

Summary
A common technique for uncovering intra-tumor genomic heterogeneity (ITH) is variant detection. However, it can be challenging to reliably characterize ITH given uneven sample quality (e.g., depth of coverage, tumor purity, and subclonality). We describe a protocol for calling point mutations and copy number alterations using sequencing of multiple related clinical patient samples across diverse tissue, optimizing for sensitivity with specificity. The ith.Variant pipeline can be run on single- or multi-region whole-genome and whole-exome sequencing.
For complete details on the use and execution of this protocol, please refer to Sun et al. (2017).1
Subject areas: Bioinformatics, Cancer, Health Sciences, Genomics, Sequencing
Graphical abstract
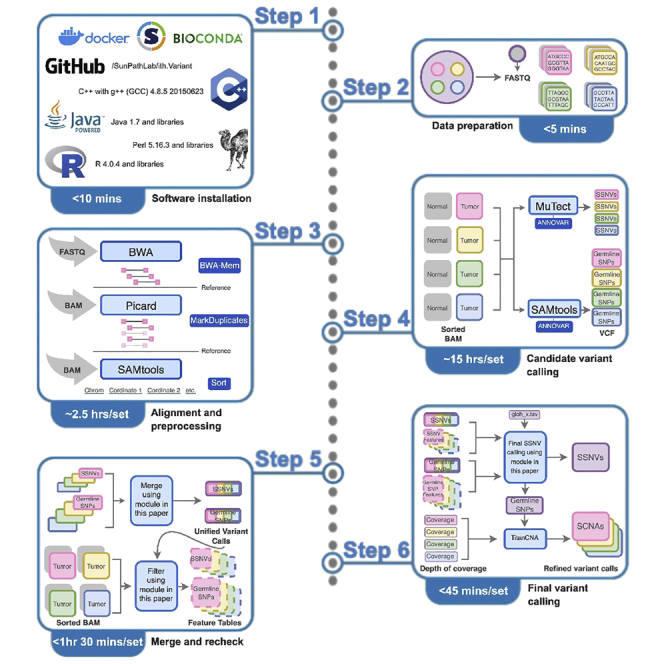
Highlights
-
•
Detects different types of variants for multi-region/stage tumor WES/WGS
-
•
Employs feature extraction and examination in calling point mutations
-
•
Merges candidate mutations across samples for ITH characterization
-
•
Applies filtered germline point mutations to copy number calling
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
A common technique for uncovering intra-tumor genomic heterogeneity (ITH) is variant detection. However, it can be challenging to reliably characterize ITH given uneven sample quality (e.g., depth of coverage, tumor purity, and subclonality). We describe a protocol for calling point mutations and copy number alterations using sequencing of multiple related clinical patient samples across diverse tissue, optimizing for sensitivity with specificity. The ith.Variant pipeline can be run on single- or multi-region whole-genome and whole-exome sequencing.
Before you begin
Timing: <3 Days
Next Generation Sequencing (NGS) introduced extensive data and initiated the era of high-throughput genomic analysis. One method of analysis is variant calling: a process of identifying variants like somatic single-nucleotide variants (SSNVs) and somatic copy number alterations (SCNAs) from sequencing data by comparing mapped reads between normal and tumor genomes. As a result of calling variants including germline single-nucleotide polymorphisms (SNPs), an individual’s genotype can be captured and matched with a known pathology like cancer for precise diagnosis and treatment.2,3,4,5 Calling SSNVs and SCNAs relative to germline SNPs is particularly challenging due to the higher potential for identifying artifacts as variants.6 One reason for this is the presence of multiple populations or clones of tumor cells within a sample, carrying their own distinct genotype.7 This intra-tumor genomic heterogeneity (ITH) is fueled by SSNVs and SCNAs that arise in spatially segregated subclones as the tumor expands. In tumor samples characterized by ITH (due to subclonality) and reduced tumor cellularity (due to impurity), SSNVs can appear at lower variant allele frequencies (VAFs). Additionally, varying depth of coverage of the sample genome due to sample preparation can increase the variance of VAF. As clinical relevance is often found in subclones harboring somatic variants with low VAFs, variant calling must entail sensitive statistical modeling and error correction techniques to ensure valid calls and downstream analysis.8 For example, correctly characterizing the genomic profiles of multiple related samples of a patient tumor can lend itself to the reliable reconstruction of its phylogeny.9,10,11 Therefore, it is imperative to design variant calling to be adaptable to the circumstances of varying coverage, impurity, and subclonal composition. By leveraging the information between tumor samples with the matched normal control from the same patient, we present ith.Variant, a unified somatic variant calling pipeline that strikes a balance in sensitivity and specificity in distinguishing SSNVs across a wide range of cellular prevalence. Our pipeline utilizes common variant analysis tools like MuTect12 and SAMtools,13 and it provides a module for refined filtration based on the mapping features of merged candidate variants. In addition, ith.Variant also detects clonal or major subclonal SCNAs (i.e., no more than two distinct SCNA states for a genomic region in a given sample) by applying TitanCNA.14 The final lists of SSNVs and SCNAs help to establish a baseline of cancer genomics, facilitating downstream analysis of genomic heterogeneity and tumor evolution. ith.Variant is an open-source package available via https://github.com/SunPathLab/ith.Variant.
The protocol below consists of user-defined steps for running the ith.Variant pipeline on multi-region WES colorectal tumor sample data. By leveraging this type of sampled sequencing, the protocol preserves ITH represented by variant calls as WES/WGS of different tumor regions captures subclonal composition and tumor phylogeny. Based on a more integrative algorithmic approach to somatic variant calling, the pipeline optimizes for sensitivity to generate a reliable list of SSNV and SCNA calls.
Hardware, software and data requirements
The ith.Variant pipeline is a command-line driven, perl-based wrapper of various tools written in C++, Java and R for somatic variant calling. It can be run on a Linux operating system. As an example, we used a specific high performance computing (HPC) cluster called Mangi by connecting to the Minnesota Supercomputer Institute (MSI) to execute the pipeline. Mangi is one of MSI’s HPC systems, featuring partitions of computing nodes that are accessible via a common queuing system called Slurm. Allocation of these computational resources using Slurm is made in the form of a Slurm script as shown in our protocol.
The input data can be raw sequencing reads in FASTQ, or unaligned or aligned BAM format generated from WES/WGS of paired tumor-normal samples. The tumor samples can be obtained by multi-region or multi-time point sampling (metastatic- or relapse-primary).15,16 Here we use multi-region single-gland and bulk paired-end WES data from colorectal tumor specimens of one patient to demonstrate the ith.Variant pipeline.1 The sequencing was performed on the Illumina platform, generating reads for tumor and normal samples. Due to the paired-end sequencing of the data, there are two read files for each sample, representing each end of the fragments. The third party tools including BWA,17 MuTect, and SAMtools necessary for ith.Variant can be installed following their own installation instructions or run in a containerized environment.
Dependencies
Before implementing our ith.Variant pipeline, there are packages in R and perl that are required. All of the prerequisite packages are listed in the key resources table. As an alternative to installing the dependencies individually or using Conda/Bioconda,18 we provide a Docker image with everything pre-installed. More information on using Conda and Docker can be found at the link (https://github.com/SunPathLab/ith.Variant/blob/main/requirements.txt) and under the section “running as a Docker container,” respectively. For manual installation, the perl packages can be installed using the CPAN command, and the majority of R packages can be installed using the install.packages() function. Here are general steps for package installation in the user’s Linux environment. Full instructions for installation are available on the protocol’s ith.Variant repository.
-
1.
Install the necessary perl packages in your working directory. The relevant perl packages and respective installation commands for our environment are shown.
> install List::Util
> install Math::CDF
> install Statistics:Basic
> install Parallel::ForkManager
> install Text::NSP::Measures::2D::Fisher::right
-
2.
Install the necessary R packages in your working directory. The relevant R libraries and respective installation commands are shown.
> if(!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
> BiocManager::install(“HMMcopy”)
> install.packages(“caTools”)
> install.packages(“KernSmooth”)
> install.packages(“doMC”)
> install.packages(“RColorBrewer”)
> install.packages(“path/to/TitanCNA.1.26.mod.tar.gz”)
Note: Here we load and execute perl modules and R packages in our remote shell environment. Ensure perl and R are installed. As many Linux distributions as well as MacOS have perl preinstalled and R binaries available for their system, the commands should successfully run. Your environment may require a different method of installing packages. The command for loading each library containing the respective package into your environment is not shown.
Downloading the pipeline
-
3.
Clone the repository.
Note: To perform this, click on the green ‘Code’ button and then copy the address link containing HTTPS (Figure 1A). This is the link to the repository which contains directories such as configurations (i.e., confs), libraries (lib), stepwise Slurm scripts (pipeline), packages (pkgs), source code (src), and utility programs (utils), as well as a ReadMe file. The directory called pipeline has the main perl program submit_slurm.pl that passes various arguments including step-specific Slurm scripts. The directory src contains the main program DTrace.pl, run in each of the commands by the main perl program submit_slurm.pl.
-
4.
Navigate to the working directory where you want the cloned directory to be deposited.
-
5.
Clone the GitHub repository using HTTPS in Git (Figure 1B).
Note: The directories and files will appear once finished (Figure 1C). An example dataset used in our protocol is shown (Figure 1D).
-
6.
Download annotation files for your genome build.
Note: The annotation files we use here are listed in the key resources table. For more details, please refer to the ith.Variant repository.
CRITICAL: Please create a separate directory where the output of the pipeline will be written to. Here we named the directory CRC_singleGland_pipeline_run as our data consists of colorectal single-gland and bulk tumor samples.
Note: Other methods for cloning the repository include “Open with Github Desktop” through the respective application and “Download ZIP” which involves placing the contents in a compressed directory on your desktop. The Git method used for the ith.Variant pipeline is relatively easy and places the repository in the user-specified directory. Depending on the computing environment, the action of using Git to download the contents of the repository locally may take a while.
Figure 1.

Clone the repository to your computing environment
(A) The pipeline is available at https://github.com/SunPathLab/ith.Variant.
(B) The shell command to remotely clone the repository to your specified directory.
(C) The contents of the cloned directory for ith.Variant.
(D) The FASTQ files are loaded in the shell environment. In total, our data consists of seven tumor samples and two normal samples.
Running as a Docker container
To ensure computational reproducibility and facilitate use, we also provide a Docker image for ith.Variant hosted on Docker Hub. Running the package as a Docker container is possible through Docker or Singularity.19 We encourage users to use Singularity in the case where an HPC system (i.e., the computing environment that has the host operating system) does not allow Docker. Here are the following steps for setting up the Docker container prior to following the “step-by-step method details.”
-
7.
Install Docker. The platform is available here: https://docs.docker.com/get-docker/.
-
8.
Pull the Docker container to your local machine. This will take 1–2 min based on your Internet connection. The second command will check if the image is already available.
$ docker pull asntech/ith.variant:v1.0
$ docker image ls
-
9.
Run the Docker image by mounting a local path using the parameter -v.
Note: The parameter -it makes the container interactive. The argument /path can be your local data and configuration directory and will be exposed to the container.
$ docker run -v /path:/path -it asntech/ith.variant:v1.0
-
10.
Run the following command to test the pipeline.
$ perl /opt/ith.Variant/pipeline/exome/submit_slurm.pl -h
Note: The ith.Variant repository is already installed and is located here (/opt/ith.Variant). All the binaries and scripts will be in the bin directory (/opt/ith.Variant/bin).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| E-MTAB-5547.idf.txt | Experimental description file under EMBL-EBI ArrayExpress archival accession number. | https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5547/ |
| E-MTAB-5547.sdrf.txt | Sample description file under EMBL-EBI ArrayExpress archival accession number. | https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5547/ |
| E-MTAB-5547 FASTQ files | Raw data files in FASTQ format under EMBL-EBI ArrayExpress archival accession number. | https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5547/samples/ |
| requirements.txt | Data dump of annotation files for the genome build used in this paper. | https://github.com/SunPathLab/ith.Variant/blob/main/requirements.txt |
| config_hg38_wxs_nrce.tsv | Configuration file used in this paper. | https://github.com/SunPathLab/ith.Variant/tree/main/confs/exome |
| config_hg38_wxs_ss.tsv | Configuration file used in this paper. | https://github.com/SunPathLab/ith.Variant/tree/main/confs/exome |
| config_hg38_wxs_ss_java6.tsv | Configuration file used in this paper. | https://github.com/SunPathLab/ith.Variant/tree/main/confs/exome |
| gloh_x.pl | Perl program to retrieve all homozygous SNPs on the X chromosome. | https://github.com/SunPathLab/ith.Variant/blob/main/src/gloh_x.pl |
| TitanCNA_1.26.0.tar.gz | Modified R package TitanCNA used in this paper. | https://github.com/SunPathLab/ith.Variant/tree/main/pkgs |
| submit_slurm.pl | Perl program for calling DTrace.pl and passing arguments for Slurm job submission. | https://github.com/SunPathLab/ith.Variant/tree/main/pipeline/exome |
| Slurm scripts (.slurm) | Slurm scripts (for jobs 0–7) used in this paper. | https://github.com/SunPathLab/ith.Variant/tree/main/pipeline/exome |
| Software and algorithms | ||
| Linux (>=3.10.0) | Linux distribution CentOS derived from Red Hat Enterprise Linux. | https://www.centos.org/centos-linux/ |
| BWA (0.7.12) | (Li and Durbin17); Command-line software package for mapping and aligning a sequence against a reference genome in a SAMtools-compatible format. | http://bio-bwa.sourceforge.net/bwa.shtml |
| SAMtools (1.5) | (Li13); Command-line software package for SNP calling and analysis. | https://www.htslib.org/ |
| VCFtools (0.1.16) | (Danecek et al.20); Command-line software package for working with files in VCF format. | http://vcftools.sourceforge.net/ |
| Java (1.7) | Object-oriented programming language commonly used for software development and networking. | https://www.java.com/en/download/help/whatis_java.html |
| MuTect (1.1.4) | (Cibulskis et al.12); Java software for reliably identifying somatic point mutations in sequencing. | https://github.com/broadinstitute/mutect |
| Picard (2.23.4) | (Broad Institute21); Java software for manipulating sequencing data including removing duplicate variant calls. | https://broadinstitute.github.io/picard/ |
| R (>=4.0.4) | Software environment for statistical computing and graphics | https://www.r-project.org/ |
| HMMcopy (1.32.0) | (Lai et al.22): R package for copy number analysis with selective removal of bias. | https://bioconductor.org/packages/release/bioc/html/HMMcopy.html |
| TitanCNA (1.26.0) | (Ha et al.14): R package that applies a Hidden Markov model to segment and predict structural variations and clonality in genomic data. | https://www.bioconductor.org/packages/release/bioc/html/TitanCNA.html |
| caTools (1.18.2) | (Tuszynski23): R package that offers multiple basic utility functions like statistical functions. | https://cran.r-project.org/web/packages/caTools/index.html |
| KernSmooth (2.23.20) | (Wand et al.24): R package that contains various techniques of probability density estimation. | https://cran.r-project.org/web/packages/KernSmooth/KernSmooth.pdf |
| doMC (1.3.8) | (Daniel et al.25): R module that provides a parallel backend. | https://cran.r-project.org/web/packages/doMC/index.html |
| RColorBrewer (1.1.2) | (Neuwirth26): R module used for colorful visualization. | https://cran.r-project.org/web/packages/RColorBrewer/index.html |
| Perl (>=5.16.3) | Software environment for general-purpose programming including web development and network programming. | https://www.perl.org/ |
| Annovar (2015Jun16) | (Wang et al.27); Perl library for functionally annotating genetic variants. | https://annovar.openbioinformatics.org/en/latest/#annovar-documentation |
| Parallel::ForkManager (2.02) | (Szabó et al.28): Perl library that offers parallel processing for forking procedures. | https://metacpan.org/pod/Parallel::ForkManager |
| List::Util (1.61) | A function from the perl package List that provides a select group of general-utility list subroutines. | https://metacpan.org/pod/List::Util |
| Math::CDF (0.1) | A perl function that uses various statistical probability functions to generate probabilistic outputs. | https://metacpan.org/pod/Math::CDF |
| Text::NSP::Measures::2D::Fisher::right (0.97) | A perl function that performs a right-sided Fisher’s exact test. | https://metacpan.org/pod/Text::NSP::Measures::2D::Fisher::right |
| Singularity (3.8.7) | (Kurtzer et al.19); Software for running containers on HPC systems. | https://docs.sylabs.io/guides/3.0/user-guide/quick_start.html |
| Other | ||
| Mangi | The HPC system used in this paper. | https://www.msi.umn.edu/mangi |
Materials and equipment
The protocol was implemented in the CentOS Linux operating system. All steps were run under the CentOS system in an HPC cluster through MSI with the computational resources listed in Table 1.
Alternatives: Other computing environments supported by a Linux system and that meet the minimum computing requirements should be sufficient to run ith.Variant.
Table 1.
Detail of compute requirements
| Computing environment | Detail |
|---|---|
| CentOS Linux | 7.9.2009 |
| RAM | 248 GB |
| CPU Vendor ID | AuthenticAMD |
| CPU(s) | 128 |
| Core(s) per socket | 64 |
| Thread(s) per core | 2 |
| CPU min MHz | 3000.0000 |
| Model | 49 |
| Model name | AMD EPYC 7702 64-Core Processor |
Step-by-step method details
We describe the stepwise methodology to perform SSNV and SCNA detection on multiple related samples with the ith.Variant pipeline. At the beginning, the repository was cloned to the specified working directory and the dependencies listed in the key resources table were either installed and loaded into the shell session in Linux or pre-installed.
Download the raw read Fastq files, create directories and establish linkages
-
1.
Create a folder for the raw data (FASTQ files).
Note: The folder does not have to be in the path of the parent directory. The folder fastq will store the raw data files.
-
2.
Download the raw data.
Note: The files we used are found at the link provided in the key resources table. They are in FASTQ format and gzipped (.fq.qz). Transfer the FASTQ files from your local machine to the folder fastq located in your remote environment.
-
3.
Create a folder for the protocol analysis. Within this folder, create a subfolder called root that will serve as your working directory. Shown below are the commands to create and navigate to the parent directory.
$ mkdir CRC_singleGland_pipeline_run/root -p
$ cd CRC_singleGland_pipeline_run
Note: Here we created CRC_singleGland_pipeline_run to be the parent directory, containing the subfolder root. Once implementation of the protocol is complete, the folder root will contain the output of the results for individual samples.
-
4.
Create a file for mapping the reads to the reference genome.
Note: This file somaticInfo.O is named using a patient identifier (i.e., Patient O) and can be in the previously made parent directory CRC_singleGland_pipeline_run. It is in tab-delimited ASCII text format and consists of two columns: one column for the name of the tumor samples, and the other column for the name of the corresponding normal samples as shown in Figure 2A.
Note: The somaticInfo.O file used in the protocol here shows varying normal samples (ON and ONs). These normal samples correspond to specific tumor samples that were originally sequenced using different exon-capture protocols. This is negligible, but the proper exome target file for a sample must be indicated in the configuration file (config.tsv, see key resources table) for analysis.
-
5.
Inside the parent directory CRC_singleGland_pipeline_run, create soft links to the pertinent files. The first soft link can be to the folder containing the raw FASTQ files. The second soft link can be to the name mapping file. The commands for both tasks are shown below.
$ ln -s /path/to/fastq/ ./
$ ln -s /path/to/somaticInfo.O ./
Figure 2.

Prepare to and run the first step of ith.Variant
(A) The somaticInfo.O file listing the tumor-normal pairing information. There must be two columns: the first for the names of the tumor samples, and the second for the respective normal samples. The names are tab-delimited.
(B) A screenshot of the options given by the perl script for submitting jobs to the Slurm queuing system. The parameters are given with their description. They are user-specified and passed when the perl script is invoked on the command-line interface.
(C) A screenshot of the output directories from mapping each sample to the reference genome. The top directory contains the raw paired-end sequencing data files for one sample with soft links to the original data files (fq.qz). The bottom directory contains the mapped and preprocessed files for the same sample.
(D) A screenshot of the output files for single-gland WES sample CRCTumorOA1 after mapping is complete. The files include the mapped and sorted file (.bam) and an index file (.bai) for random access.
Map read files against Hg38
As preparation for downstream variant calling, the sequencing read files are mapped against a human reference genome. Here we chose the human genome build 38 (hg38) with further details available on the GitHub. The reference genome hg38 is chosen as the latest developed reference genome, representing more human genomic variation, and thus leading to higher sensitivity in variant callers. The outcome of this step will be mapped reads in BAM format that can be used for further analysis.
-
6.
Check the options of the perl script wrapper for submitting jobs.
$ perl/path/to/ith.Variant/pipeline/exome/submit_slurm.pl -h
Note: The submit_slurm.pl perl script is wrapped to ease invoking the commands in ith.Variant. It can be found in the key resources table. The parameters in the perl script will be user-specified via a text editor like Emacs and Nano and are shown in Figure 2B.
-
7.Map the FASTQ data to the reference genome. The parameters are described below and are shown in Figure 2B. Edit the command for your use-case including the data being used, the pathway to the ith.Variant package, and the names of the parent and working directories. Here are the modifiable parameters that will be used to submit the Slurm job.
-
a.submit_slurm.pl is the main perl program for the pipeline. When a command is executed for each step in the protocol, it calls this perl program which passes various command-line arguments including Slurm programs and other perl programs.
-
b.--root is $YOUR_ROOT_FOLDER. This parameter is the working directory. Here we use root. It will contain the output of mapping/aligning and pre-processing each sample.
-
c.--workhard is $YOUR_SLURM_SCRIPT. This parameter is the step-specific job submission script. The mapping step corresponds to step0_mapping.slurm.
-
d.--readpool is $YOUR_FASTQ_FOLDER. This parameter is the fastq files. The FASTQ files’ names should have unique prefixes, matching the sample names shown in the somaticInfo.O file.
-
e.--somaticInfo is $SOMATICINFO_FILE. This parameter is the name mapping file.
-
f.--config is $YOUR_CONFIG_FILE. This parameter is the configuration file. Note that users can make a custom configuration file by following the same format of the example configuration file (see key resources table) to indicate the locations of executables and other required files.
-
g.--samples is $YOUR_SAMPLENAMES. This parameter is a comma-delimited list containing the names of your samples for analysis.$ perl /path/to/ith.Variant/pipeline/exome/submit_slurm.pl--root /path/to/CRC_singleGland_pipeline_run/root/--workhard /path/to/ith.Variant/pipeline/exome/step0_mapping.slurm --readpool /path/to/CRC_singleGland_pipeline_run/fastq/--somaticInfo /path/to/CRC_singleGland_pipeline_run/somaticInfo.O --config /path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsvsamplesOA,OB,CRCTumorOA1,CRCTumorOA2,CRCTumorOA3,CRCTumorOA4,CRCTumorOB1,ONNote: The FASTQ data (fq.qz) is located in the fastq folder. This step runs the Slurm script step0_mapping.slurm by which the user can submit the mapping job to the Slurm system, automating the mapping/alignment and detailed preprocessing of the read alignment files. The example Slurm script used in the protocol for this along with the other Slurm scripts can be configured to your environment and are found in the key resources table under the directory called pipeline. This step consists of the following: each sample is mapped and aligned to the hg38 genome using the efficient BWA-MEM algorithm for short-read alignment17; the resulting BAM file is sorted by SAMtools13; and duplicate reads are marked by Picard’s MarkDuplicates.21 Specifically, the BWA-MEM algorithm is applied as our Illumina paired-end reads encompass approximately 150 bp per paired-end read, falling within the algorithm’s recommended range. Upon alignment, SAMtools sorts the aligned reads by coordinates, and MarkDuplicates performs further pre-processing to retain one representative copy of each read. For instance, here we mapped seven tumor samples and two normal samples, specifying these samples in the parameter --samples. The output will be directories under each sample and include several sorted files as shown in Figures 2C and 2D, respectively.Optional: If reads are contained in pre-mapped/aligned BAM files instead of raw FASTQ files, users can re-map these reads against a new reference genome. Here we show an example where reads for the sample ONs were previously mapped and thus already in BAM format. We remap the reads from the original BAM file against a new reference genome using the command line below. This step is estimated to have a similar runtime as the mapping of FASTQ files contingent on data size. The parameters are like those of the previous step, except for a few changes including an additional parameter:
-
h.--prembamDir is $YOUR_BAM_FOLDER. This parameter is the directory containing the relevant BAM file(s) to be remapped. Note that the name of the BAM file should have a unique prefix matching the sample name stated in the somaticInfo.O file.$ perl/path/to/ith.Variant/pipeline/exome/submit_slurm.pl--root/path/to/CRC_singleGland_pipeline_run/root/--workhard/path/to/ith.Variant/pipeline/exome/step0_reMapping.slurm--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsv--prembamDir /path/to/CRC_singleGland_pipeline_run/bam/--samples ONsOptional: When running the ith.Variant pipeline, the following parameters for initiating various methods of preprocessing can be omitted. This alternative step generates relatively similar variant calls with greater efficiency. To skip the additional preprocessing, eliminate the following argument from step0_mapping.slurm:
-
i.--skipTask indelRealignment,BaseRecalibration,recalMDNote: To ensure the Slurm job is running, use the command below. If there is an error, the time will keep showing zero and no results will be generated. Confirm the parameters are correctly modified and the directories and files are in the appropriately specified locations. The following command can be executed to retrieve details related to job status.squeue -a -u <name_of_user>Note: The overall timing of this step is based on the number of samples representing input files and their data format (i.e., whether they require remapping).
-
a.
Generate candidate SSNVs and germline SNPs for each tumor sample
With the read alignment files generated from the previous step, we perform variant detection of somatic single-nucleotide variants (SSNVs) and germline single-nucleotide polymorphisms (SNPs) for Patient O. Relative to germline SNPs, somatic variants can present at low-allelic frequencies which can complicate calling. As an initial scan for candidate variants in individual samples, we apply the variant caller MuTect, leveraging its sensitivity to variants at low allelic frequencies. Similarly, we use SAMtools to generate the list of candidate germline SNPs. The outcome will be candidate SSNVs and SNPs for later processing.
-
8.Perform initial variant scanning for SSNVs with the BAM files for tumor and normal samples. The command is below.
-
a.This step will produce a directory 04_SNV (Figure 3A), containing the raw candidate SSNV calls files (Figure 3B). The primary output is the file [SampleName].mutect.genome.sorted.vcf.hg38_multianno.mod.vcf. For example, see the file for sample CRCTumorOA1 (Table 2). The file contains the candidate variant calls labeled by the functional annotation software for genetic variants known as Annovar.27 To distinguish exonic (protein-coding) variants from those that are non-coding,29,30 ith.Variant uses Annovar to perform region-based annotation on variants in WES/WGS for a more comprehensive calling.
-
b.The file CRCTumorOA1.mutect contains numerous features of the sequencing data originally returned by MuTect. It reports a table with contig position, reference and alternate alleles, and metadata including coverage power, filters, and strand bias.
-
c.The other two files (.vcf) are converted from the MuTect output table. They are in standard variant call format (VCF),20 and they contain MuTect-generated metadata like genomic coordinates and associated filtering flag (i.e., “Reject” or “Pass”).
- d.
- e.
-
a.
$ perl
/path/to/ith.Variant/pipeline/exome/submit_slurm.pl
--root /path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step1_muTect.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss_java6.tsv
--samplesOA,OB,CRCTumorOA1,CRCTumorOA2,CRCTumorOA3,CRCTumorOA4,CRCTumorOB1
Note: The variant caller is MuTect which is based on the Bayesian model of joint-allele frequencies of a tumor-normal sample pair. By framing the variant detection task as a Bayesian model of allelic frequencies, MuTect does not hold the common assumption of clonal mutations being in a pure, diploid tumor of fixed 50% allelic frequencies. The processed BAM files are used as input for the algorithm.
-
9.Perform initial scanning for germline SNPs with the previous tumor-normal sample BAM files. The command is below:
-
a.By default, the pipeline uses the SAMtools mpileup program with --ignore-RG set. This allows the program to ignore the read group tag in the BAM file and treat all reads in one BAM file as one sample.
-
b.Users can also delete the --CfiftyOff 1 option in the Slurm script step2_samtools.slurm to allow downgrading mapping quality for reads containing excessive mismatches. This will set --adjust-MQ to be 50, as recommended by SAMtools (please refer to https://samtools.github.io/hts-specs/VCFv4.2.pdfs for more details).
-
a.
$ perl /path/to/ith.Variant/pipeline/exome/submit_slurm.pl
--root/path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step2_samtools.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsv
--samplesOA,OB,CRCTumorOA1,CRCTumorOA2,CRCTumorOA3,CRCTumorOA4,CRCTumorOB1
Note: Here we use SAMtools which is based on joint genotype analysis, coupling a pair of tumor and normal samples with the assumption of a diploid genome. With this assumption, the tool calculates the log-likelihood ratio of the two genotypes with a larger ratio indicating greater confidence. SAMtools detects possible germline SNPs and small INDELS (insertions and deletions) using the previously mapped BAM files. Here the primary output is the file [SampleName].samtools.genome.sorted.vcf, consisting of a table of chromosome, position, allele, quality of read, and other metadata for each candidate SNP (please refer to https://samtools.github.io/hts-specs/VCFv4.2.pdf for more details). For example, see the file for sample CRCTumorOA1 (Table 3).
Note: The only difference between the configuration files used in steps 8 and 9 is that the former requires its configuration file to contain a path to Java resources. Please refer to the key resources table for how to run MuTect for step 8.
Figure 3.

Candidate somatic and germline calls generated by variant callers
(A) Output directory O4_SNV after running MuTect and SAMtools. This directory contains the candidate list of SSNVs and SNPs for a specific sample.
(B) Output files from MuTect and SAMtools scanning. Here is the list of candidate SSNVs generated by MuTect and candidate SNPs by SAMtools for sample CRCTumorOA1.
Table 2.
Preview of candidate SSNVs for sample CRCTumorOA1
| #Chrom | POS | ID | Ref | Alt | Qual | Filter |
|---|---|---|---|---|---|---|
| chr1 | 10247 | rs796996180 | T | C | . | REJECT |
| chr1 | 10358 | . | A | C | . | REJECT |
| chr1 | 14522 | . | G | A | . | REJECT |
| chr1 | 14653 | rs62635297 | C | T | . | REJECT |
| chr1 | 14798 | . | C | G | . | REJECT |
This is a preview of file ([SampleName].mutect.genome.sorted.vcf.hg38_multianno.mod.vcf) generated by MuTect, containing candidate SSNV calls in sample CRCTumorOA1. Here the first five genomic positions along Chromosome 1 of the reference genome hg38 are shown with the reference and alternate alleles, quality score, filter status and other information.
Table 3.
Preview of candidate germline SNPs for sample CRCTumorOA1
| #Chrom | POS | ID | Ref | Alt | Qual | Filter |
|---|---|---|---|---|---|---|
| chr1 | 14522 | . | G | A | 36.9516 | . |
| chr1 | 14542 | . | A | G | 4.0757 | . |
| chr1 | 14653 | . | C | T | 57.0491 | . |
| chr1 | 54714 | . | ttct | tt | 4.41898 | . |
| chr1 | 92750 | . | G | A | 6.98519 | . |
This is a preview of file [SampleName].samtools.genome.sorted.vcf generated by SAMtools, containing candidate germline SNPs in sample CRCTumorOA1. Here the first five genomic positions along Chromosome 1 of the reference genome hg38 are shown with the reference and alternate alleles, quality score, filter status and other information.
Merge candidate lists for variants across multiple tumor samples
To allow variant analysis across multiple tumor samples, we need to merge the candidate calls across the samples. These raw candidate calls were generated by MuTect and SAMtools in steps 8 and 9, respectively. We use a custom algorithm for bringing multiple lists of candidate variant calls from all samples into unified tables (ASCII text) for SSNVs and germline SNPs, separately. This algorithm is coded in the following perl file mergeMut.pl (found on ith.Variant). It combines all VCF files (i.e., variant calls) into one master table, containing genomic regions by samples with the table’s cells populated by delimited features. The variant annotations derived from ANNOVAR27 are incorporated in these tables as well. The merged files for SSNVs and germline SNPs can be found in the root directory.
-
10.Run the following command to merge the candidate lists.
-
a.--samples can be set to one sample. This sample is any one of the tumor samples listed in your file somaticInfo.O.
-
a.
$ perl/path/to/ith.Variant/pipeline/exome/submit_slurm.pl
--root/path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step3_mergeCalls.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsv
--samples OA
Note: The output files can be found in the parent directory. Previews of both merged candidate SSNV and germline SNPS are shown (Tables 4 and 5, respectively).
Table 4.
Unified table of candidate SSNVs across all samples
| #Chr | POS | ID | Ref | Alt | CRCTumorOA1 | CRCTumorOA2 |
|---|---|---|---|---|---|---|
| 1 | 874301 | rs76484423 | G | A | 0.067|267|39.17623,13.969898 | 0 |
| 1 | 874307 | rs80351405 | G | C | 0.066|274|35.788618,12.168915 | 0 |
| 1 | 936026 | . | C | A | 0 | 0.250|8|6.200563,1.805672 |
| 1 | 938991 | . | C | A | 0 | 0.031|64|4.291779,10.828026 |
| 1 | 944091 | . | C | A | 0 | 0.035|57|4.393723,6.919398 |
The output file is named mutect.snv.table.annotated, and it contains unified calls generated by MuTect and their annotations.
Table 5.
Unified table of candidate germline SNPs across all samples
| #Chr | POS | ID | Ref | Alt | CRCTumorOA1 | CRCTumorOA2 | CRCTumorOA3 |
|---|---|---|---|---|---|---|---|
| 1 | 822944 | rs3131955 | T | C | 0 | 1.000|178.743|1/1 | 0 |
| 1 | 899452 | rs4411087 | G | C | 0 | 1.000|999|1/1 | 0 |
| 1 | 903510 | rs28437697 | A | G | 0 | 0.800|123.245|1/1 | 0 |
| 1 | 904081 | rs78238606 | T | C | 0 | 0.667|136.211|1/1 | 0 |
| 1 | 904115 | rs113341842 | G | T | 0 | 0 | 0 |
The output file is named samtools.snv.table.annotated, and it contains unified calls generated by SAMtools and their annotations.
Extract the mapping features of the candidate calls
After collecting the candidate SSNV and germline SNPs into two separate lists, this step will extract the mapping features surrounding each candidate variant along the sample genomes. This is performed by checking the sorted BAM file for the sample. The feature extraction will be performed on all samples including normal samples, generating files with the suffix “rechecked” (Figure 4). This will prepare a feature table of the candidate SSNVs and SNPs for each sample, which will be placed under the 04_SNV folder. These mapping features will be used for the variant filtration and final variant calling.
-
11.
Extract the mapping features for the candidate SSNVs. The Slurm script indicated by the parameter --workhard reflects the name of the MuTect algorithm that had generated the calls.
$ perl /path/to/ith.Variant/pipeline/exome/submit_slurm.pl
--root/path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step4_extractFeatures_muTect.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsv
--samplesOA,OB,CRCTumorOA1,CRCTumorOA2,CRCTumorOA3,CRCTumorOA4,CRCTumorOB1,ON,ONs
Note: The SSNV features for sample CRCTumorOA1 are shown (Table 6).
-
12.
Extract the mapping features for the candidate germline SNPs. The Slurm script indicated by the parameter --workhard includes the name SAMtools.
$ perl /path/to/ith.Variant/pipeline/exome/submit_slurm.pl
--root/path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step5_extractFeatures_samtools.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsv
--samplesOA,OB,CRCTumorOA1,CRCTumorOA2,CRCTumorOA3,CRCTumorOA4,CRCTumorOB1,ON,ONs
Note: The germline SNP features for sample CRCTumorOA1 are shown (Table 7).
Figure 4.

Generated merged tumor samples and extracted mapping features
The files with suffix “rechecked” for CRCTumorOA1, containing the mapping features from SSNV and SNP candidate calls, respectively. The output is in the 04_SNV directory.
Table 6.
Preview of the mapping features for candidate SSNV calls for sample CRCTumorOA1
| 1 | 874301 | 326 | 144 | 182 | 163 | 137 | 2 | 2 | 29 | 17 | 12 |
| 1 | 874307 | 327 | 144 | 183 | 160 | 139 | 2 | 2 | 29 | 0 | 0 |
| 1 | 936026 | 9 | 2 | 7 | 7 | 1 | 0 | 0 | 0 | 0 | 0 |
| 1 | 938991 | 7 | 7 | 0 | 4 | 3 | 0 | 0 | 0 | 0 | 0 |
| 1 | 944091 | 237 | 126 | 111 | 116 | 120 | 0 | 1 | 1 | 0 | 1 |
Table 7.
Preview of the mapping features for candidate germline SNP calls for sample CRCTumorOA1
| 1 | 822944 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 899452 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 903510 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 904081 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 |
| 1 | 904115 | 3 | 2 | 1 | 1 | 2 | 1 | 2 | 3 | 0 | 0 |
Perform the final variant calling for SSNVs and SNPs
Using the merged calls and extracted mapping features, ith.Variant performs the final calling of SSNVs and SNPs across the samples.
-
13.
Generate the file gloh_x.tsv of the complete set of homozygous SNPs on the X chromosome. Run the following command to create the file.
$ perl /path/to/ith.Variant/bin/gloh_x.pl
/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
/path/to/CRC_singleGland_pipeline_run/gloh_x.tsv
Note: This step is necessary when there is no heterozygous chromosome in the case of analyzing a tumor sample from a male patient (such as Patient O). The file is shown (Table 8).
-
14.Implement the final point mutation calling. Run the following command to create the final list of SSNVs and SNPs.
-
a.Pass the gloh_x.tsv file as the argument --xloh.
-
a.
$ perl /path/to/ith.Variant/pipeline/exome/submit_slurm.pl
--root/path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step6_varSummary.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsv
--samples OA
--xloh/path/to/CRC_singleGland_pipeline_run/gloh_x.tsv
Note: The table of final SSNV and SNP calls will be placed under the user’s parent directory as MuTect and SAMtools, respectively. The latter will be simultaneously used to generate the directory titan, which will contain prerequisite files for the next step involving the algorithm TitanCNA. The final SSNV and final germline SNP mutations are shown (Tables 9 and 10, respectively).
Table 8.
Table of germline heterozygous SNPs for sample CRCTumorOA1
| Chr | POS | Ref | refCount | Alt | altCount |
|---|---|---|---|---|---|
| 1 | 942934 | G | 3 | C | 3 |
| 1 | 946247 | G | 35 | A | 28 |
| 1 | 948711 | C | 21 | G | 13 |
| 1 | 961945 | G | 92 | C | 69 |
| 1 | 962261 | C | 9 | T | 9 |
Here is the prerequisite table of germline heterozygous SNPs for sample CRCTumorOA1. This was generated in tandem with the final SSNV and germline SNP calling in step 14 and is used in the final SCNA calling.
Table 9.
Final SSNV calls
| Chr | POS | Link | ID | Ref | Alt | CRCTumorOA1maf | CRCTumorOA1d |
|---|---|---|---|---|---|---|---|
| 1 | 972310 | UCSC | rs750272925 | C | T | 0.45 | 60 |
| 1 | 1183933 | UCSC | rs761791859 | C | A | 0 | 248 |
| 1 | 1323274 | UCSC | . | C | A | 0 | 1 |
| 1 | 1339559 | UCSC | rs756210871 | C | T | 0 | 8 |
| 1 | 1395505 | UCSC | . | C | A | 0 | 356 |
Here is the generated mutect.snv.res.filtered.classified.founds.nopara.somatic.table.simplified file under the parent directory. This is a table of the final SSNV calls.
Table 10.
Final post-filtration SNP calls
| Chr | POS | ID | Ref | Alt | CRCTumorOA1 | CRCTumorOA1maf | CRCTumorOA1d |
|---|---|---|---|---|---|---|---|
| 1 | 822944 | rs3131955 | T | C | 0 | 0 | 0 |
| 1 | 899452 | rs4411087 | G | C | 0 | 0 | 0 |
| 1 | 903510 | rs28437697 | A | G | 0 | 0 | 0 |
| 1 | 904081 | rs78238606 | T | C | 0 | 1.0000|1.0000|6,6|1.0000,0,1.00000|0|2.472756|0,1,0,1|0,0|76 | 1 |
| 1 | 906633 | rs7419119 | T | G | 0 | 0 | 0 |
Here is the generated samtools.snv.res.filtered.nopara file under the parent directory. This is a table of the final post-filtration SNP calls also used to generate the prerequisite files for the tool TitanCNA under the directory titan.
Perform the final variant calling for SCNAs
After generating the final list of point mutations, the user performs the final step of calling the SCNAs. We apply the variant caller TitanCNA, a two-factor-HMM-based approach to jointly infer the genomic sequences of segmental copy number alteration (CNA) and loss of heterozygosity (LOH) events as well as their clonality.14 By leveraging an HMM, the approach retains the spatial relationship underlying segmental CNAs of contiguous SNPs along the chromosome. The hidden states are the genotype and clonal membership of CNA and LOH events with the related observable data being the log ratio of tumor-normal read depths and tumor allelic counts. By assuming a mixed population of cells (normal, tumor, and tumor with the CNA or LOH event) in a sample, the method can identify major subclonal SCNAs and improve sensitivity against other existing methods. There are two generated plots of the read-depth ratio of tumor to normal on a normalized logarithmic scale (Log R) and the B-allele frequency (BAF) across the genome, as well as clustering of the copy number states using the two measurements.
-
15.Generate wiggle or depth of coverage files for the matched tumor and normal samples to perform the SCNA calling. The output is found in the directory 03_STATS and has the suffix “wig” (Figures 5A and 5B, respectively).
-
a.Include the normal samples under the parameter --samples to produce the depth of coverage files for both tumor and normal samples, which are required by TitanCNA.
-
a.
$ perl/path/to/ith.Variant/pipeline/exome/submit_slurm.pl --root
/path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step2_stats.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsv
--samplesOA,OB,CRCTumorOA1,CRCTumorOA4,CRCTumorOB1,ON
Optional: If WGS samples were sequenced using different exome targeted sequencing kits from each other, we require the parameter --config be set to the appropriate configuration file which points to the correct target BED file. Here samples CRCTumorOA2, CRCTumorOA3 and ONs require the appropriate change. The command to generate the coverage file for these samples is below.
$ perl/path/to/ith.Variant/pipeline/exome/submit_slurm.pl
--root/path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step2_stats.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_nrce.tsv
--samples CRCTumorOA2,CRCTumorOA3,ONs
-
16.Implement the final calling. Run the following command to create the final list of SCNAs.
- a.
- b.
$ perl /path/to/ith.Variant/pipeline/exome/submit_slurm.pl
--root/path/to/CRC_singleGland_pipeline_run/root/
--workhard/path/to/ith.Variant/pipeline/exome/step7_CNA.slurm
--somaticInfo/path/to/CRC_singleGland_pipeline_run/somaticInfo.O
--config/path/to/ith.Variant/confs/exome/config_hg38_wxs_ss.tsv
--samplesOA,OB,CRCTumorOA1,CRCTumorOA4,CRCTumorOB1
Note: TitanCNA requires input of the full set of germline heterozygous SNP loci identified from the normal sample and the corresponding read depth and allele ratios at these SNP positions from the tumor. Upon completion, the step will generate SCNA calls for one clone and two-subclones solutions (i.e., allowing for up to two subclone fitting of the data) in the form of tabular text files (Tables 11 and 12) and graphical representations of the Log R and BAF across the genome (Figure 6A, upper and lower panels, respectively), as well as their correlation with each other (Figure 6B).
Optional: Here the corresponding configuration file containing the appropriate target BED file is used as the argument --config to process the rest of the tumor samples. The command that passes this argument is below.
$ perl /home/ruping/maese004/ith.Variant/pipeline/exome/submit_slurm.pl
--root/home/ruping/maese004/CRC_singleGland_pipeline_run/root/
--workhard/home/ruping/maese004/ith.Variant/pipeline/exome/step7_CNA.slurm
--somaticInfo/home/ruping/maese004/CRC_singleGland_pipeline_run/somaticInfo.O
--config/home/ruping/maese004/ith.Variant/confs/exome/config_hg38_wxs_nrce.tsv
--samples CRCTumorOA2,CRCTumorOA3
Figure 5.

Prerequisites and final calling for somatic and germline variants
(A) The list of directories under each sample, containing mapped reads, coverage files, and final variant calls.
(B) The contents of the newly made 03_STATS directory include the CRCTumorOA1.wig which is the depth of coverage file for CRCTumorOA1 to be used in the subsequent SCNA calling for the sample (right panel).
Table 11.
Features of the final SCNA calling using default settings for TitanCNA
| Sample | Chromosome | Start_Position.bp. | End_Position.bp. | Length.snp. | Median_Ratio | Median_logR | TITAN_state |
|---|---|---|---|---|---|---|---|
| CRCTumorOA1 | 1 | 946247 | 17694949 | 260 | 0.557127 | 0.317703 | 8 |
| CRCTumorOA1 | 1 | 18481403 | 39084311 | 225 | 1 | −0.649881 | 2 |
| CRCTumorOA1 | 1 | 39284212 | 153390312 | 525 | 0.540816 | 0.298852 | 8 |
| CRCTumorOA1 | 1 | 153537414 | 170552509 | 230 | 1 | 0.242671 | 6 |
| CRCTumorOA1 | 1 | 170958432 | 170958432 | 1 | 0.691824 | 0.469502 | 14 |
Here is a preview of the generated CRCTumorOA1_nclones1.TitanCNA.corrIntCNseg.txt file. It consists of the segmented genome of sample CRCTumorOA1 based on using the original TitanCNA settings for maximum integer copy number. The original value is 8, assuming the normal genome is diploid. Shown are the chromosomal and genomic coordinates for regions along with copy number state and metrics like median Log R value.
Table 12.
Features of the final SCNA calling using modified settings for TitanCNA
| Chrom | loc.start | loc.end | num.mark | seg.mean | copynumber | minor_cn | major_cn |
|---|---|---|---|---|---|---|---|
| 1 | 946247 | 18088176 | 260 | 0.332 | 4 | 2 | 2 |
| 1 | 18088177 | 3918;l4262 | 225 | −0.641 | 2 | 0 | 2 |
| 1 | 39184263 | 153463863 | 525 | 0.318 | 4 | 2 | 2 |
| 1 | 153463864 | 170755470 | 230 | 0.265 | 4 | 0 | 4 |
| 1 | 170755471 | 170958981 | 1 | 0.47 | 6 | 2 | 4 |
Here is a preview of the generated CRCTumorOA1_nclones1.TitanCNA.segments.txt file. It consists of the segmented genome of sample CRCTumorOA1 based on using the corrected settings for maximum integer copy number. It allows estimation of copy number for extremely high gains. Here the corrected integer copy number is set to 14, assuming the genome is haploid with regards to Chromosomes X and Y. Therefore, there are some regions that require correction of chromosomal state.
Figure 6.

Chromosomal events and analysis of the final copy number calls for sample CRCTumorOA1
(A) Plot of log read ratio (upper panel) and allelic ratio (lower panel) for sample CRCTumorOA1. The integer copy number (upper panel) for each genomic region is derived from the normalized log-base-2 of tumor and normal read depths. Here both copy number gains (log ratio greater than 1 in the upper panel) and losses (imbalanced BAF in the lower panel) are rampant. The allelic fraction (lower panel) is the proportion of reads matching the reference genome. Among the observable duplicated regions, Chromosomes 1 and 2 as well as 5, 6, 7 and 8 show evidence of gains on both alleles, retaining a heterozygous allelic copy number profile of 4:2. There is a suspected LOH event at Chromosomes 12 and 15 as the normal copy number is coupled with an uneven allelic fraction. Based on model selection by TitanCNA, the estimates for normal contamination (normC), average tumor ploidy (pl) and clonal cellular prevalence (cellularity) are shown.
(B) Mapping of allelic ratio to copy number profile across the genome for sample CRCTumorOA1. We set the baseline prior (i.e., log ratio of 0) according to the model complexity in explaining the log read ratio and BAF of individual SNPs. For example, on Chromosome 3, if we set the baseline to the log ratio shown, it will lead to an introduction of a subclone to explain the partial allelic imbalance of the chromosome. Similarly, on Chromosome 14, if we set the baseline to a log ratio of 0.25, it is impossible to explain the chromosome’s heterozygous state. Thus we establish a baseline of 0, reflecting a copy number profile of 2 that is holistically reasonable across chromosomes such as the adjacent Chromosome 15 which we can infer has a copy neutral loss of heterozygosity event.
Expected outcomes
The ith.Variant pipeline processes related clinical tumor samples to generate final single-nucleotide variants (SSNVs and SNPs) and somatic copy number alterations (SCNA). Applying a scalable algorithmic approach to variant calling, the user can invoke a series of commands using the command-line interface of a Linux OS on an HPC cluster, passing user-specific parameters that can be adjusted in each step (e.g., the input tumor data) for an individual use-case. The ith.Variant pipeline enhances sensitivity for low-frequency variants while maintaining specificity against artifacts. Accurate variant detection is integral for characterization of ITH and delineation of the clonal events of tumor evolution. By consolidating and filtering the candidate variant calls of multiple related tumor and normal samples, the ith.Variant pipeline generates final variant calls that reflect genomic diversity coupled with tumor evolution. With a more comprehensive profile of the genome, there is greater opportunity for accurate rendering of ITH and reconstructing of tumor phylogeny.
Limitations
As a command-line tool, the ith.Variant pipeline can be easily deployed in a HPC cluster, like a supercomputer as shown here, leveraging high-throughput processing for massive genomic data. If you find your deployment has poor performance, you can scale the amount of designated RAM or threads as needed in each Slurm script. The pipeline takes tumor-normal matched WES/WGS data derived from the same patient across multiple or single regions of the tumor. As our analysis aims to elucidate evolutionary mechanisms of cancer for a tissue type, variant calling across multiple regions is required. Our pipeline is designed for calling variants in DNA sequencing data, offering future opportunities for the adaption of our tool to long-read or RNA sequencing data. Upon finishing the final SCNA calling, the generated visualizations must be manually reviewed in a biological context. This is possible in your remote environment or locally on your PC by transferring the relevant files. Recently, a user-guided copy number clustering method called CNAViz was developed, offering the ease of a graphical-user interface in visualizing the raw Log R and BAF data and manually adjusting the copy number calling. For more details related to this visualization software, please refer to https://github.com/elkebir-group/cnaviz. Overall, ith.Variant is scalable, allowing various sequencing datasets to be reliably processed for variant detection.
Troubleshooting
Problem 1
There is no output and the job fails after running a command from a step in the ith.Variant protocol.
Potential solution
Always check the log file for the corresponding job run under the root directory. The problem may be due to an absent dependency that requires installation or an incorrect reference to the path of a relevant file in the Linux/shell command. Please install the required dependencies manually by referring to the key resources table, and then try running the specific ith.Variant command again. If there are any installation issues, please consult the documentation for each package as well as confirm your current version of R on your Linux distribution.
Problem 2
The Slurm job for running MuTect in the ith.Variant protocol fails due to a runtime error. This will likely result in a notification via email to the specified address in the corresponding Slurm job script.
Potential solution
The incorrect configuration file is being referenced in the command. For example, in steps 8 and 9 the argument --config is config_hg38_wxs_ss.java6.tsv and config_hg38_wxs_ss.tsv, respectively. The former is required in step 8 because it contains the path variable to the Java 6 executables which are the prerequisites for MuTect.
Problem 3
ith.Variant does not generate output when running the final SCNA calling.
Potential solution
The incorrect R package for TitanCNA may be in use. We slightly modified the original TitanCNA code to increase its compatibility with our pipeline. The modified R package for TitanCNA requires an alternative process of installation detailed under the section dependencies.
Problem 4
The generated plot of a logarithmic scale for copy number states over the genome is noisy and/or the CN events of the plot do not match a reasonable biological interpretation.
Potential solution
To generate a clear plot of the Log 2-based genomic copy number profile, it is necessary that the input depth of coverage files reflect high-quality samples. For instance, a poor normal sample will lead to the corresponding tumor samples yielding poor depth of coverage files, impacting the algorithm’s ability to estimate copy number. In addition, if the plot is incohesive with the allelic fraction plot, consider adjusting the initial baseline ploidy and/or the initial normal contamination of the sample. These initial parameters require prior knowledge and can be heuristically determined. The parameters for ploidy and normal contamination are --plpTitan and --ncTitan, respectively, in the Slurm script step7_CNA.slurm. Here we adjusted the ploidy from 2 to 3, generating plots consistent with biological reason.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ruping Sun (ruping@umn.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This study was completed at the invitation of STAR Protocols for a special issue dedicated to genomics. This study uses the computing resources of the Minnesota Supercomputing Institute (MSI). This work was supported by the Department of Laboratory Medicine and Pathology (https://med.umn.edu/pathology) and the Masonic Cancer Center (https://cancer.umn.edu/) at the University of Minnesota. This work was partially supported by the Karen Wyckoff Rein in Sarcoma Foundation (https://www.reininsarcoma.org/). The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Author contributions
Formal analysis, N.M., R.S.; Software, N.M., A.K., R.S.; Writing – original draft, N.M.; Writing – review & editing, N.M., A.K., R.S.; Visualization, N.M.; Supervision, R.S.
Declaration of interests
The authors declare no competing interests.
Data and code availability
The datasets and code generated during this study are available at ith.Variant: https://github.com/SunPathLab/ith.Variant. The public repository with the proper accession numbers for the data used in this study are found in the key resources table.
References
- 1.Sun R., Hu Z., Sottoriva A., Graham T.A., Harpak A., Ma Z., Fischer J.M., Shibata D., Curtis C. Between-region genetic divergence reflects the mode and tempo of tumor evolution. Nat. Genet. 2017;49:1015–1024. doi: 10.1038/ng.3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M., et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sottoriva A., Kang H., Ma Z., Graham T.A., Salomon M.P., Zhao J., Marjoram P., Siegmund K., Press M.F., Shibata D., Curtis C. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turro E., Astle W.J., Megy K., Gräf S., Greene D., Shamardina O., Allen H.L., Sanchis-Juan A., Frontini M., Thys C., et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583:96–102. doi: 10.1038/s41586-020-2434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dou Y., Gold H.D., Luquette L.J., Park P.J. Detecting somatic mutations in normal cells. Trends Genet. 2018;34:545–557. doi: 10.1016/j.tig.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao R., Davis A., McDonald T.O., Sei E., Shi X., Wang Y., Tsai P.-C., Casasent A., Waters J., Zhang H., et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016;48:1119–1130. doi: 10.1038/ng.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin H.-T., Choi Y.-L., Yun J.W., Kim N.K.D., Kim S.-Y., Jeon H.J., Nam J.-Y., Lee C., Ryu D., Kim S.C., et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat. Commun. 2017;8:1377. doi: 10.1038/s41467-017-01470-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerlinger M., Rowan A.J., Horswell S., Math M., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navin N., Kendall J., Troge J., Andrews P., Rodgers L., McIndoo J., Cook K., Stepansky A., Levy D., Esposito D., et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park S.Y., Gönen M., Kim H.J., Michor F., Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J. Clin. Invest. 2010;120:636–644. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ha G., Roth A., Khattra J., Ho J., Yap D., Prentice L.M., Melnyk N., McPherson A., Bashashati A., Laks E., et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014;24:1881–1893. doi: 10.1101/gr.180281.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A., et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding L., Ley T.J., Larson D.E., Miller C.A., Koboldt D.C., Welch J.S., Ritchey J.K., Young M.A., Lamprecht T., McLellan M.D., et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grüning B., Dale R., Sjödin A., Chapman B.A., Rowe J., Tomkins-Tinch C.H., Valieris R., Köster J., Bioconda Team Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods. 2018;15:475–476. doi: 10.1038/s41592-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurtzer G.M., Sochat V., Bauer M.W. Singularity: scientific containers for mobility of compute. PLoS One. 2017;12:e0177459. doi: 10.1371/journal.pone.0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broad Institute . Broad Institute; 2019. Picard Toolkit.https://broadinstitute.github.io/picard/ [Google Scholar]
- 22.Lai D., Ha G., Shah S. Bioconductor; 2022. Copy Number Prediction with Correction for GC and Mappability Bias for HTS Data (1.38.0) [R] [Google Scholar]
- 23.Tuszynski J. CRAN; 2021. Tools: Moving Window Statistics, GIF, Base64, ROC AUC, etc (1.18.2) [R] [Google Scholar]
- 24.Wand M., Moler C., Ripley B. CRAN; 2021. KernSmooth: Functions for Kernel Smoothing Supporting Wand & Jones (1995) (2.23-20) [Google Scholar]
- 25.Daniel F., Weston S., Revolution Analytics . CRAN; 2022. Foreach Parallel Adaptor for “Parallel” (1.3.8) [R] [Google Scholar]
- 26.Neuwirth E. CRAN; 2022. ColorBrewer Palettes (1.1-3) [R] [Google Scholar]
- 27.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabó B. CPAN; 2018. Parallel::ForkManager—A Simple Parallel Processing Fork Manager (2.02) [Perl] [Google Scholar]
- 29.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mouse Genome Sequencing Consortium. Waterston R.H., Lindblad-Toh K., Birney E., Rogers J., Abril J.F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets and code generated during this study are available at ith.Variant: https://github.com/SunPathLab/ith.Variant. The public repository with the proper accession numbers for the data used in this study are found in the key resources table.