


Abstract
Atomic-level knowledge of protein-ligand interactions allows a detailed understanding of protein functions and provides critical clues to discovering molecules regulating the functions. While recent innovative deep learning methods for protein structure prediction dramatically increased the structural coverage of the human proteome, molecular interactions remain largely unknown. A new database, HProteome-BSite, provides predictions of binding sites and ligands in the enlarged 3D human proteome. The model structures for human proteins from the AlphaFold Protein Structure Database were processed to structural domains of high confidence to maximize the coverage and reliability of interaction prediction. For ligand binding site prediction, an updated version of a template-based method GalaxySite was used. A high-level performance of the updated GalaxySite was confirmed. HProteome-BSite covers 80.74% of the UniProt entries in the AlphaFold human 3D proteome. Predicted binding sites and binding poses of potential ligands are provided for effective applications to further functional studies and drug discovery. The HProteome-BSite database is available at https://galaxy.seoklab.org/hproteome-bsite/database and is free and open to all users.
INTRODUCTION
Recent innovative deep learning methods for protein structure prediction have dramatically increased the structural coverage of the human proteome (1,2). The AlphaFold Protein Structure Database (AFDB, https://alphafold.ebi.ac.uk) allows free access to the predicted structures by AlphaFold (AF) (1) for proteosomes of 20 organisms, including human (3). AFDB, along with AF itself, has served as a resource for extensive research. For example, Kincore, a program for protein kinase structure labelling, was applied to AF-predicted human kinase domain structures (4). APPRIS, a database of principal protein isoforms annotation, was updated by including AFDB to predict the splicing effect on protein structures (5).
Many proteins function by interacting with small bioactive ligands. Knowledge of the protein-ligand interactions, i.e. the identity of ligands interacting with the given protein, ligand binding sites, and 3D binding poses, can serve as a basis for protein functional studies and drug discovery (6). Information on the interactions of protein and ligand molecules has been collected from the experimentally resolved structures in Protein Data Bank (PDB) (7) in various databases including PDBbind (8), BioLip (9), Binding MOAD (10), and sc-PDB (11). Databases like PocketDB (12) and ProBis-Dock (13) augmented the information on interaction by including predicted pockets for protein structures deposited in PDB.
Despite the continuous increase in PDB, many proteins still lack binding information. Recently, databases that provide ligand binding information on the enlarged structure database AFDB have been reported. For example, AlphaFill (14) reports structurally transplanted small molecules to AF structures from template protein-ligand complex structures selected based on sequence identity. The transplanted compounds are limited in diversity, including 400 ligands, cofactors, and metal ions that are commonly found in PDB. CavitySpace (15) provides predicted ligand binding sites of human AF model structures using a geometry-based binding site prediction method that detects potential cavities on protein surfaces. PrankWeb (16) and ScanNet (17) are examples of web servers that allow the prediction of protein binding sites on AF structures. PrankWeb also allows bulk download of precomputed predictions on AF structures. The machine-learning-based (PrankWeb) and geometry-based (CavitySpace and ScanNet) prediction methods do not provide potential binding partners. All of the previous databases and web servers use the raw model structures of AFDB. However, the structures in AFDB show varying model accuracy within each model, containing low-quality regions which can affect the reliability of binding site prediction.
Here, we present HProteome-BSite, a new database of predicted binding sites, putative ligands, and 3D binding poses for the 3D human proteome of AFDB. Binding sites and ligands were predicted by a template-based method GalaxySite (18), covering 24,691 different ligands. For more effective binding prediction, only the structural domains predicted with higher confidence were considered. This database provides ligand binding site prediction for 16,554 unique human protein UniProt entries, covering 80.74% of the human proteome. The predicted binding sites and ligands could serve as a useful resource for further functional studies or drug discovery for previously unexplored proteins (19). HProteome-BSite is freely available at https://galaxy.seoklab.org/hproteome-bsite/database with a user-friendly interface and a help page that can guide non-expert users.
METHODS FOR DATA PREPARATION
Curation of the 3D human proteome
The HProteome-BSite database provides binding information on the AlphaFold human proteome database. Since many AF structures contain low-quality regions, the structures were processed into high-quality domains before binding prediction, as shown in Figure 1. First, low-quality regions of longer than 10 consecutive residues with residue-wise pLDDT <70 (20,21) were deleted, producing multiple fragments. Second, fragments with high inter-fragment contacts compared to intra-fragment contacts were merged. Finally, high-quality fragments were assigned as domains based on size and contact density. Further details on the domain assignment process are provided in Supplementary Method SM 1. This curation of the structure database can increase the performance of binding prediction, as discussed below.
Figure 1.

Domains of high-quality structures are assigned after removing low-quality regions. In this example, three domains are assigned after deleting four low-quality regions.
Prediction of ligand binding sites
Ligand binding sites, potential ligands, and binding poses were predicted for each of the processed domain structures from the curated 3D human proteome using an updated version of GalaxySite (18), as outlined in Figure 2. Updates made to GalaxySite are described in detail in Supplementary Method SM 2. Interaction templates to the query protein are detected by both sequence- and structure-based searches on the interaction template database. This database was curated in-house from the protein-ligand complex structures in PDB, as described in SM 2. The sequence-based method used the sequence alignment score HHscore (21), and the structure-based search used the structure alignment score TM-score to select interaction templates from the database. The binding sites and ligands were taken from the top-ranking templates. Binding poses were predicted by the protein-ligand docking program GalaxyDock2 (22) using the interaction restraints extracted from the template complex structures (18).
Figure 2.
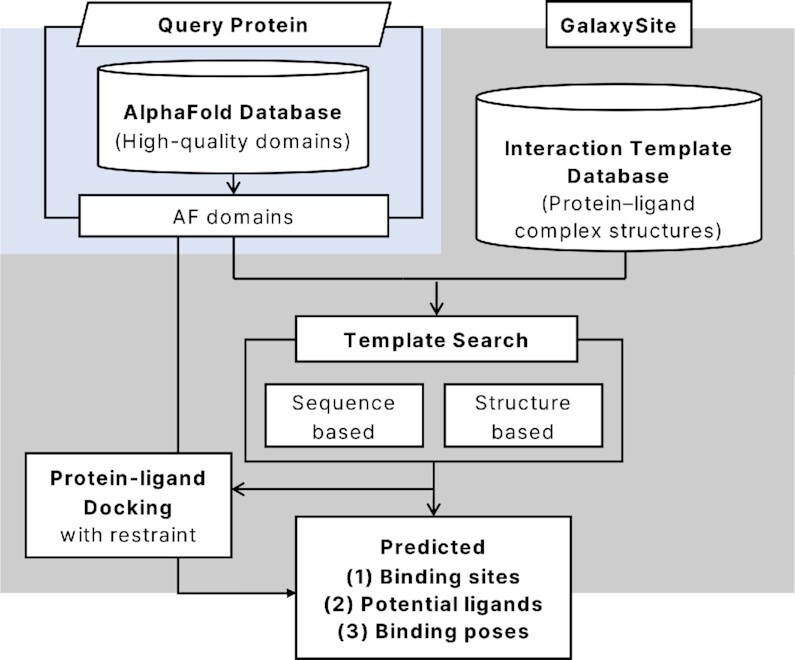
Outline of GalaxySite binding prediction on the AlphaFold human structure database.
The performance of the updated GalaxySite was tested on a benchmark set called COACH420 set (23), a subset of COACH (24,25) which consists of 420 single-chain protein targets. GalaxySite was run on the benchmark mode, discarding templates with sequence identity higher than 70% to the target from the interaction template database. The high sequence identity was chosen because a large fraction of proteins in the curated AFDB have protein-ligand complex templates of high similarity (median TMscore of 0.76, median sequence identity of 0.32). The protein crystal structures were used as input for the structure-based site prediction method in this binding site prediction test. The performance of GalaxySite was enhanced by the current update from 74% to 88% on the COACH420 set when a protein-wise success rate is considered, as shown in Supplementary Table ST 1. The performance increased slightly when the sequence- and structure-based search methods were combined.
The ligand binding poses obtained by docking can be different from those of templates, and the distribution of the ligand RMSD between the ligand pose aligned to the template and the predicted ligand pose is shown in Supplementary Figure SF 1A. An example where the ligand pose deviation after docking is large is also presented SF 1B and C.
Database statistics
Out of 20 504 UniProt entries in the AlphaFold human proteome DB, HProteome-BSite provides ligand binding site predictions for 16 554 entries, which correspond to a coverage of 80.7%. Among the entries not included in the database, 60.5% of the entries were excluded due to low model quality resulting in no structural domain with high confidence. Our database provides binding site information for 95% of the entries with at least a single assigned structural domain. The binding site information also includes possible interacting ligands with an average (median) of 41.5 (14.0) ligands per entry. The proteins with predicted interactions in HProteome-BSite cover diverse protein families with high coverage, GPCRs (98.7%), ion channels (96.9%), kinases (99.5%), nuclear hormone receptors (96.8%) and proteases (99.2%). About 70% of human drugs on the market target GPCRs, ion channels, kinases and nuclear hormone receptors (26). The predicted interactions could aid further functional studies and drug developments targeting these proteins.
UTILIZING THE DATABASE
All predictions in the HProteome-BSite database can be accessed through the website https://galaxy.seoklab.org/hproteome-bsite/database which runs on all modern browsers. The website provides a search engine for finding proteins of interest, pages containing statistics of the database, and a help section about using the database. It also supports the visualization of protein structure and binding pose of potential ligands using Mol* (27) and a WebGL-based viewer PV (https://biasmv.github.io/pv/). The viewer also runs on phone and tablet screens without the need for additional plugins.
Each entry in the database corresponds to an interaction prediction on each structural domain of the query protein. For each entry, information on the binding sites and ligands is provided, along with information on the interaction templates used for the prediction. As a measure of the reliability of the prediction, the structure similarity score (TM-score) of the query structure to the template protein structure is also given. For the top three binding sites predicted from each of the sequence-based and the structure-based search, the protein-ligand docking results of representative ligands are provided. Alternative ligands that were predicted to bind to the same binding sites are listed in addition, and a further binding pose prediction for those ligands is possible by submitting a docking run through a provided link to the docking web server within the same system.
Searching for proteins of interest
The search engine allows users to search for proteins of interest by UniProt ID or protein name. Proteins belonging to key protein families (GPCRs, proteases, kinases, nuclear hormone receptors, and ion channels) can also be browsed. The search results page shows a list of UniProt IDs for each search, as shown in Figure 3. Each UniProt ID is linked to a report page containing a list of binding predictions on individual structural domains. Each domain can be identified by a unique entry title composed of its UniProt ID, AF structure fragment number, and domain number (e.g. Q8TB40_F1_d1).
Figure 3.

A search example on the HProteome-BSite database. Proteins of interest can be searched for by UniProt ID, protein name, or protein family (left panel). If multiple targets match the search keyword, a list of all matching UniProt IDs is given on the search results page (upper right panel). Clicking on the UniProt ID leads to a report page showing a list of structural domains for which binding predictions are available (lower right panel).
Interpreting the report page
The report page for each entry provides information on predicted binding sites on the structural domain. Each page contains a 3D structure viewer that shows a protein structure (when the ‘See structural domain only’ is clicked) or a protein–ligand complex structure (when the ‘View on PV’ link of a representative ligand is clicked), as shown in the left panel of Figure 4. Detailed view of the complex structure using Mol* which allows visualization of interactions between the ligand and the protein is available on a separate page (when the ‘See Ligand Details (Mol*)’ link is clicked). The ligand details page (Figure SF 5) shows all available information on that specific ligand. The AF protein structure and the predicted protein-ligand complex structure can be downloaded in PDB and MOL2 format, respectively. A table of the maximum top 10 predicted binding sites for each of the sequence-based and structure-based searches is provided (upper right panel of Figure 4). Lists of full predictions can be downloaded in CSV format through the links below the tables (lower right panel of Figure 4). Each row of the table corresponds to a predicted binding site, with information on a representative ligand (with a predicted docking pose for the top three binding sites), templates used to predict ligands (with TM-score, which can be considered as a confidence measure), and alternative ligands. For the lower-rank representative ligands, docking poses were not pre-calculated, but docking can be run by clicking on the template name. For alternative ligands, docking can be run on the docking web server within the system (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=SITE_DOCK) using the provided information on the ligand, the protein domain, and the interaction template. A detailed explanation of how to interpret the results on the report page is also available on the website for the example shown in Figure 4. (https://galaxy.seoklab.org/hproteome-bsite/database/help_result).
Figure 4.

An example report page for the entry Q9H0K1_F1_d1 (https://galaxy.seoklab.org/hproteome-bsite/database/domains/39056).
CASE STUDIES
Salt-Inducible Kinase 2 (SIK2). Salt-inducible kinase 2 (UniProt ID Q9H0K1) is an AMP-activated protein kinase-related protein kinase that has been proposed as a target for ovarian cancer therapy (28,29). Despite its therapeutic importance, its structure has not been resolved by experiments. Two structural domains were compiled in our database, corresponding to residues 14–273 and 295–341, where the first domain was highly similar to that of the known kinase domain of SIK2 (residues 20–271). While the full protein structure deposited in the AlphaFold human proteome DB has a very low median pLDDT of 41.8, the first domain in our database shows a high median pLDDT of 95.0 (Supplementary Figure SF 2). This example illustrates that the structure processing step could retrieve functional domains with high confidence even when the overall AF structure is of low quality.
The top 1 predicted binding site for the kinase domain of SIK2 (residues 14–273) from both the structure- and sequence-based methods corresponds to the known ATP binding site of SIK2. To confirm this, a known inhibitor of SIK2 targeting the ATP binding site, HG-9–91-01, was docked using GalaxyDock3 (30) to the identified binding site. The docking results showed interactions consistent with previous findings that HG-9–91-01 targets not only the ATP-binding site of SIK2 with K49 as key residue but also a small hydrophobic pocket near the active site created by the presence of T96 at the gatekeeper site (31) (Supplementary Figure SF 3).
Diacylglycerol O-acyltransferase 2 (DGAT2). DGAT2 (UniProt ID Q96PD7) is a therapeutic target related to liver disease without any 3D structure resolved by experiments (32). This target was highlighted earlier as one of the useful predictions in the AlphaFold human proteome paper (33). In our database, the binding pocket of DGAT2 was successfully predicted by the structure-based search. When a known inhibitor, PF-06424439 (34), was docked to the binding site of the top-ranked ligand, the known key interactions between DGAT2 and the ligand involving residues H163, T194 and S244 were reproduced (33,35), as illustrated in Supplementary Figure SF 4.
CONCLUSION
A new database HProteome-BSite provides predictions of binding sites, potential ligands and binding poses of the ligands on human proteome structures predicted by AlphaFold. An updated version of GalaxySite was employed to enhance the binding site prediction performance. The 3D human proteome was curated to retrieve structural domains of high confidence to facilitate binding site prediction and ligand docking. The HProteome-Bsite database provides at least a single binding site prediction for 81% of the human proteome, accounting for 95% of moderately accurate AF structures.
Further improvements in the database would be possible through advanced protein structure prediction and binding prediction in the future. Currently, 25% of AlphaFold human protein structures are of low quality (median pLDDT < 70), and many structures are only partially resolved. The accuracy of template-based and template-free binding prediction would be improved by employing deep learning techniques, especially with increasing interaction data generated by both experiments and predictions.
Structural information on the ligand binding site of a protein can provide insights into the potential binding partners and their interactions with the protein. The information deposited in the HProteome-Bsite database would be useful for various applications, including protein functional study and drug discovery for previously unexplored proteins.
DATA AVAILABILITY
All predicted interaction information in HProteome-BSite is freely accessible at https://seoklab.galaxy.org/hproteome-bsite/database. The website is accessible and legible on both phone and tablet screens.
Supplementary Material
Contributor Information
Jiho Sim, Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea.
Sohee Kwon, Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea; Galux Inc, Gwanak-gu, Seoul 08738, Republic of Korea.
Chaok Seok, Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea; Galux Inc, Gwanak-gu, Seoul 08738, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea (NRF) grants funded by the Korean government [2020M3A9G7103933]; Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korean government (MSIT) [2021-0-02068, Artificial Intelligence Innovation Hub]; Samsung Science and Technology Foundation [SSTF-BA1901-08]. Funding for open access charge: Seoul National University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Zidek A., Potapenko A.et al.. Highly accurate protein structure prediction with alphafold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D.et al.. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021; 373:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A.et al.. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022; 50:D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Modi V., Dunbrack R.L.. Kincore: a web resource for structural classification of protein kinases and their inhibitors. Nucleic Acids Res. 2022; 50:D654–D664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rodriguez J.M., Pozo F., Cerdan-Velez D., Di Domenico T., Vazquez J., Tress M.L.. APPRIS: selecting functionally important isoforms. Nucleic Acids Res. 2022; 50:D54–D59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao J., Cao Y., Zhang L.. Exploring the computational methods for protein-ligand binding site prediction. Comput. Struct. Biotechnol. J. 2020; 18:417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burley S.K., Bhikadiya C., Bi C., Bittrich S., Chen L., Crichlow G.V., Christie C.H., Dalenberg K., Di Costanzo L., Duarte J.M.et al.. RCSB protein data bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021; 49:D437–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu Z., Li Y., Han L., Li J., Liu J., Zhao Z., Nie W., Liu Y., Wang R.. PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics. 2015; 31:405–412. [DOI] [PubMed] [Google Scholar]
- 9. Yang J., Roy A., Zhang Y.. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013; 41:D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith R.D., Clark J.J., Ahmed A., Orban Z.J., Dunbar J.B. Jr, Carlson H.A. Updates to binding MOAD (Mother of all databases): polypharmacology tools and their utility in drug repurposing. J. Mol. Biol. 2019; 431:2423–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Desaphy J., Bret G., Rognan D., Kellenberger E.. sc-PDB: a 3D-database of ligandable binding sites–10 years on. Nucleic Acids Res. 2015; 43:D399–D404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhagavat R., Sankar S., Srinivasan N., Chandra N.. An augmented pocketome: detection and analysis of small-molecule binding pockets in proteins of known 3D structure. Structure. 2018; 26:499–512. [DOI] [PubMed] [Google Scholar]
- 13. Konc J., Lesnik S., Skrlj B., Janezic D. ProBiS-Dock database: a web server and interactive web repository of small ligand-protein binding sites for drug design. J. Chem. Inf. Model. 2021; 61:4097–4107. [DOI] [PubMed] [Google Scholar]
- 14. Hekkelman M.L., de Vries I., Joosten R.P., Perrakis A.. AlphaFill: enriching the alphafold models with ligands and co-factors. 2021; bioRxiv doi:27 November 2021, preprint: not peer reviewed 10.1101/2021.11.26.470110. [DOI] [PMC free article] [PubMed]
- 15. Wang S., Lin H., Huang Z., He Y., Deng X., Xu Y., Pei J., Lai L.. CavitySpace: a database of potential ligand binding sites in the human proteome. Biomolecules. 2022; 12:967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jakubec D., Skoda P., Krivak R., Novotny M., Hoksza D. PrankWeb 3: accelerated ligand-binding site predictions for experimental and modelled protein structures. Nucleic Acids Res. 2022; 50:W593–W597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tubiana J., Schneidman-Duhovny D., Wolfson H.J.. ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nat. Methods. 2022; 19:730–739. [DOI] [PubMed] [Google Scholar]
- 18. Heo L., Shin W.H., Lee M.S., Seok C.. GalaxySite: ligand-binding-site prediction by using molecular docking. Nucleic Acids Res. 2014; 42:W210–W214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Naderi M., Lemoine J.M., Govindaraj R.G., Kana O.Z., Feinstein W.P., Brylinski M.. Binding site matching in rational drug design: algorithms and applications. Brief. Bioinform. 2019; 20:2167–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mariani V., Biasini M., Barbato A., Schwede T.. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013; 29:2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005; 21:951–960. [DOI] [PubMed] [Google Scholar]
- 22. Shin W.H., Kim J.K., Kim D.S., Seok C.. GalaxyDock2: protein-ligand docking using beta-complex and global optimization. J. Comput. Chem. 2013; 34:2647–2656. [DOI] [PubMed] [Google Scholar]
- 23. Krivak R., Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J. Cheminform. 2018; 10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roy A., Yang J., Zhang Y.. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012; 40:W471–W477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang J., Roy A., Zhang Y.. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013; 29:2588–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Santos R., Ursu O., Gaulton A., Bento A.P., Donadi R.S., Bologa C.G., Karlsson A., Al-Lazikani B., Hersey A., Oprea T.I.et al.. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017; 16:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sehnal D., Bittrich S., Deshpande M., Svobodová R., Berka K., Bazgier V., Velankar S., Burley S.K., Koča J., Rose A.S.. Mol* viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021; 49:W431–W437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Australian Ovarian Cancer Study Group Ahmed A.A., Lu Z., Jennings N.B., Etemadmoghadam D., Capalbo L., Jacamo R.O., Barbosa-Morais N., Le X.F., Vivas-Mejia P.et al.. SIK2 is a centrosome kinase required for bipolar mitotic spindle formation that provides a potential target for therapy in ovarian cancer. Cancer Cell. 2010; 18:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miranda F., Mannion D., Liu S., Zheng Y., Mangala L.S., Redondo C., Herrero-Gonzalez S., Xu R., Taylor C., Chedom D.F.et al.. Salt-Inducible kinase 2 couples ovarian cancer cell metabolism with survival at the adipocyte-rich metastatic niche. Cancer Cell. 2016; 30:273–289. [DOI] [PubMed] [Google Scholar]
- 30. Yang J., Baek M., Seok C.. GalaxyDock3: Protein–ligand docking that considers the full ligand conformational flexibility. J. Comput. Chem. 2019; 40:2739–2748. [DOI] [PubMed] [Google Scholar]
- 31. Clark K., MacKenzie K.F., Petkevicius K., Kristariyanto Y., Zhang J., Choi H.G., Peggie M., Plater L., Pedrioli P.G., McIver E.et al.. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16986–16991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Amin N.B., Carvajal-Gonzalez S., Purkal J., Zhu T., Crowley C., Perez S., Chidsey K., Kim A.M., Goodwin B.. Targeting diacylglycerol acyltransferase 2 for the treatment of nonalcoholic steatohepatitis. Sci. Transl. Med. 2019; 11:eaav9701. [DOI] [PubMed] [Google Scholar]
- 33. Tunyasuvunakool K., Adler J., Wu Z., Green T., Zielinski M., Zidek A., Bridgland A., Cowie A., Meyer C., Laydon A.et al.. Highly accurate protein structure prediction for the human proteome. Nature. 2021; 596:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Futatsugi K., Kung D.W., Orr S.T., Cabral S., Hepworth D., Aspnes G., Bader S., Bian J., Boehm M., Carpino P.A.et al.. Discovery and optimization of imidazopyridine-based inhibitors of diacylglycerol acyltransferase 2 (DGAT2). J. Med. Chem. 2015; 58:7173–7185. [DOI] [PubMed] [Google Scholar]
- 35. Wang L., Qian H., Nian Y., Han Y., Ren Z., Zhang H., Hu L., Prasad B.V.V., Laganowsky A., Yan N.et al.. Structure and mechanism of human diacylglycerol O-acyltransferase 1. Nature. 2020; 581:329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All predicted interaction information in HProteome-BSite is freely accessible at https://seoklab.galaxy.org/hproteome-bsite/database. The website is accessible and legible on both phone and tablet screens.