

Abstract
Sleep intensity is adjusted by the length of previous awake time, and under tight homeostatic control by protein phosphorylation. Here, we establish microglia as a new cellular component of the sleep homeostasis circuit. Using quantitative phosphoproteomics of the mouse frontal cortex, we demonstrate that microglia‐specific deletion of TNFα perturbs thousands of phosphorylation sites during the sleep period. Substrates of microglial TNFα comprise sleep‐related kinases such as MAPKs and MARKs, and numerous synaptic proteins, including a subset whose phosphorylation status encodes sleep need and determines sleep duration. As a result, microglial TNFα loss attenuates the build‐up of sleep need, as measured by electroencephalogram slow‐wave activity and prevents immediate compensation for loss of sleep. Our data suggest that microglia control sleep homeostasis by releasing TNFα which acts on neuronal circuitry through dynamic control of phosphorylation.
Keywords: microglia, TNFα, sleep, phosphoproteome, synapse
Subject Categories: Neuroscience, Post-translational Modifications & Proteolysis, Proteomics
This study reveals that microglia, via the release of TNFα, control phosphorylation of several proteins with known roles in sleep to maintain homeostatic sleep control.

Introduction
The neuronal circuitries and neuromodulators regulating sleep are well characterized (Scammell et al, 2017; Adamantidis et al, 2019). On the contrary, the molecular mechanisms that control sleep have only been recently investigated. In particular, protein phosphorylation has been described as a fundamental molecular process of sleep control (Ode & Ueda, 2020). The central involvement of phosphorylation in sleep regulation is supported by the fact that kinases, such as SIKs, CaMKIIα/β, and ERK1/2, have significant sleep‐inducing effects (Funato et al, 2016; Tatsuki et al, 2016; Mikhail et al, 2017; Park et al, 2020). Accordingly, hundreds of proteins, including these kinases, were found to undergo sleep‐dependent cycles of phosphorylation/dephosphorylation (Diering et al, 2017; Brüning et al, 2019). Because phosphorylation is a major form of regulation of neuronal functions, these changes in phosphorylation were proposed to adjust neuronal functions to sleep or wake brain states. In particular, the dynamics of phosphorylation were shown to principally affect synaptic proteins and to be coupled to synaptic activities (Brüning et al, 2019). Phosphorylation of synaptic proteins has also been linked to sleep homeostasis (Wang et al, 2018), the physiological process that guarantees a sufficient amount of sleep.
Homeostatic control of sleep is believed to rely on sleep‐promoting factors that accumulate in the brain proportionally to the length of the previous wake period and then induce sleep (Porkka‐Heiskanen, 2013; Franks & Wisden, 2021). Candidate factors fulfilling these criteria include adenosine, prostaglandin, tumor necrosis factor α (TNFα), and interleukin‐1 (Halassa et al, 2009; Krueger et al, 2011; Porkka‐Heiskanen, 2013; Franks & Wisden, 2021) but their molecular mechanisms of action and, in particular, their link to global phosphorylation remain to be understood.
TNFα is a signaling molecule known to control several phosphorylation pathways (Faustman & Davis, 2010; Brenner et al, 2015; Szondy & Pallai, 2017). Its role in the control of sleep has long been identified. In rodents, TNFα enhances non‐rapid eye movement (NREM) sleep when administered centrally (Shoham et al, 1987; Fang et al, 1997), while its inhibition reduces sleep (Takahashi et al, 1995). TNFα expression peaks during the light period when sleep need is the highest (Fonken et al, 2015). Additionally, TNFα can act on the cortex to modulate sleep homeostasis. Experimental modulation of TNFα levels in the cortex was shown to modulate cortical slow‐wave activity (SWA) during sleep (Yoshida et al, 2004; Taishi et al, 2007), which represents the principal marker of sleep homeostasis (Vyazovskiy et al, 2009; Franks & Wisden, 2021). In support of the active role of the cortex in the regulation of sleep homeostasis, manipulation of neuronal populations in the cortex modulates the build‐up of SWA following periods of sleep deprivation (SD) and subsequent compensatory sleep (Morairty et al, 2013; Krone et al, 2021).
In the brain parenchyma, TNFα is mostly if not exclusively expressed by microglia (Zhang et al, 2014; Zeisel et al, 2018). In turn, microglia are active sensors of brain state (Li et al, 2012; Stowell et al, 2019; Badimon et al, 2020) and have been recently proposed to participate in the regulation of sleep (Choudhury et al, 2020; Liu et al, 2021; Corsi et al, 2022). We, therefore, hypothesized that microglia‐derived TNFα is involved in phosphorylation‐based control of sleep.
In this study, we used quantitative phosphoproteomic to analyze the phosphorylation profile of the brain of mice lacking microglial TNFα. We focused on the cerebral cortex which plays an active role in the regulation of sleep homeostasis. We demonstrate that microglial TNFα massively modulates cortical phosphorylation exquisitely during the light phase, a period of maximum sleep time. We further show that phosphosubstrates of microglial TNFα comprise sleep‐promoting kinases and numerous synaptic proteins, including those controlling sleep needs (Wang et al, 2018; preprint: Taylor et al, 2021). Consistently, mice with microglial TNFα deletion display altered sleep homeostasis. Our findings reveal microglia as a new cellular actor in the homeostatic control of sleep and offer the first insights into the molecular processes downstream TNFα in sleep modulation.
Results
Daily changes in phosphorylations are microglial TNFα dependent
To assess the involvement of microglial TNFα in daily changes in protein phosphorylation, we performed quantitative phosphoproteomics using phosphopeptide enrichment coupled with label‐free quantitative mass spectrometry in transgenic control (CTL) and microglia‐specific TNFα‐deleted mice (micTNFα‐KO) (Fig 1A and Appendix Fig S1). Deletion of TNFα from microglia was accomplished in adulthood by tamoxifen‐induced recombination in CX3CR1CreERT2/+:TNFf/f mice (see Materials and Methods for details). We focused on the frontal cortex due to its high predominance of sleep SWA (Huber et al, 2000; Vyazovskiy et al, 2006), which reflects the homeostatic component of sleep (Vyazovskiy et al, 2009; Franks & Wisden, 2021). Frontal cortex lysates were analyzed at zeitgeber time 18 (ZT18) (middle of the dark period) and ZT6 (middle of the light period), in which mice spend the majority of their time (> 75%) awake and asleep, respectively (preprint: Pinto et al, 2022) (Fig 1A). As expected, the light/dark cycle was associated with changes in protein phosphorylation. In control animals, 271 phosphosites differed between ZT6 and ZT18, representing 2.3% of the phosphosites identified (Fig 1B and Dataset EV1). We then analyzed the phosphoproteome of micTNFα‐KO to assess the requirement of microglial TNFα for these daily oscillations. The majority of ZT6‐ZT18 cycling phosphosites (70.7%) did not change between ZT6 and ZT18 in these mice (Fig 1B) and are thus dependent on microglial TNFα. Moreover, half of the phosphosites that still change showed a different direction of change in CTL and micTNFα‐KO (Fig 1C), further supporting the role of microglial TNFα in their light/dark oscillation.
Figure 1. Daily oscillations in cortical phosphoproteome are microglial TNFα dependent.
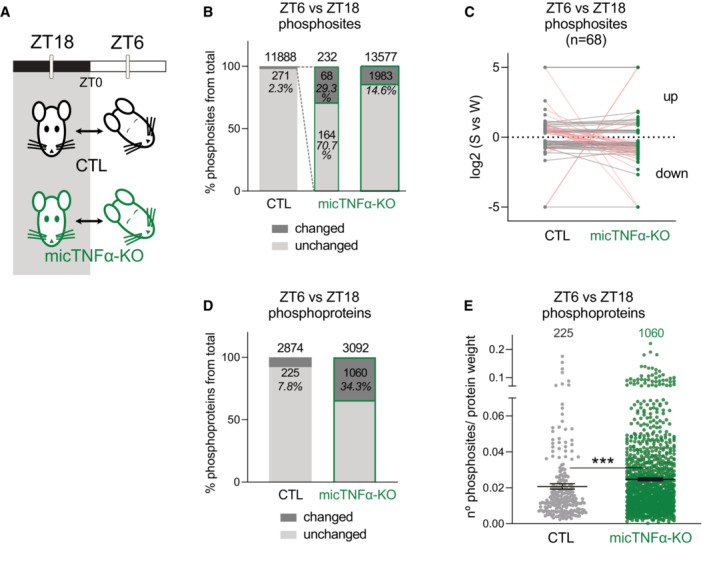
- Quantitative analysis of cortical phosphoproteome changes between ZT18 and ZT6 in controls (CTL) and microglia‐specific TNFα‐deleted mice (micTNFα‐KO). ZT18 and ZT6 correspond to the middle of the dark and light periods, during which mice spend the majority of their time in wakefulness or sleep, respectively.
- Percentage of phosphosites significantly changing in abundance (phosphosites with an adjusted P‐value ≤ 0.05 and unique phosphosites) between ZT6 and ZT18 in CTL and micTNFα‐KO mice (green border). Dashed lines represent the ZT6/ZT18‐oscillating phosphosites identified in CTL (of 271 phosphosites, 232 were identified in micTNFα‐KO ZT6 vs. ZT18 comparison, middle bar).
- ZT6 vs. ZT18 log2 (fold change) for each phosphosite that changed (phosphosites with an adjusted P‐value ≤ 0.05 and unique phosphosites) in both CTL and micTNFα‐KO mice. Datapoints above and below the dotted line show up‐ and down‐regulation at ZT6, respectively. Each dot connected by a line represents one phosphosite. Pink lines correspond to phosphosites with a different direction of change in CTL and micTNFα‐KO.
- Percentage of phosphoproteins with at least one changing phosphosite at ZT6 vs. ZT18 in CTL and micTNFα‐KO mice.
- Density of ZT6/ZT18 oscillating phosphosites per protein in CTL (225 phosphoproteins) and micTNFα‐KO mice (1,060 phosphoproteins). For each phosphoprotein (represented by a dot), the graph shows the number of significantly changed phosphosites in ZT6 vs. ZT18 comparison per protein weight. Average ± SEM is shown. n = number of proteins. ***P = 0.0002, Mann–Whitney test.
Microglial TNFα massively dampens daily variations in phosphorylations. We next compared the phosphoproteome of micTNFα‐KO cortex at ZT18 and ZT6. Unexpectedly, in contrast to the sparse changes observed in CTL, we found that 14.6% of the identified phosphosites significantly changed between ZT6 and ZT18 in micTNFα‐KO (Fig 1B and Dataset EV1). Modulation by phosphorylation can occur at multiple sites of the same protein leading us to analyze changes at the protein level. We found that 7.8% of the identified phosphorylated proteins harbor oscillating phosphosites in CTL but as much as 34.3% of the proteins displayed ZT6 vs. ZT18 modulations in micTNFα‐KO (Fig 1D). Moreover, phosphoproteins altered by lack of microglial TNFα during the light/dark cycle have a higher density of changing phosphorylations than CTL (Fig 1E), further showing that microglial TNFα acts to suppress daily changes in phosphorylations.
Altogether, these data demonstrate that microglial TNFα is a major regulator of protein phosphorylation during the light/dark cycle by acting both as a controller and buffer of changes in the cortical phosphoproteome.
Microglial TNFα modulates phosphorylation during the light period
We next investigated if the daily control of the phosphoproteome by microglial TNFα occurred preferentially during the dark or light period by comparing micTNFα‐KO and CTL mice phosphoproteomes at ZT18 and ZT6 (Fig 2A). During the dark period (ZT18), only 2.1% of 11,732 phosphosites differed between micTNFα‐KO and CTL mice (Fig 2B). In contrast, during the light period (ZT6), as much as 25.2% of 13,731 phosphosites changed between micTNFα‐KO and CTL mice (Fig 2B and Dataset EV2). Both increases and decreases in phosphosite relative abundance were observed showing that microglial TNFα bidirectionally controls the phosphoproteome (Fig 2C). Moreover, at ZT6, more than 80% of the phosphosites changed more than twofold with hundreds of sites changing more than fivefold (Fig 2C and Dataset EV2). At the protein phosphorylation level, such TNFα‐dependent regulation of phosphorylation affected 7.4 and 48.3% of phosphoproteins during dark and light periods, respectively (Fig 2D). Of note, at the protein expression level, a similar fraction of the proteome differed between micTNFα‐KO and CTL at ZT18 (7.7%) and ZT6 (6%), with no change above twofold (Appendix Fig S2A and Dataset EV3). The reduced overlap between microglial TNFα‐dependent proteome and phosphoproteome (Appendix Fig S3) further suggests that phosphomodulation by TNFα during the light period occurs independently from changes at the protein expression level.
Figure 2. Microglial TNFα acts during the light period to modulate the cortical phosphoproteome.
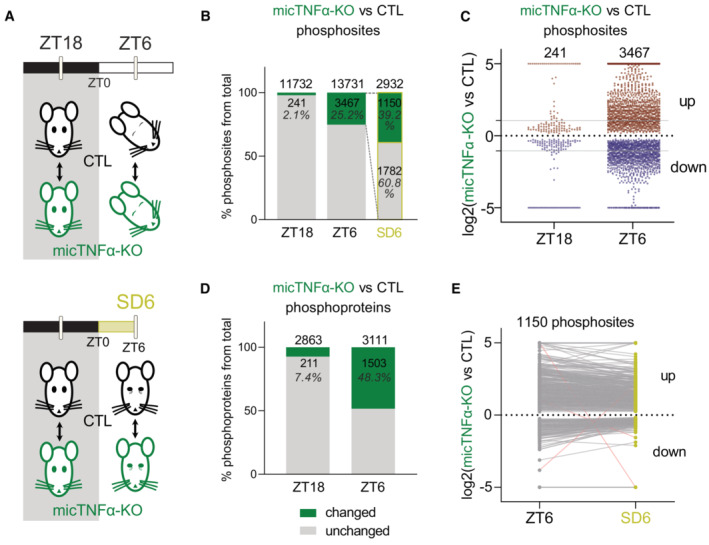
- Quantitative analysis of changes in cortical phosphoproteome between controls (CTL) and microglia‐specific TNFα‐deleted mice (micTNFα‐KO) at ZT18, ZT6, and ZT6 after sleep deprivation from ZT0 to ZT6 (SD6; yellow).
- Percentage of phosphosites significantly changing (phosphosites with an adjusted P‐value ≤ 0.05 and unique phosphosites) in micTNFα‐KO vs. CTL comparison at ZT18, ZT6, and SD6. Dashed lines represent the microglial TNFα‐modulated phosphosites at ZT6 (of 3,467 phosphosites, 2,932 were identified in CTL vs. micTNFα‐KO comparison at SD6).
- Log2(fold change) of significantly different phosphosites in micTNFα‐KO vs. CTL comparison (phosphosites with an adjusted P‐value ≤ 0.05 and unique phosphosites) at ZT18 and ZT6. Each point represents one phosphosite. Data points above and below the dotted line show up‐ and downregulation in micTNFα‐KO, respectively. Gray lines indicate twofold change.
- Percentage of phosphoproteins with at least one changing phosphosite in micTNFα‐KO vs. CTL comparison at ZT18 and ZT6.
- Comparison of micTNFα‐KO vs. CTL log2(fold change) for phosphosites changing (phosphosites with an adjusted P‐value ≤ 0.05 and unique phosphosites) at both ZT6 and SD6. Data points above and below the dotted line show up‐ and downregulation in micTNFα‐KO, respectively. Each dot connected by a line represents one phosphosite. Pink lines correspond to phosphosites with a different direction of change at ZT6 and SD6.
Sleep has been shown to trigger daily cycles of phosphorylations (Brüning et al, 2019). On the other hand, TNFα expression in microglia is upregulated during the light phase (Fonken et al, 2015). We, therefore, asked whether modulation of the phosphoproteome by microglial TNFα at the light period is dependent on sleep mechanisms or is driven by the light/dark cycle. This was assessed by SD experiments. CTL or micTNFα‐KO mice were sleep‐deprived for 6 h (from ZT0 to ZT6: SD6) and differences in their cortical phosphoproteome were compared to those of mice with ad libitum sleep at the same time of day (ZT6; Fig 2A). From the pool of phosphosites that differed between micTNFα‐KO and CTL mice at ZT6 (green in Fig 2B), 60.8% (1,782 of 2,932) did not change between micTNFα‐KO and CTL upon SD6 (Fig 2B). This demonstrates that modulation of these phosphosites by microglial TNFα is sleep dependent. The remaining microglial TNFα‐dependent phosphosites still differed between micTNFα‐KO and CTL upon SD6 (Fig 2B) with similar amplitudes and directions of change (Fig 2E). This demonstrates that part of the TNFα‐dependent modulation of phosphorylation events (39.2%—1,150 of 2,932 phosphosites) is not dependent on sleep but is rather influenced by the circadian cycle. Taken together, these data show that during the light period, microglial TNFα modulates the cortical phosphoproteome both in a manner dependent on the light/dark cycle and, in addition, sleep‐intrinsic mechanisms.
Kinases and phosphatases acting downstream microglial TNFα during the light period
To dissect the molecular mechanisms downstream of microglial TNFα in the light/dark cycle, we first assessed the activation of well‐established targets of TNFα signaling, namely the NF‐κB and apoptotic pathways (Brenner et al, 2015). At both ZT18 and ZT6, lack of microglial TNFα does not alter phosphorylation of NF‐κB p65 at Ser563 and does not induce cleavage of the apoptotic effector caspase‐3 (Appendix Fig S4), indicating no changes in the activation of the NF‐κB pathway or apoptosis downstream microglial TNFα.
The large‐scale regulation of the phosphoproteome by microglial TNFα during the light period implies active involvement of kinases and phosphatases. We, therefore, analyzed the protein levels and phosphorylation of kinases and phosphatases in the proteome of CTL and micTNFα‐KO mice during the dark (ZT18) and light (ZT6) periods. First, we confirmed that the abundance of kinases and phosphatases minimally differed between these mice at ZT18 and ZT6 (Appendix Fig S5A). Then, we showed that at ZT18, only 3.1% of phosphatases and 6.7% of kinases displayed differential phosphorylation status between micTNFα‐KO and CTL mice (Fig 3A). In contrast, at ZT6, more than half of phosphatases (56.5%) and kinases (52.5%) were differentially phosphorylated in micTNFα‐KO and CTL mice illustrating TNFα‐dependent phosphorylations (Fig 3A and Appendix Fig S5B). For instance, the phosphatases Plppr4 (a lipid phosphatase) and Ppm1h (a metal‐dependent protein phosphatase) have a high density of phosphorylation sites modified by microglial TNFα and so likely prone to its regulation during the light period (Fig 3B and Dataset EV4). Furthermore, several regulatory subunits of phosphoprotein phosphatases were identified (e.g., Ppp1r1b and Ppp1r1a; Fig 3B and Dataset EV4), which possibly account for an indirect modulation of phosphatases' activity (Yger & Girault, 2011).
Figure 3. Kinases involved in microglial TNFα phosphomodulation during the light period.
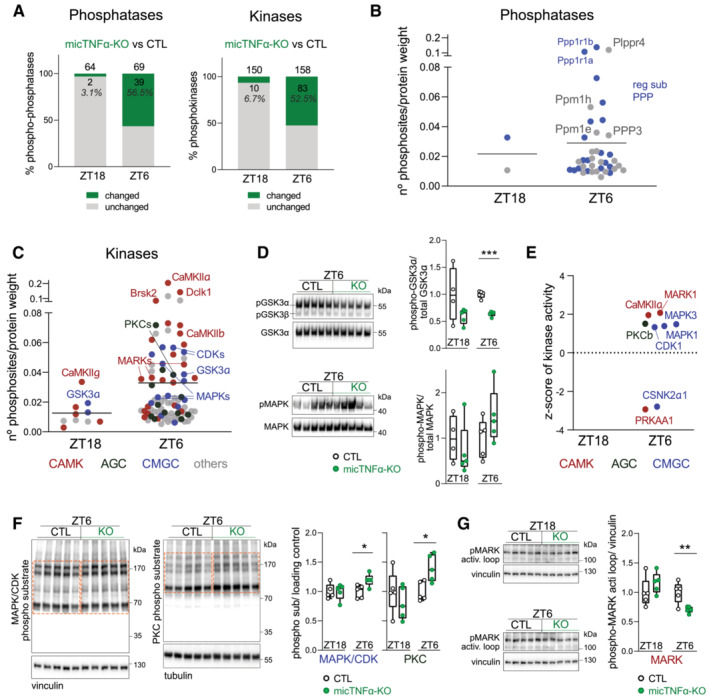
-
APhosphomodulation of phosphatases and kinases by microglial TNFα. Graphs show the percentage of phospho‐phosphatases and phosphokinases with at least one changing phosphosite in micTNFα‐KO vs. CTL comparison (phosphosites with an adjusted P‐value ≤ 0.05 and unique phosphosites) at ZT18 and ZT6.
-
B, CThe density of phosphosites on (B) phosphatases and (C) kinases modulated by microglial TNFα at ZT18 and ZT6. Graphs show the number of significantly changed phosphosites in micTNFα‐KO vs. CTL comparison per protein weight. Each dot is one phosphoprotein. (B) Phosphatases are shown in gray. Regulatory subunits of phosphoprotein phosphatases (reg sub PPP) in blue. (C) Kinases are color‐coded according to the major families: calcium/calmodulin‐regulated kinases (CAMK, red); CDK, MAPK, GSK3, and CLK kinases families (GMGC, blue); protein kinase A, G, and C families (AGC, dark green); and kinases belonging to other families (others, gray).
-
DValidation by western blot of changes in specific phosphorylation events on kinases. Left, Immunoblots of cortical lysates of CTL and micTNFα‐KO at ZT6 for: (top) phosphorylated GSK3α/β (pGSK3α at Ser21 and pGSK3β at Ser9) and total GSK3α; and (bottom) phosphorylated MAPK (pMAPK and MAPK3 at Thr203/Tyr205 and MAPK1 at Thr183/Tyr185) and total MAPK. Right, Ratio between phosphorylated and total kinase normalized to CTL at ZT18 and ZT6. Immunoblots at ZT18 in Appendix Fig S5C. n = 5 mice per group. ***P = 0.000009 (GSK3α), multiple t‐test with Benjamini–Hochberg correction.
-
EKinases with altered activity between CTL and micTNF‐KOs predicted by robust kinase activity inference (RoKAI) at ZT18 and ZT6. The graph shows the z‐score value of each kinase assigned by RoKAI. Kinases above and below the dotted line represent the prediction of up‐ and downregulated activity in micTNFα‐KO, respectively. Kinases are color coded according to the kinase group as described in (C).
-
F, GValidation by western blot of altered activity of predicted kinases between CTL and micTNF‐KOs. (F) Left, Immunoblots of cortical lysates of CTL and micTNFα‐KO at ZT6 using antibodies specific to target phosphorylation motifs of MAPK/CDK (PXS*P, S*PX(K/R)) and PKC ((K/R)XS*X(K/R)). Dashed boxes indicate quantified signals. Right, Ratio between phosphorylated substrates and loading control normalized to CTL at ZT18 and ZT6. Immunoblots at ZT18 in Appendix Fig S5D. (G) Left, Immunoblots show signal of phosphorylated threonine in MARKs activation loop. Right, Ratio between phosphorylated MARK activation loop and loading control normalized to CTL at ZT18 and ZT6. (F, G) n = 5 mice per group. *P = 0.022 (MAPK/CDK phospho‐sub), *P = 0.014 (PKC phospho‐sub), and **P = 0.0091 (MARK), multiple t‐test with Benjamini–Hochberg correction.
Among the kinases with phosphorylations modified by microglial TNFα during the light period, we found known sleep‐promoting kinases like CaMKIIα/β (Tatsuki et al, 2016) and MAPKs (Vanderheyden et al, 2013; Mikhail et al, 2017) and the sleep need‐associated kinases MARKs (Wang et al, 2018) (Fig 3C and Dataset EV4). We found a substantial number of members of the CAMK family (e.g., CaMKIIα/β, Dclk1, Brsk1/2, and MARK1‐4) and the CMGC family (including CDKs, MAPKs, and GSK3α) displaying multiple sites of microglial TNFα phosphomodulation (Fig 3C and Dataset EV4). We also found that phosphorylation sites known to control the activity of kinases are altered by microglial TNFα during the light period, such as Thr286 on CaMKIIα, Tyr205/Tyr185 on MAPK3/1 (positive log2‐fold changes indicative of upregulation in micTNFα‐KO), and Ser21 on GSK3α (negative log2‐fold change indicative of downregulation in micTNFα‐KO) (Dataset EV2) (Fang et al, 2000; Bayer & Schulman, 2019). To validate the phosphoproteomic data, we performed a western blot using phospho‐specific antibodies. In agreement with the obtained fold change in the micTNFα‐KO vs. CTL comparison at ZT6 (Dataset EV2), a decrease in GSK3α Ser21 phosphorylation and a trend for an increase in MAPK phosphorylation were observed in micTNFα‐KO (Fig 3D). These results emphasize the uncharted potential of microglial TNFα to modulate the activity of kinases and phosphatases through phosphorylation during the light period.
We then used RoKAI, an unbiased kinase activity inference tool, to identify kinases affected by microglial TNFα at ZT18 and ZT6 based on changes in the phosphorylation profile of their substrates (Yılmaz et al, 2021). The set of phosphosites significantly altered in the micTNFα‐KO vs. CTL comparison and the correspondent adjusted log2‐fold change (Dataset EV2) was used as input. RoKAI identified mostly members of the CAMK and CMGC families (Fig 3E). Notably, MARK1, CaMKIIα, PRKAA1, CDK1, MAPK1/3, and CSNK2α1 were identified as kinases with altered activity at ZT6 in micTNFα‐KO as compared to CTL (Fig 3E). PKCβ, a member of the AGC family was also identified as a kinase putatively upregulated by loss of microglial TNFα at ZT6. Of note, no significant hits were obtained at ZT18 (empty panel in Fig 3E), suggesting a minimal effect of microglial TNFα in the activity of kinases at this time of day. We then validated the RoKAI predictions by western blot analysis and we chose MAPKs/CDKs, PKCs, and MARKs as test cases. In accordance with the RoKAI‐predicted upregulation of activities, an increase in the phosphorylation of substrates of MAPK/CDK and PKC was observed on micTNFα‐KOs frontal cortex proteins in comparison to CTL at ZT6 but not at ZT18 (Fig 3F). MARKs are activated by phosphorylation of a conserved threonine in the activation loop (Timm et al, 2003). Changes in the activity of MARKs were evaluated by quantifying the phosphorylation of their activation loop. Similar to MAPK/CDK and PKC, reduced phosphorylation of the activation loop of MARKs only occurs at ZT6 in micTNFα‐KO (Fig 3G). We further verified that the phosphorylation of substrates of PKA, AKT, and AMPK, whose activities were not predicted to be affected (Fig 3E), did not display changes in micTNFα‐KO as compared to CTL mice (Appendix Fig S6). Finally, we used brain organotypic slices to demonstrate the acute effects of TNFα on the candidate kinases. This confirmed that short‐term neutralization of TNFα mimicked the changes in MAPK/CDK and PKC signaling pathways observed on micTNFα‐KOs in vivo (Appendix Fig S7). Together, these observations support a direct effect of TNFα on the above‐mentioned kinases.
Phosphorylation of sleep‐related synaptic proteins is regulated by microglial TNFα
To gain further insight into the roles of microglial TNFα‐mediated phosphorylation during the light period, we used the Reactome Pathway Analysis (Milacic et al, 2012; Croft et al, 2014), an unbiased pathways analysis tool, to characterize the functions enriched in the substrates of TNFα phosphomodulation (Fig 2C and Dataset EV5). Strikingly, the enriched pathways were almost exclusively related to synaptic functions both at the presynaptic and postsynaptic level (Fig 4A). In agreement, we found that 34.7% (521 of 1,503) of the phosphoproteins that were differentially phosphorylated between micTNFα‐KO and CTL mice at ZT6 is annotated as synaptic. Moreover, several of these synaptic proteins undergo modulation by microglial TNFα at multiple phosphorylation sites, particularly during the light period (ZT6) (Fig 4B and Dataset EV5). These include presynaptic (e.g., synapsins, RIMs, Syngap2, Bassoon, and Piccolo) and postsynaptic proteins (e.g., NMDARs, Shanks, Ank2/3, Dlgap1, and Agap2), as well as synaptically localized kinases (e.g., CaMKIIα, Dclk1, and MARKs). Finally, no synaptic pathways were enriched when considering the proteins whose abundance is altered by microglial TNFα at ZT6 (Appendix Fig S2B).
Figure 4. Sleep‐associated synaptic proteins are major targets of microglial TNFα phosphomodulation during the light period.
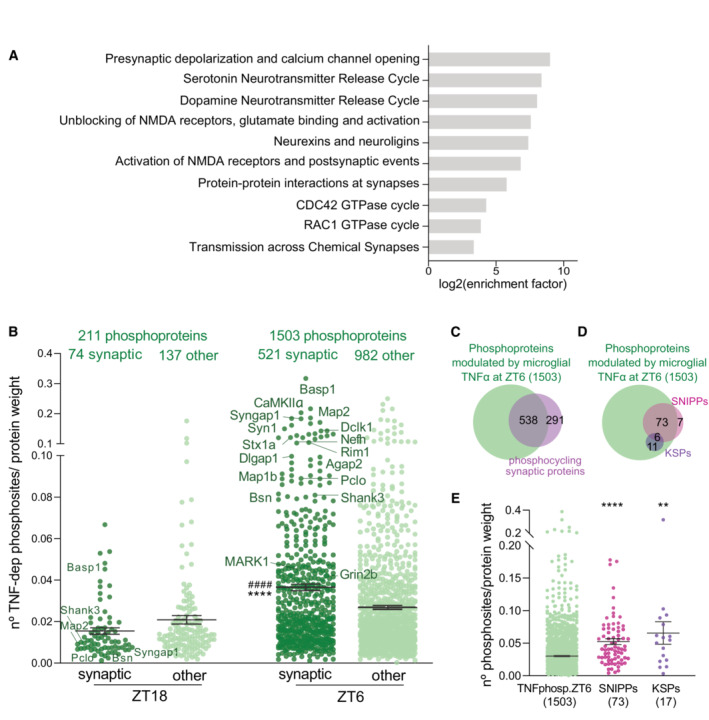
- Pathway analysis of substrates modulated by microglial TNFα through phosphorylation during the light period.
- Density of phosphosites on synaptic and non‐synaptic proteins (other) modulated by microglial TNFα at ZT18 and ZT6. Graph shows the number of significantly changed phosphosites in micTNFα‐KO vs. CTL comparison per protein weight. Each dot corresponds to one phosphoprotein. n = number of proteins. ****P < 0.0001 compared to ZT18 synaptic and #### P < 0.0001 compared to ZT6 other, Kruskal–Wallis test followed by Dunn's multiple‐comparison test.
- Venn diagram shows high overlap of TNFα‐modulated phosphoproteins at ZT6 and synaptic proteins harboring daily phosphorylation changes (Brüning et al, 2019).
- Venn diagram of microglial TNFα‐modulated phosphoproteins at ZT6 and sleep‐associated phosphoproteins SNIPPs and KSPs.
- Density of phosphosites modulated by microglial TNFα at ZT6 on all TNFα phosphosubstrates, SNIPPs and KSPs. The graph shows the number of significantly changed phosphosites in micTNFα‐KO vs. CTL comparison per protein weight. Each dot corresponds to one phosphoprotein. n = number of proteins. ****P < 0.0001 and **P = 0.0016 compared to TNFphosp.ZT6, Kruskal–Wallis test followed by Dunn's multiple‐comparison test.
Several studies have demonstrated that the phosphorylation of proteins, in particular, synaptic proteins, controls the duration of sleep (Funato et al, 2016; Tatsuki et al, 2016; Wang et al, 2018; preprint: Taylor et al, 2021). For instance, sleep/wake cycles were shown to specifically drive the phosphorylation of more than 800 synaptic proteins (Brüning et al, 2019). Remarkably, we found that 65% of these synaptic proteins are differentially phosphorylated between micTNFα‐KO and CTL mice at ZT6 (Fig 4C). Homeostatic regulation of sleep relies on internal mechanisms that encode sleep need according to the previous time spent awake. The phosphorylation–dephosphorylation of 80 synaptic proteins identified as the synaptic sleep need index phosphoproteins (SNIPPs) has been proposed to code sleep need (Wang et al, 2018). Phosphorylation of another set of synaptic proteins identified as keystone sleep phosphoproteins (KSPs) has been causally linked to sleep induction (preprint: Taylor et al, 2021). Remarkably, 91% of SNIPPs and 100% of KSPs are differentially phosphorylated between micTNFα‐KO and CTL mice at ZT6 and are thus substrates of microglial TNFα phosphomodulation (Fig 4D). Furthermore, the high density of phosphosites modulated by microglial TNFα on SNIPPs and KSPs highlights the prime ability of microglial TNFα to regulate their phosphorylation status (Fig 4E and Dataset EV5). Taken together, our results identify microglial TNFα as a major modulator of phosphorylation of sleep‐related synaptic proteins and therefore a putative upstream regulator of sleep homeostasis.
Homeostatic control of sleep requires microglial TNFα
Sleep homeostasis sets the intensity of sleep according to the duration of the prior wake. Actually, sleep need increases during spontaneous or enforced wake and then dissipates during the following sleep period. It has been proposed that the phosphorylation of SNIPPs determines sleep need and is therefore part of the regulatory mechanism of sleep homeostasis (Wang et al, 2018). Because we have shown that microglial TNFα massively controls the phosphorylation status of SNIPPs, we hypothesized that microglial TNFα is a regulator of sleep need. This was first shown by measuring dynamic changes in cortical EEG SWA in NREM sleep, a reliable indicator of sleep need (Vyazovskiy et al, 2009; Franks & Wisden, 2021), in micTNFα‐KO and CTL mice throughout a 24‐h sleep–wake cycle (baseline recording) (Fig 5A). As expected, in CTL mice, SWA increased during the dark (wake) period reflecting the build‐up of sleep need, and decreases progressively during the light (sleep) period when sleep need dissipates (Fig 5A). Strikingly, at the onset of the light (sleep) period, the amount of SWA was significantly lower in micTNFα‐KO mice as compared to CTL mice (Fig 5A).
Figure 5. Microglial TNFα modulates sleep homeostasis.
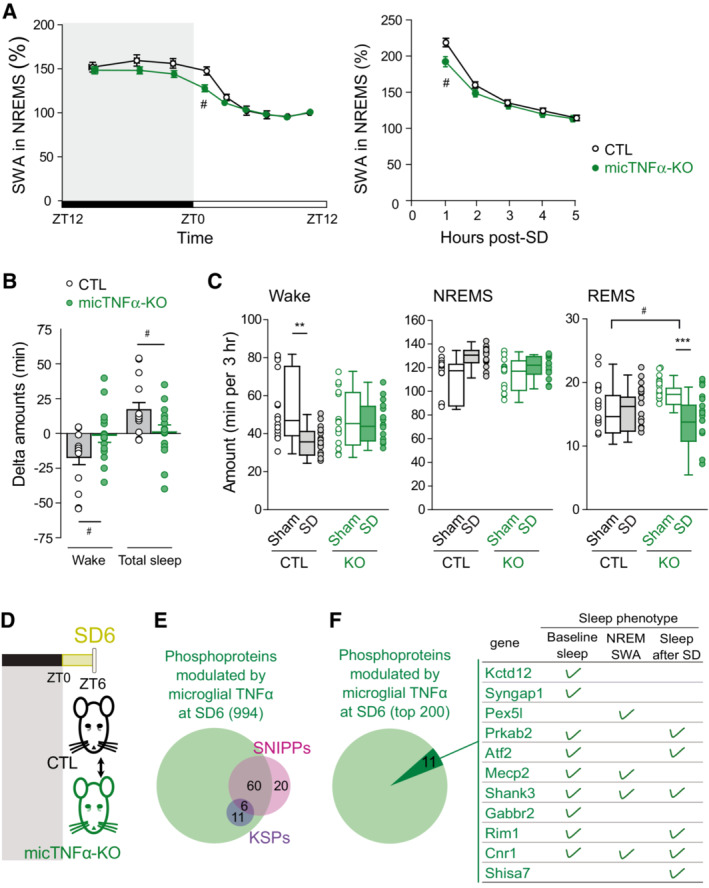
- Time course of SWA during NREM sleep across a 24 h baseline sleep–wake cycle (left) and in the first 5 h after sleep deprivation (SD; right) in controls CTL (black) and micTNFα‐KO (green) mice. Mean SWA values normalized to the mean SWA during ZT8 to ZT12 of the baseline recording, a period during which sleep need is lowest (see Methods). SWA in baseline sleep: CTL, 14 mice; micTNFα‐KO, 15 mice. Two‐way rANOVA; genotype, F(1,27) = 2.170, P = 0.1523; time: F(3.003,81.09) = 117.8, P < 0.0001; interaction F(8,216) = 2.257, P = 0.0246; SWA after sleep deprivation: CTL, 12 mice; micTNFα‐KO, 15 mice. Two‐way rANOVA; genotype, F(1,25) = 1.902, P = 0.1801; time: F(2.489,62.23) = 415.8, P < 0.0001; interaction F(4,100) = 4.196, P = 0.0035. Post hoc test with Sidak's multiple‐comparisons tests, # P < 0.05 CTL vs. micTNFα‐KO.
- Delta change in wake and total sleep amounts between SD and sham in the first 3 h of the recovery phase. Unpaired t‐test (two tailed): wake, # P = 0.0355; total sleep, # P = 0.0359.
- Wake, NREM sleep, and REM sleep amounts in the first 3 h of the recovery phase after sleep deprivation (SD, plain) or sham condition (Sham, empty) in CTL and micTNFα‐KO mice. SD: mice were sleep deprived for 6 h starting at ZT0; Sham condition: each mouse was gently awakened at ZT6, 2 days prior to the SD procedure (matched control recording). CTL, 15 mice; micTNFα‐KO, 15 mice. Data are represented as mean ± SEM. Two‐way rANOVA; wake: genotype, F(1,28) = 0.5302, P = 0.4726; SD: F(1,28) = 5.863, P = 0.0222; interaction F(1,28) = 6.105, P = 0.0198; NREM sleep, F(1,28) = 0.7292, P = 0.4004; SD: F(1,28) = 11.40, P = 0.0022; interaction F(1,28) = 2.657, P = 0.1143, REM sleep, F(1,28) = 0.2423, P = 0.6264; SD: F(1,28) = 8.273, P = 0.0076; interaction F(1,28) = 12.91, P = 0.0012. Post hoc test with Sidak's multiple‐comparisons tests, **P < 0.01 and ***P < 0.001 Sham vs. SD and # P < 0.05 CTL vs. micTNFα‐KO.
- Quantitative analysis of changes in cortical phosphoproteome between CTL and micTNFα‐KO after sleep deprivation from ZT0 to ZT6 (SD6; yellow).
- Identified sleep need phosphoproteins are modulated by microglial TNFα following sleep deprivation. The 994 phosphoproteins that had at least one phosphosite changing in micTNFα‐KO vs. CTL comparison at SD6 (phosphosites with an adjusted P‐value ≤ 0.05 and unique phosphosites; Dataset EV5) comprise all KSPs and 60 of 80 SNIPPS.
- Phosphosubstrates of microglial TNFα during sleep deprivation are causally linked to sleep regulation. Venn diagram shows the 200 phosphoproteins with a higher density of TNFα‐modulated phosphosites at SD6. Dark green highlights proteins whose mutation and/or knockdown leads to sleep phenotypes in baseline sleep, NREM SWA, and/or sleep rebound after SD (green tick) (refer to Dataset EV5 for a list of relevant citations). Proteins are listed in descending order of density of TNFα‐modulated phosphosites at SD6.
To further demonstrate the role of microglial TNFα as a regulator of sleep need, sleep homeostasis in micTNFα‐KO mice was then challenged by SD. Sleep deprivation substantially increases sleep need and triggers a compensatory response by boosting subsequent SWA in NREM sleep and sleep duration (Borbély et al, 1981; Huber et al, 2000; Franken et al, 2001). CTL and micTNFα‐KO mice were forced to stay awake during their normal sleep period (SD from ZT0 to ZT6) and the consequences of SD were assessed in the subsequent recovery phase. As expected, during the first hours of recovery following SD, CTL mice displayed a sharp increase in SWA in NREM sleep (Fig 5A), reflecting elevated sleep need (Vyazovskiy et al, 2009; Franks & Wisden, 2021). Moreover, SD elicited a sleep rebound (+9.4% of total sleep time in CTL) at the expense of wake levels during the following recovery phase (Fig 5B and C). Remarkably, at the beginning of the recovery period, micTNFα‐KO mice displayed less SWA as compared to CTL mice (Fig 5A) consistent with a compromised build‐up of sleep need following extended periods of wake. In contrast to CTL mice, micTNFα‐KO mice lacked total sleep rebound and concomitant decrease in wake amounts during the recovery phase following SD indicative of impaired sleep homeostasis (Fig 5B and C). Noteworthy, the amounts of NREM sleep after SD were not different between CTL and micTNFα‐KO mice (Fig 5C). Yet, the mean duration of NREM sleep bouts was longer after SD as compared to the sham condition in CTL mice but not in micTNFα‐KO mice (Table EV1). Additionally, rapid eye movement (REM) sleep amounts in micTNFα‐KO mice were increased as compared to CTL in the sham condition and decreased following SD (Fig 5C and Table EV1).
Brain oscillations at theta and gamma frequencies during wake have been proposed to drive sleep need (Vassalli & Franken, 2017). We thus analyzed these oscillatory activities during SD in CTL and micTNFα‐KO mice. We found that during SD, waking theta, low‐ and high‐gamma activities were not different in CTL and micTNFα‐KO mice (Appendix Fig S8). This suggests that microglial TNFα modulation of sleep need is not linked to waking oscillatory activities.
Finally, to gain insights into how microglia regulate sleep need, we analyzed the microglial TNFα‐targeted substrates and pathways upon SD (Fig 5D), a state of maximal accumulation of sleep need. As expected, proteins harboring phosphorylations modulated by microglial TNFα at SD (1,818 phosphosites in a total of 994 phosphoproteins, listed in Datasets EV2 and EV5, respectively) include most SNIPPs and all KSPs (Fig 5E and Dataset EV5). In addition, a literature search of the 200 phosphoproteins showing a higher density of TNFα‐modulated phosphosites during SD revealed 11 genes that have already been causally linked to sleep phenotypes (Fig 5F and Dataset EV5). Noteworthy, these include proteins that control homeostatic sleep rebounds after SD, such as the β2 subunit of AMPK kinase (Prkab2), the cyclic AMP‐dependent transcription factor (Atf2) and synaptic proteins (e.g., the postsynaptic proteins Shank3 and Shisa7 and the presynaptic protein Rim1—Fig 5F). Noteworthy, we also tested the kinases shown to be modified by microglial TNFα in the light period (GSK3α, MAPK/CDK, PKC, and MARK; Fig 3). We found that none of these kinases were differently phosphorylated, nor their phosphorylated substrates in CTL and micTNFα‐KO upon SD (Appendix Fig S9). This suggests that a different set of kinases acts downstream microglial TNFα at conditions of high accumulation of sleep pressure.
Discussion
Sleep is under tight homeostatic control but the cellular and molecular mechanisms of such control are still largely unknown. We now demonstrate that microglial TNFα extensively modulates cortical phosphoproteome during the light period, a phase of maximal sleep (Fig 2), including many kinases, phosphatases (Fig 3), and synaptic proteins (Fig 4). A set of these microglial TNFα‐targeted substrates are SNIPPs and KSPs (Fig 4), proteins whose phosphorylation status has been proposed to encode sleep need and control sleep duration, respectively (Wang et al, 2018; preprint: Taylor et al, 2021). In agreement, we showed that microglial TNFα is required for the expression of sleep need and concomitantly controls homeostatic sleep rebound (Fig 5). We further identified several phosphorylated substrates as potential candidates acting downstream microglial TNFα in the regulation of sleep need (Fig 5).
Homeostatic regulation of sleep by microglia
The homeostatic regulation of sleep sets the intensity of sleep as a function of the preceding time spent awake. Such control relies on the build‐up of sleep need during wake and subsequent translation into sleep drive. One currently held belief is that sleep need arises in discrete neuronal networks driven by the accumulation of sleep‐promoting substances following prolonged periods of neuronal activity during extended wake (Porkka‐Heiskanen, 2013; Franks & Wisden, 2021). Astrocytes have been implicated in sleep homeostasis via the release of adenosine (Halassa et al, 2009; Bjorness et al, 2016; Ingiosi et al, 2020), which acts on the basal forebrain and hypothalamus to induce sleep (Porkka‐Heiskanen et al, 1997; Lazarus et al, 2019). Despite work in Drosophila suggesting a connection between glia‐derived immune factors and homeostatic sleep (Vanderheyden et al, 2018; Blum et al, 2021), the involvement of microglia has remained elusive. Our work now provides functional evidence that microglia are active players in sleep homeostasis via TNFα. It is worth noting that homeostatic sleep control by microglia may not be limited to the secretion of TNFα, as these cells control the levels of adenosine (Badimon et al, 2020) and can release other known sleep‐promoting substances such as interleukin‐1 and prostaglandins (Giulian et al, 1986; Ikeda‐Matsuo et al, 2005).
The somnogenic effect of TNFα has long been described. Central or peripheral injections of TNFα increase NREM sleep (Shoham et al, 1987; Fang et al, 1997; Kubota et al, 2002), while neutralization of TNFα signaling attenuates spontaneous sleep (Takahashi et al, 1995) and homeostatic sleep rebound in rabbits (Takahashi et al, 1996). In the brain parenchyma, TNFα expression is restricted to microglia (Buttini et al, 1997; Zhang et al, 2014; Zeisel et al, 2018). However, peripheral TNFα can cross the blood–brain barrier and play an active role in the neuronal circuitry (Garré et al, 2017; Paouri et al, 2017). Assessing the relative contribution of central and peripheral TNFα‐producing cells to sleep control is thus an important issue. Herein, we prove that, despite not being involved in the control of basal sleep amounts (preprint: Pinto et al, 2022), TNFα of microglial origin participates in the initial sleep recovery when sleep homeostasis is challenged by SD (Fig 5B and C).
In support of the role of microglial TNFα in regulating sleep need, we herein show that slow‐wave activity during NREM sleep, which is an established index of sleep need (Vyazovskiy et al, 2009; Franks & Wisden, 2021), is reduced in micTNFα‐KO mice both at baseline sleep and after SD (Fig 5A). Features of individual slow waves during NREM sleep are also modulated by sleep need with slow waves characterized by sharper slopes and shorter duration following prolonged wake (Hubbard et al, 2020). Abnormal sleep homeostasis in mice lacking TNFα is further suggested by our previous findings showing that loss of microglial TNFα reduces the slope and increases the duration of individual slow waves during NREM sleep (preprint: Pinto et al, 2022). Moreover, and in agreement with previous findings on constitutive knock‐out mice with altered TNFα signaling (Deboer et al, 2002; Kapás et al, 2008), we also reported an enhanced lower‐range SWA in baseline NREM sleep in micTNFα‐KO mice (preprint: Pinto et al, 2022). Several studies have shown that both slower‐ and faster‐range SWA during NREM sleep are differentially regulated according to the duration of prior wake (Huber et al, 2000; Vyazovskiy et al, 2006; Hubbard et al, 2020) and by wake‐promoting neurotransmitters (Cirelli et al, 2005; Vassalli & Franken, 2017). These studies yield opposite findings about which of these two SWA activities best reflects sleep–wake history. Therefore, whether the shift in SWA toward lower‐frequency activities in micTNFα‐KO mice additionally reflects a blunted sleep homeostasis remains to be investigated.
The involvement of microglia in homeostatic sleep control is consistent with current models of sleep regulation and the well‐known ATP‐driven microglia behavior. The former posits that the accumulation of ATP due to enhanced brain activity during prolonged wake triggers the release of cytokines which in turn promote sleep (Clinton et al, 2011; Ingiosi et al, 2013). This concept fits with the ability of microglia to react to neuronal activity via ATP‐mediated changes in their behavior (Li et al, 2012; Badimon et al, 2020; preprint: Pinto et al, 2022). Indeed, ATP triggers the release of TNFα, interleukin‐1, and prostaglandin by microglial cells (Hide et al, 2000; Bianco et al, 2005; Anrather et al, 2011). Collectively, our work and these findings support the idea that microglia sense extended periods of wake via ATP and respond by releasing proportional amounts of TNFα, which in turn feedback onto the neuronal networks to control sleep. Alternatively, upon extended wake microglia could be recruited by wake‐promoting neuromodulators (such as noradrenaline, serotonin, dopamine, and histamine), which modulate microglia function including triggering the release of sleep substances like TNFα (Lewitus et al, 2016; Stowell et al, 2019; Albertini et al, 2020).
The molecular mechanisms linking sleep‐promoting substances and sleep need transformation into sleep drive are poorly known. We now propose that one of the molecular determinants of microglial TNFα on homeostatic sleep regulation is protein phosphorylation in the cortex. Such phosphorylation‐mediated control can occur at two different levels: either contributing to the molecular coding of sleep need and/or transforming sleep need into sleep drive. In support of the former possibility, microglial TNFα controls (de)phosphorylation events in SNIPPs (Figs 4 and 5), proteins whose phosphorylation status determines sleep need (Wang et al, 2018). For instance, mGluR5 was previously shown to contribute to sleep need (Holst et al, 2017) and our data identifies TNFα‐dependent phosphorylation sites that affect mGluR5 signaling (Ser839 and Thr840 on mGluR5; Dataset EV2) (Kim et al, 2005). In agreement, lack of microglial TNFα leads to reduced slow‐wave activity, a classic measure of sleep need, at times of high sleep pressure (Fig 5). In support of the latter, microglial TNFα also adjusts the phosphorylation of the sleep‐inducing phosphoproteins KSPs (preprint: Taylor et al, 2021) (Fig 4), of proteins with a causal link to sleep control (Fig 5 and Dataset EV5), and of sleep‐promoting kinases like CaMKIIα/β (Tatsuki et al, 2016) and MAPKs (Vanderheyden et al, 2013; Mikhail et al, 2017) (Fig 3). Phosphosites identified on the mentioned kinases are known to control their activity (Dataset EV2 and Fig 3), suggesting that microglial TNFα could trigger sleep rebound after SD by controlling the activity of specific kinases via phosphorylation.
The phosphoproteomic analysis presented in this study was performed in the frontal cortex. Our work thus confirms the role attributed to the cortex in sleep homeostasis (Morairty et al, 2013; preprint: Tossell et al, 2020; Krone et al, 2021). Microglia, via TNFα‐mediated phosphomodulation, likely act in synchrony with discrete cortical circuits to regulate homeostatic sleep. Nevertheless, combined action of TNFα at different brain regions needs to be considered for the maintenance of sleep homeostasis. In fact, we deleted microglial TNFα in all the brain regions and not only in the cortex. Actually, injection of TNFα in different brain regions affects different aspects of sleep: while cortical TNFα injection enhances SWA (Yoshida et al, 2004; Taishi et al, 2007), TNFα locally applied to the hypothalamus changes the amount of NREM sleep (Kubota et al, 2002). It is therefore likely that microglial TNFα controls sleep homeostasis by acting in a brain region‐specific manner.
A potential role of microglial TNFα in synaptic plasticity during sleep
Importantly, despite the fact that phosphorylation of hundreds of proteins is under the control of microglial TNFα during the light phase (Dataset EV5), the period of maximal sleep, the structure of baseline sleep is not disrupted upon loss of microglial TNFα (preprint: Pinto et al, 2022). This observation favors a model in which microglial TNFα‐dependent phosphorylation is not required to keep sleep time and duration at steady‐state conditions but instead supports molecular mechanisms underlying sleep functions. For instance, we show that microglial TNFα acts during the sleep period to buffer daily changes in the cortical phosphoproteome (Fig 1). This may constitute a homeostatic mechanism to counteract wake‐induced changes in protein phosphorylation. On the other hand, protein phosphorylation has a pivotal role in controlling synaptic plasticity (Lee, 2006; Bayer & Schulman, 2019; Coba, 2019), which occurs during sleep to shape neuronal networks (Vyazovskiy et al, 2008; Diering et al, 2017; Bellesi & de Vivo, 2020). Thus, our data further suggest a promising role of microglial TNFα in orchestrating sleep‐associated synaptic plasticity.
Indeed, an unbiased pathway analysis of the substrates of microglial TNFα phosphomodulation at the light period reveals enrichment of synaptic functions (Fig 4). Notably, many of these substrates change in phosphorylation upon induction of homeostatic downscaling (Appendix Fig S10) (Desch et al, 2021), a form of synaptic plasticity believed to occur during sleep to weaken excitatory synapses (Vyazovskiy et al, 2008; Diering et al, 2017). Furthermore, predictive and experimental validation of microglial TNFα‐targeted kinases allowed the identification of PKCs and MAPKs (Fig 3), which are known to regulate synaptic plasticity (Thomas & Huganir, 2004; de Jong & Fioravante, 2014; Woolfrey & Dell'Acqua, 2015).
In further support of the prominent role of microglial TNFα at synapses during sleep, several known functional phosphosites on pre‐ and postsynaptic proteins were identified as altered by TNFα in the light period (Fig 4 and Dataset EV2). For instance, the postsynaptic NMDAR 2A subunit (Grin2a) displays changes in the phosphorylations of Ser929 and Ser1459, residues involved in the trafficking and synaptic presentation of the receptor (Yong et al, 2021; Zhou et al, 2021), while NMDAR 2B subunit (Grin2b) becomes dephosphorylated at Ser1303 known to control its binding to CaMKII (O'Leary et al, 2011). The latter is a central kinase in postsynaptic plasticity whose activity is controlled by phosphorylation at Thr286 (Bayer & Schulman, 2019), also shown to be modified by TNFα. Another target phosphosite is Ser1539 on the postsynaptic scaffold Shank3, recently shown to control synaptic availability of the protein and thereby expression of homeostatic plasticity (preprint: Wu et al, 2021). Examples can also be drawn on the regulation of presynaptic proteins by specific phosphorylations, herein identified as microglial TNFα‐dependent during the light period (Fig 4 and Dataset EV2). For instance, the release and turnover of synaptic vesicles are controlled by phosphorylation of syntaxins at Ser14 and synapsins at Ser62 (Chi et al, 2003; Shi et al, 2021) and long‐term plasticity is supported by enhanced presynaptic release via Rim1 phosphorylation at Ser413 (Lonart et al, 2003). Together, our data favor active participation of microglial cells in synaptic remodeling during basal sleep via TNFα‐controlled phosphorylation. Supporting findings have recently been reported on sleep‐dependent plasticity of inhibitory synapses (preprint: Pinto et al, 2022).
Finally, we showed that modulation of the phosphoproteome by microglial TNFα occurs specifically during the light period downstream a joint action of sleep and circadian processes (Fig 2). Future work is needed to unravel the mechanisms linking microglia sensing of brain states, like sleep, and its translation into molecular processes that tune the neuronal network. Another aspect that deserves attention in future studies is the role of microglia in female sleep regulation. The phosphoproteomic analyses and sleep recordings described in this study were exclusively performed on male mice. It is now acknowledged that microglia display sex‐specific functions (e.g., VanRyzin et al, 2019) and the mechanisms that are described here may not extrapolate to female brains.
Materials and Methods
Animals and housing
Animal experiments were performed in accordance with the European Committee Council Directive 86/609/EEC and procedures approved by the local Charles Darwin Ethical Committee (Ce5‐2014‐001; 1339‐2015073113467359 and 2018022121466547). CX3CR1GFP (Jung et al, 2000), CX3CR1CreERT2 (Yona et al, 2013), and TNFflox (Grivennikov et al, 2005) mouse lines were housed at the animal facility of Institut de Biologie de l'ENS or the animal facility of Institut de Biologie Paris Seine (Paris, France). CX3CR1GFP and CX3CR1CreERT2 were kindly provided by Sonia Garel and TNFflox by Etienne Audinat. All mice used in this study were 4‐ to 5‐month‐old males at the time of tissue collection and sleep recordings, housed under standard conditions (12 h light/dark cycle; lights on at 7:00 a.m.) with food and water ad libitum. Appendix Fig S11 provides a general outline of the experiments performed in this study.
Conditional microglia‐specific TNFα deletion
Proteomic/phosphoproteomic experiments and sleep recordings were performed on mice following the conditional deletion of TNFα on microglia cells. To accomplish so, CX3CR1GFP/+:TNFf/f (transgenic controls, CTL) and CX3CR1CreERT2/+:TNFf/f (microglia TNFα‐depleted mice, micTNFα‐KO) mice were fed with tamoxifen‐containing food (1,000 mg tamoxifen citrate in 1 kg of chow, formulated by SSNIFF, A115‐T71000) for 6 days. All experiments were performed 5–6 weeks after tamoxifen feeding, which allows for the repopulation of short‐lived peripheral CX3CR1+ cells thus restricting recombination of the TNFα locus to microglia (Parkhurst et al, 2013). To confirm microglia depletion of TNFα, adult primary microglia were isolated and cultured as previously described (Lee & Tansey, 2013). In brief, PBS‐perfused adult mouse brains were dissected and dissociated in 30–40 U papain (Worthington, LK003176), 7.2 U dispase II (Sigma, D4693), and 10 mg/ml DNAse (Sigma, DN25). Following mechanical dissociation, microglia were isolated using a Percoll gradient (Sigma, P4937) and plated in DMEM/F12 (GIBCO, 31331‐028) supplemented with 10% heat‐inactivated fetal bovine serum (GIBCO, 10500‐064), 10 U/ml penicillin, and 10 μg/ml streptomycin. Cells were stimulated with 10 μg/ml LPS for 10 h to induce TNFα release. The medium was collected and centrifuged for 5 min at 10,000 g at 4°C. Levels of TNFα in the supernatant were determined by the mouse TNFα uncoated ELISA kit (Invitrogen, 88‐7324).
Tissue collection and sample preparation for mass spectrometry
Before tissue collection mice were put in individual cages for 2 weeks. Age‐matched CTL and micTNFα‐KO mice were randomly assigned to each group, guaranteeing a similar number of individuals from one litter in each experimental group. At ZT6 and ZT18, which correspond to the middle of the light and dark phases, mice spend most of their time asleep and awake, respectively. Sleep deprivation was accomplished by the presentation of novel objects and gentle cage tapping for 6 h starting at light phase onset (from ZT0 to ZT6). Sleep‐deprived animals were not allowed to sleep before euthanasia at ZT6. At the indicated time points, mice (5 per group for each genotype) were sacrificed by cervical dislocation, their brains rapidly dissected and rinsed in ice‐cold PBS, the frontal cortex collected into an ice‐cold tube, and flash frozen in liquid nitrogen. Samples were stored at −80°C until further processing (all samples were prepared at the same time).
Frozen cortex tissues were lysed in freshly prepared 200 μl of urea lysis buffer [8 M urea, 50 Mm NH4HCO3 supplemented with protease (Roche, 05056489001), and phosphatase inhibitor cocktail (Roche, 04906837001)] at room temperature by mechanical dissociation with a pipette. Cortex lysates were sonicated on ice to avoid heating and centrifuged at 17,000 g for 10 min at 4°C. The supernatant was collected and protein concentration was determined by the Pierce BCA protein assay kit (Thermo Fisher, 23225). 250 μg of each cell lysate was reduced with 5 mM DTT for 1 h at 37°C and alkylated with 10 mM iodoacetamide for 30 min at room temperature in the dark. The samples were then diluted in 200 mM ammonium bicarbonate to reach a final concentration of 1 M urea, and digested overnight at 37°C with Trypsin/Lys‐C (Promega, V5071) at a ratio of 1/50. Digested peptide lysates were acidified with formic acid to a final concentration of 5% formic acid. Samples were centrifuged at 1500 g and loaded onto homemade SepPak C18 Tips packed by stacking three AttractSPE disks (Affinisep, SPE‐Disks‐Bio‐C18‐100.47.20) and 2 mg beads (Cartridge Waters, 186004621 SepPak C18) into a 200 μl micropipette tip for desalting. Peptides were eluted and 90% of the starting material was enriched using Titansphere™ Phos‐TiO kit centrifuge columns (GL Sciences, 5010‐21312) as described by the manufacturer. After elution from the spin tips, the phosphopeptides and the remaining 10% eluted peptides were vacuum concentrated to dryness and reconstituted in 0.1% formic acid prior to LC–MS/MS phosphoproteome and proteome analyses.
LC–MS/MS analysis
Online chromatography was performed with an RSLCnano system (Ultimate 3000, Thermo Scientific) coupled to an Orbitrap Exploris 480 mass spectrometer (Thermo Scientific). Peptides were trapped on a C18 column (75 μm inner diameter × 2 cm; nanoViper Acclaim PepMap™ 100, Thermo Scientific) with buffer A (2/98 MeCN/H2O in 0.1% formic acid) at a flow rate of 3.0 μl/min over 4 min. Separation was performed on a 50 cm × 75 μm C18 column (nanoViper Acclaim PepMap™ RSLC, 2 μm, 100 Å, Thermo Scientific) regulated to a temperature of 40°C with a linear gradient of 3–29% buffer B (100% MeCN in 0.1% formic acid) at a flow rate of 300 nl/min over 91 min for the phosphoproteome analyses and a linear gradient of 3–32% buffer B over 211 min for the proteome analyses. MS full scans were performed in the ultrahigh‐field Orbitrap mass analyzer in ranges m/z 375–1,500 with a resolution of 120,000 (at m/z 200). The top 20 most intense ions were subjected to Orbitrap for phosphoproteomes and the top 30 for proteomes for further fragmentation via high‐energy collision dissociation (HCD) activation and a resolution of 15,000 with the AGC target set to 100%. We selected ions with a charge state from 2+ to 6+ for screening. Normalized collision energy (NCE) was set at 30 and the dynamic exclusion to 40 s.
Proteomic and phosphoproteomic analyses
For identification, the data were searched against the Mus Musculus UP000000589 database (downloaded 03/2020) using Sequest HT through Proteome Discoverer (version 2.4). Enzyme specificity was set to trypsin and a maximum of two missed cleavage sites were allowed. Oxidized methionine, N‐terminal acetylation, methionine loss, and acetylated‐methionine loss were set as variable modifications. Phosphoserine, threonine, and tyrosines were also set as variable modifications in phosphoproteome analyses. The maximum allowed mass deviation was set to 10 ppm for monoisotopic precursor ions and 0.02 Da for MS/MS peaks. False‐discovery rate (FDR) was calculated using Percolator (The et al, 2016) and was set to 1% at the peptide level for the whole study. The resulting files were further processed using myProMS v.3.9.3 (Poullet et al, 2007) (https://github.com/bioinfo‐pf‐curie/myproms). Label‐free quantification was performed using peptide‐extracted ion chromatograms (XICs), computed with MassChroQ v.2.2.1 (Valot et al, 2011). For protein quantification, XICs from proteotypic peptides shared between compared conditions (TopN matching for proteome setting and simple ratios for phosphoproteome) were used, including peptides with missed cleavages. Median and scale normalization at the peptide level were applied to the total signal to correct the XICs for each biological replicate (N = 5). The phosphosite localization accuracy was estimated by using the PtmRS node in PD (Proteome Discoverer version 2.4) and PhosphoRS mode only. Phosphosites with a localization site probability greater than 75% were quantified at the peptide level. To estimate the significance of the change in protein abundance, a linear model inspired by the LIMMA algorithm (Kammers et al, 2015) (adjusted on peptides and biological replicates) was performed, and P‐values were adjusted using the Benjamini–Hochberg FDR procedure (van Iterson et al, 2010).
For proteome analyses, proteins were considered in the analysis only when they were found with at least three total peptides across three biological replicates per group. Then, proteins with an adjusted P‐value ≤ 0.05 were considered significantly enriched in sample comparisons. For phosphoproteomic analyses, the phosphoproteome of all analyzed groups was corrected to changes in the proteome before analysis. The peptides threshold was decreased to one peptide across three biological replicates of a group (meaning it was identified in at least three of the five replicates) to consider a phosphosite in the downstream analysis. Then, phosphosites (in one or more phosphopeptides) with an adjusted P‐value ≤ 0.05 were considered significantly different in sample comparisons. Unique phosphosites were also included when identified in at least three biological replicates in only one of the groups in each comparison. As unique phosphosites are only present in one of the groups in a pairwise comparison, they do not have a fold‐change value. For analysis of the amplitude of change in all changing phosphosites in a given comparison (phosphosites and unique phosphosites), the log2 value of the fold change of unique phosphosites was imputed to 5 and −5 (representing up‐ and downregulation, respectively). These arbitrary values were chosen as 99.8% of the significantly different phosphosites herein identified have log2‐fold change values that fall within the 5 to −5 range. The density of changed phosphosites per phosphoprotein was determined as the number of significantly changed phosphosites (adjusted P‐value ≤ 0.05) and unique phosphosites identified in one comparison normalized to protein weight (kDa). Proteins and phosphoproteins identified with these criteria were further subjected to Reactome pathway analysis (FDR ≤ 10%) within myProMS (Milacic et al, 2012; Croft et al, 2014).
For the analysis of proteomic and phosphoproteomic changes on kinases and phosphatases, a list of mouse kinases and phosphatases was generated from UniProt database by filtering with the following criteria (“kinase” or “phosphatase” and organism “Mus musculus” and “Reviewed”). The lists of kinases and phosphatases obtained were imported into the platform myProMS using their UniProt accession number. Kinases and phosphatases were manually annotated to the different kinase and phosphatases groups using kinhub.org and http://phanstiel‐lab.med.unc.edu/coralp/, respectively. Only kinases and phosphatases annotated to the major groups were considered for the analysis. Prediction of kinase activity was done by the web‐based tool robust kinase activity inference (RoKAI) at http://rokai.io (Yılmaz et al, 2021). From the phosphoproteomic data comparing micTNF‐KO and controls at the light or dark period, a list of all altered phosphosites, the corresponding protein, and the adjusted log2‐fold change value was created and used as input to RoKAI. Analysis was done using the mouse database as reference, PhosphoSitePlus (PSP) and Signor as kinase substrates databases, and the combined KS + PPI + SD + CoEV RoKAI network. From the list of kinases' outputs obtained, only kinases with four or more substrates and a P‐value < 0.2 were considered.
The lists of SNIPPs and KSPs were imported into myProMS platform using their UniProt accession number and used for the analysis of phosphoproteome changes. Synaptic proteins were annotated according to the synaptome database at http://metamoodics.org/SynaptomeDB/index.php (Pirooznia et al, 2012).
Brain organotypic slices
C57BL/6J pregnant females were obtained from Janvier Labs and P3–P5 pups used for the preparation of organotypic slices as previously described (Hill et al, 2014). Brains were dissected, hemispheres separated along the midline, and the cerebellum was removed with a razor blade in ice‐cold dissection medium (33 mM glucose in PBS). Whole‐brain coronal slices (350 μm) were cut using a McIllwain tissue chopper (Mickle Laboratory). Slices were separated and 6–8 from each hemisphere (excluding the most posterior and anterior regions of the brain) were plated on Millicell cell culture inserts (Millipore, PICM03050) and maintained in culture medium [MEM (GIBCO, 21090‐022) supplemented with 20% heat‐inactivated horse serum (GIBCO, 16050‐122), 2 mM glutamine, 10 mM glucose, 20 mM HEPES, 10 U/ml penicillin, and 10 μg/ml streptomycin] at 37°C in 5% CO2/air. The culture medium was changed three times per week. Stimulation of organotypic slices with TNFα‐neutralizing antibody (1 μg/ml, R&D systems, MAB4101) was performed at DIV18‐21 in prewarmed aCSF (aCSF; 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, and 33 mM glucose, pH 7.3). For western blot analysis, organotypic slices (three slices per insert for each condition) were homogenized in cell extraction buffer [10 mM Tris, 100 mM NaCl, 1 mM EDTA, 1% triton‐X100, 10% glycerol, and 0.1% SDS supplemented with protease (Roche, 05056489001) and phosphatase inhibitor cocktail (Roche, 04906837001)] by mechanical dissociation, centrifuged for 10 min at 16,000 g at 4°C, and the supernatant collected. Total protein concentration was determined by the Pierce BCA protein assay kit (Thermo Fisher, 23225).
Western blot
Equal amounts of protein samples from cortex lysates and organotypic slice lysates were used for immunoblotting. Samples were denatured at 95°C for 5 min in denaturating buffer (125 mM Tris–HCl, 10% glycerol, 2% SDS, bromophenol blue, and 5% β‐mercaptoethanol added fresh), resolved by SDS–PAGE in 4–15% Tris‐glycine mini‐PROTEAN gels (BioRad, 4561086) or homemade 6% polyacrylamide gel (for staining with anti‐phospho‐MARK activation loop antibody), and transferred to PVDF membranes. Membranes were blocked in 5% dry milk, and incubated with primary antibody at 4°C overnight and HRP‐conjugated secondary antibody at RT for 1 h. Phosphorylated proteins were visualized by chemiluminescence using Lumi‐Light Western Blotting substrate (Roche, 12015196001) or SuperSignal West Femto Maximum Sensitivity substrate (Thermo Scientific, 34095). Membranes were scanned on a ImageQuant LAS 4000 imaging system (GE Healthcare). Membranes were stripped with 0.2 M NaOH for 40 min and reprobed for loading control. Quantification was performed on Image J. Primary antibodies used were as follows: anti‐phospho‐MAPK/CDK substrate motif (PXS*P, S*PX(K/R)) (1:1,000, Cell Signaling, 2325), anti‐phospho‐PKC substrate motif ((K/R)XS*X(K/R)) (1:1,000, Cell Signaling, 6967), anti‐phospho‐PKA substrate motif ((K/R)(K/R)X(S*/T*)) (1:1,000, Cell Signaling, 9624), anti‐phospho‐AKT substrate motif (RXX(S*/T*)) (1:1,000, Cell Signaling, 9614), anti‐phospho‐AMPK substrate motif (LXRXX(S*/T*)) (1:1,000, Cell Signaling, 5759), anti‐phospho‐MARK family activation loop (MARK1/2/3 at Thr215/208/234) (1:1,000, Cell Signaling, 4836), anti‐phospho‐GSK3α/β at Ser21/9 (1:1,000, Cell Signaling, 9331), anti‐GSK3α (1:1,000, Cell Signaling, 4818), anti‐phospho‐p44/42 MAPK at Thr202/Tyr204 (1:1,000, Cell Signaling, 4370), anti‐p44/42 MAPK (Erk1/2) (1:2,000, Cell Signaling, 4696), anti‐phospho‐NF‐κB p65 at Ser563 (1:1,000, Cell Signaling, 3033), anti‐NF‐κB p65 (1:1,000, Cell Signaling, 8242), anti‐caspase3 (1:1,000, Cell Signaling, 9662), anti‐vinculin (1:5,000, Cell Signaling, 13901), anti‐tubulin (1:10,000, Millipore, 05‐829), and anti‐transferrin receptor (1:1,000, Invitrogen, 13‐6800). Caspase‐3 control cell extracts (Cell Signaling, 9663) were used as negative and positive controls for apoptosis.
Electrode implantation
Under ketamine/xylazine anesthesia, male mice were fixed in a stereotaxic apparatus and implanted with electrodes (made of enameled nichrome wire; diameter, 150 μm) for polygraphic sleep monitoring (Henderson et al, 2017). Briefly, two electroencephalogram (EEG) electrodes were positioned onto the dura through holes made into the skull over the left frontal cortex and cerebellum (2 mm lateral to midline and 2 mm anterior to bregma; and at midline, 2 mm posterior to lambda, respectively). Using this configuration, the EEG signal is acquired as a monopolar derivation with the frontal electrode compared to a neutral (cerebellum) reference electrode (Mang & Franken, 2012). Two electromyogram (EMG) electrodes were inserted into the neck muscles. All electrodes were anchored to the skull with Superbond (GACD) and acrylic cement and were soldered to a miniconnector also embedded in cement. Mice were transferred to individual recording cages (20 × 20 × 30 cm) and allowed to recover for 10 days under standard conditions. Control and mutant mice were randomly assigned to recording chambers. They were habituated to the recording cables for at least 4 days before recordings were started.
Sleep recording and sleep deprivation protocol
Mice were recorded at 4–5 months of age. EMG and EEG signals were recorded with Somnologica software (Medcare, Reykjavik, Iceland), amplified, analog‐to‐digital converted (2 kHz), and down‐sampled at 100 Hz (EMG) or 200 Hz (EEG), and digitalized by an AddLife A/D Module. Undisturbed spontaneous sleep–wake patterns were first examined by recording mice for 24 h starting at dark onset (baseline recordings). Then, a 6‐h SD protocol starting at light onset (ZT0) was achieved by removing the nest, gently moving the mouse, and adding new bedding material or novel objects as soon as EEG signs of sleep were detected by the experimenter. In the last hour of the protocol (ZT5‐6), mice were placed in a new cage to promote exploratory behavior. At ZT6, mice were put back in their home cage and allowed to recover. Matched control recordings (Sham condition) were obtained the day before by gently waking‐up mice at ZT6. The recording order (baseline sleep, sham condition, and SD) was chosen to end the protocol with the most severe procedure. Three independent studies were conducted each including control and mutant mice.
Sleep analysis
Polygraphic recordings were visually scored every 10 s (baseline sleep) or 4 s (SD) epoch as wake (W), NREM sleep, or REM sleep using the Somnologica software (Medcare, Reykjavik, Iceland). The scorer was blind to the mouse genotype. Briefly, wake was defined by low‐amplitude/high‐frequency EEG signal and high EMG activity; NREM sleep by high‐amplitude/low‐frequency (< 4 Hz) EEG signal and reduced EMG activity and REM sleep by dominant theta oscillations (5–10 Hz) on EEG signal and a flat EMG activity with occasional muscle twitches.
The amounts of time spent on each vigilance state during recovery after SD or sham condition were expressed as minutes per 3 h intervals. The sleep architecture was assessed by calculating the mean duration and frequency of vigilance states bouts (a bout could be as short as one epoch).
The EEG signal was processed for power spectrum analysis. Consecutive epochs were subjected to a fast Fourier transformation routine (FFT), yielding power spectra between 0.4 and 50 Hz, with a 0.4 Hz frequency and a 10 s (baseline sleep) or 4 s (SD) time resolution. For analysis of the SWA spectrum, one control mouse for baseline sleep and three control mice for the SD experiment were excluded from the spectral analysis as they showed EEG recording artifacts affecting more than 20% of the recording time. Similarly, two control mice and one mutant mouse were excluded from the analysis of waking oscillatory activities during SD. For each animal, a mean SWA spectrum corresponding to the 0.5–4.5 Hz frequency band was then obtained by averaging the SWA spectra of an equal number of 10 s or 4 s epochs of NREM sleep, referred to as quantiles as described in Mang & Franken (2012). Mean theta (6.0–9.5 Hz), low‐gamma (32–45 Hz), and high‐gamma (55–80 Hz) values over 6 h of SD were obtained by averaging the waking theta and low‐ and high‐gamma spectra of an equal number of 4 s epochs of wake. The recording sessions were subdivided into three quantiles for the baseline dark period, six quantiles for the baseline light period, six quantiles for the SD, and five quantiles for the recovery light period immediately following SD. The corresponding mean SWA, theta, and low‐gamma and high‐gamma were normalized to the mean SWA, theta, and low‐gamma and high‐gamma during ZT8 to ZT12 of the baseline recording day (Mang & Franken, 2012).
Statistical analysis
For western blot data, statistical significance between CTL and micTNFα‐KO mice at each condition (ZT18, ZT6, or SD6) was assessed by multiple t‐test corrected with the original FDR method of Benjamini–Hochberg with an FDR cut‐off of 5% using Prism 8.0 (GraphPad software).
Statistical analyses for sleep data were performed using Prism 9.3 (GraphPad Software). Normality was verified prior to the use of any parametric tests (D'Agostino–Pearson normality test). Data violating normality was log transformed. Vigilance state amounts, number, and duration of bouts, as well as time‐course dynamics of SWA during NREM sleep and oscillatory activities during wake were assessed using two‐way repeated‐measures analysis of variance (rANOVA). When appropriate, rANOVAs were followed by the Sidak's multiple comparisons. Delta wake and total sleep changes were followed by two‐tailed unpaired t‐tests. Statistical significance was considered as P < 0.05, and all results are given as mean values ± SEM.
Author contributions
Maria J Pinto: Conceptualization; software; formal analysis; investigation; writing – original draft; writing – review and editing. Léa Cottin: Investigation. Florent Dingli: Software. Victor Laigle: Software. Luís F Ribeiro: Investigation. Antoine Triller: Conceptualization; funding acquisition; writing – review and editing. Fiona Henderson: Investigation. Damarys Loew: Software; writing – original draft; writing – review and editing. Véronique Fabre: Conceptualization; formal analysis; funding acquisition; investigation; writing – review and editing. Alain Bessis: Conceptualization; formal analysis; funding acquisition; investigation; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Acknowledgements
We gratefully acknowledge Clément Léna for his great help with the SWA analysis. We are grateful to Amandine Delecourt, Eleonore Touzalin, Deborah Souchet, and the Ibens animal core facility as well as the IBPS Behavioral Core Facility for excellent technical assistance. We thank Patrick Poullet from the bioinformatics platform of the Institut Curie U900 for the continuous development of myProMS and Stéphane Liva for the kinase activity analysis made. Agence nationale pour la recherche: SYNTRACK ‐R17096DJ (AT); European Research Council: PLASTINHIB Project No: 322821 and MICROCOPS (AT); Human Brain Project: HBP SGA2‐OPE‐2018‐0017 (AT); EMBO fellowship: ALTF 362‐2017 (MJP); Région Île‐de‐France and Fondation pour la Recherche Médicale (DL); Marie Skłodowska‐Curie Action Individual Fellowship (101031398) from European Commission and National funds through the Portuguese Science and Technology Foundation FCT: PTDC/BIA‐CEL/2286/2020 (LFR).
The EMBO Journal (2023) 41: e111485
Contributor Information
Maria J Pinto, Email: pinto@bio.ens.psl.eu.
Véronique Fabre, Email: veronique.fabre@inserm.fr.
Alain Bessis, Email: alain.bessis@bio.ens.psl.eu.
Data availability
The mass spectrometry proteomics and phosphoproteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the data set identifier PXD030568 (http://www.ebi.ac.uk/pride/archive/projects/PXD030568). All other data are available in the main text or the Supplementary Materials.
References
- Adamantidis AR, Gutierrez Herrera C, Gent TC (2019) Oscillating circuitries in the sleeping brain. Nat Rev Neurosci 20: 746–762 [DOI] [PubMed] [Google Scholar]
- Albertini G, Etienne F, Roumier A (2020) Regulation of microglia by neuromodulators: modulations in major and minor modes. Neurosci Lett 733: 135000 [DOI] [PubMed] [Google Scholar]
- Anrather J, Gallo EF, Kawano T, Orio M, Abe T, Gooden C, Zhou P, Iadecola C (2011) Purinergic signaling induces cyclooxygenase‐1‐dependent prostanoid synthesis in microglia: roles in the outcome of excitotoxic brain injury. PLoS One 6: e25916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, Hwang P, Chan AT, Graves SM, Uweru JO et al (2020) Negative feedback control of neuronal activity by microglia. Nature 586: 417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, Schulman H (2019) CaM kinase: still inspiring at 40. Neuron 103: 380–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellesi M, de Vivo L (2020) Structural synaptic plasticity across sleep and wake. Curr Opin Physio 15: 74–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco F, Pravettoni E, Colombo A, Schenk U, Möller T, Matteoli M, Verderio C (2005) Astrocyte‐derived ATP induces vesicle shedding and IL‐1 beta release from microglia. J Immunol 174: 7268–7277 [DOI] [PubMed] [Google Scholar]
- Bjorness TE, Dale N, Mettlach G, Sonneborn A, Sahin B, Fienberg AA, Yanagisawa M, Bibb JA, Greene RW (2016) An adenosine‐mediated glial‐neuronal circuit for homeostatic sleep. J Neurosci 36: 3709–3721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum ID, Keleş MF, Baz ES, Han E, Park K, Luu S, Issa H, Brown M, Ho MCW, Tabuchi M et al (2021) Astroglial calcium signaling encodes sleep need in Drosophila . Curr Biol 31: 150–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbély AA, Baumann F, Brandeis D, Strauch I, Lehmann D (1981) Sleep deprivation: effect on sleep stages and EEG power density in man. Electroencephalogr Clin Neurophysiol 51: 483–493 [DOI] [PubMed] [Google Scholar]
- Brenner D, Blaser H, Mak TW (2015) Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15: 362–374 [DOI] [PubMed] [Google Scholar]
- Brüning F, Noya SB, Bange T, Koutsouli S, Rudolph JD, Tyagarajan SK, Cox J, Mann M, Brown SA, Robles MS (2019) Sleep‐wake cycles drive daily dynamics of synaptic phosphorylation. Science 366: eaav3617 [DOI] [PubMed] [Google Scholar]
- Buttini M, Mir A, Appel K, Wiederhold KH, Limonta S, Gebicke‐Haerter PJ, Boddeke HW (1997) Lipopolysaccharide induces expression of tumour necrosis factor alpha in rat brain: inhibition by methylprednisolone and by rolipram. Br J Pharmacol 122: 1483–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA (2003) Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38: 69–78 [DOI] [PubMed] [Google Scholar]
- Choudhury ME, Miyanishi K, Takeda H, Islam A, Matsuoka N, Kubo M, Matsumoto S, Kunieda T, Nomoto M, Yano H et al (2020) Phagocytic elimination of synapses by microglia during sleep. Glia 68: 44–59 [DOI] [PubMed] [Google Scholar]
- Cirelli C, Huber R, Gopalakrishnan A, Southard TL, Tononi G (2005) Locus ceruleus control of slow‐wave homeostasis. J Neurosci 25: 4503–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton JM, Davis CJ, Zielinski MR, Jewett KA, Krueger JM (2011) Biochemical regulation of sleep and sleep biomarkers. J Clin Sleep 7: S38–S42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coba MP (2019) Regulatory mechanisms in postsynaptic phosphorylation networks. Curr Opin Struct Biol 54: 86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsi G, Picard K, di Castro MA, Garofalo S, Tucci F, Chece G, del Percio C, Golia MT, Raspa M, Scavizzi F et al (2022) Microglia modulate hippocampal synaptic transmission and sleep duration along the light/dark cycle. Glia 70: 89–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, Caudy M, Garapati P, Gillespie M, Kamdar MR et al (2014) The Reactome pathway knowledgebase. Nucleic Acids Res 42: D481–D487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deboer T, Fontana A, Tobler I (2002) Tumor necrosis factor (TNF) ligand and TNF receptor deficiency affects sleep and the sleep EEG. J Neurophysiol 88: 839–846 [DOI] [PubMed] [Google Scholar]
- Desch K, Langer JD, Schuman EM (2021) Dynamic bi‐directional phosphorylation events associated with the reciprocal regulation of synapses during homeostatic up‐ and down‐scaling. Cell Rep 36: 109583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A, Huganir RL (2017) Homer1a drives homeostatic scaling‐down of excitatory synapses during sleep. Science 355: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Wang Y, Krueger JM (1997) Mice lacking the TNF 55 kDa receptor fail to sleep more after TNFalpha treatment. J Neurosci 17: 5949–5955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Yu SX, Lu Y, Bast RC, Woodgett JR, Mills GB (2000) Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A 97: 11960–11965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustman D, Davis M (2010) TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov 9: 482–493 [DOI] [PubMed] [Google Scholar]
- Fonken LK, Frank MG, Kitt MM, Barrientos RM, Watkins LR, Maier SF (2015) Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behav Immun 45: 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken P, Chollet D, Tafti M (2001) The homeostatic regulation of sleep need is under genetic control. J Neurosci 21: 2610–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP, Wisden W (2021) The inescapable drive to sleep: overlapping mechanisms of sleep and sedation. Science 374: 556–559 [DOI] [PubMed] [Google Scholar]
- Funato H, Miyoshi C, Fujiyama T, Kanda T, Sato M, Wang Z, Ma J, Nakane S, Tomita J, Ikkyu A et al (2016) Forward‐genetics analysis of sleep in randomly mutagenized mice. Nature 539: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garré JM, Silva HM, Lafaille JJ, Yang G (2017) CX3CR1+ monocytes modulate learning and learning‐dependent dendritic spine remodeling via TNF‐α. Nat Med 23: 714–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Baker TJ, Shih LCN, Lachman LB (1986) Interleukin 1 of the central nervous system is produced by ameboid microglia. J Exp Med 164: 594–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, Murakami T, Drutskaya LN, Förster I, Clausen BE et al (2005) Distinct and nonredundant in vivo functions of TNF produced by T cells and macrophages/neutrophils: protective and deleterious effects. Immunity 22: 93–104 [DOI] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG (2009) Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61: 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson F, Vialou V, El Mestikawy S, Fabre V (2017) Effects of social defeat stress on sleep in mice. Front Behav Neurosci 11: 227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, Nakata Y (2000) Extracellular ATP triggers tumor necrosis factor‐alpha release from rat microglia. J Neurochem 75: 965–972 [DOI] [PubMed] [Google Scholar]
- Hill RA, Medved J, Patel KD, Nishiyama A (2014) Organotypic slice cultures to study oligodendrocyte dynamics and myelination. J Vis Exp 90: e51835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst SC, Sousek A, Hefti K, Saberi‐Moghadam S, Buck A, Ametamey SM, Scheidegger M, Franken P, Henning A, Seifritz E et al (2017) Cerebral mGluR5 availability contributes to elevated sleep need and behavioral adjustment after sleep deprivation. Elife 6: e28751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard J, Gent TC, Hoekstra MMB, Emmenegger Y, Mongrain V, Landolt HP, Adamantidis AR, Franken P (2020) Rapid fast‐delta decay following prolonged wakefulness marks a phase of wake‐inertia in NREM sleep. Nat Commun 11: 3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Deboer T, Tobler I (2000) Topography of EEG dynamics after sleep deprivation in mice. J Neurophysiol 84: 1888–1893 [DOI] [PubMed] [Google Scholar]
- Ikeda‐Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y (2005) Microglia‐specific expression of microsomal prostaglandin E2 synthase‐1 contributes to lipopolysaccharide‐induced prostaglandin E2 production. J Neurochem 94: 1546–1558 [DOI] [PubMed] [Google Scholar]
- Ingiosi AM, Opp MR, Krueger JM (2013) Sleep and immune function: glial contributions and consequences of aging. Curr Opin Neurobiol 23: 806–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingiosi AM, Hayworth CR, Harvey DO, Singletary KG, Rempe MJ, Wisor JP, Frank MG (2020) A role for astroglial calcium in mammalian sleep and sleep regulation. Curr Biol 30: 4373–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Iterson M, Boer JM, Menezes RX (2010) Filtering, FDR and power. BMC Bioinformatics 11: 450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong APH, Fioravante D (2014) Translating neuronal activity at the synapse: presynaptic calcium sensors in short‐term plasticity. Front Cell Neurosci 8: 356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20: 4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammers K, Cole RN, Tiengwe C, Ruczinski I (2015) Detecting significant changes in protein abundance. EuPA Open Proteom 7: 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapás L, Bohnet SG, Traynor TR, Majde JA, Szentirmai É, Magrath P, Taishi P, Krueger JM (2008) Spontaneous and influenza virus‐induced sleep are altered in TNF‐alpha double‐receptor deficient mice. J Appl Physiol 105: 1187–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Braud S, Isaac JTR, Roche KW (2005) Protein kinase C phosphorylation of the metabotropic glutamate receptor mGluR5 on Serine 839 regulates Ca2+ oscillations. J Biol Chem 280: 25409–25415 [DOI] [PubMed] [Google Scholar]
- Krone LB, Yamagata T, Blanco‐Duque C, Guillaumin MCC, Kahn MC, van der Vinne V, McKillop LE, Tam SKE, Peirson SN, Akerman CJ et al (2021) A role for the cortex in sleep‐wake regulation. Nat Neurosci 24: 1210–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Clinton JM, Winters BD, Zielinski MR, Taishi P, Jewett KA, Davis CJ (2011) Involvement of cytokines in slow wave sleep. Prog Brain Res 193: 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Li N, Guan Z, Brown RA, Krueger JM (2002) Intrapreoptic microinjection of TNF‐alpha enhances non‐REM sleep in rats. Brain Res 932: 37–44 [DOI] [PubMed] [Google Scholar]
- Lazarus M, Chen JF, Huang ZL, Urade Y, Fredholm BB (2019) Adenosine and Sleep. Handb Exp Pharmacol 253: 359–381 [DOI] [PubMed] [Google Scholar]
- Lee HK (2006) Synaptic plasticity and phosphorylation. Pharmacol Ther 112: 810–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Tansey MG (2013) Microglia isolation from adult mouse brain. Methods Mol Biol 1041: 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D (2016) Microglial TNF‐α suppresses cocaine‐induced plasticity and behavioral sensitization. Neuron 90: 483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Du X, Liu C, Wen Z, Du J (2012) Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo . Dev Cell 23: 1189–1202 [DOI] [PubMed] [Google Scholar]
- Liu H, Wang X, Chen L, Chen L, Tsirka SE, Ge S, Xiong Q (2021) Microglia modulate stable wakefulness via the thalamic reticular nucleus in mice. Nat Commun 12: 4646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ (2003) Phosphorylation of RIM1alpha by PKA triggers presynaptic long‐term potentiation at cerebellar parallel fiber synapses. Cell 115: 49–60 [DOI] [PubMed] [Google Scholar]
- Mang GM, Franken P (2012) Sleep and EEG phenotyping in mice. Curr Protoc Mouse Biol 2: 55–74 [DOI] [PubMed] [Google Scholar]
- Mikhail C, Vaucher A, Jimenez S, Tafti M (2017) ERK signaling pathway regulates sleep duration through activity‐induced gene expression during wakefulness. Sci Signal 10: eaai9219 [DOI] [PubMed] [Google Scholar]
- Milacic M, Haw R, Rothfels K, Wu G, Croft D, Hermjakob H, D'Eustachio P, Stein L (2012) Annotating cancer variants and anti‐cancer therapeutics in reactome. Cancer 4: 1180–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morairty SR, Dittrich L, Pasumarthi RK, Valladao D, Heiss JE, Gerashchenko D, Kilduff TS (2013) A role for cortical nNOS/NK1 neurons in coupling homeostatic sleep drive to EEG slow wave activity. Proc Natl Acad Sci U S A 110: 20272–20277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ode KL, Ueda HR (2020) Phosphorylation hypothesis of sleep. Front Psychol 11: 575328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary H, Liu WH, Rorabaugh JM, Coultrap SJ, Bayer KU (2011) Nucleotides and phosphorylation bi‐directionally modulate Ca2+/calmodulin‐dependent protein kinase II (CaMKII) binding to the N‐methyl‐D‐aspartate (NMDA) receptor subunit GluN2B. J Biol Chem 286: 31272–31281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paouri E, Tzara O, Kartalou G‐I, Zenelak S, Georgopoulos S (2017) Peripheral tumor necrosis factor‐alpha (TNF‐α) modulates amyloid pathology by regulating blood‐derived immune cells and glial response in the brain of AD/TNF transgenic mice. J Neurosci 37: 5155–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Miyoshi C, Fujiyama T, Kakizaki M, Ikkyu A, Honda T, Choi J, Asano F, Mizuno S, Takahashi S et al (2020) Loss of the conserved PKA sites of SIK1 and SIK2 increases sleep need. Sci Rep 10: 8676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, Hempstead BL, Littman DR, Gan WB (2013) Microglia promote learning‐dependent synapse formation through brain‐derived neurotrophic factor. Cell 155: 1596–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto MJ, Bizien L, Fabre JM, Ðukanović N, Lepetz V, Henderson F, Pujol M, Sala RW, Tarpin T, Popa D et al (2022) Microglial TNFα controls GABAAR plasticity, slow waves and memory consolidation during sleep. bioRxiv 10.1101/2022.02.21.481254 [PREPRINT] [DOI]
- Pirooznia M, Wang T, Avramopoulos D, Valle D, Thomas G, Huganir RL, Goes FS, Potash JB, Zandi PP (2012) SynaptomeDB: an ontology‐based knowledgebase for synaptic genes. Bioinforma Oxf Engl 28: 897–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka‐Heiskanen T (2013) Sleep homeostasis. Curr Opin Neurobiol 23: 799–805 [DOI] [PubMed] [Google Scholar]
- Porkka‐Heiskanen T, Strecker RE, Thakkar M, Bjørkum AA, Greene RW, McCarley RW (1997) Adenosine: a mediator of the sleep‐inducing effects of prolonged wakefulness. Science 276: 1265–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poullet P, Carpentier S, Barillot E (2007) myProMS, a web server for management and validation of mass spectrometry‐based proteomic data. Proteomics 7: 2553–2556 [DOI] [PubMed] [Google Scholar]
- Scammell TE, Arrigoni E, Lipton JO (2017) Neural circuitry of wakefulness and sleep. Neuron 93: 747–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi V, Craig TJ, Bishop P, Nakamura Y, Rocca D, Wilkinson KA, Henley JM (2021) Phosphorylation of Syntaxin‐1a by casein kinase 2α regulates pre‐synaptic vesicle exocytosis from the reserve pool. J Neurochem 156: 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoham S, Davenne D, Cady AB, Dinarello CA, Krueger JM (1987) Recombinant tumor necrosis factor and interleukin 1 enhance slow‐wave sleep. Am J Physiol 253: R142–R149 [DOI] [PubMed] [Google Scholar]
- Stowell RD, Sipe GO, Dawes RP, Batchelor HN, Lordy KA, Whitelaw BS, Stoessel MB, Bidlack JM, Brown E, Sur M et al (2019) Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat Neurosci 22: 1782–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szondy Z, Pallai A (2017) Transmembrane TNF‐alpha reverse signaling leading to TGF‐beta production is selectively activated by TNF targeting molecules: therapeutic implications. Pharmacol Res 115: 124–132 [DOI] [PubMed] [Google Scholar]
- Taishi P, Churchill L, Wang M, Kay D, Davis CJ, Guan X, De A, Yasuda T, Liao F, Krueger JM (2007) TNFalpha siRNA reduces brain TNF and EEG delta wave activity in rats. Brain Res 1156: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Kapás L, Fang J, Krueger JM (1995) An anti‐tumor necrosis factor antibody suppresses sleep in rats and rabbits. Brain Res 690: 241–244 [DOI] [PubMed] [Google Scholar]
- Takahashi S, Kapás L, Seyer JM, Wang Y, Krueger JM (1996) Inhibition of tumor necrosis factor attenuates physiological sleep in rabbits. Neuroreport 7: 642–646 [DOI] [PubMed] [Google Scholar]
- Tatsuki F, Sunagawa GAA, Shi S, Susaki EAA, Yukinaga H, Perrin D, Sumiyama K, Ukai‐Tadenuma M, Fujishima H, Ohno RI et al (2016) Involvement of Ca(2+)‐dependent hyperpolarization in sleep duration in mammals. Neuron 90: 70–85 [DOI] [PubMed] [Google Scholar]
- Taylor L, Palumaa T, Reardon PK, Walsh S, Liberatori S, Hasan S, Clark K, Cohen P, Vasudevan S, Peirson S et al (2021) Light regulated SIK1 remodels the synaptic phosphoproteome to induce sleep. bioRxiv 10.1101/2021.09.28.462159 [PREPRINT] [DOI]
- The M, MacCoss MJ, Noble WS, Käll L (2016) Fast and accurate protein false discovery rates on large‐scale proteomics data sets with Percolator 3.0. J Am Soc Mass Spectrom 27: 1719–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004) MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5: 173–183 [DOI] [PubMed] [Google Scholar]
- Timm T, Li XY, Biernat J, Jiao J, Mandelkow E, Vandekerckhove J, Mandelkow EM (2003) MARKK, a Ste20‐like kinase, activates the polarity‐inducing kinase MARK/PAR‐1. EMBO J 22: 5090–5101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tossell K, Yu X, Soto BA, Vicente M, Miracca G, Giannos P, Miao A, Hsieh B, Ma Y, Yustos R et al (2020) Sleep deprivation triggers somatostatin neurons in prefrontal cortex to initiate nesting and sleep via the preoptic and lateral hypothalamus. bioRxiv 10.1101/2020.07.01.179671 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Valot B, Langella O, Nano E, Zivy M (2011) MassChroQ: a versatile tool for mass spectrometry quantification. Proteomics 11: 3572–3577 [DOI] [PubMed] [Google Scholar]
- Vanderheyden WM, Gerstner JR, Tanenhaus A, Yin JC, Shaw PJ (2013) ERK phosphorylation regulates sleep and plasticity in Drosophila . PLoS One 8: e81554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderheyden WM, Goodman AG, Taylor RH, Frank MG, Van Dongen HPA, Gerstner JR (2018) Astrocyte expression of the Drosophila TNF‐alpha homologue, Eiger, regulates sleep in flies. PLoS Genet 14: e1007724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRyzin JW, Marquardt AE, Argue KJ, Vecchiarelli HA, Ashton SE, Arambula SE, Hill MN, McCarthy MM (2019) Microglial phagocytosis of newborn cells is induced by endocannabinoids and sculpts sex differences in juvenile rat social play. Neuron 102: 435–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassalli A, Franken P (2017) Hypocretin (orexin) is critical in sustaining theta/gamma‐rich waking behaviors that drive sleep need. Proc Natl Acad Sci U S A 114: E5464–E5473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Ruijgrok G, Deboer T, Tobler I (2006) Running wheel accessibility affects the regional electroencephalogram during sleep in mice. Cereb Cortex 16: 328–336 [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister‐Genskow M, Faraguna U, Tononi G (2008) Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci 11: 200–208 [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Lazimy YM, Faraguna U, Esser SK, Williams JC, Cirelli C, Tononi G (2009) Cortical firing and sleep homeostasis. Neuron 63: 865–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ma J, Miyoshi C, Li Y, Sato M, Ogawa Y, Lou T, Ma C, Gao X, Lee C et al (2018) Quantitative phosphoproteomic analysis of the molecular substrates of sleep need. Nature 558: 435–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, Dell'Acqua ML (2015) Coordination of protein phosphorylation and dephosphorylation in synaptic plasticity. J Biol Chem 290: 28604–28612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C‐H, Tatavarty V, Beltran PM, Guerrero A, Keshishian H, Krug K, MacMullan MA, Li L, Carr SA, Cottrell JR et al (2021) A bidirectional switch in the Shank3 phosphorylation state biases synapses toward up or down scaling. bioRxiv 10.1101/2021.10.03.462942 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Yger M, Girault JA (2011) DARPP‐32, jack of all trades… master of which? Front Behav Neurosci 5: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yılmaz S, Ayati M, Schlatzer D, Çiçek AE, Chance MR, Koyutürk M (2021) Robust inference of kinase activity using functional networks. Nat Commun 12: 1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona S, Kim K‐W, Wolf Y, Mildner A, Varol D, Breker M, Strauss‐Ayali D, Viukov S, Guilliams M, Misharin A et al (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38: 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong XLH, Zhang L, Yang L, Chen X, Tan JZA, Yu X, Chandra M, Livingstone E, Widagdo J, Vieira MM et al (2021) Regulation of NMDA receptor trafficking and gating by activity‐dependent CaMKIIα phosphorylation of the GluN2A subunit. Cell Rep 36: 109338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Peterfi Z, García‐García F, Kirkpatrick R, Yasuda T, Krueger JM (2004) State‐specific asymmetries in EEG slow wave activity induced by local application of TNFα. Brain Res 1009: 129–136 [DOI] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G et al (2018) Molecular architecture of the mouse nervous system. Cell 174: 999–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N et al (2014) An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou MH, Chen SR, Wang L, Huang Y, Deng M, Zhang J, Zhang J, Chen H, Yan J, Pan HL (2021) Protein kinase C‐mediated phosphorylation and α2δ‐1 interdependently regulate NMDA receptor trafficking and activity. J Neurosci 41: 6415–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]