

Abstract
Background
Malignant gliomas, the most common malignant brain tumors in adults, represent a heterogeneous group of diseases with poor prognosis. Retroviruses can cause permanent genetic alterations that modify genes close to the viral integration site.
Methods
Here we describe the use of a high-throughput pipeline coupled to the commonly used tissue-specific retroviral RCAS-TVA mouse tumor model system. Utilizing next-generation sequencing, we show that retroviral integration sites can be reproducibly detected in malignant stem cell lines generated from RCAS-PDGFB-driven glioma biopsies.
Results
A large fraction of common integration sites contained genes that have been dysregulated or misexpressed in glioma. Others overlapped with loci identified in previous glioma-related forward genetic screens, but several novel putative cancer-causing genes were also found. Integrating retroviral tagging and clinical data, Ppfibp1 was highlighted as a frequently tagged novel glioma-causing gene. Retroviral integrations into the locus resulted in Ppfibp1 upregulation, and Ppfibp1-tagged cells generated tumors with shorter latency on orthotopic transplantation. In human gliomas, increased PPFIBP1 expression was significantly linked to poor prognosis and PDGF treatment resistance.
Conclusions
Altogether, the current study has demonstrated a novel approach to tagging glioma genes via forward genetics, validating previous results, and identifying PPFIBP1 as a putative oncogene in gliomagenesis.
Keywords: forward genetics screen, glioblastoma, liprin-beta-1, PDGFB, RCAS
Key Points.
Next-generation sequencing of hybridized probes can annotate RCAS virus integrations.
PDGF collaborates with tagged genes to promote tissue-specific glioma progression.
PPFIBP1 tagged by RCAS viruses promotes GBM malignancy and PDGF treatment resistance.
Importance of the Study.
Malignant gliomas are common primary brain tumors with high morbidity and mortality. Their intertumoral differences suggest they arise from distinct cells of origin and their strong resistance to standard therapy proposes a large variety of potential driver genes. Platelet-derived growth factor (PDGF) signaling is frequently upregulated in gliomas but PDGF-targeted therapies have not yet proven successful in clinical trials. Here we use a retroviral forward genetic screen with a high-throughput sequencing approach to identify genes potentially collaborating in PDGF-driven glioma initiated from tissue-specific promoters in mice. The screen identified putative cancer genes that correlate with poor prognosis in glioma patients with elevated PDGF pathway signatures but also genes that are glioma specific or that define distinct brain cell types.
Gliomas comprise a broad group of diseases that display a wide range of genetic alterations and pathological phenotypes. Classified as grade IV astrocytoma, the highest malignancy grade among gliomas, Glioblastoma (GBM) represents the most frequent and also most aggressive type of glioma.1
Based on transcriptional profiling, GBM has been further divided into three distinct molecular subtypes, referred to as proneural (PN), classical (CL), and mesenchymal (MS).2 Some of the most commonly mutated genes across all GBM subtypes include tumor suppressor genes like PTEN, TP53, and RB1,3 but also receptor tyrosine kinases such as EGFR and PDGFRA. PGDFRA amplification can be found in all GBM subtypes but is most abundant within the PN subtype. In addition, PDGFRA activation by PDGF ligands (A–C) is an early event in the GBM initiation,4 while PDGF ligands B and D can also stimulate PDGFRB. Autocrine stimulation with the PDGFB-chain can induce murine gliomas morphologically resembling human GBM.5 Furthermore, PDGFRA activation in distinct cell types like astrocytes or oligodendrocyte precursor cells creates a pool of glioma initiating cells.6,7
Yet, despite extensive research into the underlying tumor biology, GBM remains still essentially incurable, with current treatment options, including combinations of surgery, radio-, and chemotherapy, leaving patients only with a dismal <2-year median survival prognosis, prompting the exploration of alternative therapeutic avenues such as targeted therapy.8 However, personalized medicine based on target cancer genes in turn requires knowledge about the individual cancer genes and their interacting roles in tumor development.
One valuable group of methods for detecting collaborating cancer genes, beyond the molecular profiling of GBM biopsies from patients, is referred to as forward genetic screens.9 Particularly, the technique is based on the use of mutagenic agents, such as retroviruses10,11 or transposon systems,12 which allow the overexpression of a particular oncogene in a host cell, while at the same time causing insertional mutations through integration into the host’s genome. Mutations that are beneficial to tumor development are then expected to be enriched due to the growth advantage conveyed to the respective host cells, thus enabling the identification, ie tagging, of putative cancer genes as those genomic loci hit more frequently than expected by chance.13
Utilizing a recombinant PDGFB-encoding Moloney murine leukemia virus (MMLV) to establish a mouse glioma model, we previously demonstrated the usefulness of the forward genetic screen for tagging putative glioma-related genes.10,14 However, as different families of retroviruses might display diverging insertional preferences,15 it appears relevant to validate the results of such an effort via another mutagenic agent. Furthermore, many early studies, including our own screen, identified the retroviral integration sites in the host genome via comparatively cumbersome and less high-throughput workflows involving eg PCR-based protocols for sequence enrichment/amplification followed by directed/Sanger sequencing, and finally a BLASTn run to map the identified sequences.10,16–18 Yet, with the increasing availability of next-generation sequencing (NGS) techniques, much more high-throughput and automated strategies of annotating such genomic loci have become possible.19
Accordingly, the current study aimed to further advance our previous forward genetic screen in glioma by studying retroviral tagging using a different retrovirus, ie, the replication-competent Avian sarcoma-leukosis virus (ASLV) long terminal repeat (LTR) with a Splice acceptor (RCAS). The RCAS/TVA model is a frequently used genetically engineered model systems developed to follow tumor development in mice.20 The system is particularly useful in delineating functional drivers of malignant brain cancer from tissue-specific promoters active in neural,21 astrocytic,21 and oligodendrocyte progenitors.22 To the best of our knowledge, RCAS has not yet been extensively studied as a mutagenic agent in forward genetic screens. In addition, while the previous MMLV-PDGFB-based forward genetic screen was tissue-unspecific, the RCAS-based system is allowing the targeting of distinct cell types during glioma development,23 thus enabling us to compare tissue-specific integration preferences. Finally, the current study presents a novel pipeline for detecting and processing the tagged genomic loci using NGS methods. In summary, the results of this study provide an independent validation of our previous forward genetics screen and a more robust and streamlined approach to detecting candidate glioma genes collaborating with PDGFB.
Methods
A flowchart of the overall procedure pursued in the current study is depicted in Figure 1, the individual steps of which will be briefly described below. For a more detailed account of the utilized materials and methods, the reader is referred to the Supplementary Material. A list of primers used for different validation steps can be found in Supplementary Tables 3 and 4.
Fig. 1.

Flowchart depicting the methodological strategy employed in the current study. (A) Mouse gliomas were induced by PDGFB using RCAS/tv-a brain tumor models, after which cell lines are established and genomic DNA is extracted. (B) DNA fragments including the proviral sequence are pulled down using a hybridization probe capture protocol, sequenced, and mapped against the mouse genome. (C) Putative retroviral integration sites are detected as characteristic alignment peaks of softclipped reads. (D) The preliminary list of integration sites is filtered to remove potential false positive hits. (E) The integration direction of each retroviral insertion site is determined by analyzing the alignment of the softclipped read parts against the proviral sequence. (F) Integration sites are annotated against RefSeq genes. (G) Tagged, putative cancer genes are detected as common insertion sites across cell lines. (H) Tagged genes and loci are characterized in vitro and evaluated in the context of human GBM patient clinical data.
Animal Experiments and Tumor Cell Lines
All animal experiments were conducted according to the Swedish Animal Welfare Act and were approved by the Uppsala Animal Research Board (ethical permit C114/13). Methods used were in accordance with standards set with national guidelines. Mouse gliomas were induced via an RCAS-based system encoding for PDGFB5,7,23–25 or from glioma cell lines already established from biopsies of generated brain tumors as previously described.7,23–27 Genomic DNA was extracted as described in the Supplementary Material and used in NGS (Figure 1A).
Hybridization-Based Capture and Sequencing
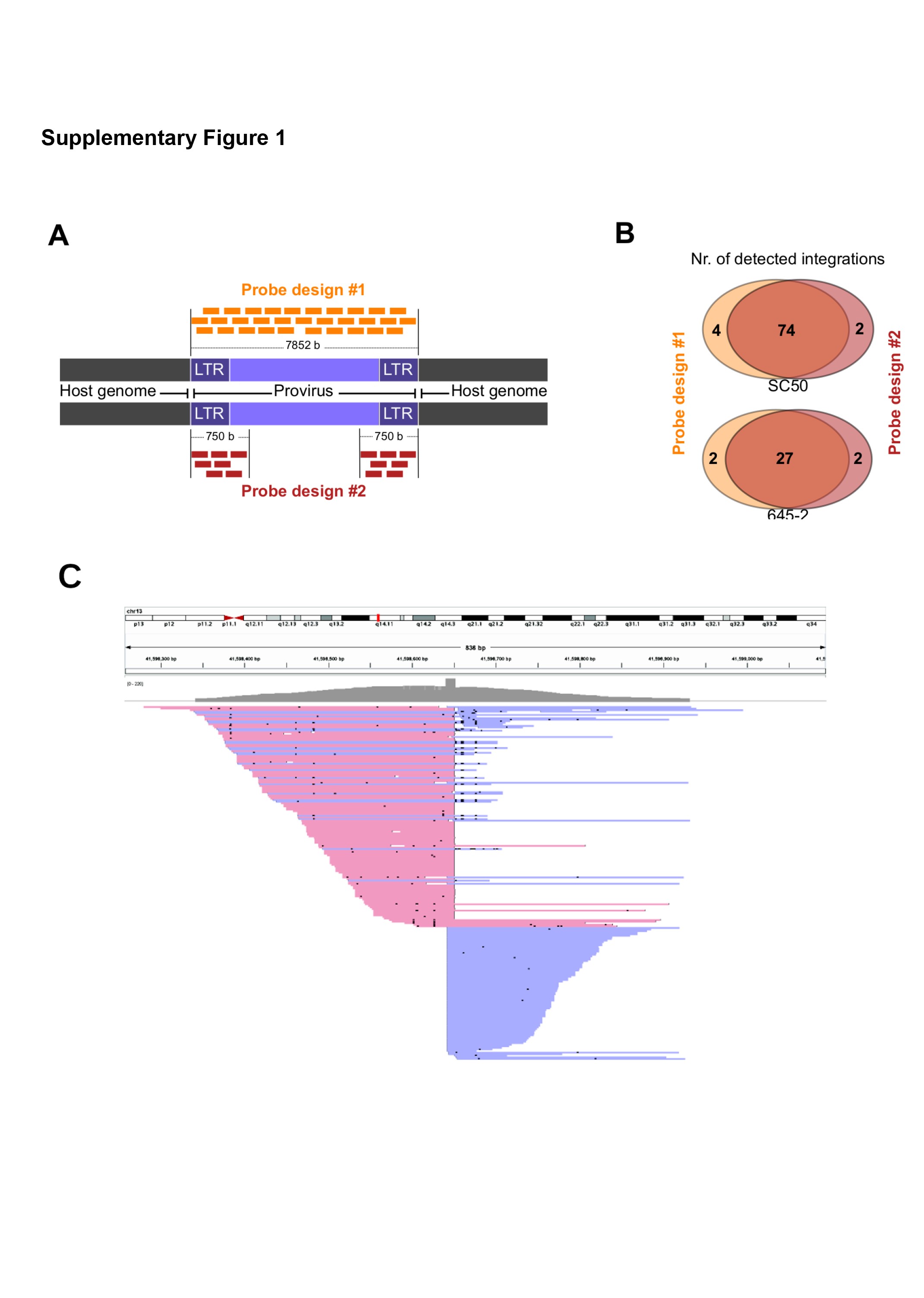
A hybridization-based capture protocol was employed, utilizing probes specific to the RCAS proviral sequence to pull down DNA fragments including only the integrand or integrand-host junctions. NGS of this enriched pool of DNA fragments followed by an alignment to the mouse genome would then produce a list of softclipped reads, ie reads with one part mapping to the mouse genome, while the remainder is unmapped, ie potentially mapping to the proviral sequence (Figure 1B). Two different probe designs were employed, ie an initial design targeting the entire proviral sequence and a later design targeting only the start and end of the sequence including the LTR sequences (Supplementary Figure 1A). As the two designs produced highly comparable downstream results (Supplementary Figure 1B), the data from both were utilized equivalently throughout the study.
Integration Site DetectoR (InSiDeR)
Assuming a mapping result with softclipped reads, a viral integration site (IS) is expected to produce a very specific alignment pattern, ie a peak consisting of reads that are all softclipped at the position where the virus is integrated into the genome (Figure 1C, Supplementary Figure 1C). To identify ISs, we developed a Perl program (InSiDeR; Integration Site DetectoR; https://github.com/UppsalaGenomeCenter/InSiDeR) that extracted the genomic coordinates at which such alignment peaks occurred in the sequencing results. InSiDeR works both for short-read and long-read NGS data and has previously been used also for detection of CRISPR-Cas9 off-target cleavage sites.28
Removal of Putative False-Positive Integration Sites
After the initial identification of putative ISs, further mapping analyses against both mouse genome and RCAS proviral sequence were performed to filter potential false positives (Figure 1D). Reads originating from a true IS would be expected to exhibit a part that unambiguously maps to the mouse genome and a softclipped sequence that consistently maps against the proviral sequence. The initial list of reads contained numerous cases in which the mouse-mapping sequence was comparatively short and displayed a high mapping score to the RCAS sequence (Supplementary Figure 2B), suggesting putative false positives (Supplementary Figure 2B and C). Conversely, in other cases, only a small fraction of the softclipped sequence was mapped to the RCAS sequence (Supplementary Figure 2D and E), representing other putative false positives.
Detection of Integration Direction
In retroviruses, the 5′ and 3′ LTRs have been shown to exhibit differences in their enhancer/promoter functions,29 and thus the orientation of the retroviral integration into the host genome has been considered an important factor determining how gene transcription can be altered.11,13,30 Specifically, to exert a promoter activation on a gene, the insertion must be located upstream and in sense orientation, while enhancer activations are usually considered to occur from antisense integrations upstream or sense integrations downstream of genes.11
To determine the direction for each previously identified IS, we followed the rationale proposed by Sarver et al.31 Specifically, knowing from which site of an integration each softclipped read originated, based on the alignment against the genome, the direction was determined by requiring that the softclipped sequences consistently mapped against either the forward or the reverse strand of the proviral sequence (Figure 1E, Supplementary Figure 2E). Of note, for some integration sites the integration direction could not be determined, as the associated pool of reads displayed inconsistent alignment orientations (Supplementary Figure 2E), typically with softclipped sequences from one site of the integration showing a different orientation than sequences from the other site. In some of these cases, the entire pool of reads displayed some junctions of RCAS sequences with opposite strand orientations, suggesting the potential presence of tandem integrations (Supplementary Figure 2F).
Integration Site Annotation
All individual ISs were annotated against a list of RefSeq genes (Figure 1F). Specifically, integrations were either (i) annotated with all genes with transcriptional start sites (TSSs) at most 100 kb from the IS (to inspect the integration environment), (ii) with the single gene with the most proximate TSS (in order to study retroviral integration location preferences), or (iii) with any gene directly hit by the IS or with a TSS or transcriptional end at most 50 kb distant from the IS (for extracting IS-related genes).
Detection of Common Insertion Sites
Putative glioma loci were identified as common insertion sites (CISs), ie genomic regions that harbored more integration sites across different cell lines than expected by random chance (Figure 1G). For sake of comparison with our previous forward genetic screen, we used the same Monte Carlo-based definition of a CIS, requiring two integrations within a 30 kb genomic window, three integrations within a 50 kb window, or at least four integrations inside a 100 kb window.10,18 The obtained RCAS-PDGFB CISs were compared to the CIS landscapes of various other forward genetic screens, including our previous MMLV-PDGFB system,10 utilizing raw integration coordinates from the RTCGD database32 and an integrative approach (Supplementary Figure 3) previously described.33
Clinical and Functional Characterization of Selected Loci
To rank/score CISs prior to functional validation, various clinical and molecular aspects of the tagged target genes were inspected in the context of a human GBM dataset from The Cancer Genome Atlas (TCGA) (Figure 1H). Specifically, for each tagged gene that could be mapped to a human ortholog, analyses were conducted to estimate (i) correlation of gene expression values with PDGFB, PDGFRA, or PDGFRB, within or across GBM subtypes, (ii) differential gene expression between GBM subtypes, (iii) overall survival difference between patients with high and low expression of the tagged gene, either across all samples or within PDGFB-high, PDGFRA-high, or PDGFRB-high groups, respectively, and (iv) frequencies of mutations, amplifications, and deletions of the tagged gene in GBM patients.
Results
Establishment of Cell Lines From PDGFB-Driven Gliomas
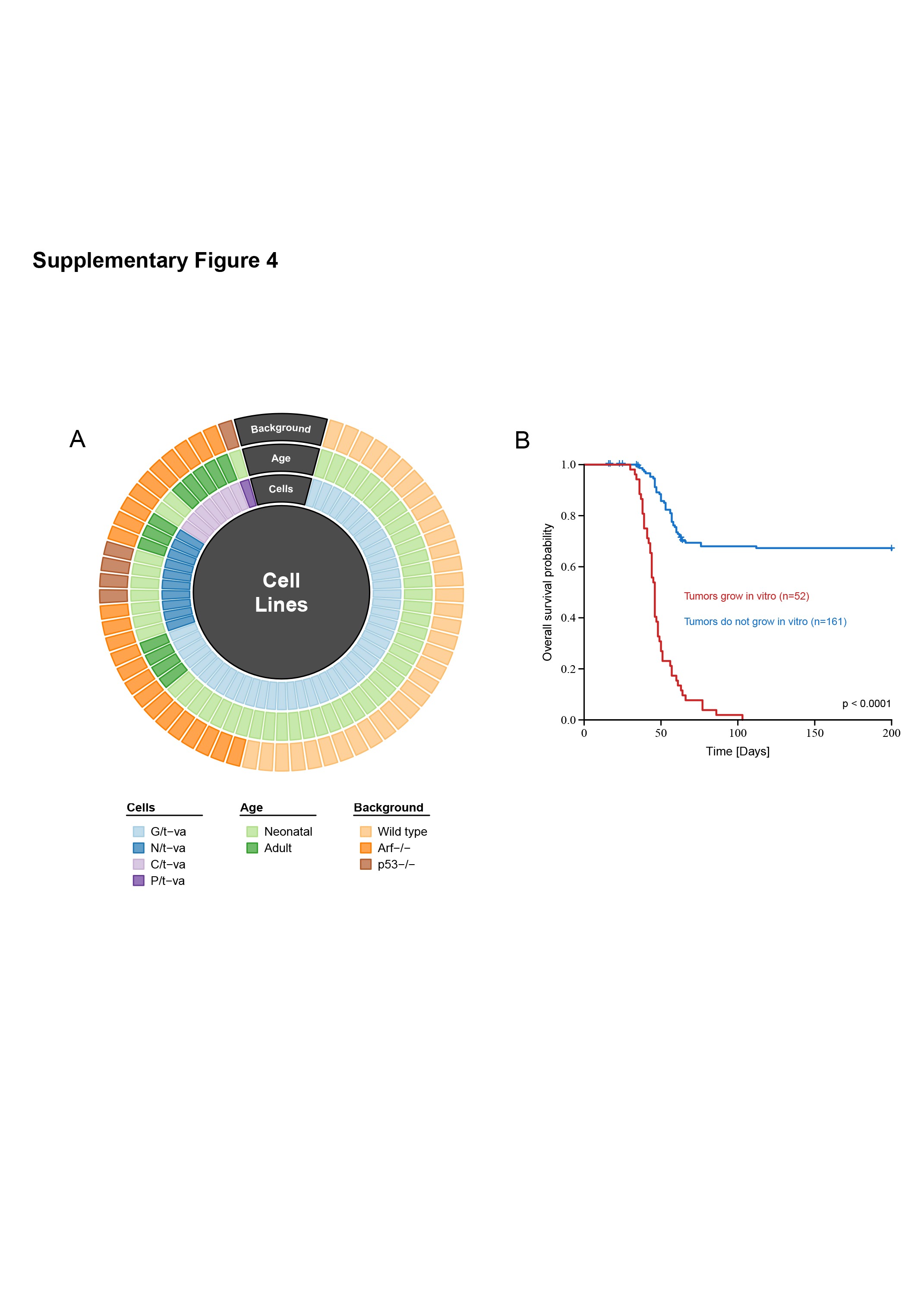
Mouse gliomas were induced in transgenic animals through a cell type-specific oncogene transfer using the RCAS-PDGFB system. By expressing the avian retroviral receptor tumor virus A (tv-a) under the respective transgene, malignant tumor transformation was driven from GFAP expressing cells (G/tv-a),6,34 Nestin expressing cells (N/tv-a),6,35 CNPase expressing cells (C/tv-a),7,24 or Pax-3 expressing cells (P/tv-a)26 (Supplementary Figure 4A). Beyond the particular transgene, tumor models also differed in age (neonatal or adult) and biological background (wild type, Ink4A/Arf deficient, or p53 deficient) of the mice (Supplementary Figure 4A). There was a clear link between the aggressiveness of the tumors and the ability to derive cell lines from biopsies of such tumors. Specifically, the tumors that could be cultured in glioma stem cell conditions7,27 to produce a glioma cell line typically also exhibited a shorter latency in mice as compared to the tumors that did not grow in vitro (Supplementary Figure 4B).
Characterization of RCAS Integration Patterns
The described targeted whole-genome sequencing pipeline provided not only a highly automated solution for identifying retroviral integration sites but also allowed the characterization of the RCAS integration pattern.
After removal of putative false positives, 3418 insertion sites were retained, of which 938 were found across the 63 cell lines and 2480 were identified across 14 biopsies. Thus, when compared to the tumor biopsies, the tumor-derived cell lines showed on average significantly fewer insertion sites (Figure 2A), suggesting that only the most malignant clones are able to grow in vitro, while many retrovirally induced passenger mutations might have been lost. In addition, the number of proviral integrations found in the mouse genome correlated with PDGFB levels (Figure 2B). When compared between the different transgenes, mouse age groups, and genetic backgrounds (Supplementary Figure 5A–C), only the division by background corresponded to a significant (P < .05) difference in integration numbers among cell lines, with the WT lines receiving significantly fewer integrations than the Arf−/− and p53−/− lines. Since there was a strong overrepresentation of WT cases among the G/tv-a and newborn samples, another comparison was conducted including only Arf−/− and p53−/− cell lines (Supplementary Figure 5D and E), which instead suggested even a significant difference in integration numbers between transgenes (significantly more integrations in G/tv-a as compared to C/tv-a) but not between mouse age groups.
Fig. 2.

Characterization of RCAS integration patterns. (A) Strip chart displaying the number of integration sites detected per cell line or tumor biopsy. Solid black lines indicate median numbers. The P-value indicates the result of a two-tailed Wilcoxon rank sum test. (B) Scatterplot illustrating the expression of PDGFB (normalized fold change relative to Gapdh) with respect to the number of RCAS-PDGFB integrations detected in that cell line. (C) Heatmap depicting the number/percentages of integrations found within a certain vicinity (y-axis; integrations falling inside an intron/exon were considered to have a distance of 0 b) of the TSSs of a certain number of genes (x-axis). (D) Histogram displaying the number of integration sites occurring with a certain distance to the closest TSS. (E) Pie chart illustrating the distribution of integrations across introns and exons, within the promoter (<20 kbp), or further upstream/downstream of the gene, with each IS coordinate considered relative only to the single gene annotation that exhibits the TSS closest to the IS. (F) Barplot depicting the percentages of integrations with forward and reverse orientation for each combination of gene orientation and relative IS location. Only one comparison was significant at P < .05 (indicated with P-value, Fisher’s exact test).
Gene set overlap analyses also indicated that RCAS integrations might target different groups of genes in the various transgene models (Supplementary Figure 5G–I). Consistent with the previously published observation that RCAS-PDGFB-induced gliomas driven from G/tv-a cells appeared more stem cell-like and self-renewing as compared to the more differentiated N/tv-a and C/tv-a models,23 IS-related genes in the G/tv-a model showed an enrichment of stem cell signatures (Supplementary Figure 5G and J), while IS-associated genes in the N/tv-a and C/tv-a models displayed an enrichment of more differentiated, astrocyte-, glial-, and oligodendrocyte-related gene sets (Supplementary Figure 5H–J). When combining the models in a single gene ontology (GO) analysis, the IS-related genes demonstrated a clear enrichment of (neural) developmental pathways (Supplementary Table 1).
A large fraction (~46%) of all integration sites lay within 5 kb distance of the TSS (or directly within the intron/exon) of just a single gene, and even when inspecting distances of up to 100 kb around ISs, the majority of insertions still resided only in the vicinity of a single gene (Figure 2C). In fact, ISs displayed a clear tendency to occur predominantly in close proximity to TSSs (Figure 2D), which is reminiscent of the integration preferences of other retroviruses such as MLV.15 When considering only the relative position to the gene with the closest TSS, a majority (~34%) of integration sites fell into the intron regions of genes, while virtually no integrations occurred in exons (Figure 2E). In fact, when considered not only with respect to the most proximate TSS, 42% of all ISs fell into at least one intron region of a gene, while 13% of all ISs were found in the first intron of a gene (Supplementary Figure 5F).
Finally, no clear connection between the orientation of the closest TSS and the location and integration direction of the RCAS integration sites was observed (Figure 2F), suggesting no clear preference between promoter and enhancer activities among ISs.
CIS-tagging Identifies Glioma-Related Genes
When comparing the charted RCAS-PDGFB integration landscape to various other forward genetic screens, the by far greatest similarity, significantly higher than that with other models, was observed with the RCAS-MMLV landscape (Figure 3A, Supplementary Figure 7A), validating our previous screen. In contrast, the comparison with a Sleeping Beauty (SB) transposon-driven glioma model36 did reveal a comparatively low number of shared CIS (Figure 3A, Supplementary Figure 7A). However, an inspection of individual CISs between the models suggested that similar members in the same gene families were targeted (eg Pdgfc/Pdgfa, Ppfibp2/Ppfibp1, and Nfia/Nfix). In order to further inspect this possibility, we conducted another GO analysis, which indicated that similar to the RCAS-PDGFB (Supplementary Table 1), the SB integrations (Supplementary Table 2) targeted (neural) developmental pathways. Ultimately, both approaches likely lead to a tagging of similar brain/glioma-related gene families/pathways.
Fig. 3.

Characterization of RCAS-PDGFB CISs. (A) Box-and-Whisker plot comparing the number of shared CISs between the RCAS-PDGFB model and six other forward genetic screens. Each Box-and-Whisker object represents 100 values, each obtained by randomly selecting 100 insertion sites from the respective model and computing the corresponding number of shared CISs upon integration with all 938 RCAS-PDGFB insertions. (B) Circos plot visualizing for each identified CIS between RCAS-PDGFB and MMLV-PDGFB the number of ISs in the RCAS and MMLV screens, respectively, and the number of genes in the vicinity. Only CISs with at least one IS in the current RCAS-PDGFB screen were considered. (C) Molecular and clinical characterization of human orthologs for 13 top candidate genes from (B) using GBM data. The displayed correlations indicate Pearson’s correlation coefficient of the tagged gene expression with PDGFB, PDGFRA, or PDGFRB, respectively, with the value representing the maximum absolute value observed when computing the correlation either among all patients or within each subtype separately. The AOV column indicates the P-value of an ANOVA of the gene expression values between GBM subtypes. The OS columns indicate the log-rank P-value and direction of a survival difference between samples with high and low expression of the tagged gene, either across all samples or within PDGFB-high, PDGFRA-high, or PDGFRB-high subsets. See also Supplementary Figures 7–11. Genomic alteration data was obtained from the GBM dataset in the TCGA portal (from 421 patient samples) and visualized frequencies were calculated as described in the Supplementary Material under GBM Patient Data.
In the integrated RCAS-MMLV landscape, a total of 61 CISs with at least one IS in the RCAS-PDGFB screen were identified, six of which constituted CISs that were identified independently in the two studies (Figure 3B, Supplementary Table 5). Furthermore, additional ISs provided by the novel screen allowed the identification of 27 CISs previously tagged by single ISs in the MMLV-PDGFB screen, while 21 CISs were also located in entirely novel loci (Figure 3B). In total, 142 genes were found in the vicinity of these 61 CISs (Figure 3B), some of which comprised known glioma-related genes such as Pdgfa,37Pik3ca,38Sox10,39 and Pick1.40 Furthermore, in the CIS-ID 61 with most (7) integrations, the ISs were found closely downstream of a known tumor-suppressive miR-29a/b1 cluster (Supplementary Figure 8E) that are often suppressed by other oncogenes. Strikingly, mir-29a has previously been identified as a regulator of competing endogenous RNAs in PDGFRA signaling in GBM38 and was recently found to specifically target and down-regulate PDGF ligands in GBMs by binding to their 3′UTR regions.41 The observed abundance of known glioma-related genes in our CISs constitutes a proof-of-concept of the power of this type of forward genetic screen in tagging glioma-related genes. In addition, when subjecting a subset of the CIS-contributing cell lines to an experimental validation, the presence of the ISs could be confirmed in almost all (37/40) cases (Supplementary Figure 6).
Out of the 142 CIS-tagged genes, clinical and molecular data were available for 76 human orthologs, allowing a further characterization (Figure 3C, Supplementary Figure 7). None of the genes exhibited a strong (R ≥ 0.5) gene expression correlation with PDGFB, PDGFRA, or PDGFRB, while most genes displayed a significant differential expression between subtypes, rendering these analyses obsolete as a way to score CISs (Figure 3C, Supplementary Figure 7). Yet, the expression of numerous genes also showed clear prognostic survival effects and/or increased mutation/amplification/deletion frequencies (Figure 3C, Supplementary Figure 7, Supplementary Figures 9–11). Ultimately, focusing on a combination of how strong the tagging and how clinically relevant a gene was, 13 top candidate genes were selected, which exhibited either (i) a very strong (P < .001) overall survival difference between high and low expressing GBM samples, or (ii) at least three ISs in the corresponding CIS coupled to a significant (P < .05) survival difference (Figure 3C).
Ppfibp1 Constitutes a Glioma Oncogene
Of the aforementioned candidate genes, PPFIBP1 was of particular interest due to its clinical properties and the strength of support from retroviral tagging. Specifically, retroviral tagging of PPFIBP1 appeared rather robust and unambiguous, with the locus having been identified as an independent CIS in both the RCAS- and MMLV-driven models (Figure 3B), and all CIS-related ISs falling within introns of the gene (Supplementary Figure 8F). In addition, among the top candidate genes, PPFIBP1 also displayed the strongest association between high expression and worse patient outcome in the PDGFB-high group (Figure 3C, Supplementary Figure 11F), which was of particular interest as the model is driven through PGDFB overexpression.
When comparing murine glioma cell lines with or without viral integrations in this locus, IS-harboring cell lines displayed a significantly increased Ppfibp1 gene expression (Figure 4A) and increased protein levels (Figure 4B and C). These findings are consistent with the results from our previous study showing drastically higher levels of Ppfibp1 in MMLV-PDGFB induced tumors compared to neonatal brains.14
Fig. 4.

PPFIBP1 represents a putative glioma oncogene. (A) Barplot comparing the Ppfibp1 gene expression between murine glioma lines with (purple; P1–P2) and without (gray; N1–N4) RCAS-PDGFB integration site in the Ppfibp1 locus. The error bars indicate the standard deviation across triplicates. (B, C) A western blot (B) and immunohistochemical staining (C) comparing the Ppfibp1 protein levels between one murine glioma model with (P2, cell line 110_9_24) and one without (N2) an RCAS-PDGFB integration site in the Ppfibp1 locus. (D) Pie chart displaying the percentage of clones with and without a retroviral tag in the Ppfibp1 locus when clonally expanding the P2 cell line. (E) Kaplan-Meier plot comparing the tumor latency of two P2 clones, one with an integration (C2) and one without an integration (C35) in the Ppfibp1 locus, when transplanted back into mice. The P-value reflects the result of a log-rank test. (F) Boxplot displaying the PPFIBP1 expression in HGCC samples across GBM subtypes. (G–I) Scatterplots comparing PPFIBP1 gene expression and mean cell viability in HGCC samples across different drug treatments: Temozolomide (G), Sunitinib (H), and Vatalanib (I). R-values and P-values indicate the Pearson’s correlation coefficient and significance, respectively.
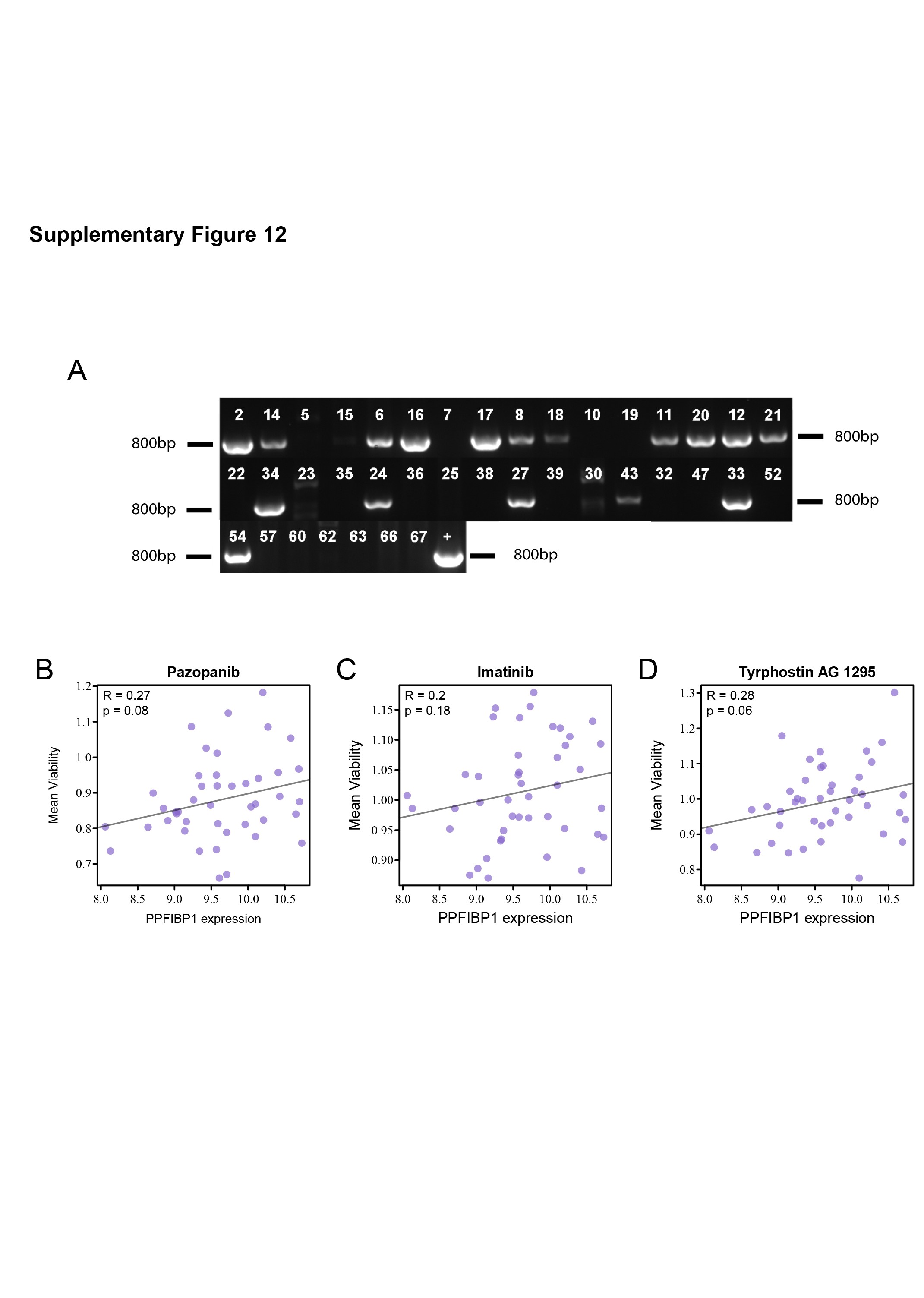
When clonally expanding one Ppfibp1-tagged murine glioma cell line, it was found that there are two populations of clones, one which harbored retroviral integrations in Ppfibp1 and one which did not (Figure 4D, Supplementary Figure 12A). Interestingly, when then retransplanting two clones in syngeneic mice, the clone with a Ppfibp1 retroviral tag demonstrated a significantly shorter latency compared to the clone that lacked the retroviral integration in the Ppfibp1 locus (Figure 4E).
Human glioma cell line data (HGCC data portal42) suggested that PPFIPB1 expression diverged significantly between GBM subtypes with highest average expression observed in the MS subtype (Figure 4F); and while PPFIBP1 expression levels did not appear associated with the outcome of Temozolomide treatment in these cell lines (Figure 4G), high PPFIBP1 expression displayed a positive correlation with treatment resistance in various PDGF-related drug treatments (Figure 4H and I, Supplementary Figure 12B–D). Collectively, our data suggest an oncogene role of PPFIBP1 in PDGF-driven gliomagenesis.
Discussion
Malignant gliomas are complex diseases characterized by alterations in several different genes.38 Although many GBM-promoting genes have been identified, current treatment options still leave the patients with a very dismal prognosis. More targeted therapeutic strategies represent a potential avenue to overcome such treatment failures,8 but would require a better understanding of individual glioma-related genes and their role in gliomagenesis. Accordingly, our work focused on the identification of genes that together with commonly mutated pathways, like the PDGF pathway, would contribute to tumor progression.
We previously demonstrated a tumorigenic role of PDGFB overexpression in murine models of glioma utilizing different retroviral systems as tools for gene delivery.5,7,10,23,24 It is commonly accepted that oncogenic retroviruses alone can drive tumorigenesis in various models.13 However, a single alteration in an oncogene, like PDGFB overexpression in nonviral transgenic models,43 is probably not enough to drive complete tumor formation. Forward genetic screens based on retroviral insertions, each introducing a traceable insertional mutation in the cell’s DNA, thus represent a valuable platform to identify genomic loci and genetic alterations that collaborate with the driver oncogene in promoting tumor development.9
We already described the potential of such a retroviral tagging approach using a MMLV-PDGFB-driven glioma model.10 The current study strove to provide an independent validation and further improvement of these initial efforts, by (i) utilizing a different ASLV-based retrovirus (RCAS) able to infect nondividing cells44 coupled to (ii) a transgene system enabling us to target the retrovirus to specific cell types,5,7,23,24 and (iii) a more high-throughput and automated NGS approach to retroviral integration site detection and processing.
Our analysis of RCAS integration sites and preferences suggested significant differences in the number of integrations observed based on the transgene and the genetic background. For instance, PDGFB overexpression with inactivation of Ink4/Arf or p53 generated more integrations instead of fewer ones. The latter would perhaps have been expected given that these tumors would likely require fewer secondary hits to provide a selective growth advantage or a more malignant phenotype. With respect to the differences between backgrounds, one possible explanation could be that the combination of PDGFB overexpression with inactivation of these important tumor suppressor genes might allow more cells to give rise to a fully blown malignant tumor. Consequently, the tumors driven from these backgrounds could be more polyclonal and present with an increased number of total integrations as compared to PDGFB as a single driver.
In addition, we found that the RCAS integrations, likely directed into active genes and open chromatin regions, created a landscape that is closely linked to the identities of the infected cell. Consistent with the phenotypes previously attributed to the different transgene models,23 stem cell-like genes were found enriched among the RCAS integration-related genes in G/tv-a, while N/tv-a and C/tv-a displayed an enrichment of more differentiated glial lineage signatures among retrovirally tagged genes. The potential link between retroviral gene tagging and cell identity was further emphasized when comparing the integrations in the RCAS-PDGFB gliomas to various other forward genetic models, indicating that the highest similarity existed with the MMLV-PDGFB glioma model. When comparing RCAS integration preferences to those of SB transposons, which do not show any insertion preference and can even access tightly packed chromatin,45 the number of shared ISs was not that great. The differences in the number of shared CIS between the RCAS-PDGFB glioma model and the MMLV-glioma or SB-glioma, respectively, might thus be a consequence of the different integration preferences between retroviruses and transposons.
While several novel genes were tagged by the RCAS-PDGFB system, many CISs were shared with the MMLV-PDGFB, strengthening the support for linking the identified loci to brain tumor development. Many of the tagged loci harbored genes that were already well known for their role in glioma. Ppfibp1 belonged to one of the most robustly tagged loci and was identified as an independent CIS in both model systems. PPFIBP1 is a protein tyrosine phosphatase-interacting protein that has been found fused with ALK in lung carcinoma patients46 and suggested as a target of the Metastasis-associated protein S100A4 (Mts-1),47 but if and how PPFIBP1 regulates metastasis is not clear. The current study demonstrated that cell lines with integrations in the PPFIBP1 locus displayed an increased expression of the gene coupled to shorter tumor latency, suggesting PPFIBP1 as a novel oncogene in PDGF-regulated gliomas. As further shown in the current study, there also appears to be a link between PPFIBP1 and more progressive GBM.2 Considering the problem with PDGF treatment resistance48 and that specific inhibitors that include PDGFR as a target (eg imatinib and sunitinib) have not shown beneficial activity for GBM patients, it is interesting to note that our results suggest an association between PPFIBP1 and PDGF treatment resistance.
GBM is still an incurable disease. It is important to find novel, potentially druggable, targets that are involved in driving malignant glioma progression and therapy resistance. The RCAS/TVA system is a frequently used model to study cancer development from distinct cell types in mice. We here show that the common insertions generated by the retroviruses in this system are causing alterations that partake and contribute to PDGF-driven brain tumor maintenance and in therapy resistance.
Funding
This work was generously sponsored by the European Research Council under Horizon 2020 (Project No. 640275, Medulloblastoma), the Swedish Cancer Society, the Swedish Childhood Cancer Fund, the Swedish Research Council, and the Ragnar Söderberg’s Foundation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We acknowledge technical support from the National Genomics Infrastructure (NGI)/Uppsala Genome Center and UPPMAX for providing assistance in massive parallel sequencing, and computational infrastructure, funded by RFI/VR and the Science for Life Laboratory, Sweden. The computations and data handling were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at UPPMAX partially funded by the Swedish Research Council. Some of the results shown in this study are based upon data generated and provided by the TCGA Research Network (https://www.cancer.gov/tcga). Finally, we would like to thank Ines Neves and Anders Sundström for technical assistance.
Contributor Information
Holger Weishaupt, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Matko Čančer, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Gabriela Rosén, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Karl O Holmberg, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Susana Häggqvist, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Ignas Bunikis, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Yiwen Jiang, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Smitha Sreedharan, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Ulf Gyllensten, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Oren J Becher, Department of Pediatrics and Biochemistry and Molecular Genetics, Northwestern University, Chicago, Illinois, USA; Department of Pediatrics and Oncological Sciences, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
Lene Uhrbom, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Adam Ameur, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Fredrik J Swartling, Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
Conflict of interest statement. The authors declare no conflict of interest.
Authorship statement. HW, MC, and FJS designed the study. HW, MC, AA, and FJS wrote the manuscript with help from LU. HW, MC, AA, and IB designed the hybridization probes. SH conducted the NGS library preparation and sequencing. AA and IB developed the InSiDeR program for integration site detection together with UG. HW performed all other bioinformatic analyses. FJS, LU, and OJB provided tumor material and established cell lines. MC and GR performed cell culture and genomic DNA preparation. MC, GR, YJ, and SS conducted transgenic animal infections and cell line establishment. MC, GR, and KHO performed H&E staining, IHC and WB. MC conducted the common integration site validation and RT-qPCR experiments.
References
- 1. Ostrom QT, Patil N, Cioffi G, et al. . CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. 2020;22(Supplement_1):iv1–iv96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang Q, Hu B, Hu X, et al. . Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer cell 2017;32(1):42–56.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parsons DW, Jones S, Zhang X, et al. . An integrated genomic analysis of human glioblastoma multiforme. science 2008;321(5897):1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002;62(13):3729–3735. [PubMed] [Google Scholar]
- 5. Uhrbom L, Hesselager G, Nistér M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58(23):5275–5279. [PubMed] [Google Scholar]
- 6. Dai C, Celestino JC, Okada Y, et al. . PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & Dev. 2001;15(15):1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang Y, Boije M, Westermark B, Uhrbom L. PDGF-B Can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia 2011;13(6):492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan AC, Ashley DM, López GY, et al. . Management of glioblastoma: State of the art and future directions. CA Cancer J Clin. 2020;70(4):299–312. [DOI] [PubMed] [Google Scholar]
- 9. Moresco EMY, Li X, Beutler B. Going forward with genetics: recent technological advances and forward genetics in mice. Am J Pathol. 2013;182(5):1462–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johansson FK, Brodd J, Eklöf C, et al. . Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci USA. 2004;101(31):11334–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kool J, Berns A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat Rev Cancer. 2009;9(6):389–399. [DOI] [PubMed] [Google Scholar]
- 12. Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nat Rev Cancer. 2010;10(10):696–706. [DOI] [PubMed] [Google Scholar]
- 13. Ranzani M, Annunziato S, Adams DJ, Montini E. Cancer gene discovery: exploiting insertional mutagenesis. Mol Cancer Res. 2013;11(10):1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johansson FK, Göransson H, Westermark B. Expression analysis of genes involved in brain tumor progression driven by retroviral insertional mutagenesis in mice. Oncogene 2005;24(24):3896–3905. [DOI] [PubMed] [Google Scholar]
- 15. Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003;300(5626):1749–1751. [DOI] [PubMed] [Google Scholar]
- 16. Lund AH, Turner G, Trubetskoy A, et al. . Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat Genet. 2002;32(1):160–165. [DOI] [PubMed] [Google Scholar]
- 17. Mikkers H, Allen J, Knipscheer P, et al. . High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet. 2002;32(1):153–159. [DOI] [PubMed] [Google Scholar]
- 18. Suzuki T, Shen H, Akagi K, et al. . New genes involved in cancer identified by retroviral tagging. Nat Genet. 2002;32(1):166–174. [DOI] [PubMed] [Google Scholar]
- 19. Wang Q, Jia P, Zhao Z. VirusFinder: software for efficient and accurate detection of viruses and their integration sites in host genomes through next generation sequencing data. PLoS One. 2013;8(5):e64465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Federspiel MJ, Bates P, Young J, Varmus HE, Hughes SH. A system for tissue-specific gene targeting: transgenic mice susceptible to subgroup A avian leukosis virus-based retroviral vectors. Proc Natl Acad Sci USA. 1994;91(23):11241–11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12(23):3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009;28(23):2266–2275. [DOI] [PubMed] [Google Scholar]
- 23. Jiang Y, Marinescu VD, Xie Y, et al. . Glioblastoma cell malignancy and drug sensitivity are affected by the cell of origin. Cell Rep. 2017;18(4):977–990. [DOI] [PubMed] [Google Scholar]
- 24. Lindberg N, Jiang Y, Xie Y, et al. . Oncogenic signaling is dominant to cell of origin and dictates astrocytic or oligodendroglial tumor development from oligodendrocyte precursor cells. J Neurosci. 2014;34(44):14644–14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sreedharan S, Maturi NP, Xie Y, et al. . Mouse models of pediatric supratentorial high-grade glioma reveal how cell-of-origin influences tumor development and phenotype. Cancer Res. 2017;77(3):802–812. [DOI] [PubMed] [Google Scholar]
- 26. Halvorson KG, Barton KL, Schroeder K, et al. . A high-throughput in vitro drug screen in a genetically engineered mouse model of diffuse intrinsic pontine glioma identifies BMS-754807 as a promising therapeutic agent. PLoS One. 2015;10(3):e0118926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Swartling FJ, Savov V, Persson AI, et al. . Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012;21(5):601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Höijer I, Johansson J, Gudmundsson S, et al. . Amplification-free long-read sequencing reveals unforeseen CRISPR-Cas9 off-target activity. Genome Biol. 2020;21(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boerkoel CF, Kung H-J. Transcriptional interaction between retroviral long terminal repeats (LTRs): mechanism of 5’LTR suppression and 3’LTR promoter activation of c-Myc in avian B-cell lymphomas. J Virol. 1992;66(8):4814–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johansson Swartling F. Identifying candidate genes involved in brain tumor formation. Ups J Med Sci. 2008;113(1):1–38. [DOI] [PubMed] [Google Scholar]
- 31. Sarver AL, Erdman J, Starr T, Largaespada DA, Silverstein KA. TAPDANCE: an automated tool to identify and annotate transposon insertion CISs and associations between CISs from next generation sequence data. BMC Bioinf. 2012;13(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32(suppl_1):D523–D527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weishaupt H, Cancer M, Engström C, Silvestrov S, Swartling FJ. Comparing the landcapes of common retroviral insertion sites across tumor models. Paper presented at: AIP Conference Proceedings, Vol. 1798, No. 1, p. 020173; 2017. In: Sivasundaram S, ed. 11th International Conference on Mathematical problems in Engineering, Aerospace, and Sciences ICNPAA 2016, La Rochelle, France, 04–08 July 2016.
- 34. Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci USA. 1998;95(3):1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uhrbom L, Kastemar M, Johansson FK, Westermark B, Holland EC. Cell type-specific tumor suppression by Ink4a and Arf in Kras-induced mouse gliomagenesis. Cancer Res. 2005;65(6):2065–2069. [DOI] [PubMed] [Google Scholar]
- 36. Vyazunova I, Maklakova VI, Berman S, et al. . Sleeping Beauty mouse models identify candidate genes involved in gliomagenesis. PLoS One. 2014;9(11):e113489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Westermark B, Heldin CH, Nistér M. Platelet-derived growth factor in human glioma. Glia 1995;15(3):257–263. [DOI] [PubMed] [Google Scholar]
- 38. Brennan CW, Verhaak RG, McKenna A, et al. . The somatic genomic landscape of glioblastoma. Cell 2013;155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu Y, Fletcher M, Gu Z, et al. . Glioblastoma epigenome profiling identifies SOX10 as a master regulator of molecular tumour subtype. Nat Commun. 2020;11(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cockbill LM, Murk K, Love S, Hanley JG. Protein interacting with C kinase 1 suppresses invasion and anchorage-independent growth of astrocytic tumor cells. Mol Biol Cell. 2015;26(25):4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang Y, Dodbele S, Park T, et al. . MicroRNA-29a inhibits glioblastoma stem cells and tumor growth by regulating the PDGF pathway. J Neurooncol. 2019;145(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johansson P, Krona C, Kundu S, et al. . A patient-derived cell atlas informs precision targeting of glioblastoma. Cell Rep. 2020;32(2):107897. [DOI] [PubMed] [Google Scholar]
- 43. Hede SM, Hansson I, Afink GB, et al. . GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia 2009;57(11):1143–1153. [DOI] [PubMed] [Google Scholar]
- 44. Hatziioannou T, Goff SP. Infection of nondividing cells by Rous sarcoma virus. J Virol. 2001;75(19):9526–9531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yoshida J, Akagi K, Misawa R, et al. . Chromatin states shape insertion profiles of the piggyBac, Tol2 and Sleeping Beauty transposons and murine leukemia virus. Sci Rep. 2017;7(1):43613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takeuchi K, Soda M, Togashi Y, et al. . Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1–ALK: reappraisal of anti-ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin Cancer Res. 2011;17(10):3341–3348. [DOI] [PubMed] [Google Scholar]
- 47. Kriajevska M, Fischer-Larsen M, Moertz E, et al. . Liprin β1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1). J Biol Chem. 2002;277(7):5229–5235. [DOI] [PubMed] [Google Scholar]
- 48. Bolcaen J, Nair S, Driver CH, et al. . Novel receptor tyrosine kinase pathway inhibitors for targeted radionuclide therapy of glioblastoma. Pharmaceuticals 2021;14(7):626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.