

Abstract
Jackfruit (Artocarpus heterophyllus Lam.) is the national fruit of Bangladesh and produces fruit in the summer season only. However, jackfruit is not commercially grown in Bangladesh because of an extremely high variation in fruit quality, short seasonal fruiting (June-August) and susceptibility to abiotic stresses. Conversely, a year-round high yielding (ca. 4-fold higher than the seasonal variety) jackfruit variety, BARI Kanthal-3 developed by the Bangladesh Agricultural Research Institute (BARI) derived from a wild accession found in Ramgarh of Chattogram Hiltracts of Bangladesh, provides fruits from September to June. This study aimed to generate a draft whole-genome sequence (WGS) of BARI Kanthal-3 to obtain molecular insights including genes associated with year-round fruiting trait of this important unique variety. The estimated genome size of BARI Kanthal-3 was 1.04-gigabase-pair (Gbp) with a heterozygosity rate of 1.62%. De novo assembly yielded a scaffolded 817.7 Mb genome while a reference-guided approach, yielded 843 Mb of genome sequence. The estimated GC content was 34.10%. Variant analysis revealed that BARI Kanthal-3 included 5.7 M (35%) and 10.4 M (65%) simple and heterozygous single nucleotide polymorphisms (SNPs), and about 90% of all these polymorphisms are in inter-genic regions. Through BUSCO assessment, 97.2% of the core genes were represented in the assembly with 1.3% and 1.5% either fragmented or missing, respectively. By comparing identified orthologous gene groups in BARI Kanthal-3 with five closely and one distantly related species of 10,092 common orthogroups were found across the genomes of the six species. The phylogenetic analysis of the shared orthogroups showed that A. heterophyllus was the closest species to BARI Kanthal-3 and orthogroups related to flowering time were found to be more highly prevalent in BARI Kanthal-3 compared to the other Arctocarpus spp. The findings of this study will help better understanding the evolution, domestication, phylogenetic relationships, year-round fruiting of this highly nutritious fruit crop as well as providing a resource for molecular breeding.
Keywords: BARI Kanthal-3, genome sequencing and assembly, heterozygosity, SCOS, SNPs, scaffolds, flowering gene orthologues
1. Introduction
Jackfruit (Artocarpus heterophyllus Lam), which belongs to the Moraceae family, has attracted the attention of food experts and technologists due to its nutritional health benefits (Zhang et al., 2022). It is a tropical evergreen tree, which produces the largest edible single fruit in the world (up to 50 kg/fruit) (Naik, 1949; Simmonds and Preedy, 2015; Lin et al., 2022). The place of origin of this fruit tree is still unclear, but it is widely grown in tropical countries including China, India, Malaysia, Thailand, Indonesia, Philippines and Bangladesh (Zhang et al., 2021b), and parts of central and eastern Africa, Florida (USA), Latin America and the Caribbean (Rahman et al., 1999; Sidhu, 2012; Sahu et al., 2020). Jackfruit (popularly known as ‘Kanthal’) is the national fruit of Bangladesh. Its demand is increasing gradually due to its low price, high nutritious value, diversified uses and potential for commercial cultivation (Sidhu, 2012). The crop is commonly referred to as “poor man’s food” due to its lower market price as well as high abundance in the summer season (Rahman et al., 1995; Sahu et al., 2020). Jackfruit flesh, the main edible portion, has a unique aroma and contains high levels of sugars (mainly sucrose, fructose and glucose), carboxylic acids, minerals, vitamins, and dietary fiber (Ong et al., 2006; Lin et al., 2022). The flesh is used as an ingredient in salads, or made into ice cream, jams, nectars, fruit bars, juices, chutney, cakes, jelly and fermented beverages (Zhang et al., 2021a). Notably, jackfruit seeds are rich in starch (60–80% based on dry matter), protein, vitamins and minerals, which may be boiled or roasted and eaten, or boiled and preserved in syrup like chestnuts (Anaya-Esparza et al., 2018).
Bangladesh is one of the largest producers of jackfruit and accounts for about 21% of total fruit production of the country, second only to Mango as the principal fruit crop. During 2019-20, Bangladesh produced 1.1 million tons of jackfruit covering 16,592 hectares area (Statistics., 2020). Despite its numerous advantages, jackfruit trees are not commercially grown as a crop because of an extremely high variation in fruit quality, which is due to its cross-pollinated nature, seed-mediated propagation, short seasonal fruiting and susceptibility to abiotic stresses (Sidhu, 2012). Therefore, the potential of this unique nutritious fruit crop has not yet been utilized in Bangladesh for ensuring food and nutritional security through commercial cultivation and industrial processing. Genetic improvement of existing germplasm to overcome these problems will accelerate jackfruit to become a commercial crop in Bangladesh (Dhar, 1998). A series of earlier studies evaluated the yield, quality and genetic diversity of jackfruit of Bangladesh but none of these studies are systematic and comprehensive (Hossain, 1996; Saha et al., 1996; Rahman et al., 2016) and the underlying molecular mechanisms of the trait diversity in jackfruit is largely unknown. The harvesting period of jackfruit is short (June-August) resulting in a large wastage of this fruit amounting to 20-30% of the crop or even more during some seasons. The Bangladesh Agricultural Research Institute (BARI) developed a year-round jackfruit variety namely, BARI Kanthal-3 in 2014. The number of fruits per plant per year ranges between 219-245 (average = 232), fruits are medium in size (averaging 5.43 kg each) and yield is 1,334.6 (ranges between 1165-1504.2) kg fruit/plant ( Table 1 ). The ripened edible portion contains 35.06 mg/g ß- carotene and 23.6% of total soluble sugar (TSS) (Azad et al., 2007).
Table 1.
Tree and fruit characteristics of Artocarpus heterophyllus L. (Year-round vs seasonal jackfruit).
| Parameters | Features/traits | Reference (Seasonal jackfruit) | |
|---|---|---|---|
| Year-round (BARI Kanthal-3) | Seasonal jackfruit | ||
| Average No. of fruits/plant | 232 | 57 | (Rahman et al., 2016) |
| Average fruit yield/plant/year (kg) | 1,334 | 308 | (Rahman et al., 2016) |
| Average fruit weight (kg) | 5.43 | 5.8 | (Rahman et al., 2016) |
| Harvesting period | September-June | June-August | (Goswami and Chacrabati, 2016) |
| Edible portion (%) | 52.5 | 53.43 | (Rahman et al., 2016) |
| pH | 5.21 | 6.0 | (Goswami and Chacrabati, 2016) |
| TSS (%) | 23.6 | 19.87 | (Rahman et al., 2016) |
| Vitamin C (mg/100g) | 2.0 | 6.46 | (Goswami and Chacrabati, 2016) |
| Total sugar (%) | 21.74 | 14.95 | (Goswami and Chacrabati, 2016) |
| Reducing sugar (%) | 6.02 | 6.43 | (Goswami and Chacrabati, 2016) |
| Nonreducing sugar (%) | 15.72 | 8.52 | (Goswami and Chacrabati, 2016) |
| Moisture content (%) | 69.10 | 81.6 | (Goswami and Chacrabati, 2016) |
| Dry matter | 30.90 | 18.4 | (Goswami and Chacrabati, 2016) |
Whole-genome sequencing (WGS) provides complete coverage of the coding and noncoding regions of the genome (Galperin and Koonin, 2010), which allows a comprehensive assessment of the genome of any organism including those of plants (Chen et al., 2019). It provides a genetic foundation that enables a greater efficiency to identify genetic diversity at key genes that can be used to enhance, reduce or add certain features to a plant phenotype (Chen et al., 2019). Since the first WGS of the model plant, Arabidopsis thaliana in 2000, a large number of plants from diverse taxonomic groups have been sequenced, and genes responsible for various plant traits have been characterized and cloned (Gardner et al., 2016; Chen et al., 2019; Hübner et al., 2019). Recently, WGS of economically important plants and animals such as jute, Corchorus spp. (Islam et al., 2017), hilsa (Tenualosa ilisha) (Das et al., 2018), and goat (Capra hircus) (Siddiki et al., 2019) have created huge public interest in Bangladesh. Recent advances in genomic analyses have revealed large numbers of single nucleotide polymorphisms (SNPs) as the most common form of DNA sequence variation between alleles in several plant species (Morgil et al., 2020). Because of their high abundance, significant information content, when associated with genes, SNPs have gained the center stage as the principal markers of choice for molecular genetics studies. This includes their application in shortening the time of breeding new varieties in many crops through marker assisted selection (Mammadov et al., 2012; Morgil et al., 2020). SNPs have also been applied for several years to assess diversity in specific genes or genomic regions, revealing the phylogenetic relationships between species. However, the emergence of high throughput sequencing technologies allows the SNP-based genetic diversity studies to be carried out at scale and can be useful in conserving diversity in domesticated populations. Plant phylogenetic and evolutionary studies are conventionally based on variation that exist at genes, and hence the knowledge of SNPs in these regions is essential for this analysis (Lasky et al., 2012). It is also important to know the location of SNPs in the whole genome, because if a SNP is present in the coding or regulatory region of a gene, it can greatly affect the functional activity of the resulting protein, such as an enzyme in a biosynthetic pathway (Somerville and Koornneef, 2002) by affecting gene expression and transcriptional and translational promoter activities. Therefore, SNPs can often be responsible for phenotypic variations that exits between individuals and be utilized as selectable genetic markers for improving agronomic traits.
However until now, only a limited amount of genomic information has been made available for the genus of A. heterophyllus (Laricchia et al., 2018; Sahu et al., 2020; Lin et al., 2022). Although the development of a year-round fruiting variety BARI Kanthal-3 from a wild accession offers an opportunity for commercial cultivation and processing of the jackfruit, nothing is known about the underlying molecular mechanism of its year-round fruiting characteristics and other beneficial traits. Molecular understanding of the extremely high phenotypic variabilities in jackfruit would facilitate the future development of high yielding, year-round fruiting, biotic and abiotic stress (e.g., flood, saline, drought, pathogen and pest) tolerant jackfruit varieties through molecular breeding, which is essential for establishing a jackfruit-based processing industry in Bangladesh and elsewhere. We report here an annotated Whole-genome assembly of the year-round fruiting A. heterophyllus cv. BARI Kanthal-3 for first time. The results of the promising phenotypic characteristics of the BARI Kanthal-3 variety, together with both the de novo and reference-guided assemblies, the identified SNPs, and copy number variation in genes that impact flowering time sheds light on the genetic diversity that exists within the A. heterophyllus genome.
2. Materials and methods
2.1. Collection of phenotypic data
The mean values (n = 5) of the phenotypic and biochemical data on tree and fruit characteristics of Artocarpus heterophyllus (BARI Kanthal-3) were obtained using the standard protocols, and compared with the published data of seasonal jackfruit (Goswami and Chacrabati, 2016; Rahman et al., 2016).
2.2. Collection of leaf samples, genomic DNA extraction, WGS library construction, and sequencing
The year-round jackfruit cultivar ‘BARI Kanthal-3’, a superior variety of jackfruit of Bangladesh, was used for genome sequencing. We collected fresh leaf samples from germplasm repository at BARI, Joydebpur, Bangladesh. The samples were identified as jackfruit by Professor Md. Abdul Baset Mia of Department of Crop Botany of Bangabandhu Sheikh Mujibur Rahman Agricultural University (BSMRAU) in Bangladesh. A voucher specimen was deposited in the herbarium of the Department of Crop Botany of BSMRAU with an accession No. VS (HM) 005/2021. The origin of this germplasm is from a wild accession found in Chattogram Hiltracts (Ramgarh) of Bangladesh. Genomic DNA was extracted from freshly harvested leaves using the QIAGEN DNeasy Plant Mini Kit (QIAGEN, Valencia, California, USA) following the manufacturer’s protocol. The quality of the DNA was visually inspected by 1% agarose gel electrophoresis. The quantity of the DNA was assessed by a Qubit Fluorometer (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. Whole-genome sequencing (WGS) library preparation was performed using Nextera XT DNA library preparation kit (Illumina Inc., San Diego, CA, USA) according to the manufacturer’s protocol. Briefly, after normalization, DNA samples were fragmented and tagged by tagmentation in a single-tube reaction (Hoque et al., 2019). The tagmented DNA was amplified through a limited-cycle PCR program using a unique combination of barcode primers, the Index 1 (i7), Index 2 (i5) and full adapter sequences required for cluster generation. Amplification was followed by a cleanup step that purified the library DNA, and removed small library fragments by using Agencourt AMPure XP beads (Beckman Coulter, Inc.). Finally, prepared libraries were loaded onto a reagent cartridge, clustered on the NextSeq 550 System, and paired-end sequencing (2×150 bp) was performed using the Illumina NextSeq 550 High-Output Kit on the NextSeq 550 desktop sequencer.
2.3. Retrieval of data from the GenBank
In addition to the WGS data of BARI Kanthal-3, we also collected the genomic data (WGS) of five related and one distantly related species viz., Artocarpus heterophyllus (family Moraceae) i.e., A. heterophyllus, A. altilis, Morus notabilis, Arabidopsis thaliana and Ficus carica from the AOCC ORCAE platform and the National Center for Biotechnology Information (NCBI) under GenBank accession numbers of CNGB CNP0000486 (https://bioinformatics.psb.ugent.be/orcae/aocc/overview/Arthe), CNGB CNP0000715 (https://bioinformatics.psb.ugent.be/orcae/aocc/overview/Artal), NCBI ASM41409v2 (https://www.ncbi.nlm.nih.gov/genome/?term=ASM41409v2), NCBI TAIR10.1 (https://www.ncbi.nlm.nih.gov/genome/?term=TAIR10.1) and, NCBI Bioproject PRJNA565858, respectively.
2.4. De novo genome assembly and annotation
The generated WGS data were filtered through Trimmomatic v0.38 (Bolger et al., 2014) with option “LEADING:20 TRAILING:20 SLIDINGWINDOW:4:15 MINLEN:50” parameters to remove Illumina adapter, known Illumina artifacts, phiX, and low-quality regions. The processed reads were assembled by SOAPdenovo2 v2.04 (Luo et al., 2012) with k-mer=39 and subsequently scaffolded using a reference guided approach by RAGTAG (Alonge et al., 2019) software with default parameters. GapCloser v1.12 (Luo et al., 2012) with default parameters (“-l 150 -t 32 -p 31”) was utilized for gap closing using the pair-end data. The assembled genome was analyzed by EDTA (Ou et al., 2019) software to create a non-redundant transposable element database which was then used to soft-mask the genome sequence. Genome annotation was performed by using Braker2 (Brůna et al., 2021) software with coding-gene sequence evidence from A. heterophyllus.
2.5. Genome assembly validation
The scaffolded sequences were compared against the reference genome by nucmer v4.0.0rc1 (Kurtz et al., 2004) using the default parameters. The genome assembly completeness was assessed using BUSCO (Benchmarking Universal Single-Copy Orthologues), v4.1.4 (Seppey et al., 2019) to evaluate the presence of conserved plant orthologs with the Embryophyta database 10 lineage.
2.6. Genome size estimation
The high-quality data (~50X depth) were provided to Jellyfish v2.2.6 (Marçais and Kingsford, 2011) with “-C -m 21 -s 5G –min-quality=25” parameters to generate k-mer (K=21) frequency distribution. The output histograms were analyzed using GenomeScope (Vurture et al., 2017) to estimate the genome size, heterozygosity level, error rates, and repeat fraction.
2.7. Variant calling
Processed Illumina data were aligned against the draft A. heterophyllus genome (Sahu et al., 2020) using Burrows-Wheeler Aligner (BWA) v 0.7.17 (Li and Durbin, 2009). The aligned reads mapped with a quality score 30 or greater were analyzed by samtools/bcftools (Li, 2011) to produce the raw variant calls. Simple single nucleotide polymorphisms (SNPs) were defined as SNPs with greater than 90% ALT allele frequency observed in at least 30 high quality reads (MAPQ>30). Following variant calling, we used snpEff to annotate variants and predict their effects on genes using a custom database generated using the A. heterophylus genome and annotation from https://bioinformatics.psb.ugent.be/gdb/aocc/arthe/ (Sahu et al., 2020).
2.8. Orthologous gene analysis
Gene orthology and orthogroups were inferred by Orthofinder (Emms and Kelly, 2019) based on protein sequence similarity searches using Diamond (Buchfink et al., 2015) software. 306 genes involved in A. thaliana flowering-time gene networks were downloaded from the Flowering-Interactive Database (FLOR-ID) (Bouché et al., 2016). Gene expansion or contraction were assessed based on relative gene count within each orthogroup relative to A. thaliana.
3. Results and discussion
3.1. Source and phenotypic features of the BARI Kanthal-3 variety
To develop the variety of BARI Kanthal-3, germplasm was collected from locations all over the Bangladesh including in the Chattogram Hiltracts, such as Ramgarh of Khagrachari. In 2014, an accession of Ramgarh was certified for cultivation in Bangladesh with a varietal name of BARI Kanthal-3, representing a new and unique variety of jackfruit in Bangladesh that bear fruits for ten months of the calendar year (September to June) while the seasonal plant gives fruit only for three months (June to August) each year. This year-round fruiting variety produces more than 4-fold higher average number of fruits per plant and fruit yield per plant per year compared to seasonal jackfruit. A mature plant produces an average of 232 (range 219-245) fruits per plant yielding about 1,334.6 kg (1165 -1504.2 kg) fruit/plant/year ( Table 1 ). The fruits of BARI Kanthal-3 were medium (average 5.43 kg each) and average yield was 133.2 t/ha/year. This variety was not affected by any sort of infectious pathogens or pests (data not shown). The tree is erect and medium bushy, and the pulp of the fruit is medium soft, slightly yellow, medium juicy, highly sweet and aromatic ( Figure 1 ). The amounts of ß- carotene and total soluble solid (TSS) in fruits were 35.06 mg/g and 23.6%, respectively. The edible portion of the fruit was 52.5% ( Table 1 ). A large body of literature has revealed that jackfruit is a rich source of carbohydrates, minerals, carboxylic acids, dietary fiber, vitamins and minerals and bioactive compounds (Azad et al., 2007; Khan et al., 2021). Clearly, BARI Kanthal-3 is an extremely high yielding (ca. 4-fold higher than average fruit yield of a seasonal jackfruit per year) and high quality jackfruit.
Figure 1.

Phenotypic characteristics of BARI Kanthal-3. (A) Tree in situ, (B) Leaf, (C) Young fruits from the tree, (D) Mature fruit, (E) Kernel (flesh), and (F) Seed.
3.2. Genome sequencing, assembly and annotation
A total of 472 million raw Illumina pair-end sequencing reads from the extracted DNA of A. heterophyllus cv. BARI Kanthal-3 leaf tissues were generated. The genome size of BARI Kanthal-3 was estimated to be 1.04 Gb with a heterozygosity rate of 1.62% based on K-mer analysis of the short read data ( Figure 2 ). The estimated size is similar to the recently reported 1.01Gb genome size of seasonal A. heterophyllus (Sahu et al., 2020), and is consistent with the c-value of 1.20 pg (Ohri and Kumar, 1986). BARI Kanthal-3 has a higher heterozygosity rate compared to the available reference genomes of seasonal jackfruit recently published, at 0.90 and 0.91, respectively (Sahu et al., 2020; Lin et al., 2022).
Figure 2.
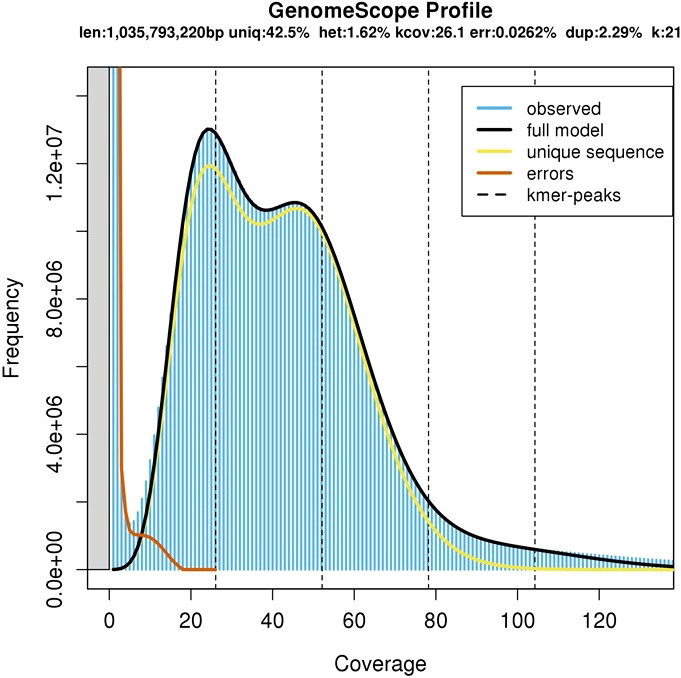
Genome size prediction of Artocarpus heterophyllus cv. BARI Kanthal-3. The X-axis represents the coverage of the genome while the Y-axis represents the frequency levels.
After quality filtering of the short reads using Trimmomatic, 439 million clean reads were obtained ( Table 2 ). The high-quality reads assembled into different contigs using SOAPdenovo2, which ultimately yielded a base assembly of 1.36 M scaffolds, totaling 817.7 Mb. The N50s of scaffolds were 1.8 Kb ( Table 2 ). The BARI Kanthal-3 contigs were then further scaffolded together using the reference guided approach using the existing published draft reference genome of jackfruit (Sahu et al., 2020) and the software RAGTAG, and finally gaps in the scaffolds were filled using the same pair-end Illumina data and Gapcloser software. In this case, SOAPdenovo2 + RAGTAG + GapCloser produced a base assembly of N50 size = 425 Kb in 218,562 scaffolds ( Table 2 ). The mapping of paired-end short read data against the de novo scaffolded assembly and out of 439 million read pairs, 400 million (91.1%) aligned to the assembly were properly paired indicating a high proportion of the original data is represented in the assembly. The GC content of BARI Kanthal-3 was 34.10% which is comparable to the GC content of a seasonal A. heterophyllus from Indian and Chinese origins that were recorded at 32.9% and 34.9%, respectively ( Table S1 ) (Sahu et al., 2020; Lin et al., 2022).
Table 2.
Result of de novo and reference guided genome assembly of Artocarpus heterophyllus cv. BARI Kanthal-3.
| Assembly | N50 number | N50 size (Kb) | N90 number | N90 size (Kb) | Longest Sequence (Kb) | Num Sequence | Total Bases (Mb) |
|---|---|---|---|---|---|---|---|
| SOAPdenovo2 | 88,402 | 1.8 | 720,032 | 0.2 | 65 | 1,364,052 | 817.7 |
| SOAPdenovo2 + RAGTAG + Gapcloser | 561 | 425 | 14,076 | 1.4 | 2,611 | 218,562 | 843 |
We annotated 41,088 protein-coding genes in BARI-Kanthal-3 assembly using the Braker2 gene prediction pipeline. The gene number is comparable to the two existing A. heterophyllus genome assemblies with 35,858 (Sahu et al., 2020) and 41,997 (Lin et al., 2022) annotated genes, respectively.
3.3. Genome assembly validation
To assess the representation of a complete conserved core gene set in the BARI Kanthal-3, assembly, an analysis was carried out to assess the quality and completeness of the draft genome using the Benchmarking Universal Single-Copy Orthologs (BUSCO) datasets and an orthologue data base ( Figure 3 ). We identified 1,614 single copy orthologs (SCOs). Among these SCOs, 97.2% (1569/1614) were complete (single copy = 1094, and duplicated = 475), whereas 21 and 24 were fragmented and missing, respectively ( Figure 3 ). Our present findings aligned with the recently reported findings of Sahu et al. (2020), who reported that out of 1440 BUSCO ortholog groups searched in the A. heterophyllus assembly, 95% (1369/1440) were complete BUSCOs, 932 (64.7%) were “complete single-copy”, 437 (30.3%) were “complete duplicated”, and 15 (1%) were “fragmented”, and 56 (4%) were “missing”.
Figure 3.
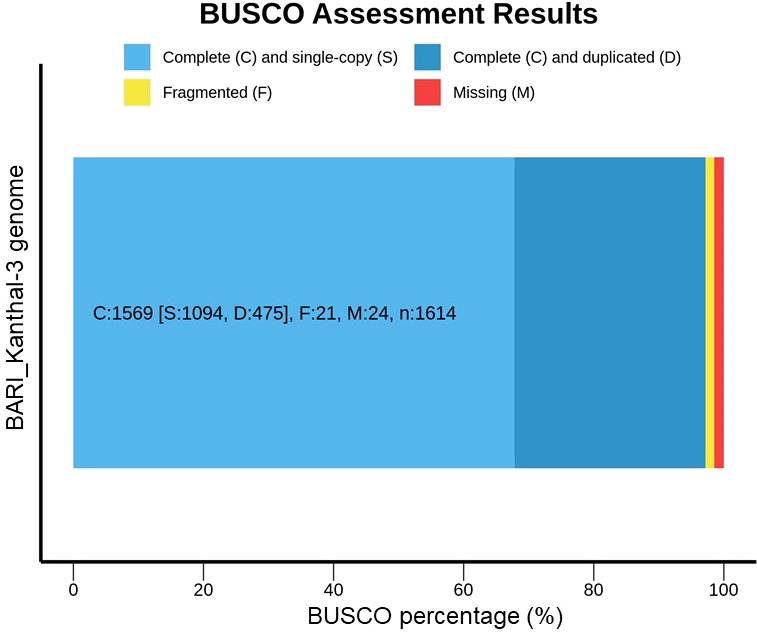
BUSCO (v.4.0.4) assessment results of BARI Kanthal-3. BUSCO completeness in Artocarpus heterophyllus cv. BARI Kanthal-3 is high indicating that the genome assemblies are of high gene space completeness.
We then carried out an orthologous gene analysis across the BARI Kanthal-3 genome and the genomes of five other species (A. heterophyllus, A. altilis, F. carica, M. notabilis, A. thaliana) based upon a protein similarity search using Diamond software and the inference of gene orthology and orthogroups using Orthofinder software. In this study, 10,924 orthogroups were found to be shared across the genomes of the six species ( Figure 4A ) with 399 found to be unique for BARI Kanthal-3 genome. Phylogenetic analysis was performed across the 10,924 shared orthogroups using the rooted gene tree method in Orthofinder ( Figure 4B ). The analysis revealed that the two genomes of A. heterophylus (BARI Kanthal-3 and A. heterophyllus) clustered more closely related to the other three Moraceae genomes with A. thaliana as an outgroup in the phylogenetic tree ( Figure 4B ). Finally, to assess if there was a relationship between differences in flowering time between BARI Kanthal-3 and seasonal A. heterophyllus and differences in the representation of orthogroups related to flowering time between them, we identified the relative numbers of these orthogroups in each of the Moraceae genomes compared to Arabidopsis ( Figure 4C and Table S2 ). This identified large scale expansion in the Artocarpus spp. likely reflecting the whole genome duplication event previously identified in their evolution (Lin et al., 2022). However, a higher number of duplications were identified in the BARI Kanthal-3 genome compared to the other Artocarpus spp. indicating the longer season nature of the accession could be explained by an overall expansion of these gene families, although the more fractured nature of the genome assembly could also be impacting this observation. The availability of more contiguous long read genome assemblies for BARI Kanthal-3 and other Artocarpus spp. would enable this observation to be confirmed.
Figure 4.

Distribution of orthogroups in the genomes of five species (Artocarpus heterophyllus, A. altilis, Ficus carica, Morus notabilis, Arabidopsis thaliana and BARI Kanthal-3). (A) diagram showing unique and shared orthogroups in three genomes of Artocarpus species, M. notabilis, F. carica and Arabidopsis. Each number represents the number of orthogroups in common between each pairing. (B) Inferred phylogenetic tree constructed with common orthogroups from A. heterophyllus, A. altilis, M. notabilis, F. carica, A. thaliana and BARI Kanthal-3 (A. heterophyllus) genomes. The tree was constructed using rooted gene tree method in OrthoFinder. (C) Relative numbers of flowering gene orthogroups in each of the Moraceae genomes compared to Arabidopsis thaliana.
3.4. Variant analysis
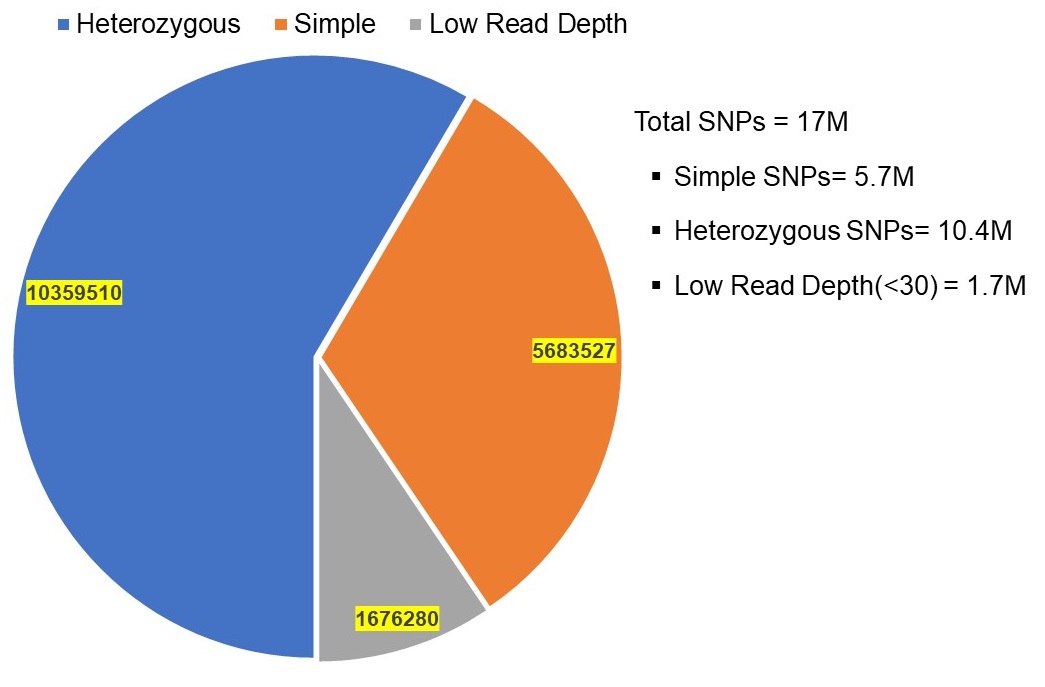
The processed WGS reads were aligned against the A. heterophyllus draft assembly. Out of a total of 439 M reads, 417 M (95.1%) were found to be aligned in exact pairs. A total of 16 million single-nucleotide polymorphisms (SNPs) were called from the dataset including 5.7 M (35.0%) simple and 10.4 M (65.0%) heterozygous SNPs ( Table 3 and Figure S1 ). Approximately, 90% of all polymorphisms are located in intergenic regions. In this study, 144,787 (2.5%) and 426,997 (7.5%) of the simple SNPs, and 250,715 (2.4%) and 739,288 (7.1%) of the heterozygous SNPs, were found in the exons and introns, respectively ( Table 3 ). We further predicted the effects of variants on genes. As expected, large fraction of the variants was in the intergenic (64.5%), intronic (5%) and up/down-stream regions (29%) of the genes ( Figure 5A ). There are 232,587 missense mutations and 4,750 gained stop codons suggesting an altered protein function in BARI Kanthal-3 ( Figure 5B ). One of the important findings of this study is the high level of heterozygosity in the year-round fruiting jackfruit genome. The high level of heterozygosity in A. heterophyllus genome raises the question of which allele, for each heterozygous locus, is represented by the reference genome ( Table S2 ). Therefore, the inherent differences between individual plants should always be considered when utilizing the reference genome to detect SNP variants (Hawkins et al., 2016).
Table 3.
Results of variant analysis of Artocarpus heterophyllus cv. BARI Kanthal-3.
| Genomic region | Simple SNPs | Heterozygous SNPs |
|---|---|---|
| Exonic | 144,787 | 250,715 |
| Intronic | 426,997 | 739,288 |
| Inter-genic | 5,111,742 | 9,369,507 |
| Total | 5,683,526 | 10,359,510 |
Figure 5.

Variant annotation and effects of variant on genes. (A) Distribution and abundance of variants in the genome of BARI Kanthal-3, and (B) Number of effects by type and region of the gene variants.
SNPs have been indicated as the major factors in the creation of phenotypic variation and their effect on functional changes of genes is used as a tool in functional genomics of organisms (Hirakawa et al., 2013). In plants, many traits of interest have been linked with SNPs (Mammadov et al., 2012; Huq et al., 2016; Zhang et al., 2018), including roles in metabolism, cellular processes and signaling, that could have an impact on breeding. This study for the first time identified the SNPs in the genome of a year-round fruiting jackfruit cultivar, which promises the development of genetic markers associated with the important traits of this economically important species including the genes regulating flowering and fruit development. The availability of SNPs within the coding and regulatory sequences also offers the prospect of identifying the causative variations influencing these processes (Varshney, 2010). One of the hallmark findings of this study is that majority of the SNPs (47.29%) of BARI Kanthal-3 were localized in the intergenic regions in the proximity of genes (including 5′ UTR, 3′ UTR, and introns). Approximately, 25% of the intergenic SNPs were detected within the region spanning 10 kb upstream of the gene start site and 10 kb downstream of the gene end site (Yamamoto et al., 2007), implying the possibility that some of these SNPs affect the expression of the nearest neighboring genes. It has been reported that a high frequency of genetic variants in the noncoding regions likely results from less selection pressure from natural selection and/or domestication (Barreiro et al., 2008). However, DNA polymorphisms in these regions have been reported to play important roles during evolution and domestication. For example, a mutation in the 5′ regulatory region of the qSH1 gene, an ortholog of the Arabidopsis homeobox gene REPLUMLESS (RPL) results in the absence of abscission zone formation and thus loss of seed shattering in a subset of temperate japonica cultivar of rice (Konishi et al., 2006). Similarly, a considerable number of mutations in introns in pre-harvest sprouting (PHS) genes lead to PHS in rice plant (Lee et al., 2017). Among the 12 PHS mutants (phs), mutations in genes encoding major enzymes of the carotenoid biosynthesis pathway, cause photo-oxidation and ABA-deficiency phenotypes, of which the latter is a major factor controlling the PHS trait in rice (Fang et al., 2008). Interestingly, in jackfruit, MADS-box genes and carotenoid biosynthesis genes, were the primary targets for domestication (Laricchia et al., 2018). However, the role of inter-genic SNPs in A. heterophylus in domestication of this horticultural plant needs to be explored further.
SNP markers have become extremely popular in plant molecular genetics due to their genome-wide abundance and amenability for high-throughput detection platforms. For example, SNPs regulating various Quantitative Trait Loci (QTL) responsible for cold and disease resistance such as such as blight, bacterial canker and gray mold have been reported (Zhang et al., 2003; Coaker and Francis, 2004), and SNPs associated with flowering in Raphanus sativus have recently been discovered by transcriptome sequencing and computational analysis (Kim et al., 2019). An additional transcriptomics study in BARI Kanthal-3 is needed for the identification of genes associated with flowering time and year-round fruiting.
4. Conclusions
Although genomic information has been made available for seasonal jackfruit, this is the first report of an assembled and annotated genome for a year-round fruiting jackfruit variety. The annotation yielded a very similar number of annotated genes to a recent genome assembly of a seasonal jackfruit together with a high representation of core othologous genes. An orthologous gene analysis with other Moraceae gennomes confirmed the close realtionship between BARI Kanthal-3 and other Arctocarpus spp. but also indicated a higher number of genes related to flowering time were present in the year-round fruiting variety.
This study also reported the distribution of a large collection of nucleotide variation identified across the genome that can be used to identify new functional genes and their regulatory activities specific to BARI Kanthal-3. Furthermore, the genomic data and the identified SNPs of this year-round fruiting jackfruit cultivar could facilitate further genomics and post-genomics studies for detecting other trait-specific genes that are essential for molecular breeding of jackfruit. One of the limitations of this study is lack of information on the role of inter-genic SNPs in BARI Kanthal-3 and the impact on gene function (i.e., changes in protein-coding genes, differential gene expression, and the specific functions of protein-coding genes). To further understand the underlying molecular mechanisms of unique traits of this new variety, a high quality long read genome assembly of BARI-Kanthal-3 and comparative transcriptome analysis with seasonal jackfruit are needed.
The fruit nutritional quality, yield and year-round fruiting properties of BARI Kanthal-3 indicate a unique and valuable genetic material for the improvement of commercial cultivation and development of jackfruit-based processing industry in Bangladesh. The genomic data, associated orthogroups, and SNPs identified in this research will be useful for characterization of trait-specific genes and development of markers for molecular breeding for the improvement of jackfruit, and provides an opportunity to develop this underutilized crop for ensuring food and nutritional security for the increasing population of Bangladesh and other tropical countries.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at https://www.ncbi.nlm.nih.gov/nuccore/JAIQDR000000000.1, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA686208.
Ethics statement
Permission to collect leaf sample of Artocarpus heteropyllus Lam. was obtained.
Author contributions
All authors contributed intellectually to this study. TI conceived the study, designed the experiment, coordinated the project, provided reagents and laboratory support, interpreted the results, wrote and revised the manuscript. NA collected the plant samples, extracted DNA, and prepared the draft manuscript; CK assembled and annotated the sequenced data, interpreted results and wrote the manuscript; MNH, interpreted the results, wrote and revised the manuscript; MJH, provided plant samples, collected and interpreted phenotypic and biochemical data and wrote manuscript; NUM and DRG, conducted experiments and prepared library for the DNA sequencing using Illumina Nextseq 550; AAN and RI, analyzed sequenced data, prepared phylogenetic tree and wrote the manuscript; PKB, wrote and revised the manuscript; AGS, coordinated, wrote and revised the manuscript; All authors read, revised, edited and approved the final manuscript.
Acknowledgments
We are thankful to the ‘BSMRAU Physical Facility and Research Capacity Strengthening Project’ under the Ministry of Education of the People’s Republic of Bangladesh for funding this research. The Global Institute for Food Security (GIFS), Canada and BDREN of UGC, Bangladesh also deserve our thanks for offering facilities for bioinformatics analyses of the genomic data of jackfruit cv. BARI Kanthal-3.
Funding
This project was funded by the ‘BSMRAU Physical Facility and Research Capacity Strengthening Project’ under the Ministry of Education of the People’s Republic of Bangladesh. Bioinformatics support and analysis was supported with funds from the Bangabandhu Research Chair in Food Security held by AGS at GIFS and supported by GIFS and the Bangladesh Agriculture Research Council.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1044420/full#supplementary-material
Comparison between the previously reported Artocarpus heterophyllus sequence and BARI Kanthal-3 sequence.
Flowering locus C (FLC). Listing of flowering gene orthologues (Enclosed in a separate file).
Categories of candidate SNPs identified in the genome of BARI_Kanthal-3.
{kind=link}
References
- Alonge M., Soyk S., Ramakrishnan S., Wang X., Goodwin S., Sedlazeck F. J., et al. (2019). RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 1–17. doi: 10.1186/s13059-019-1829-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaya-Esparza L. M., González-Aguilar G. A., Domínguez-Ávila J. A., Olmos-Cornejo J. E., Pérez-Larios A., Montalvo-González E. (2018). Effects of minimal processing technologies on jackfruit (Artocarpus heterophyllus lam.) quality parameters. Food Bioprocess. Technol. 11, 1761–1774. doi: 10.1007/s11947-018-2136-z [DOI] [Google Scholar]
- Azad A., Jones J., Haq N. (2007). Assessing morphological and isozyme variation of jackfruit (Artocarpus heterophyllus lam.) in Bangladesh. Agroforestry Syst. 71, 109–125. doi: 10.1007/s10457-007-9039-8 [DOI] [Google Scholar]
- Barreiro L. B., Laval G., Quach H., Patin E., Quintana-Murci L. (2008). Natural selection has driven population differentiation in modern humans. Nat. Genet. 40, 340–345. doi: 10.1038/ng.78 [DOI] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouché F., Lobet G., Tocquin P., Périlleux C. (2016). FLOR-ID: an interactive database of flowering-time gene networks in arabidopsis thaliana. Nucleic Acids Res. 44, D1167–D1171. doi: 10.1093/nar/gkv1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brůna T., Hoff K. J., Lomsadze A., Stanke M., Borodovsky M. (2021). BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR. Genomics Bioinf. 3, lqaa108. doi: 10.1093/nargab/lqaa108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., Huson D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- Chen F., Song Y., Li X., Chen J., Mo L., Zhang X., et al. (2019). Genome sequences of horticultural plants: past, present, and future. Horticult. Res. 6, 1–23. doi: 10.1038/s41438-019-0195-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coaker G., Francis D. M. (2004). Mapping, genetic effects, and epistatic interaction of two bacterial canker resistance QTLs from lycopersicon hirsutum. Theor. Appl. Genet. 108, 1047–1055. doi: 10.1007/s00122-003-1531-6 [DOI] [PubMed] [Google Scholar]
- Das A., Ianakiev P., Baten A., Nehleen R., Ehsan T., Ahmed O., et al. (2018). Genome of tenualosa ilisha from the river padma, Bangladesh. BMC Res. Notes 11, 1–3. doi: 10.1186/s13104-018-4028-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar M. (1998). Techniques of vegetative and in vitro propagation of jackfruit, Salna, Gazipur Bangladesh:Institute Postgraduate Studies in Agriculture. Ph D Thesis, pp. 27–31 [Google Scholar]
- Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14. doi: 10.1186/s13059-019-1832-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J., Chai C., Qian Q., Li C., Tang J., Sun L., et al. (2008). Mutations of genes in synthesis of the carotenoid precursors of ABA lead to pre-harvest sprouting and photo-oxidation in rice. Plant J. 54, 177–189. doi: 10.1111/j.1365-313X.2008.03411.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin M. Y., Koonin E. V. (2010). From complete genome sequence to ‘complete’understanding? Trends Biotechnol. 28, 398–406. doi: 10.1016/j.tibtech.2010.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner E. M., Johnson M. G., Ragone D., Wickett N. J., Zerega N. J. (2016). Low-coverage, whole-genome sequencing of artocarpus camansi (Moraceae) for phylogenetic marker development and gene discovery. Appl. Plant Sci. 4, 1600017. doi: 10.3732/apps.1600017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C., Chacrabati R. (2016). ““Jackfruit (Artocarpus heterophylus),,” in MSJ. Simmonds and V. R. Preedy (Eds) Nutritional composition of fruit cultivars (Elsevier; ), 317–335. doi: 10.1016/B978-0-12-408117-8.00014-3 [DOI] [Google Scholar]
- Hawkins C., Caruana J., Schiksnis E., Liu Z. (2016). Genome-scale DNA variant analysis and functional validation of a SNP underlying yellow fruit color in wild strawberry. Sci. Rep. 6, 1–11. doi: 10.1038/srep29017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa H., Shirasawa K., Ohyama A., Fukuoka H., Aoki K., Rothan C., et al. (2013). Genome-wide SNP genotyping to infer the effects on gene functions in tomato. DNA Res. 20, 221–233. doi: 10.1093/dnares/dst005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M. N., Istiaq A., Clement R. A., Sultana M., Crandall K. A., Siddiki A. Z., et al. (2019). Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci. Rep. 9, 1–14. doi: 10.1038/s41598-019-49468-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain A. (1996). Status report on genetic resources of jackfruit in Bangladesh (Singapore: International Plant Genetic Resources Institute Regional Office; ). [Google Scholar]
- Hübner S., Bercovich N., Todesco M., Mandel J. R., Odenheimer J., Ziegler E., et al. (2019). Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 5, 54–62. doi: 10.1038/s41477-018-0329-0 [DOI] [PubMed] [Google Scholar]
- Huq M. A., Akter S., Nou I. S., Kim H. T., Jung Y. J., Kang K. K. (2016). Identification of functional SNPs in genes and their effects on plant phenotypes. J. Plant Biotechnol. 43, 1–11. doi: 10.5010/JPB.2016.43.1.1 [DOI] [Google Scholar]
- Islam M. S., Saito J. A., Emdad E. M., Ahmed B., Islam M. M., Halim A., et al. (2017). Comparative genomics of two jute species and insight into fibre biogenesis. Nat. Plants 3, 1–7. doi: 10.1038/nplants.2016.223 [DOI] [PubMed] [Google Scholar]
- Khan A. U., Ema I. J., Faruk M., Tarapder S. A., Khan A. U., Noreen S., et al. (2021). A review on importance of artocarpus heterophyllus L. (Jackfruit) J. Multidiscip. Appl. Natural Science 1 (2), 106–116. doi: 10.47352/jmans.v1i2.88 [DOI] [Google Scholar]
- Kim J., Manivannan A., Kim D.-S., Lee E.-S., Lee H.-E. (2019). Transcriptome sequencing assisted discovery and computational analysis of novel SNPs associated with flowering in raphanus sativus in-bred lines for marker-assisted backcross breeding. Horticult. Res. 6, 1–12. doi: 10.1038/s41438-019-0200-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi S., Izawa T., Lin S. Y., Ebana K., Fukuta Y., Sasaki T., et al. (2006). An SNP caused loss of seed shattering during rice domestication. Science 312, 1392–1396. doi: 10.1126/science.1126410 [DOI] [PubMed] [Google Scholar]
- Kurtz S., Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5, 1–9. doi: 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laricchia K. M., Johnson M. G., Ragone D., Williams E. W., Zerega N. J., Wickett N. J. (2018). A transcriptome screen for positive selection in domesticated breadfruit and its wild relatives (Artocarpus spp.). Am. J. Bot. 105, 915–926. doi: 10.1002/ajb2.1095 [DOI] [PubMed] [Google Scholar]
- Lasky J. R., Des Marais D. L., Mckay J. K., Richards J. H., Juenger T. E., Keitt T. H. (2012). Characterizing genomic variation of arabidopsis thaliana: the roles of geography and climate. Mol. Ecol. 21, 5512–5529. doi: 10.1111/j.1365-294X.2012.05709.x [DOI] [PubMed] [Google Scholar]
- Lee G.-A., Jeon Y.-A., Lee H.-S., Hyun D. Y., Lee J.-R., Lee M.-C., et al. (2017). New genetic loci associated with preharvest sprouting and its evaluation based on the model equation in rice. Front. Plant Sci. 8, 1393. doi: 10.3389/fpls.2017.01393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., Feng C., Lin T., Harris A., Li Y., Kang M. (2022). Jackfruit genome and population genomics provide insights into fruit evolution and domestication history in China. Horticult. Res. 9, 1–13. doi: 10.1093/hr/uhac173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. doi: 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammadov J., Aggarwal R., Buyyarapu R., Kumpatla S. (2012). SNP markers and their impact on plant breeding. Int. J. Plant Genomics, 728398. doi: 10.1155/2012/728398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G., Kingsford C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgil H., Gercek Y. C., Tulum I. (2020). “Single nucleotide polymorphisms (SNPs) in plant genetics and breeding,” In The recent topics in genetic polymorphisms Çalışkan M., et al. eds. (IntechOpen; ). doi: 10.5772/intechopen.91886 [DOI] [Google Scholar]
- Naik K. C. (1949) South Indian fruits and their culture. South Indian fruits their culture P. Varadachary & Co., Madras. p. 335 [Google Scholar]
- Ohri D., Kumar A. (1986). Nuclear DNA amounts in some tropical hardwoods. Caryologia 39, 303–307. doi: 10.1080/00087114.1986.10797792 [DOI] [Google Scholar]
- Ong B., Nazimah S., Osman A., Quek S., Voon Y., Hashim D. M., et al. (2006). Chemical and flavour changes in jackfruit (Artocarpus heterophyllus lam.) cultivar J3 during ripening. Postharvest. Biol. Technol. 40, 279–286. doi: 10.1016/j.postharvbio.2006.01.015 [DOI] [Google Scholar]
- Ou S., Su W., Liao Y., Chougule K., Agda J. R., Hellinga A. J., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 1–18. doi: 10.1186/s13059-019-1905-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A. M., Huq E., Mian A., Chesson A. (1995). Microscopic and chemical changes occurring during the ripening of two forms of jackfruit (Artocarpus heterophyllus l.). Food Chem. 52, 405–410. doi: 10.1016/0308-8146(95)93290-8 [DOI] [Google Scholar]
- Rahman M. A., Nahar N., Mian A. J., Mosihuzzaman M. (1999). Variation of carbohydrate composition of two forms of fruit from jack tree (Artocarpus heterophyllus l.) with maturity and climatic conditions. Food Chem. 65, 91–97. doi: 10.1016/S0308-8146(98)00175-7 [DOI] [Google Scholar]
- Rahman M., Patwary M. A., Barua H., Nahar S., Ahmmed A. N. F. (2016). Evaluation of yield and quality of three jackfruit (Artocarpus heterophyllus l.) genotypes. Agriculturists. 14, 107–111. doi: 10.3329/agric.v14i1.29108 [DOI] [Google Scholar]
- Saha M., Saha M., Rahman M., Nazrul M., Quasem A., Halder N., et al. (1996). “Variability in jackfruit,” in Proceedings of the Internal Research Review Workshop (Gazipur, Bangladesh: Horticulture Research Center, BARI; ), 1–4. [Google Scholar]
- Sahu S. K., Liu M., Yssel A., Kariba R., Muthemba S., Jiang S., et al. (2020). Draft genomes of two artocarpus plants, jackfruit (A. heterophyllus) and breadfruit (A. altilis). Genes 11, 27. doi: 10.3390/genes11010027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppey M., Manni M., Zdobnov E. M. (2019). BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245. doi: 10.1007/978-1-4939-9173-0_14 [DOI] [PubMed] [Google Scholar]
- Siddiki A. Z., Baten A., Billah M., Alam M., Shawrob K. S. M., Saha S., et al. (2019). The genome of the black Bengal goat (Capra hircus). BMC Res. Notes 12, 1–3. doi: 10.1186/s13104-019-4400-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A. S. (2012). Jackfruit improvement in the Asia-pacific region: A status report (APAARI) FAO Regional Office for Asia & the Pacific (FAO RAP) Maliwan Mansion, 39, Phra Atit Road Bangkok 10200. [Google Scholar]
- Simmonds M., Preedy V. R. (2015). Nutritional composition of fruit cultivars (Academic Press). (San Diego, USA: Academic Press; ). [Google Scholar]
- Somerville C., Koornneef M. (2002). A fortunate choice: the history of arabidopsis as a model plant. Nat. Rev. Genet. 3, 883–889. doi: 10.1038/nrg927 [DOI] [PubMed] [Google Scholar]
- Statistics. Y. O. A. (2020). Bangladesh Bureau of statistics (BBS), statistics and informatics division (SID), ministry of planning, government of the people’s republic of Bangladesh (Government of the People’s Republic of Bangladesh; ). [Google Scholar]
- Varshney R. K. (2010). “Gene-based marker systems in plants: high throughput approaches for marker discovery and genotyping,” in Molecular techniques in crop improvement (Dordrecht: Springer; ), 119–142. doi: 10.1007/978-90-481-2967-6_5 [DOI] [Google Scholar]
- Vurture G. W., Sedlazeck F. J., Nattestad M., Underwood C. J., Fang H., Gurtowski J., et al. (2017). GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y. Y., Ichida H., Matsui M., Obokata J., Sakurai T., Satou M., et al. (2007). Identification of plant promoter constituents by analysis of local distribution of short sequences. BMC Genomics 8, 1–23. doi: 10.1186/1471-2164-8-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin G., Nino-Liu D., Foolad M. R. (2003). Mapping QTLs conferring early blight (Alternaria solani) resistance in a lycopersicon esculentum× l. hirsutum cross by selective genotyping. Mol. Breed. 12, 3–19. doi: 10.1023/A:1025434319940 [DOI] [Google Scholar]
- Zhang Y., Li B., Xu F., He S., Zhang Y., Sun L., et al. (2021. a). Jackfruit starch: Composition, structure, functional properties, modifications and applications. Trends Food Sci. Technol. 107, 268–283. doi: 10.1016/j.tifs.2020.10.041 [DOI] [Google Scholar]
- Zhang W., Mirlohi S., Li X., He Y. (2018). Identification of functional single-nucleotide polymorphisms affecting leaf hair number in brassica rapa. Plant Physiol. 177, 490–503. doi: 10.1104/pp.18.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wang Q., Zhang Y., Wu G., Tan L., Zhang Z. (2022). Effects of moisture content on digestible fragments and molecular structures of high amylose jackfruit starch prepared by improved extrusion cooking technology. Food Hydrocolloids. 133, 108023. doi: 10.1016/j.foodhyd.2022.108023 [DOI] [Google Scholar]
- Zhang Y., Zuo H., Xu F., Zhu K., Tan L., Dong W., et al. (2021. b). The digestion mechanism of jackfruit seed starch using improved extrusion cooking technology. Food Hydrocolloids. 110, 106154. doi: 10.1016/j.foodhyd.2020.106154 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison between the previously reported Artocarpus heterophyllus sequence and BARI Kanthal-3 sequence.
Flowering locus C (FLC). Listing of flowering gene orthologues (Enclosed in a separate file).
Categories of candidate SNPs identified in the genome of BARI_Kanthal-3.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at https://www.ncbi.nlm.nih.gov/nuccore/JAIQDR000000000.1, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA686208.