been clearly demonstrated. 173 , 176 The mechanism by which YAP/TAZ activity is promoted in invasive melanoma cells compared with non‐invasive cells is currently unclear. 174 , 176
7. TARGETING YAP AND TAZ FOR SKIN CANCER TREATMENT
In the clinic, the biggest challenge for skin cancers remains treatment of patients with advanced or metastatic disease. 3 , 4 , 8 , 29 , 36 , 179 , 180 There are several excellent comprehensive reviews on current treatment options. 5 , 180 , 181 , 182 Since Hippo signalling acts as a tumor suppressor pathway and aberrant YAP/TAZ activity is implicated in various types of skin cancers, targeting Hippo/YAP/TAZ signalling offers potential opportunities for cancer therapy. Below, we highlight current strategies for therapeutic intervention, some of which are showing promise in initial pre‐clinical studies.
7.1. Targeting YAP/TAZ nuclear shuttling
As transcription co‐regulators, YAP/TAZ exert their activity in the nucleus. Consequently, interfering with the nuclear localization YAP/TAZ could be one approach to inhibit YAP/TAZ activity (Table 1). The photosensitizer verteporfin was found to have significant anti‐tumour effects. 183 , 184 Several reports have indicated that verteporfin can block YAP‐TEAD activity, either through disruption of YAP‐TEAD binding, 185 or through increased cytoplasmic sequestration of YAP by 14–3‐3σ. 186 However, verteporfin‐mediated anti‐tumor effects may not be specific to only inhibiting YAP‐TEAD complexes, as verteporfin has also been reported to have proteotoxic effects. 187 , 188 , 189 A35, a synthetic inhibitor of DNA topoisomerase II, was found to decrease YAP nuclear localization by activating its phosphorylation. 190 Likewise, dichloroacetate, a small molecule metabolic regulator used for treating mitochondrial genetic diseases and lactic acid poisoning, was shown to promote the nuclear‐cytoplasmic translocation of YAP. 191 However, as in the case of verteporfin, the reported anti‐tumour effects of these compounds are likely not YAP/TAZ‐specific.
TABLE 1.
YAP/TAZ nuclear shuttling inhibitors
| Molecule | Other name(s) | Inhibitory action | Binding validation | Activity validation | Binding affinity (KD) | IC50 | Reference(s) | Vendor |
|---|---|---|---|---|---|---|---|---|
| Verteporfin | Visudyne | Inhibitor of YAP nuclear localization | NR | Co‐IP, TEAD luciferase reporter, PLA, YAP‐TEAD target gene qPCR | NR | 8 nM–11.16 μM (Cell line‐dependent) | 184, 185, 195 |
Selleckchem (#S1786) Tocris (#5305) |
| A35 | CHEMBL3041188 | YAP S127 phosphorylation agonist | NR | WB | NR |
<1 μM (Cell line‐dependent) |
190 | |
| Dichloroacetate | Dichloroacetic acid salt | Inhibitor of YAP nuclear localization (Hippo‐dependent) | NR | IFM, WB, RNA‐seq, TEAD luciferase reporter, YAP‐TEAD target gene qPCR | NR | 80–183 μM | 191 |
Selleckchem (#S8615) |
| Dasatinib | Sprycel | Inhibitor of YAP nuclear localization (tyrosine kinase dependent) | NR | IFM, TEAD luciferase reporter, WB, YAP‐TEAD target gene qPCR | NR | ~1 nM | 130 |
Selleckchem (#S1021) Merck (#SML2589) |
| Pazopanib | YAP phosphorylation agonist and promotes proteasomal degradation | NR | IFM, TEAD luciferase reporter, WB | NR | <15 μg/ml | 199 |
Merck (#SML3104) |
|
| CA3 | CIL56 | Inhibitor of YAP expression | NR | IFM, TEAD luciferase reporter, WB, YAP‐TEAD target gene qPCR | NR |
<1 μM (Cell line dependent) |
194, 195 |
Selleckchem (#S8661) |
|
Statins: Cerivastatin Mevastatin Simvastatin Fluvastatin |
Brand names: Baycol Compactin Zocor Lescol |
Inhibitors of YAP nuclear localization via mevalonate pathway (HMG‐CoA reductase) | NR | IFM, TEAD luciferase reporter, WB, YAP‐TEAD target gene qPCR | NR |
<10 μM ~26 nM ~6 μM >0.8 μM (Cell line‐dependent) |
198, 199, 200, 239 |
Merck (#SML0005) (#M2537) (#S6196) (#SML0038) |
|
PI3K‐AKT/PDK1 signalling inhibitors: LY294002 – PI3Ki AKT inhibitor V – AKTi PDK1 inhibitor II – PDK1i Wortmannin – PI3Ki AKT inhibitor VIII – AKTi Calphostin C – PKCi Gö 6983 – PKCi BX795 – PDK1i, TBK1i |
Triciribine | YAP phosphorylation agonists via PDK1‐Hippo‐dependent activity | NR | IFM, YAP‐TEAD target gene qPCR | NR |
<10 μM <20 μM 6 nM 2–4 nM 0.058–2.12 μM 50 nM 6–60 nM 6 nM |
136, 138, 204 |
Merck (#L9908) (#124038) (#521276) (#W1628) (#124017) (#C6303) (#G1918) Santa‐Cruz (#sc‐281 689) |
Abbreviations: Co‐IP, co‐immunoprecipitation; IFM, immunofluorescence microscopy; NR, not reported; PLA, proximity ligation assay; WB, Western blotting.
Dasatinib, a pharmacological inhibitor of SFKs, could have potential as a YAP‐targeting therapy for cSCC. In orthotopic mouse xenograft models, dasatinib treatment caused prominent inhibition of tumour growth through interference with SFK‐induced YAP activation. 130 Of note, topical application of dasatinib onto murine cSCC was found to induce tumour regression with less side effects when compared to treatment with 5‐fluorouracil, one of the standard chemotherapies for cSCC. 192 Suppression of YAP/TAZ activity by SFK inhibition might also be beneficial for the treatment of BRAF inhibitor‐resistant metastatic melanomas, in which YAP‐induced PD‐L1 expression drives immune evasion. 131 , 193
CA3, a small molecule with anti‐tumour activity in various cancers, notably including head and neck SCC and cSCC, appears to act by reducing YAP expression, probably at the level of YAP1 gene transcription. 172 , 194 , 195 , 196 , 197
Statins, a class of drugs used to lower the level of low‐density lipoprotein (LDL) cholesterol in the blood, block YAP/TAZ nuclear localization and activity through Rho‐GTPases. 198 , 199 , 200 Combined EGFR and YAP inhibition (with statins) prolongs survival in lung cancer patients. 201 Consequently, a similar targeted approach could potentially be applied to treat invasive BRAF inhibitor‐resistant melanoma with EGFR overexpression 199 , 202 , 203 and YAP/TAZ dependency. 178 , 193 Other interesting compounds with documented YAP/TAZ inhibition capacity that could be used for combination therapies (together with SFK or statin inhibitors) are multi‐tyrosine kinase inhibitors such as pazopanip and PI3K‐AKT inhibitors. 136 , 138 , 199 , 204
7.2. Targeting the YAP/TAZ‐TEAD interface
YAP/TAZ are natively unfolded proteins, making it a challenge to target them directly. However, YAP/TAZ become structured upon binding to TEADs. 205 , 206 , 207 This enables targeting of the YAP/TAZ‐TEAD binding interface by protein–protein binding disruptors (PPBDs). 208 Crystal structures of the YAP‐TEAD complex have revealed that the C‐terminal YAP‐binding domain of the TEADs comprises an immunoglobulin‐like β‐sandwich fold structure and two helix‐turn‐helix motifs. 205 , 207 , 209 , 210 , 211 The N‐terminal TEAD‐binding motif of YAP adopts a helix–loop–helix structure, with helix α1 and helix α2 forming the main hydrophobic and hydrogen‐bond interactions with TEADs. 205 , 207 , 209 , 210 , 211 , 212 The loop region in YAP is relatively long and also forms interactions with TEADs. Consequently, TAZ‐TEAD binding differs slightly from YAP‐TEAD binding because of a shorter loop region. 212 Interestingly, the TAZ‐TEAD crystal structure revealed two possible binding modes of the co‐regulator with its transcription factor (1:1 and 2:2 complexes), with implications for target gene expression. 212 However, despite these structural differences, PPBDs able to disrupt the YAP‐TEAD interaction should have the ability to also disrupt the TAZ‐TEAD interaction.
The interaction between the N‐terminal motif of YAP/TAZ and the C‐terminal YAP/TAZ‐binding domain in TEADs involves three highly conserved interfaces, with the third interface being the major energetic determinant of high‐affinity binding. 205 , 207 , 209 , 210 , 211 , 212 The crystal structures of YAP‐TEAD complexes revealed clear surface pockets at the second and third interfaces that might enable the rational design of PPBDs 163 , 208 (Table 2). Of note, residues within the YAP/TAZ‐binding TEAD pocket at the third binding interface are highly conserved across all TEAD family members, 163 , 207 , 210 , 211 suggesting that targeting this interface could offer possible pan‐TEAD ligands. Indeed, various efforts to target the second and third YAP‐TEAD interface have yielded promising peptides and small molecules, 208 , 213 which are summarized in Table 2. Another pocket in the centre of the C‐terminal YAP‐binding domain of TEADs is also accessible to small molecules. 209 Palmitate is the natural ligand that binds to the central pocket. 214 , 215 Several central pocket binders have been identified that inhibit TEAD palmitoylation, 208 , 213 with some of them acting as allosteric PPBDs by disrupting YAP‐TEAD interaction, while others are not (Table 3). The permeability, specificity, efficacy, in vivo impact and, ultimately, safety of all these inhibitors remains to be determined.
TABLE 2.
YAP/TAZ‐TEAD inhibitors of protein–protein interaction, surface binding
| Molecule | Other name(s) | Inhibitory action | Binding validation | Activity validation | Interface | Binding affinity (KD) | IC50 | References | Vendor |
|---|---|---|---|---|---|---|---|---|---|
| TB1G1/TB1G2 | Cysteine‐dense peptide (CDP) | iPPI | Rosetta protein design, mammalian surface display, SPR, Co‐IP, PLA | TEAD luciferase reporter, competitive mammalian surface display | 2 |
TB1G1: 31 ± 2 nM TB1G2: 368 ± 4 pM |
NR | 240 | |
| Fragment 1 | iPPI scaffold | Co‐crystal structure | ITC, TEAD luciferase reporter | 2 | ~300 μM | NR | 241 | Chemspace (#CSSB00000239375) | |
| Peptide 17 | YAP‐TEAD inhibitor 1 | iPPI | Molecular docking, co‐crystal structure, pull‐down assay | Pull‐down assay | 3 | 15 nM | 25 nM | 242 |
Selleckchem (#S8164) AdooQ (#A15858) APExBIO (#A1149) MedChemExpress (#HY‐P2244) |
| Peptide 10 | iPPI | Co‐crystal structure, SPR | Pull‐down assay | 3 | 289.5 nM | 297 nM | 243 | ||
| Peptides 9, 10 | iPPI | Co‐crystal structure | TR‐FRET | 3 |
Peptide 9: 25 nM Peptide 10: NR |
Peptide 9: 16 ± 5 nM Peptide 10: 9 ± 2 nM |
244 | ||
| TEAD‐binding fragment | MFCD00187673 | iPPI scaffold | NMR | ITC | 3 | 77 nM | NR | 245 |
Chemspace (#CSC000003413) |
| Dioxo‐benzoisothiazole Example 22 | iPPI | NMR | TEAD luciferase reporter, AlphaLISA® | 3 | NR | 83 nM | 246 | patented: WO2017064277A1 | |
| 1,2,3‐Triazole‐4‐carbohydrazide derivatives (hit 2) | iPPI | Molecular docking, TSA | MST, TSA, TEAD luciferase reporter, YAP‐TEAD target gene qPCR | 3 | 650 μM | 6.5 μM | 247 |
MCule (#MCULE‐3696035303) MolPort (# MolPort‐002‐604‐580) |
|
| Compound 3.1 |
Pyrazidol, Pirlindol, Pirlindolum |
iPPI | Molecular docking, STD NMR | Co‐IP, TEAD luciferase reporter, pull‐down assay, YAP‐TEAD target gene qPCR | 3 | ~12 μM | 33–44 μM (TEAD isoform‐dependent) | 248 |
Chemspace (#CSC020600808) |
| Super‐TDU | iPPI | Molecular modelling | Co‐IP, mutational studies | 2&3 | NR | 57.9 ng/ml | 249 |
Selleckchem (#S8554) MedChemExpress (#HY‐P1727) |
Abbreviations: AlphaLISA, amplified luminescent proximity homogenous assay (linked immunosorbent assay); Co‐IP, co‐immunoprecipitation; iPPI, inhibitor of protein–protein interaction; ITC, isothermal titration calorimetry; MST, microscale thermophoresis; NMR, nuclear magnetic resonance; NR, not reported; PLA, proximity ligation assay; SPR, surface plasmon resonance; STD NMR, saturation transfer difference nuclear magnetic resonance; TR‐FRET, time‐resolved fluorescence resonance energy transfer; TSA, thermal shift assay.
TABLE 3.
YAP/TAZ‐TEAD inhibitors of protein–protein interaction, central pocket binding
| Molecule | Other name(s) | Inhibitory action | Binding validation | Activity validation | Interface | Binding affinity (KD) | IC50 | Reference(s) | Vendor |
|---|---|---|---|---|---|---|---|---|---|
| Flufenamic acid |
Paraflu, Parlef, Ristogen, Sastridex, Tecramine |
API | Co‐crystal structure, molecular modelling, STD NMR | ITC, TEAD luciferase reporter | Central pocket & interface 3 (weak affinity) | 73 μM | NR | 209 |
Tocris (#4522) Merck (#151300) |
| Niflumic acid |
Donalgin, Niflam, Forenol |
API | Co‐crystal structure, molecular modelling, STD NMR | ITC, TEAD luciferase reporter | Central pocket & interface 3 (weak affinity) | 28 μM | NR | 209 |
Tocris (#4112) Merck (#N0630) |
| TED‐347 (compound 2) | API—covalent | Co‐crystal structure, molecular docking, molecular dynamics simulation, protein mass spectrometry, FP |
FP, Biolayer interferometry, Co‐IP, TEAD luciferase reporter, YAP‐TEAD target gene qPCR |
Central pocket | NR | 5.9 μM | 250 |
Selleckchem (#S8951) MedChemExpress (#HY‐125269) |
|
| MYF‐01‐037 | API –covalent | Molecular docking | YAP‐TEAD split Gaussia luciferase assay, YAP‐TEAD target gene qPCR | Central pocket | NR | 0.8 μM | 251 |
Selleckchem (#S8950) Cambridge Bioscience (#B3298‐5) |
|
| Non‐fused tricyclic compound 42 | NR | NR | TEAD luciferase reporter | Central pocket | NR | <0.1 μM | 252 | Patented: WO2018204532A1 | |
| MGH‐CP1 | API | Co‐crystal structure | Co‐IP, TEAD luciferase reporter | Central pocket | NR | 672–710 nM (TEAD isoform‐dependent) | 253 |
Selleckchem (#S9735) |
|
| Indole incorporated triazine derivatives (e.g compound 9) | API | Molecular docking, NanoDSF, FP | NanoDSF, FP, YAP‐TEAD target gene qPCR | Central pocket | NR | 6.75 μM | 254 | ||
| Dihydropyrazolo pyramidines (e.g. compound 7) | NR | NR | TR‐FRET | Central pocket | NR | 18–87 nM (TEAD isoform‐dependent) | 255 | Patented: WO2019232216A1 | |
| K‐975 | K975 | API | Co‐crystal structure, SPR | SPR, pull down assay, YAP‐TEAD target gene qPCR | Central pocket | NR | NR | 256 |
MedChemExpress (#HY‐138565) |
| Kojic acid‐derived Betti bases (e.g. compound 19) | API | FP, whole‐protein ESI–MS spectrometry, NMR | Thiol conjugation assay, FP, cellular thermal shift assay | Central pocket | 28 nM | 0.2 ± 0.04 μM | 257 | ||
| Compound 2 | API | Co‐crystal structure, FP, SPR | FP, TR‐FRET, Co‐IP, SPR | Central pocket | 229 nM | 31.8 nM | 258 | ||
| DC‐TEADin02 | DCTEADin02 | API—covalent | Molecular docking, NMR, SPR, mass spectrometry | Pull‐down assay, TEAD luciferase reporter, YAP‐TEAD target gene qPCR | Central pocket | NR | 197 ± 19 nM | 259 |
MedKoo (#463183) |
| Quinolinol Q2 | API | Molecular docking, molecular dynamics simulation | TEAD luciferase reporter, RNA‐seq | Central pocket | 2.6 ± 0.3 μM | 2.6 μM | 260 |
Hit2Lead (#5926377) Mcule (#MCULE‐5191032439) MolPort (#MolPort‐003‐183‐526) |
Abbreviations: API, autopalmitoylation inhibitor; Co‐IP, co‐immunoprecipitation; ESI‐MS, electrospray ionization mass spectrometry; FP, fluorescence polarization; ITC, isothermal titration calorimetry; MST, microscale thermophoresis; NMR, nuclear magnetic resonance; Nano‐DSF, nano differential scanning fluorimetry; NR, not reported; PLA, proximity ligation assay; SPR, surface plasmon resonance; STD NMR, saturation transfer difference nuclear magnetic resonance; TR‐FRET, time‐resolved fluorescence resonance energy transfer; TSA, thermal shift assay.
7.3. Targeting the YAP/TAZ‐associated transcriptional machinery
Once in the nucleus, YAP/TAZ execute their biological functions by regulating gene transcription through their association with various TFs, as well as chromatin remodelling protein complexes including the switch/sucrose nonfermentable (SWI/SNF) and nucleosome remodelling and deacetylase (NuRD) complexes (reviewed in 216 ). Constitutive nuclear YAP/TAZ expression in cancer cells promotes hyper‐transcription at YAP/TAZ target genes and a dependency on YAP/TAZ‐driven transcriptional programmes. 78 , 217 Such transcriptional dependencies are amenable to inhibition with small molecules. 218
For example, in triple‐negative breast cancer cells, YAP/TAZ‐driven transcriptional addiction is mediated through association with the bromodomain and extraterminal (BET)‐coactivator protein BRD4, and consequently YAP/TAZ pro‐tumorigenic activity was shown to be vulnerable to treatment with the BET inhibitor JQ1. 78
Gene repressive activity by YAP/TAZ in breast epithelial cells was shown to depend on recruitment of the NuRD complex, which has both nucleosome‐remodelling and histone deacetylase (HDAC) activity, to render chromatin inaccessible. 219 Therefore, HDAC inhibitors may represent a potential therapeutic avenue for specifically targeting the NuRD‐mediated co‐repressor functions of YAP/TAZ. Of note, the HDAC inhibitor vorinostat is FDA approved for the treatment of cutaneous T‐cell lymphoma and is being investigated for the topical treatment of cutaneous malignancies 220 and in BRAFV600E melanoma. 221
The functions of SWI/SNF complexes in squamous cell cancers are ambiguous, as both pro‐tumorigenic and tumour‐suppressive roles have been identified in studies focusing on head and neck SCC. 222 , 223 , 224 A key factor in this puzzle appears to be the SWI/SNF subunit ACTL6A, which, when overexpressed, stoichiometrically assembles into BAF‐type SWI/SNF complexes which then drive chromatin loading of TEAD‐YAP complexes. 222 Should a similar mechanism operate in cSCC, targeting the catalytic subunits of SWI/SNF complexes could be used as therapeutic strategy. 225
Cooperation of YAP/TAZ‐TEAD with AP1 TFs has been documented in both BCC and cSCC cells. 12 , 89 , 165 Multiple AP‐1 inhibitors including small molecules and peptides have been assessed in preclinical and clinical trials, 226 , 227 , 228 therefore providing a toolbox to potentially interfere with the YAP/TAZ‐TEAD‐AP‐1 complex.
High resolution mapping of DNA double–strand breaks (DSBs) in cancer and non‐tumorigenic cells revealed a transcription‐coupled DNA damage repair mechanism at oncogenic super‐enhancers. 229 This mechanism involves high transcriptional activity mediated by YAP‐TEAD and AP‐1 factors, which leads to DNA topoisomerase (TOP1)‐mediated induction of DSBs, which are repaired through the homologous recombination DNA damage repair pathway. Depletion of TEAD4 or RAD51 increased DSBs at RAD51/TEAD4 common binding sites within super‐enhancers and decreased expression of related genes, which are mostly oncogenes. 229 These findings therefore suggest that, at least in certain cancer types, targeting YAP/TAZ‐TEAD could help to selectively reduce transcription of oncogenes.
8. CONCLUSIONS/MAJOR OPEN QUESTIONS
Genetic studies in mice have unequivocally demonstrated that YAP/TAZ are essential for BCC and cSCC initiation and progression. 12 , 14 , 152 , 165 However, due to the inherent differences between mouse and human skin and limitations of genetic mouse models of skin cancers, 230 , 231 , 232 it remains to be vigorously tested if this is true also in the human skin cancer context. That said, we must also acknowledge that there are limitations to the use of human cells/tissues as the correct physiological context (including an appropriate microenvironment) is difficult (if not impossible) to reproduce in vitro. So far, only few studies have assessed the roles of YAP/TAZ in vitro in human skin cancer models, the majority focusing on melanoma. 35 , 59 , 131 , 167 , 174 , 176 , 178 Additional human cell/tissue‐focused studies are therefore required to address several open questions and to complement studies done in mice and other animal model systems: (1) Is there genetic evidence for YAP/TAZ hyperactivation in human skin cancers? SCCs of the lung, oesophagus and head and neck display the highest levels of YAP1 or WWTR1 gene amplifications across multiple cancer types, often in a mutually exclusive manner. 94 , 161 Indeed, a comprehensive survey of the genomic landscape of human cSCC was able to detect evidence of YAP1 amplification in a small set of in situ and invasive cSCC samples. 32 , 233 Another study focusing on BCC detected inactivating mutations in LATS1 and PTPN14 in 16% and 23% of BCC samples, respectively, as well as missense mutations in LATS2 (12% of analysed BCCs). 234 Clearly, a more targeted analysis of genomic alterations of Hippo/YAP/TAZ components across various skin cancer types is therefore warranted. (2) What are the distinct roles and regulatory mechanisms of YAP/TAZ in skin cancer? As discussed in section 5, the mechanisms controlling YAP/TAZ nuclear localization and transcriptional activity in skin cancer are still poorly understood. The majority of past studies on YAP/TAZ focused on either one of the paralogues or had simply assumed similar functions between them. However, emerging evidence demonstrates that YAP and TAZ have distinct roles where they partner with different transcription factors, drive different transcriptional programs and also modulate the tumour microenvironment distinctively. 69 , 87 , 91 , 94 , 235 , 236 This question can be addressed with comprehensive RNAi or CRISPR‐based gene knock‐down/knock‐out studies, ideally performed across a panel of human skin cancer models and employing 3D organotypic skin cancer models to test for physiological relevance. 237 , 238 Such studies could also provide dermatologists with prognostic cSCC and BCC‐specific YAP/TAZ signatures, as exists already for melanoma. 176 These gene expression signatures could be particularly important if—similar to melanoma—parameters such as YAP/TAZ expression or their nuclear localization turn out to serve as poor predictors of YAP/TAZ activity. 176 (3) Do different types of CM display different dependencies on Hippo/YAP/TAZ signalling? To the best of our knowledge, a comprehensive investigation, using primary cells/cell lines and human tissue sample stratified according to the current WHO classification of human CM has not been performed. (4) How can we target YAP/TAZ‐dependencies in skin cancer? In addition to the studies that comprehensively investigate the cellular functions of YAP/TAZ and their transcriptional outputs, translational efforts will also benefit from focused proteomics studies aimed at characterizing the YAP/TAZ interactome in skin cancer cells. This could lead to the discovery of YAP/TAZ‐associated proteins that can possibly be targeted by existing compounds.
AUTHOR CONTRIBUTIONS
GW and UJ conceptualized the manuscript. AH prepared figures and Tables. AH, JB, BF, SB, UJ and GW performed literature review and wrote different paragraphs.
FUNDING INFORMATION
This work was supported by grants from the Biotechnology and Biological Sciences Research Council (grant BB/T012978/1), the Academy of Medical Sciences (grant SBF005\1005) and the British Skin Foundation (grant 036/S/18)(to Gernot Walko).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We thank Prof Randy Mrsny (Department of Pharmacy and Pharmacology, University of Bath) for critical feedback on our draft manuscript. We apologize to those authors we were unable to cite within this review due to space constraints.
Howard A, Bojko J, Flynn B, Bowen S, Jungwirth U, Walko G. Targeting the Hippo/YAP/TAZ signalling pathway: Novel opportunities for therapeutic interventions into skin cancers. Exp Dermatol. 2022;31:1477‐1499. doi: 10.1111/exd.14655
Alexander Howard and Jodie Bojko contributed equally to the work.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Gonzales KAU, Fuchs E. Skin and its regenerative powers: an alliance between stem cells and their niche. Dev cell. 2017;43(4):387‐401. doi: 10.1016/j.devcel.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Belokhvostova D, Berzanskyte I, Cujba AM, et al. Homeostasis, regeneration and tumour formation in the mammalian epidermis. Int J Dev Biol. 2018;62(6‐7‐8):571‐582. doi: 10.1387/ijdb.170341fw [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fernandes AR, Santos AC, Sanchez‐Lopez E, et al. Neoplastic multifocal skin lesions: biology, etiology, and targeted therapies for nonmelanoma skin cancers. Skin Pharmacol Physiol. 2018;31(2):59‐73. doi: 10.1159/000479529 [DOI] [PubMed] [Google Scholar]
- 4. Didona D, Paolino G, Bottoni U, Cantisani C. Non melanoma skin cancer pathogenesis overview. Biomedicines. 2018;6(1):6. doi: 10.3390/biomedicines6010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kasakovski D, Skrygan M, Gambichler T, Susok L. Advances in targeting cutaneous melanoma. Cancers (Basel). 2021;13(9):2090. doi: 10.3390/cancers13092090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Apalla Z, Nashan D, Weller RB, Castellsague X. Skin cancer: epidemiology, disease burden, pathophysiology, diagnosis, and therapeutic approaches. Dermatol Ther (Heidelb). 2017;7(Suppl 1):5‐19. doi: 10.1007/s13555-016-0165-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Curti BD, Faries MB. Recent advances in the treatment of melanoma. N Engl J Med. 2021;384(23):2229‐2240. doi: 10.1056/NEJMra2034861 [DOI] [PubMed] [Google Scholar]
- 8. Rognoni E, Walko G. The roles of YAP/TAZ and the Hippo pathway in healthy and diseased skin. Cells. 2019;8(5):411. doi: 10.3390/cells8050411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29(6):783‐803. doi: 10.1016/j.ccell.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andl T, Zhou L, Yang K, Kadekaro AL, Zhang Y. YAP and WWTR1: new targets for skin cancer treatment. Cancer Lett. 2017;396:30‐41. doi: 10.1016/j.canlet.2017.03.001 [DOI] [PubMed] [Google Scholar]
- 11. Moon S, Yeon Park S, Woo PH. Regulation of the Hippo pathway in cancer biology. Cell Mol Life Sci. 2018;75(13):2303‐2319. doi: 10.1007/s00018-018-2804-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zanconato F, Forcato M, Battilana G, et al. Genome‐wide association between YAP/TAZ/TEAD and AP‐1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17(9):1218‐1227. doi: 10.1038/ncb3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elbediwy A, Vincent‐Mistiaen ZI, Spencer‐Dene B, et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development. 2016;143(10):1674‐1687. doi: 10.1242/dev.133728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Debaugnies M, Sanchez‐Danes A, Rorive S, et al. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep. 2018;19(7):e45809. doi: 10.15252/embr.201845809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dekoninck S, Blanpain C. Stem cell dynamics, migration and plasticity during wound healing. Nat Cell Biol. 2019;21(1):18‐24. doi: 10.1038/s41556-018-0237-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rognoni E, Watt FM. Skin cell heterogeneity in development, wound healing, and cancer. Trends Cell Biol. 2018;28(9):709‐722. doi: 10.1016/j.tcb.2018.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schepeler T, Page ME, Jensen KB. Heterogeneity and plasticity of epidermal stem cells. Development. 2014;141(13):2559‐2567. doi: 10.1242/dev.104588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flora P, Ezhkova E. Regulatory mechanisms governing epidermal stem cell function during development and homeostasis. Development. 2020;147(22):dev194100. doi: 10.1242/dev.194100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirsch T, Rothoeft T, Teig N, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551(7680):327‐332. doi: 10.1038/nature24487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Rosa L, Latella MC, Secone Seconetti A, et al. Toward combined cell and gene therapy for genodermatoses. Cold Spring Harb Perspect Biol. 2020;12(5):a035667. doi: 10.1101/cshperspect.a035667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guan Y, Yang YJ, Nagarajan P, Ge Y. Transcriptional and signalling regulation of skin epithelial stem cells in homeostasis, wounds and cancer. Exp Dermatol. 2021;30(4):529‐545. doi: 10.1111/exd.14247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN Estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209‐249. doi: 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 23. Green AC, Olsen CM. Cutaneous squamous cell carcinoma: an epidemiological review. Br J Dermatol. 2017;177(2):373‐381. doi: 10.1111/bjd.15324 [DOI] [PubMed] [Google Scholar]
- 24. Kasper M, Jaks V, Hohl D, Toftgard R. Basal cell carcinoma ‐ molecular biology and potential new therapies. J Clin Invest. 2012;122(2):455‐463. doi: 10.1172/JCI58779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peterson SC, Eberl M, Vagnozzi AN, et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell stem cell. 2015;16(4):400‐412. doi: 10.1016/j.stem.2015.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grachtchouk M, Pero J, Yang SH, et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. J Clin Invest. 2011;121(5):1768‐1781. doi: 10.1172/JCI46307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Colmont CS, Benketah A, Reed SH, et al. CD200‐expressing human basal cell carcinoma cells initiate tumor growth. Proc Natl Acad Sci USA. 2013;110(4):1434‐1439. doi: 10.1073/pnas.1211655110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ratushny V, Gober MD, Hick R, Ridky TW, Seykora JT. From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J Clin Invest. 2012;122(2):464‐472. doi: 10.1172/JCI57415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burton KA, Ashack KA, Khachemoune A. Cutaneous squamous cell carcinoma: a review of high‐risk and metastatic disease. Am J Clin Dermatol. 2016;17(5):491‐508. doi: 10.1007/s40257-016-0207-3 [DOI] [PubMed] [Google Scholar]
- 30. Sanchez‐Danes A, Blanpain C. Deciphering the cells of origin of squamous cell carcinomas. Nat Rev Cancer. 2018;18(9):549‐561. doi: 10.1038/s41568-018-0024-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hofbauer GF, Bouwes Bavinck JN, Euvrard S. Organ transplantation and skin cancer: basic problems and new perspectives. Exp Dermatol. 2010;19(6):473‐482. doi: 10.1111/j.1600-0625.2010.01086.x [DOI] [PubMed] [Google Scholar]
- 32. Inman GJ, Wang J, Nagano A, et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat Commun. 2018;9(1):3667. doi: 10.1038/s41467-018-06027-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Gruijl FR, Tensen CP. Pathogenesis of skin carcinomas and a stem cell as focal origin. Front Med (Lausanne). 2018;5:165. doi: 10.3389/fmed.2018.00165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thieu K, Ruiz ME, Owens DM. Cells of origin and tumor‐initiating cells for nonmelanoma skin cancers. Cancer Lett. 2013;338(1):82‐88. doi: 10.1016/j.canlet.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pastushenko I, Mauri F, Song Y, et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature. 2021;589(7842):448‐455. doi: 10.1038/s41586-020-03046-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schadendorf D, van Akkooi ACJ, Berking C, et al. Melanoma. Lancet. 2018;392(10151):971‐984. doi: 10.1016/S0140-6736(18)31559-9 [DOI] [PubMed] [Google Scholar]
- 37. Shain AH, Bastian BC. From melanocytes to melanomas. Nat Rev Cancer. 2016;16(6):345‐358. doi: 10.1038/nrc.2016.37 [DOI] [PubMed] [Google Scholar]
- 38. Elder DE, Bastian BC, Cree IA, Massi D, Scolyer RA. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: detailed analysis of 9 distinct subtypes defined by their evolutionary pathway. Arch Pathol Lab Med. 2020;144(4):500‐522. doi: 10.5858/arpa.2019-0561-RA [DOI] [PubMed] [Google Scholar]
- 39. Leonardi GC, Falzone L, Salemi R, et al. Cutaneous melanoma: from pathogenesis to therapy (Review). Int J Oncol. 2018;52(4):1071‐1080. doi: 10.3892/ijo.2018.4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marzagalli M, Ebelt ND, Manuel ER. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin Cancer Biol. 2019;59:236‐250. doi: 10.1016/j.semcancer.2019.08.002 [DOI] [PubMed] [Google Scholar]
- 41. Arozarena I, Wellbrock C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer. 2019;19(7):377‐391. doi: 10.1038/s41568-019-0154-4 [DOI] [PubMed] [Google Scholar]
- 42. Ma S, Meng Z, Chen R, Guan KL. The Hippo pathway: biology and pathophysiology. Annu Rev Biochem. 2019;88:577‐604. doi: 10.1146/annurev-biochem-013118-111829 [DOI] [PubMed] [Google Scholar]
- 43. Moya IM, Halder G. Hippo‐YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2018;20(4):211‐226. doi: 10.1038/s41580-018-0086-y [DOI] [PubMed] [Google Scholar]
- 44. Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30(1):1‐17. doi: 10.1101/gad.274027.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hansen CG, Moroishi T, Guan KL. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25(9):499‐513. doi: 10.1016/j.tcb.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Callus BA, Verhagen AM, Vaux DL. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C‐terminal coiled‐coil domains, leads to its stabilization and phosphorylation. FEBS J. 2006;273(18):4264‐4276. doi: 10.1111/j.1742-4658.2006.05427.x [DOI] [PubMed] [Google Scholar]
- 47. Deng Y, Pang A, Wang JH. Regulation of mammalian STE20‐like kinase 2 (MST2) by protein phosphorylation/dephosphorylation and proteolysis. J Biol Chem. 2003;278(14):11760‐11767. doi: 10.1074/jbc.M211085200 [DOI] [PubMed] [Google Scholar]
- 48. Praskova M, Xia F, Avruch J. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr Biol. 2008;18(5):311‐321. doi: 10.1016/j.cub.2008.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. The Ste20‐like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24(12):2076‐2086. doi: 10.1038/sj.onc.1208445 [DOI] [PubMed] [Google Scholar]
- 50. Hergovich A, Schmitz D, Hemmings BA. The human tumour suppressor LATS1 is activated by human MOB1 at the membrane. Biochem Biophys Res Commun. 2006;345(1):50‐58. doi: 10.1016/j.bbrc.2006.03.244 [DOI] [PubMed] [Google Scholar]
- 51. Meng Z, Moroishi T, Mottier‐Pavie V, et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun. 2015;6:8357. doi: 10.1038/ncomms9357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zheng Y, Wang W, Liu B, Deng H, Uster E, Pan D. Identification of Happyhour/MAP4K as alternative Hpo/Mst‐like kinases in the Hippo Kinase Cascade. Dev Cell. 2015;34(6):642‐655. doi: 10.1016/j.devcel.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154(6):1342‐1355. doi: 10.1016/j.cell.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sharif AAD, Hergovich A. The NDR/LATS protein kinases in immunology and cancer biology. Semin Cancer Biol. 2018;48:104‐114. doi: 10.1016/j.semcancer.2017.04.010 [DOI] [PubMed] [Google Scholar]
- 55. Liu CY, Zha ZY, Zhou X, et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}‐TrCP E3 ligase. J Biol Chem. 2010;285(48):37159‐37169. doi: 10.1074/jbc.M110.152942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747‐2761. doi: 10.1101/gad.1602907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kwon H, Kim J, Jho EH. Role of the Hippo pathway and mechanisms for controlling cellular localization of YAP/TAZ. FEBS J. 2021. doi: 10.1111/febs.16091. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 58. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta‐TRCP). Genes Dev. 2010;24(1):72‐85. doi: 10.1101/gad.1843810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Walko G, Woodhouse S, Pisco AO, et al. A genome‐wide screen identifies YAP/WBP2 interplay conferring growth advantage on human epidermal stem cells. Nat Commun. 2017;8:14744. doi: 10.1038/ncomms14744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Papaspyropoulos A, Bradley L, Thapa A, et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat Commun. 2018;9(1):424. doi: 10.1038/s41467-017-02786-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Das A, Fischer RS, Pan D, Waterman CM. YAP nuclear localization in the absence of cell‐cell contact is mediated by a filamentous actin‐dependent, myosin II‐ and phospho‐YAP‐independent pathway during extracellular matrix mechanosensing. J Biol Chem. 2016;291(12):6096‐6110. doi: 10.1074/jbc.M115.708313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Elbediwy A, Vanyai H, Diaz‐de‐la‐Loza MD, Frith D, Snijders AP, Thompson BJ. Enigma proteins regulate YAP mechanotransduction. J Cell Sci. 2018;131(22):jcs221788. doi: 10.1242/jcs.221788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Q, Zhang N, Xie R, et al. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev. 2015;29(12):1285‐1297. doi: 10.1101/gad.264234.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moroishi T, Park HW, Qin B, et al. A YAP/TAZ‐induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015;29(12):1271‐1284. doi: 10.1101/gad.262816.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. He C, Lv X, Huang C, et al. YAP1‐LATS2 feedback loop dictates senescent or malignant cell fate to maintain tissue homeostasis. EMBO Rep. 2019;20(3):e44948. doi: 10.15252/embr.201744948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kofler M, Speight P, Little D, Di Ciano‐Oliveira C, Szaszi K, Kapus A. Mediated nuclear import and export of TAZ and the underlying molecular requirements. Nat Commun. 2018;9(1):4966. doi: 10.1038/s41467-018-07450-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ege N, Dowbaj AM, Jiang M, et al. Quantitative analysis reveals that actin and src‐family kinases regulate nuclear YAP1 and its export. Cell Syst. 2018;6(6):692‐708. doi: 10.1016/j.cels.2018.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Manning SA, Dent LG, Kondo S, Zhao ZW, Plachta N, Harvey KF. Dynamic fluctuations in subcellular localization of the Hippo pathway effector Yorkie in vivo. Curr Biol. 2018;28(10):1651‐1660. doi: 10.1016/j.cub.2018.04.018 [DOI] [PubMed] [Google Scholar]
- 69. Reggiani F, Gobbi G, Ciarrocchi A, Sancisi V. YAP and TAZ are not identical twins. Trends Biochem Sci. 2021;46(2):154‐168. doi: 10.1016/j.tibs.2020.08.012 [DOI] [PubMed] [Google Scholar]
- 70. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. 2018;20(8):888‐899. doi: 10.1038/s41556-018-0142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Currey L, Thor S, Piper M. TEAD family transcription factors in development and disease. Development. 2021;148(12):dev196675. doi: 10.1242/dev.196675 [DOI] [PubMed] [Google Scholar]
- 72. Oh H, Slattery M, Ma L, et al. Genome‐wide association of Yorkie with chromatin and chromatin‐remodeling complexes. Cell Rep. 2013;3(2):309‐318. doi: 10.1016/j.celrep.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oh H, Slattery M, Ma L, White KP, Mann RS, Irvine KD. Yorkie promotes transcription by recruiting a histone methyltransferase complex. Cell Rep. 2014;8(2):449‐459. doi: 10.1016/j.celrep.2014.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ikmi A, Gaertner B, Seidel C, Srivastava M, Zeitlinger J, Gibson MC. Molecular evolution of the Yap/Yorkie proto‐oncogene and elucidation of its core transcriptional program. Mol Biol Evol. 2014;31(6):1375‐1390. doi: 10.1093/molbev/msu071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Galli GG, Carrara M, Yuan WC, et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell. 2015;60(2):328‐337. doi: 10.1016/j.molcel.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stein C, Bardet AF, Roma G, et al. YAP1 exerts its transcriptional control via TEAD‐mediated activation of enhancers. PLoS Genet. 2015;11(8):e1005465. doi: 10.1371/journal.pgen.1005465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yuan Y, Park J, Feng A, et al. YAP1/TAZ‐TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat Commun. 2020;11(1):1472. doi: 10.1038/s41467-020-15301-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zanconato F, Battilana G, Forcato M, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med. 2018;24(10):1599‐1610. doi: 10.1038/s41591-018-0158-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Koontz LM, Liu‐Chittenden Y, Yin F, et al. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped‐mediated default repression. Dev Cell. 2013;25(4):388‐401. doi: 10.1016/j.devcel.2013.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Guo T, Lu Y, Li P, et al. A novel partner of Scalloped regulates Hippo signaling via antagonizing Scalloped‐Yorkie activity. Cell Res. 2013;23(10):1201‐1214. doi: 10.1038/cr.2013.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Murakami M, Nakagawa M, Olson EN, Nakagawa O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt‐Oram syndrome. Proc Natl Acad Sci USA. 2005;102(50):18034‐18039. doi: 10.1073/pnas.0509109102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rosenbluh J, Nijhawan D, Cox AG, et al. beta‐Catenin‐driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151(7):1457‐1473. doi: 10.1016/j.cell.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vitolo MI, Anglin IE, Mahoney WM Jr, et al. The RUNX2 transcription factor cooperates with the YES‐associated protein, YAP65, to promote cell transformation. Cancer Biol Ther. 2007;6(6):856‐863. [DOI] [PubMed] [Google Scholar]
- 84. Jang JW, Kim MK, Lee YS, et al. RAC‐LATS1/2 signaling regulates YAP activity by switching between the YAP‐binding partners TEAD4 and RUNX3. Oncogene. 2017;36(7):999‐1011. doi: 10.1038/onc.2016.266 [DOI] [PubMed] [Google Scholar]
- 85. Strano S, Monti O, Pediconi N, et al. The transcriptional coactivator Yes‐associated protein drives p73 gene‐target specificity in response to DNA damage. Mol Cell. 2005;18(4):447‐459. doi: 10.1016/j.molcel.2005.04.008 [DOI] [PubMed] [Google Scholar]
- 86. Strano S, Munarriz E, Rossi M, et al. Physical interaction with Yes‐associated protein enhances p73 transcriptional activity. J Biol Chem. 2001;276(18):15164‐15173. doi: 10.1074/jbc.M010484200 [DOI] [PubMed] [Google Scholar]
- 87. He L, Pratt H, Gao M, Wei F, Weng Z, Struhl K. YAP and TAZ are transcriptional co‐activators of AP‐1 proteins and STAT3 during breast cellular transformation. eLife. 2021;10:e67312. doi: 10.7554/eLife.67312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhao R, Fallon TR, Saladi SV, et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self‐renewal of stem cells. Dev Cell. 2014;30(2):151‐165. doi: 10.1016/j.devcel.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yuan Y, Salinas Parra N, Chen Q, Iglesias‐Bartolome R. Oncogenic Hedgehog‐smoothened signaling depends on YAP1‐TAZ/TEAD transcription to restrain differentiation in basal cell carcinoma. J Invest Dermatol. 2021;142(1):65‐76. doi: 10.1016/j.jid.2021.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu X, Li H, Rajurkar M, et al. Tead and AP1 coordinate transcription and motility. Cell Rep. 2016;14(5):1169‐1180. doi: 10.1016/j.celrep.2015.12.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Plouffe SW, Lin KC, Moore JL 3rd, et al. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J Biol Chem. 2018;293(28):11230‐11240. doi: 10.1074/jbc.RA118.002715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sun C, De Mello V, Mohamed A, et al. Common and distinctive functions of the Hippo effectors Taz and Yap in skeletal muscle stem cell function. Stem cells. 2017;35(8):1958‐1972. doi: 10.1002/stem.2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shreberk‐Shaked M, Dassa B, Sinha S, et al. A division of labor between YAP and TAZ in non‐small cell lung cancer. Cancer Res. 2020;80(19):4145‐4157. doi: 10.1158/0008-5472.CAN-20-0125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chai AWY, Yee PS, Price S, et al. Genome‐wide CRISPR screens of oral squamous cell carcinoma reveal fitness genes in the Hippo pathway. eLife. 2020;9:e57761. doi: 10.7554/eLife.57761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Aragona M, Panciera T, Manfrin A, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin‐processing factors. Cell. 2013;154(5):1047‐1059. doi: 10.1016/j.cell.2013.07.042 [DOI] [PubMed] [Google Scholar]
- 96. Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138(18):3907‐3914. doi: 10.1242/dev.070987 [DOI] [PubMed] [Google Scholar]
- 97. Bao M, Xie J, Piruska A, WTS H. 3D microniches reveal the importance of cell size and shape. Nat Commun. 2017;8(1):1962. doi: 10.1038/s41467-017-02163-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179‐183. doi: 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- 99. Furukawa KT, Yamashita K, Sakurai N, Ohno S. The epithelial circumferential actin belt regulates YAP/TAZ through nucleocytoplasmic shuttling of Merlin. Cell Rep. 2017;20(6):1435‐1447. doi: 10.1016/j.celrep.2017.07.032 [DOI] [PubMed] [Google Scholar]
- 100. Schlegelmilch K, Mohseni M, Kirak O, et al. Yap1 acts downstream of alpha‐catenin to control epidermal proliferation. Cell. 2011;144(5):782‐795. doi: 10.1016/j.cell.2011.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Silvis MR, Kreger BT, Lien WH, et al. Alpha‐catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal. 2011;4(174):ra33. doi: 10.1126/scisignal.2001823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kim NG, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK‐Src‐PI3K pathway. J Cell Biol. 2015;210(3):503‐515. doi: 10.1083/jcb.201501025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kim NG, Koh E, Chen X, Gumbiner BM. E‐cadherin mediates contact inhibition of proliferation through Hippo signaling‐pathway components. Proc Natl Acad Sci USA. 2011;108(29):11930‐11935. doi: 10.1073/pnas.1103345108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26(1):54‐68. doi: 10.1101/gad.173435.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. De Rosa L, Secone Seconetti A, De Santis G, et al. Laminin 332‐dependent YAP dysregulation depletes epidermal stem cells in junctional epidermolysis bullosa. Cell Rep. 2019;27(7):2036‐2049. doi: 10.1016/j.celrep.2019.04.055 [DOI] [PubMed] [Google Scholar]
- 106. Biswas R, Banerjee A, Lembo S, et al. Mechanical instability of adherens junctions overrides intrinsic quiescence of hair follicle stem cells. Dev Cell. 2021;56(6):761‐780. doi: 10.1016/j.devcel.2021.02.020 [DOI] [PubMed] [Google Scholar]
- 107. Martin D, Degese MS, Vitale‐Cross L, et al. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat Commun. 2018;9(1):2372. doi: 10.1038/s41467-018-04590-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Block MR, Brunner M, Ziegelmeyer T, et al. The mechano‐sensitive response of beta1 integrin promotes SRC‐positive late endosome recycling and activation of Yes‐associated protein. J Biol Chem. 2020;295(39):13474‐13487. doi: 10.1074/jbc.RA120.013503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev cell. 2010;18(2):288‐299. doi: 10.1016/j.devcel.2009.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chen CL, Gajewski KM, Hamaratoglu F, et al. The apical‐basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc Natl Acad Sci USA. 2010;107(36):15810‐15815. doi: 10.1073/pnas.1004060107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sun G, Irvine KD. Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev Biol. 2011;350(1):139‐151. doi: 10.1016/j.ydbio.2010.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zhou B, Flodby P, Luo J, et al. Claudin‐18‐mediated YAP activity regulates lung stem and progenitor cell homeostasis and tumorigenesis. J Clin Invest. 2018;128(3):970‐984. doi: 10.1172/JCI90429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Szymaniak AD, Mahoney JE, Cardoso WV, Varelas X. Crumbs3‐mediated polarity directs airway epithelial cell fate through the Hippo pathway effector Yap. Dev Cell. 2015;34(3):283‐296. doi: 10.1016/j.devcel.2015.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Narimatsu M, Samavarchi‐Tehrani P, Varelas X, Wrana JL. Distinct polarity cues direct Taz/Yap and TGFbeta receptor localization to differentially control TGFbeta‐induced Smad signaling. Dev Cell. 2015;32(5):652‐656. doi: 10.1016/j.devcel.2015.02.019 [DOI] [PubMed] [Google Scholar]
- 115. Yang CC, Graves HK, Moya IM, et al. Differential regulation of the Hippo pathway by adherens junctions and apical‐basal cell polarity modules. Proc Natl Acad Sci USA. 2015;112(6):1785‐1790. doi: 10.1073/pnas.1420850112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tilston‐Lunel A, Mazzilli S, Kingston NM, et al. Aberrant epithelial polarity cues drive the development of precancerous airway lesions. Proc Natl Acad Sci USA. 2021;118(18):e2019282118. doi: 10.1073/pnas.2019282118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Cheong SS, Akram KM, Matellan C, et al. The planar polarity component VANGL2 is a key regulator of mechanosignaling. Front Cell Dev Biol. 2020;8:577201. doi: 10.3389/fcell.2020.577201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cho YS, Jiang J. Hippo‐independent regulation of Yki/Yap/Taz: a non‐canonical view. Front Cell Dev Biol. 2021;9:658481. doi: 10.3389/fcell.2021.658481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cai X, Wang KC, Meng Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front Cell Dev Biol. 2021;9:673599. doi: 10.3389/fcell.2021.673599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Miller E, Yang J, DeRan M, et al. Identification of serum‐derived sphingosine‐1‐phosphate as a small molecule regulator of YAP. Chem Biol. 2012;19(8):955‐962. doi: 10.1016/j.chembiol.2012.07.005 [DOI] [PubMed] [Google Scholar]
- 121. Yu FX, Zhao B, Panupinthu N, et al. Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell. 2012;150(4):780‐791. doi: 10.1016/j.cell.2012.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zindel D, Mensat P, Vol C, et al. G protein‐coupled receptors can control the Hippo/YAP pathway through Gq signaling. FASEB J. 2021;35(7):e21668. doi: 10.1096/fj.202002159R [DOI] [PubMed] [Google Scholar]
- 123. Iglesias‐Bartolome R, Torres D, Marone R, et al. Inactivation of a Galpha(s)‐PKA tumour suppressor pathway in skin stem cells initiates basal‐cell carcinogenesis. Nat Cell Biol. 2015;17(6):793‐803. doi: 10.1038/ncb3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Azzolin L, Panciera T, Soligo S, et al. YAP/TAZ incorporation in the beta‐catenin destruction complex orchestrates the Wnt response. Cell. 2014;158(1):157‐170. doi: 10.1016/j.cell.2014.06.013 [DOI] [PubMed] [Google Scholar]
- 125. Park HW, Kim YC, Yu B, et al. Alternative Wnt signaling activates YAP/TAZ. Cell. 2015;162(4):780‐794. doi: 10.1016/j.cell.2015.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lim SK, Lu SY, Kang SA, et al. Wnt signaling promotes breast cancer by blocking ITCH‐mediated degradation of YAP/TAZ transcriptional coactivator WBP2. Cancer Res. 2016;76(21):6278‐6289. doi: 10.1158/0008-5472.CAN-15-3537 [DOI] [PubMed] [Google Scholar]
- 127. Cai J, Maitra A, Anders RA, Taketo MM, Pan D. beta‐Catenin destruction complex‐independent regulation of Hippo‐YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015;29(14):1493‐1506. doi: 10.1101/gad.264515.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lee Y, Kim NH, Cho ES, et al. Dishevelled has a YAP nuclear export function in a tumor suppressor context‐dependent manner. Nat Commun. 2018;9(1):2301. doi: 10.1038/s41467-018-04757-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Guillermin O, Angelis N, Sidor CM, et al. Wnt and Src signals converge on YAP‐TEAD to drive intestinal regeneration. EMBO J. 2021;40(13):e105770. doi: 10.15252/embj.2020105770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Li P, Silvis MR, Honaker Y, Lien WH, Arron ST, Vasioukhin V. alphaE‐catenin inhibits a Src‐YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016;30(7):798‐811. doi: 10.1101/gad.274951.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Lamar JM, Xiao Y, Norton E, et al. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J Biol Chem. 2019;294(7):2302‐2317. doi: 10.1074/jbc.RA118.004364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yui S, Azzolin L, Maimets M, et al. YAP/TAZ‐dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell. 2018;22(1):35‐49. doi: 10.1016/j.stem.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Si Y, Ji X, Cao X, et al. SRC inhibits the Hippo tumor suppressor pathway through tyrosine phosphorylation of Lats1. Cancer Res. 2017;77(18):4868‐4880. doi: 10.1158/0008-5472.CAN-17-0391 [DOI] [PubMed] [Google Scholar]
- 134. Taniguchi K, Wu LW, Grivennikov SI, et al. A gp130‐Src‐YAP module links inflammation to epithelial regeneration. Nature. 2015;519(7541):57‐62. doi: 10.1038/nature14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pefani DE, Pankova D, Abraham AG, et al. TGF‐beta targets the Hippo pathway scaffold RASSF1A to facilitate YAP/SMAD2 nuclear translocation. Mol Cell. 2016;63(1):156‐166. doi: 10.1016/j.molcel.2016.05.012 [DOI] [PubMed] [Google Scholar]
- 136. Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3‐kinase and phosphoinositide‐dependent kinase‐1. Proc Natl Acad Sci USA. 2013;110(7):2569‐2574. doi: 10.1073/pnas.1216462110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes‐associated protein, YAP, to induce interaction with 14‐3‐3 and attenuation of p73‐mediated apoptosis. Mol Cell. 2003;11(1):11‐23. [DOI] [PubMed] [Google Scholar]
- 138. Borreguero‐Munoz N, Fletcher GC, Aguilar‐Aragon M, Elbediwy A, Vincent‐Mistiaen ZI, Thompson BJ. The Hippo pathway integrates PI3K‐Akt signals with mechanical and polarity cues to control tissue growth. PLoS Biol. 2019;17(10):e3000509. doi: 10.1371/journal.pbio.3000509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Djiane A, Zaessinger S, Babaoglan AB, Bray SJ. Notch inhibits Yorkie activity in Drosophila wing discs. PloS one. 2014;9(8):e106211. doi: 10.1371/journal.pone.0106211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Slemmons KK, Crose LES, Riedel S, Sushnitha M, Belyea B, Linardic CM. A novel Notch‐YAP circuit drives stemness and tumorigenesis in embryonal Rhabdomyosarcoma. Mol Cancer Res. 2017;15(12):1777‐1791. doi: 10.1158/1541-7786.MCR-17-0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Totaro A, Castellan M, Battilana G, et al. YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat Commun. 2017;8:15206. doi: 10.1038/ncomms15206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tariki M, Dhanyamraju PK, Fendrich V, Borggrefe T, Feldmann G, Lauth M. The Yes‐associated protein controls the cell density regulation of Hedgehog signaling. Oncogenesis. 2014;3:e112. doi: 10.1038/oncsis.2014.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Akladios B, Mendoza Reinoso V, Cain JE, et al. Positive regulatory interactions between YAP and Hedgehog signalling in skin homeostasis and BCC development in mouse skin in vivo. PloS One. 2017;12(8):e0183178. doi: 10.1371/journal.pone.0183178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhang H, Pasolli HA, Fuchs E. Yes‐associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci USA. 2011;108(6):2270‐2275. doi: 10.1073/pnas.1019603108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Beverdam A, Claxton C, Zhang X, James G, Harvey KF, Key B. Yap controls stem/progenitor cell proliferation in the mouse postnatal epidermis. J Invest Dermatol. 2013;133(6):1497‐1505. doi: 10.1038/jid.2012.430 [DOI] [PubMed] [Google Scholar]
- 146. Akladios B, Mendoza‐Reinoso V, Samuel MS, et al. Epidermal YAP2‐5SA‐DeltaC drives beta‐catenin activation to promote keratinocyte proliferation in mouse skin in vivo. J Invest Dermatol. 2017;137(3):716‐726. doi: 10.1016/j.jid.2016.10.029 [DOI] [PubMed] [Google Scholar]
- 147. Lee MJ, Byun MR, Furutani‐Seiki M, Hong JH, Jung HS. YAP and TAZ regulate skin wound healing. J Invest Dermatol. 2014;134(2):518‐525. doi: 10.1038/jid.2013.339 [DOI] [PubMed] [Google Scholar]
- 148. Aragona M, Sifrim A, Malfait M, et al. Mechanisms of stretch‐mediated skin expansion at single‐cell resolution. Nature. 2020;584(7820):268‐273. doi: 10.1038/s41586-020-2555-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Wong SY, Reiter JF. Wounding mobilizes hair follicle stem cells to form tumors. Proc Natl Acad Sci USA. 2011;108(10):4093‐4098. doi: 10.1073/pnas.1013098108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Brakebusch C, Grose R, Quondamatteo F, et al. Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. EMBO J. 2000;19(15):3990‐4003. doi: 10.1093/emboj/19.15.3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E. Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol. 2000;150(5):1149‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Vincent‐Mistiaen Z, Elbediwy A, Vanyai H, et al. YAP drives cutaneous squamous cell carcinoma formation and progression. eLife. 2018;7:e33304. doi: 10.7554/eLife.33304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22(4):356‐360. doi: 10.1038/11926 [DOI] [PubMed] [Google Scholar]
- 154. Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6(3):331‐343. [DOI] [PubMed] [Google Scholar]
- 155. Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci USA. 1987;84(8):2302‐2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Nishio M, Hamada K, Kawahara K, et al. Cancer susceptibility and embryonic lethality in Mob1a/1b double‐mutant mice. J Clin Invest. 2012;122(12):4505‐4518. doi: 10.1172/JCI63735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Hatterschide J, Castagnino P, Kim HW, et al. YAP1 activation by human Papillomavirus E7 promotes basal cell identity in squamous epithelia. eLife. 2022;11:e75466. doi: 10.7554/eLife.75466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Fujiwara H, Ferreira M, Donati G, et al. The basement membrane of hair follicle stem cells is a muscle cell niche. Cell. 2011;144(4):577‐589. doi: 10.1016/j.cell.2011.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Margadant C, Charafeddine RA, Sonnenberg A. Unique and redundant functions of integrins in the epidermis. FASEB J. 2010;24(11):4133‐4152. doi: 10.1096/fj.09-151449 [DOI] [PubMed] [Google Scholar]
- 160. Walko G, Castanon MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015;360(2):363‐378. doi: 10.1007/s00441-014-2061-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang Y, Xu X, Maglic D, et al. Comprehensive molecular characterization of the hippo signaling pathway in cancer. Cell Rep. 2018;25(5):1304‐1317. doi: 10.1016/j.celrep.2018.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zanconato F, Battilana G, Cordenonsi M, Piccolo S. YAP/TAZ as therapeutic targets in cancer. Curr Opin Pharmacol. 2016;29:26‐33. doi: 10.1016/j.coph.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19(7):480‐494. doi: 10.1038/s41573-020-0070-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Thompson BJ. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. BioEssays. 2020;42(5):e1900162. doi: 10.1002/bies.201900162 [DOI] [PubMed] [Google Scholar]
- 165. Maglic D, Schlegelmilch K, Dost AF, et al. YAP‐TEAD signaling promotes basal cell carcinoma development via a c‐JUN/AP1 axis. EMBO J. 2018;37(17):e98642. doi: 10.15252/embj.201798642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Quan T, Xu Y, Qin Z, et al. Elevated YAP and its downstream targets CCN1 and CCN2 in basal cell carcinoma: impact on keratinocyte proliferation and stromal cell activation. Am J Pathol. 2014;184(4):937‐943. doi: 10.1016/j.ajpath.2013.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Jia J, Li C, Luo S, et al. Yes‐associated protein contributes to the development of human cutaneous squamous cell carcinoma via activation of RAS. J Invest Dermatol. 2016;136(6):1267‐1277. doi: 10.1016/j.jid.2016.02.005 [DOI] [PubMed] [Google Scholar]
- 168. Cappellesso R, Bellan A, Saraggi D, Salmaso R, Ventura L, Fassina A. YAP immunoreactivity is directly related to pilomatrixoma size and proliferation rate. Arch Dermatol Res. 2015;307(4):379‐383. doi: 10.1007/s00403-014-1530-2 [DOI] [PubMed] [Google Scholar]
- 169. Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi‐stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009;4(9):1350‐1362. doi: 10.1038/nprot.2009.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Boumahdi S, Driessens G, Lapouge G, et al. SOX2 controls tumour initiation and cancer stem‐cell functions in squamous‐cell carcinoma. Nature. 2014;511(7508):246‐250. doi: 10.1038/nature13305 [DOI] [PubMed] [Google Scholar]
- 171. Fisher ML, Kerr C, Adhikary G, et al. Transglutaminase interaction with alpha6/beta4‐integrin stimulates YAP1‐dependent DeltaNp63alpha stabilization and leads to enhanced cancer stem cell survival and tumor formation. Cancer Res. 2016;76(24):7265‐7276. doi: 10.1158/0008-5472.CAN-16-2032 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 172. Mickle M, Adhikary G, Shrestha S, Xu W, Eckert RL. VGLL4 inhibits YAP1/TEAD signaling to suppress the epidermal squamous cell carcinoma cancer phenotype. Mol Carcinog. 2021;60(7):497‐507. doi: 10.1002/mc.23307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Nallet‐Staub F, Marsaud V, Li L, et al. Pro‐invasive activity of the Hippo pathway effectors YAP and TAZ in cutaneous melanoma. J Invest Dermatol. 2014;134(1):123‐132. doi: 10.1038/jid.2013.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Zhang X, Tang JZ, Vergara IA, et al. Somatic hypermutation of the YAP oncogene in a human cutaneous melanoma. Mol Cancer Res. 2019;17(7):1435‐1449. doi: 10.1158/1541-7786.MCR-18-0407 [DOI] [PubMed] [Google Scholar]
- 175. Feng Q, Guo P, Kang S, Zhao F. High expression of TAZ/YAP promotes the progression of malignant melanoma and affects the postoperative survival of patients. Pharmazie. 2018;73(11):662‐665. doi: 10.1691/ph.2018.8499 [DOI] [PubMed] [Google Scholar]
- 176. Zhang X, Yang L, Szeto P, et al. The Hippo pathway oncoprotein YAP promotes melanoma cell invasion and spontaneous metastasis. Oncogene. 2020;39(30):5267‐5281. doi: 10.1038/s41388-020-1362-9 [DOI] [PubMed] [Google Scholar]
- 177. Verfaillie A, Imrichova H, Atak ZK, et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun. 2015;6:6683. doi: 10.1038/ncomms7683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Kim MH, Kim J, Hong H, et al. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 2016;35(5):462‐478. doi: 10.15252/embj.201592081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Puig S, Berrocal A. Management of high‐risk and advanced basal cell carcinoma. Clin Transl Oncol. 2015;17(7):497‐503. doi: 10.1007/s12094-014-1272-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Henriques V, Martins T, Link W, Ferreira BI. The emerging therapeutic landscape of advanced melanoma. Curr Pharm Des. 2018;24(5):549‐558. doi: 10.2174/1381612824666180125093357 [DOI] [PubMed] [Google Scholar]
- 181. Chong K, Daud A, Ortiz‐Urda S, Arron ST, Program UHRSC. Cutting edge in medical management of cutaneous oncology. Semin Cutan Med Surg. 2012;31(2):140‐149. doi: 10.1016/j.sder.2012.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Spallone G, Botti E, Costanzo A. Targeted therapy in nonmelanoma skin cancers. Cancers (Basel). 2011;3(2):2255‐2273. doi: 10.3390/cancers3022255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Wei C, Li X. The role of photoactivated and non‐photoactivated verteporfin on tumor. Front Pharmacol. 2020;11:557429. doi: 10.3389/fphar.2020.557429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Gibault F, Corvaisier M, Bailly F, Huet G, Melnyk P, Cotelle P. Non‐photoinduced biological properties of verteporfin. Curr Med Chem. 2016;23(11):1171‐1184. doi: 10.2174/0929867323666160316125048 [DOI] [PubMed] [Google Scholar]
- 185. Liu‐Chittenden Y, Huang B, Shim JS, et al. Genetic and pharmacological disruption of the TEAD‐YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26(12):1300‐1305. doi: 10.1101/gad.192856.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Wang C, Zhu X, Feng W, et al. Verteporfin inhibits YAP function through up‐regulating 14‐3‐3sigma sequestering YAP in the cytoplasm. Am J Cancer Res. 2016;6(1):27‐37. [PMC free article] [PubMed] [Google Scholar]
- 187. Dasari VR, Mazack V, Feng W, Nash J, Carey DJ, Gogoi R. Verteporfin exhibits YAP‐independent anti‐proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget. 2017;8(17):28628‐28640. doi: 10.18632/oncotarget.15614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Zhang H, Ramakrishnan SK, Triner D, et al. Tumor‐selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal. 2015;8(397):ra98. doi: 10.1126/scisignal.aac5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Konstantinou EK, Notomi S, Kosmidou C, et al. Verteporfin‐induced formation of protein cross‐linked oligomers and high molecular weight complexes is mediated by light and leads to cell toxicity. Sci Rep. 2017;7:46581. doi: 10.1038/srep46581 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 190. Zhao W, Liu H, Wang J, Wang M, Shao R. Cyclizing‐berberine A35 induces G2/M arrest and apoptosis by activating YAP phosphorylation (Ser127). J Exp Clin Cancer Res. 2018;37(1):98. doi: 10.1186/s13046-018-0759-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Su D, Lin Z. Dichloroacetate attenuates the stemness of hepatocellular carcinoma cells via promoting nucleus‐cytoplasm translocation of YAP. Environ Toxicol. 2021;36(5):975‐983. doi: 10.1002/tox.23098 [DOI] [PubMed] [Google Scholar]
- 192. Yang X, Daifallah AEM, Shankar S, et al. Topical kinase inhibitors induce regression of cutaneous squamous cell carcinoma. Exp Dermatol. 2019;28(5):609‐613. doi: 10.1111/exd.13902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kim MH, Kim CG, Kim SK, et al. YAP‐induced PD‐L1 expression drives immune evasion in BRAFi‐resistant melanoma. Cancer Immunol Res. 2018;6(3):255‐266. doi: 10.1158/2326-6066.CIR-17-0320 [DOI] [PubMed] [Google Scholar]
- 194. Qi L, Shi C, Li J, et al. Yes‐associated protein promotes cell migration via activating Wiskott‐Aldrich syndrome protein family member 1 in oral squamous cell carcinoma. J Oral Pathol Med. 2019;48(4):290‐298. doi: 10.1111/jop.12833 [DOI] [PubMed] [Google Scholar]
- 195. Morice S, Mullard M, Brion R, et al. The YAP/TEAD axis as a new therapeutic target in osteosarcoma: effect of verteporfin and CA3 on primary tumor growth. Cancers (Basel). 2020;12(12):3847. doi: 10.3390/cancers12123847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Li F, Xu Y, Liu B, et al. YAP1‐mediated CDK6 activation confers radiation resistance in esophageal cancer ‐ rationale for the combination of YAP1 and CDK4/6 inhibitors in esophageal cancer. Clin Cancer Res. 2019;25(7):2264‐2277. doi: 10.1158/1078-0432.CCR-18-1029 [DOI] [PubMed] [Google Scholar]
- 197. Song S, Xie M, Scott AW, et al. A novel YAP1 inhibitor targets CSC‐enriched radiation‐resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol Cancer Ther. 2018;17(2):443‐454. doi: 10.1158/1535-7163.MCT-17-0560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Sorrentino G, Ruggeri N, Specchia V, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16(4):357‐366. doi: 10.1038/ncb2936 [DOI] [PubMed] [Google Scholar]
- 199. Oku Y, Nishiya N, Shito T, et al. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio. 2015;5:542‐549. doi: 10.1016/j.fob.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Hao F, Xu Q, Wang J, et al. Lipophilic statins inhibit YAP nuclear localization, co‐activator activity and colony formation in pancreatic cancer cells and prevent the initial stages of pancreatic ductal adenocarcinoma in KrasG12D mice. PloS One. 2019;14(5):e0216603. doi: 10.1371/journal.pone.0216603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Lee TF, Tseng YC, Nguyen PA, Li YC, Ho CC, Wu CW. Enhanced YAP expression leads to EGFR TKI resistance in lung adenocarcinomas. Sci Rep. 2018;8(1):271. doi: 10.1038/s41598-017-18527-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Billing O, Holmgren Y, Nosek D, Hedman H, Hemmingsson O. LRIG1 is a conserved EGFR regulator involved in melanoma development, survival and treatment resistance. Oncogene. 2021;40(21):3707‐3718. doi: 10.1038/s41388-021-01808-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118‐122. doi: 10.1038/nature13121 [DOI] [PubMed] [Google Scholar]
- 204. Hao F, Xu Q, Zhao Y, et al. Insulin receptor and GPCR crosstalk stimulates YAP via PI3K and PKD in pancreatic cancer cells. Mol Cancer Res. 2017;15(7):929‐941. doi: 10.1158/1541-7786.MCR-17-0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Mesrouze Y, Bokhovchuk F, Meyerhofer M, et al. Dissection of the interaction between the intrinsically disordered YAP protein and the transcription factor TEAD. eLife. 2017;6:e25068. doi: 10.7554/eLife.25068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Mesrouze Y, Bokhovchuk F, Izaac A, et al. Adaptation of the bound intrinsically disordered protein YAP to mutations at the YAP:TEAD interface. Protein Sci. 2018;27(10):1810‐1820. doi: 10.1002/pro.3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Tian W, Yu J, Tomchick DR, Pan D, Luo X. Structural and functional analysis of the YAP‐binding domain of human TEAD2. Proc Natl Acad Sci USA. 2010;107(16):7293‐7298. doi: 10.1073/pnas.1000293107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Pobbati AV, Rubin BP. Protein‐Protein interaction disruptors of the YAP/TAZ‐TEAD transcriptional complex. Molecules. 2020;25(24):6001. doi: 10.3390/molecules25246001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Pobbati AV, Han X, Hung AW, et al. Targeting the central pocket in human transcription factor TEAD as a potential cancer therapeutic strategy. Structure. 2015;23(11):2076‐2086. doi: 10.1016/j.str.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Chen L, Chan SW, Zhang X, et al. Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes Dev. 2010;24(3):290‐300. doi: 10.1101/gad.1865310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Li Z, Zhao B, Wang P, et al. Structural insights into the YAP and TEAD complex. Genes Dev. 2010;24(3):235‐240. doi: 10.1101/gad.1865810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Kaan HYK, Chan SW, Tan SKJ, et al. Crystal structure of TAZ‐TEAD complex reveals a distinct interaction mode from that of YAP‐TEAD complex. Sci Rep. 2017;7(1):2035. doi: 10.1038/s41598-017-02219-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Iftikhar R, Zahoor AF, Irfan M, Rasul A, Rao F. Synthetic molecules targeting yes associated protein activity as chemotherapeutics against cancer. Chem Biol Drug Des. 2021;98(6):1025‐1037. doi: 10.1111/cbdd.13960 [DOI] [PubMed] [Google Scholar]
- 214. Noland CL, Gierke S, Schnier PD, et al. Palmitoylation of TEAD transcription factors is required for their stability and function in Hippo pathway signaling. Structure. 2016;24(1):179‐186. doi: 10.1016/j.str.2015.11.005 [DOI] [PubMed] [Google Scholar]
- 215. Chan P, Han X, Zheng B, et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat Chem Biol. 2016;12(4):282‐289. doi: 10.1038/nchembio.2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Hillmer RE, Link BA. The roles of Hippo signaling transducers Yap and Taz in chromatin remodeling. Cells. 2019;8(5):502. doi: 10.3390/cells8050502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Lavado A, Park JY, Paré J, et al. The Hippo pathway prevents YAP/TAZ‐driven hypertranscription and controls neural progenitor number. Dev Cell. 2018;47(5):576‐591. doi: 10.1016/j.devcel.2018.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell. 2017;168(4):629‐643. doi: 10.1016/j.cell.2016.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Kim M, Kim T, Johnson Randy L, Lim D‐S. Transcriptional co‐repressor function of the Hippo pathway transducers YAP and TAZ. Cell Rep. 2015;11(2):270‐282. doi: 10.1016/j.celrep.2015.03.015 [DOI] [PubMed] [Google Scholar]
- 220. Sallam MA, Prakash S, Krishnan V, Todorova K, Mandinova A, Mitragotri S. Hyaluronic acid conjugates of vorinostat and bexarotene for treatment of cutaneous malignancies. Adv Ther. 2020;3(10):2000116. doi: 10.1002/adtp.202000116 [DOI] [Google Scholar]
- 221. Huijberts S, Wang L, de Oliveira RL, et al. Vorinostat in patients with resistant BRAF(V600E) mutated advanced melanoma: a proof of concept study. Future Oncol. 2020;16(11):619‐629. doi: 10.2217/fon-2020-0023 [DOI] [PubMed] [Google Scholar]
- 222. Chang CY, Shipony Z, Lin SG, et al. Increased ACTL6A occupancy within mSWI/SNF chromatin remodelers drives human squamous cell carcinoma. Mol Cell. 2021;81(24):4964‐4978. doi: 10.1016/j.molcel.2021.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Luo Q, Wu X, Chang W, et al. ARID1A hypermethylation disrupts transcriptional homeostasis to promote squamous cell carcinoma progression. Cancer Res. 2020;80(3):406‐417. doi: 10.1158/0008-5472.CAN-18-2446 [DOI] [PubMed] [Google Scholar]
- 224. Luo Q, Wu X, Chang W, et al. ARID1A prevents squamous cell carcinoma initiation and chemoresistance by antagonizing pRb/E2F1/c‐Myc‐mediated cancer stemness. Cell Death Differ. 2020;27(6):1981‐1997. doi: 10.1038/s41418-019-0475-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Wanior M, Kramer A, Knapp S, Joerger AC. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene. 2021;40(21):3637‐3654. doi: 10.1038/s41388-021-01781-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP‐1) miniperspective. J Med Chem. 2014;57(16):6930‐6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Brennan A, Leech JT, Kad NM, Mason JM. Selective antagonism of cJun for cancer therapy. J Exp Clin Cancer Res. 2020;39(1):184. doi: 10.1186/s13046-020-01686-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Zhao Q, Zhang K, Li Y, et al. OLFML2A is necessary for anti‐triple negative breast cancer effect of selective activator protein‐1 inhibitor T‐5224. Transl Oncol. 2021;14(8):101100. doi: 10.1016/j.tranon.2021.101100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Hazan I, Monin J, Bouwman BAM, Crosetto N, Aqeilan RI. Activation of Oncogenic Super‐Enhancers Is Coupled with DNA Repair by RAD51. Cell Rep. 2019;29(3):560‐572. doi: 10.1016/j.celrep.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Khavari PA. Modelling cancer in human skin tissue. Nat Rev Cancer. 2006;6(4):270‐280. doi: 10.1038/nrc1838 [DOI] [PubMed] [Google Scholar]
- 231. Popis MC, Wagner RE, Constantino‐Casas F, Blanco S, Frye M. Considerations for skin carcinogenesis experiments using inducible transgenic mouse models. BMC Res Notes. 2018;11(1):67. doi: 10.1186/s13104-018-3182-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Rebecca VW, Somasundaram R, Herlyn M. Pre‐clinical modeling of cutaneous melanoma. Nat Commun. 2020;11(1):2858. doi: 10.1038/s41467-020-15546-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Mitsui H, Suarez‐Farinas M, Gulati N, et al. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL‐24‐driven MMP‐7. J Invest Dermatol. 2014;134(5):1418‐1427. doi: 10.1038/jid.2013.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Bonilla X, Parmentier L, King B, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. 2016;48(4):398‐406. doi: 10.1038/ng.3525 [DOI] [PubMed] [Google Scholar]
- 235. Callus BA, Finch‐Edmondson ML, Fletcher S, Wilton SD. YAPping about and not forgetting TAZ. FEBS Lett. 2019;593(3):253‐276. doi: 10.1002/1873-3468.13318 [DOI] [PubMed] [Google Scholar]
- 236. Janse van Rensburg HJ, Azad T, Ling M, et al. The Hippo pathway component TAZ promotes immune evasion in human cancer through PD‐L1. Cancer Res. 2018;78(6):1457‐1470. doi: 10.1158/0008-5472.CAN-17-3139 [DOI] [PubMed] [Google Scholar]
- 237. Berning M, Pratzel‐Wunder S, Bickenbach JR, Boukamp P. Three‐dimensional in vitro skin and skin cancer models based on human fibroblast‐derived matrix. Tissue Eng Part C Methods. 2015;21(9):958‐970. doi: 10.1089/ten.TEC.2014.0698 [DOI] [PubMed] [Google Scholar]
- 238. Hill DS, Robinson ND, Caley MP, et al. A novel fully humanized 3D skin equivalent to model early melanoma invasion. Mol Cancer Ther. 2015;14(11):2665‐2673. doi: 10.1158/1535-7163.MCT-15-0394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Yun MR, Choi HM, Lee YW, et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK‐rearranged lung cancer. EMBO Mol Med. 2019;11(12):e10581. doi: 10.15252/emmm.201910581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Crook ZR, Sevilla GP, Friend D, et al. Mammalian display screening of diverse cystine‐dense peptides for difficult to drug targets. Nat Commun. 2017;8(1):2244. doi: 10.1038/s41467-017-02098-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Kaan HYK, Sim AYL, Tan SKJ, Verma C, Song H. Targeting YAP/TAZ‐TEAD protein‐protein interactions using fragment‐based and computational modeling approaches. PLoS One. 2017;12(6):e0178381. doi: 10.1371/journal.pone.0178381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Zhang Z, Lin Z, Zhou Z, et al. Structure‐based design and synthesis of potent cyclic peptides inhibiting the YAP‐TEAD protein‐protein interaction. ACS Med Chem Lett. 2014;5(9):993‐998. doi: 10.1021/ml500160m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Zhou Z, Hu T, Xu Z, et al. Targeting Hippo pathway by specific interruption of YAP‐TEAD interaction using cyclic YAP‐like peptides. FASEB J. 2015;29(2):724‐732. doi: 10.1096/fj.14-262980 [DOI] [PubMed] [Google Scholar]
- 244. Furet P, Salem B, Mesrouze Y, et al. Structure‐based design of potent linear peptide inhibitors of the YAP‐TEAD protein‐protein interaction derived from the YAP omega‐loop sequence. Bioorg Med Chem Lett. 2019;29(16):2316‐2319. doi: 10.1016/j.bmcl.2019.06.022 [DOI] [PubMed] [Google Scholar]
- 245. Li Y, Liu S, Ng EY, et al. Structural and ligand‐binding analysis of the YAP‐binding domain of transcription factor TEAD4. Biochem J. 2018;475(12):2043‐2055. doi: 10.1042/BCJ20180225 [DOI] [PubMed] [Google Scholar]
- 246. Barth M, Contal S, Montalbetti C, Spitzer L, inventors; New compounds inhibitors of the yap/taz‐tead interaction and their use in the treatment of malignant mesothelioma; 2017. patent WO2017064277A1.
- 247. Gibault F, Coevoet M, Sturbaut M, et al. Toward the discovery of a novel class of YAP(−)TEAD interaction inhibitors by virtual screening approach targeting YAP(−)TEAD Protein(−)Protein interface. Cancers (Basel). 2018;10(5):140. doi: 10.3390/cancers10050140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Smith SA, Sessions RB, Shoemark DK, et al. Antiproliferative and antimigratory effects of a novel YAP‐TEAD interaction inhibitor identified using in silico molecular docking. J Med Chem. 2019;62(3):1291‐1305. doi: 10.1021/acs.jmedchem.8b01402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Jiao S, Wang H, Shi Z, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25(2):166‐180. doi: 10.1016/j.ccr.2014.01.010 [DOI] [PubMed] [Google Scholar]
- 250. Bum‐Erdene K, Zhou D, Gonzalez‐Gutierrez G, et al. Small‐molecule covalent modification of conserved cysteine leads to allosteric inhibition of the TEADYap Protein‐Protein interaction. Cell Chem Biol. 2019;26(3):378‐389. doi: 10.1016/j.chembiol.2018.11.010 [DOI] [PubMed] [Google Scholar]
- 251. Kurppa KJ, Liu Y, To C, et al. Treatment‐induced tumor dormancy through YAP‐mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell. 2020;37(1):104‐122. doi: 10.1016/j.ccell.2019.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Konradi A, Lin T, inventors. Non‐fused tricyclic compounds; 2018. patent WO/2018/204532.
- 253. Li Q, Sun Y, Jarugumilli GK, et al. Lats1/2 sustain intestinal stem cells and Wnt activation through TEAD‐dependent and independent transcription. Cell Stem Cell. 2020;26(5):675‐692. doi: 10.1016/j.stem.2020.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Kunig VBK, Potowski M, Akbarzadeh M, et al. TEAD‐YAP Interaction Inhibitors and MDM2 Binders from DNA‐Encoded Indole‐Focused Ugi Peptidomimetics. Angew Chem Int Ed Engl. 2020;59(46):20338‐20342. doi: 10.1002/anie.202006280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Zbieg J, Beroza P, Crawford J, inventors. Therapeutic compounds; 2019. patent application PCT/US2019/034660.
- 256. Kaneda A, Seike T, Danjo T, et al. The novel potent TEAD inhibitor, K‐975, inhibits YAP1/TAZ‐TEAD protein‐protein interactions and exerts an anti‐tumor effect on malignant pleural mesothelioma. Am J Cancer Res. 2020;10(12):4399‐4415. [PMC free article] [PubMed] [Google Scholar]
- 257. Karatas H, Akbarzadeh M, Adihou H, et al. Discovery of covalent inhibitors targeting the transcriptional enhanced associate domain central pocket. J Med Chem. 2020;63(20):11972‐11989. doi: 10.1021/acs.jmedchem.0c01275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Holden JK, Crawford JJ, Noland CL, et al. Small molecule dysregulation of TEAD lipidation induces a dominant‐negative inhibition of Hippo pathway signaling. Cell Rep. 2020;31(12):107809. doi: 10.1016/j.celrep.2020.107809 [DOI] [PubMed] [Google Scholar]
- 259. Lu W, Wang J, Li Y, et al. Discovery and biological evaluation of vinylsulfonamide derivatives as highly potent, covalent TEAD autopalmitoylation inhibitors. Eur J Med Chem. 2019;184:111767. doi: 10.1016/j.ejmech.2019.111767 [DOI] [PubMed] [Google Scholar]
- 260. Pobbati AV, Mejuch T, Chakraborty S, et al. Identification of quinolinols as activators of TEAD‐dependent transcription. ACS Chem Biol. 2019;14(12):2909‐2921. doi: 10.1021/acschembio.9b00786 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.