

Abstract
Summary
In recent years, there has been an increasing interest in bacteriophages, which has led to growing numbers of bacteriophage genomic sequences becoming available. Consequently, there is a need for a rapid and consistent genomic annotation tool dedicated for bacteriophages. Existing tools either are not designed specifically for bacteriophages or are web- and email-based and require significant manual curation, which makes their integration into bioinformatic pipelines challenging. Pharokka was created to provide a tool that annotates bacteriophage genomes easily, rapidly and consistently with standards compliant outputs. Moreover, Pharokka requires only two lines of code to install and use and takes under 5 min to run for an average 50-kb bacteriophage genome.
Availability and implementation
Pharokka is implemented in Python and is available as a bioconda package using ‘conda install -c bioconda pharokka’. The source code is available on GitHub (https://github.com/gbouras13/pharokka). Pharokka has been tested on Linux-64 and MacOSX machines and on Windows using a Linux Virtual Machine.
1 Introduction
As the number of bacteriophage (phage) sequences increases, there is a need for bioinformatic tools that enable fast, consistent and scalable genomic annotation (Cook et al., 2021). Existing tools such as RAST (Aziz et al., 2008; Davis et al., 2020), PHASTER (Arndt et al., 2016) and CPT Galaxy (Ramsey et al., 2020) are web-server based which may be laborious in particular when multiple phage genomes require annotation (Shen and Millard, 2021). On the other hand, currently available customizable bioinformatics pipelines such as multiPhATE2 require significant understanding of bioinformatics to implement and have lengthy run times (Ecale Zhou et al., 2021). Furthermore, command-line programs designed for viral discovery in metagenomic datasets such as Cenote-Taker 2 (Tisza et al., 2021), Hecatomb (Roach et al., 2022) and MetaPhage (Pandolfo et al., 2022) require significant computational resources and storage for database installation.
As a result, one-line prokaryotic genome annotation tools such as Prokka (Seemann, 2014) are often used for phages, especially where tens or hundreds of phages need to be annotated simultaneously (Beamud et al., 2022; Nordstrom et al., 2022). However, such tools implement prokaryotic gene prediction tools that are based on models that are not designed for phage genomes. In addition, phage genes are often lacking in their default databases, resulting in incomplete or hypothetical functional annotation of phage genes.
Inspired by Prokka, here we created Pharokka as a one-line tool tailored to phages that provides annotations in a fast, scalable and consistent fashion. Pharokka identifies predicted coding sequences (CDS), transfer RNAs (tRNAs), transfer-messenger RNAs (tmRNAs) and clustered regularly interspaced short palindromic repeats (CRISPRs), providing functional annotation for CDS using the PHROGs database (Terzian et al., 2021). With accessible bioconda installation, Pharokka is easily integrated into complex bioinformatic pipelines such as those created with Snakemake (Mölder et al., 2021) or Nextflow (Di Tommaso et al., 2017).
2 Materials and methods
The Pharokka workflow is outlined in Figure 1.
Fig. 1.
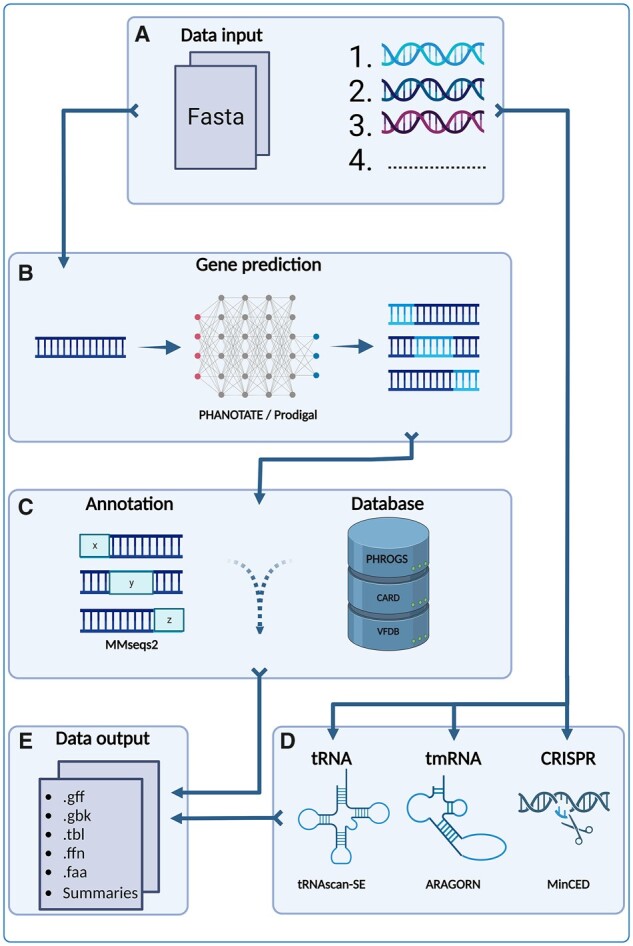
Pharokka workflow. (A) An input phage complete assembly or input phage contigs are loaded. (B) CDS are predicted with PHANOTATE (default) or Prodigal. (C) CDS are functionally annotated by matching them to the PHROGs database with mmseqs2. The CDS are then matched to the CARD and the VFDB. (D) tRNAs, tmRNAs and CRISPRs are detected using tRNAscan-SE, Aragorn and MinCED. (E) All annotations are amalgamated into standards compliant output formats
2.1 Input
Pharokka requires assembled DNA sequences in FASTA format (Fig. 1A). For phage isolates, this usually consists of one complete contig, but Pharokka can also annotate incomplete assemblies or metavirome samples with multiple contigs in the multiFASTA format. Furthermore, metagenomically assembled phage genomes and genomic contigs, derived using programs such as Virstorter2 (Guo et al., 2021), Hecatomb (Roach et al., 2022) and Cenote-taker 2 (Tisza et al., 2021), are also suitable to be annotated using Pharokka using meta mode.
2.2 Feature prediction
Pharokka uses PHANOTATE by default to predict CDS, as it is the only existing tool that is designed to predict genes in phage genomes (McNair et al., 2019; Fig. 1B). PHANOTATE considers unique features of phage genomes such as small gene size, high coding density and alternative start codons (McNair et al., 2019). Moreover, PHANOTATE predicts significantly more genes that are on average smaller than other gene prediction tools (McNair et al., 2019). Small phage genes are more prevalent than predicted based on existing non-phage specific annotation tools and may encode for anti-CRISPR and antimicrobial resistance proteins (Fremin et al., 2022). Alternatively, Pharokka users can specify Prodigal, a gene predictor designed for prokaryotic genes, be used for CDS prediction (Hyatt et al., 2010; Fig. 1B). Prodigal may be useful if users wish to annotate large metavirome datasets quickly using meta mode (Table 4). In addition, Pharokka employs tRNAscan-SE 2 (Chan et al., 2021) to predict tRNAs, Aragorn (Laslett and Canback, 2004) to predict tmRNAs and MinCED to predict CRISPRs (Bland et al., 2007; Fig. 1D). When meta mode is specified, Pharokka runs PHANOTATE on each contig separately, leading to considerable speed improvement when multiple threads are specified.
Table 4.
Benchmarking 673 crAss-like metagenome assembled phage genomes (Yutin et al., 2021)
| Pharokka PHANOTATE meta mode | Pharokka Prodigal meta mode | Prokka with PHROGs | |
|---|---|---|---|
| Time (min) | 106.55 | 11.88 | 252.33 |
| Time gene prediction (min) | 96.21 | 3.4 | 5.12 |
| Time tRNA prediction (min) | 1.25 | 1.08 | 0.30 |
| Time database searches (min) | 6.75 | 5.58 | 238.77 |
| CDS | 138 628 | 90 497 | 89 802 |
| contig min coding density (%) | 66.01 | 46.18 | 46.13 |
| Contig max coding density (%) | 98.86 | 97.85 | 97.07 |
| Annotated function CDS | 9341 | 9228 | 14 461 |
| Unknown function CDS | 129 287 | 81 269 | 75 341 |
2.3 Functional gene annotation
Functional assignment of predicted CDS is conducted using the PHROGs database (Terzian et al., 2021; Fig. 1C). The PHROGs database contains 38 880 PHROGs (protein orthologous groups) containing 868 340 proteins from 17 473 complete genomes of viruses infecting bacteria or archaea that are grouped together using remote homology detection. Each PHROG is assigned to one of nine functional categories. Each gene within each PHROG has a specific product description if known. All predicted CDS are translated into a protein sequence and assigned to the closest matching protein and accompanying PHROG in the PHROGs database using the protein searching tool mmseqs2 (Steinegger and Söding, 2017). An e-value threshold of 10−5 is used by default. Users can also specify their own e-value if desired. This is particularly useful if the phage is novel with few or no similar sequences in the PHROGs database, as a lower threshold can be used to detect less similar matches. If no PHROG match is found, then the CDS is annotated as ‘No PHROG’ and ‘hypothetical protein’. All CDS that are annotated as ‘terminase large subunit’ are extracted into separate output files, as the terminase large subunit is commonly used for phylogenetic analysis (Al-Shayeb et al., 2020).
2.4 Virulence factor and antimicrobial resistance gene detection
While virulence factors are commonly found on prophages (Fortier and Sekulovic, 2013), lytic phages rarely encode antibiotic resistance genes (Enault et al., 2017; Cook et al., 2021). Nonetheless, screening is required for all phages that are intended to be used for phage therapy (Shen and Millard, 2021). Pharokka adds antimicrobial resistance and virulence gene detection using the Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2020) and the Virulence Factor Database (VFDB) (Liu et al., 2019; Fig. 1C). All protein CDS are assigned to the closest matching protein in each database with mmseqs2 if the match passes strict thresholds of 80% identity over 40% coverage recommended by Enault et al. (2017).
3 Output
Pharokka’s output files are outlined in Table 1. The primary output of Pharokka is a .gff file that is suitable for use in downstream pan-genome programs such as Roary (Page et al., 2015) and panaroo (Tonkin-Hill et al., 2020; Fig. 1E). Other files include a .tbl file, which is a flat-file table suitable to be uploaded to the NCBI's Bankit, a cds_functions.tsv file, which includes counts of CDS, tRNAs, CRISPRs and tmRNAs and CDS within each functional category for each contig in the input FASTA file, a length_gc_cds_density.tsv file, which outputs the length, GC percentage and CDS coding density for each contig, a cds_final_merged_output.tsv, which gives the combined parsed output from mmseqs2 searches against the PHROGs, VFDB and CARD databases, and terL.faa and terL.ffn output files that contain the amino acid and nucleotide sequences of any predicted terminase large subunit genes. When run on metavirome input, Pharokka’s contig-level summary output allows users to identify specific contigs within the metavirome that possess unusual features such as virulence factors, antimicrobial resistance genes or potential stop codon reassignment as indicated by low CDS coding density (Peters et al., 2022).
Table 1.
Description of Pharokka output files
| Output files | Description of file contents |
|---|---|
| .gff | Gff3 feature file with all genomic annotations |
| .gbk | GenBank formatted feature file |
| .faa | FASTA file of predicted CDS (amino acid) |
| .ffn | FASTA file of predicted CDS (nucleotide) |
| minced_spacers.txt and minced.gff | MinCED CRISPR spacers and parsed CRISPR annotations |
| aragorn.txt and aragorn.gff | Aragorn tmRNA annotations |
| trnascan_out.gff | tRNAscan-SE 2 tRNA annotations |
| cds_functions.tsv | Functional summary counts of CDS |
| cds_final_merged_output.tsv | Combined output from PHROGs, VFDB and CARD |
| length_gc_cds_density.tsv | Summary of genome length, GC percentage, coding density |
| .tbl | Feature table for NCBI (BankIt) submission |
| top_hits_card.tsv and top_hits_vfdb.tsv | Top hits from CARD and VFDB annotations. Will contain no rows if there are no hits |
| terL.faa and terL.ffn | Predicted terminase large subunit sequences |
4 Results
To test the performance of Pharokka, we compared the run-time and annotations of Enterobacteria phage lambda (Genbank accession J02459), Staphylococcus phage SAOMS1 (Genbank accession MW460250) and 673 crAss-like metagenome-assembled phage genomes from the human gut (Yutin et al., 2021) with default Pharokka v1.1.0 using PHANOTATE as a gene predictor, Pharokka v1.1.0 specifying Prodigal as gene predictor and Prokka v1.14.6 using a version of the PHROGs HMM database that has been reformatted for use with Prokka (Millard, 2021https://millardlab.org/2021/11/21/phage-annotation-with-phrogs/). For the 673 crAss-like phage genomes, Pharokka’s meta mode was employed. Benchmarking was conducted on an Intel® Xeon® CPU E5-4610 v2 @ 2.30 GHz specifying 16 threads for Pharokka and 16 cpus for Prokka.
Tables 2 and 3 show that for phages Lambda and SAOMS1, Pharokka is slower than Prokka but still fast, finishing within 5 min regardless of gene predictor used, with PHANOTATE predicting more CDS with higher coding density than Prodigal. Table 4 shows that for the crAss-like phage genomes, Pharokka is considerably faster than using Prokka regardless of gene predictor, due to Pharokka using mmseqs2 for database searching rather than HMMER3 (Finn et al., 2011) employed by Prokka (Mirdita et al., 2019). As the size of the input increases, the extra-time cost incurred by Pharokka is due to the gene prediction and tRNA calling, rather than the database searching. For extremely large metavirome datasets, Pharokka in Prodigal meta mode is therefore recommended.
Table 2.
Benchmarking Enterobacteria phage Lambda (48 052 bp)
| Pharokka PHANOTATE | Pharokka Prodigal | Prokka with PHROGs | |
|---|---|---|---|
| Time (min) | 4.19 | 3.88 | 0.27 |
| CDS | 88 | 61 | 62 |
| Coding density (%) | 94.55 | 83.69 | 84.96 |
| Annotated function CDS | 43 | 37 | 45 |
| Unknown function CDS | 45 | 24 | 17 |
Table 3.
Benchmarking Staphylococcus phage SAOMS1 (140 315 bp)
| Pharokka PHANOTATE | Pharokka Prodigal | Prokka with PHROGs | |
|---|---|---|---|
| Time (min) | 4.26 | 3.89 | 0.93 |
| CDS | 246 | 212 | 212 |
| Coding density (%) | 92.27 | 89.69 | 89.31 |
| Annotated function CDS | 92 | 93 | 92 |
| Unknown function CDS | 154 | 119 | 120 |
In addition, for the crAss-like phage genomes, the low coding density output of some contigs identified by Pharokka indicates that stop codon reassignment may be occurring in these contigs (Peters et al., 2022; Yutin et al., 2021).
Acknowledgements
The authors thank Ronen Hazan, Paulina Detpula, Camilo Garcia, Brady Cress, Ryan Cook, Bhavya Nalagampalli Papudeshi, Susanna Grigson, Ryan Dan Hesse and Robert Edwards who provided bug-reports, ideas and advice about how to improve Pharokka and Torsten Seemann for inspiring its creation.
Funding
This research was supported by the National Health and Medical Research Council Investigator Grant (grant number 1196832) awarded to P.-J.W.
Conflict of Interest: none declared.
Contributor Information
George Bouras, Adelaide Medical School, Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5070, Australia; Department of Surgery—Otolaryngology Head and Neck Surgery, University of Adelaide and the Basil Hetzel Institute for Translational Health Research, Central Adelaide Local Health Network, Adelaide, SA 5070, Australia.
Roshan Nepal, Adelaide Medical School, Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5070, Australia; Department of Surgery—Otolaryngology Head and Neck Surgery, University of Adelaide and the Basil Hetzel Institute for Translational Health Research, Central Adelaide Local Health Network, Adelaide, SA 5070, Australia.
Ghais Houtak, Adelaide Medical School, Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5070, Australia; Department of Surgery—Otolaryngology Head and Neck Surgery, University of Adelaide and the Basil Hetzel Institute for Translational Health Research, Central Adelaide Local Health Network, Adelaide, SA 5070, Australia.
Alkis James Psaltis, Adelaide Medical School, Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5070, Australia; Department of Surgery—Otolaryngology Head and Neck Surgery, University of Adelaide and the Basil Hetzel Institute for Translational Health Research, Central Adelaide Local Health Network, Adelaide, SA 5070, Australia.
Peter-John Wormald, Adelaide Medical School, Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5070, Australia; Department of Surgery—Otolaryngology Head and Neck Surgery, University of Adelaide and the Basil Hetzel Institute for Translational Health Research, Central Adelaide Local Health Network, Adelaide, SA 5070, Australia.
Sarah Vreugde, Adelaide Medical School, Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5070, Australia; Department of Surgery—Otolaryngology Head and Neck Surgery, University of Adelaide and the Basil Hetzel Institute for Translational Health Research, Central Adelaide Local Health Network, Adelaide, SA 5070, Australia.
Data availability
All benchmarking input FASTA and output files, including the python script calc_gff_coding_density_prokka.py script, is available at https://doi.org/10.5281/zenodo.7227091.
References
- Alcock B.P. et al. (2020) CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res., 48, D517–D525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shayeb B. et al. (2020) Clades of huge phages from across earth’s ecosystems. Nature, 578, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt D. et al. (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res., 44, W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz R.K. et al. (2008) The RAST server: rapid annotations using subsystems technology. BMC Genomics, 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beamud B. et al. (2022) Genetic determinants of host tropism in Klebsiella phages. 10.1101/2022.06.01.494021. [DOI] [PMC free article] [PubMed]
- Bland C. et al. (2007) CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics, 8, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P.P. et al. (2021) tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res., 49, 9077–9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook R. et al. (2021) INfrastructure for a PHAge REference database: identification of large-scale biases in the current collection of cultured phage genomes. Phage (New Rochelle), 2, 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.J. et al. (2020) The PATRIC bioinformatics resource center: expanding data and analysis capabilities. Nucleic Acids Res., 48, D606–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tommaso P. et al. (2017) Nextflow enables reproducible computational workflows. Nat. Biotechnol., 35, 316–319. [DOI] [PubMed] [Google Scholar]
- Ecale Zhou C.L. et al. (2021) MultiPhATE2: code for functional annotation and comparison of phage genomes. G3 (Bethesda), 11, jkab074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enault F. et al. (2017) Phages rarely encode antibiotic resistance genes: a cautionary tale for virome analyses. ISME J., 11, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D. et al. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res., 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortier L.-C., Sekulovic O. (2013) Importance of prophages to evolution and virulence of bacterial pathogens. Virulence, 4, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremin B.J. et al. ; Global Phage Small Open Reading Frame (GP-SmORF) Consortium (2022) Thousands of small, novel genes predicted in global phage genomes. Cell Rep., 39, 110984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. et al. (2021) VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome, 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. et al. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D., Canback B. (2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res., 32, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B. et al. (2019) VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res., 47, D687–D692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNair K. et al. (2019) PHANOTATE: a novel approach to gene identification in phage genomes. Bioinformatics, 35, 4537–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard A. (2021) PHAGE ANNOTATION WITH PHROGS. Millardlab.
- Mirdita M. et al. (2019) MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics, 35, 2856–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölder F. et al. (2021) Sustainable data analysis with Snakemake. F1000Res., 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom H.R. et al. (2022) Genomic characterization of lytic bacteriophages targeting genetically diverse Pseudomonas aeruginosa clinical isolates. iScience, 25, 104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfo M. et al. (2022) MetaPhage: an automated pipeline for analyzing, annotating, and classifying bacteriophages in metagenomics sequencing data. mSystems, 7, e0074122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page A.J. et al. (2015) Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics, 31, 3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters S.L. et al. (2022) Experimental validation that human microbiome phages use alternative genetic coding. Nat. Commun., 13, 5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey J. et al. (2020) Galaxy and apollo as a biologist-friendly interface for high-quality cooperative phage genome annotation. PLoS Comput. Biol., 16, e1008214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach M. et al. (2022) Hecatomb: an end-to-end research platform for viral metagenomics. 10.1101/2022.05.15.492003. [DOI]
- Seemann T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics, 30, 2068–2069. [DOI] [PubMed] [Google Scholar]
- Shen A., Millard A. (2021) Phage genome annotation: where to begin and end. Phage (New Rochelle), 2, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M., Söding J. (2017) MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol., 35, 1026–1028. [DOI] [PubMed] [Google Scholar]
- Terzian P. et al. (2021) PHROG: families of prokaryotic virus proteins clustered using remote homology. NAR Genom. Bioinform., 3, lqab067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisza M.J. et al. (2021) Cenote-Taker 2 democratizes virus discovery and sequence annotation. Virus Evol., 7, veaa100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkin-Hill G. et al. (2020) Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol., 21, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutin N. et al. (2021) Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features. Nat. Commun., 12, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All benchmarking input FASTA and output files, including the python script calc_gff_coding_density_prokka.py script, is available at https://doi.org/10.5281/zenodo.7227091.