

Abstract
Epigenetic regulators are proteins involved in controlling gene expression. Information about the epigenetic regulators within the Fagaceae, a relevant family of trees and shrubs of the northern hemisphere ecosystems, is scarce. With the intent to characterize these proteins in Fagaceae, we searched for orthologs of DNA methyltransferases (DNMTs) and demethylases (DDMEs) and Histone modifiers involved in acetylation (HATs), deacetylation (HDACs), methylation (HMTs), and demethylation (HDMTs) in Fagus, Quercus, and Castanea genera. Blast searches were performed in the available genomes, and freely available RNA‐seq data were used to de novo assemble transcriptomes. We identified homologs of seven DNMTs, three DDMEs, six HATs, 11 HDACs, 32 HMTs, and 21 HDMTs proteins. Protein analysis showed that most of them have the putative characteristic domains found in these protein families, which suggests their conserved function. Additionally, to elucidate the evolutionary history of these genes within Fagaceae, paralogs were identified, and phylogenetic analyses were performed with DNA and histone modifiers. We detected duplication events in all species analyzed with higher frequency in Quercus and Castanea and discuss the evidence of transposable elements adjacent to paralogs and their involvement in gene duplication. The knowledge gathered from this work is a steppingstone to upcoming studies concerning epigenetic regulation in this economically important family of Fagaceae.
1. INTRODUCTION
Fagaceae is an ecological and economically important family of deciduous and persistent trees and shrubs, spread throughout the Northern hemisphere, comprising of more than 900 species belonging to eight genera (Rogers, 2004). Among these, Fagus, Castanea, and Quercus are the wider dispersed genera present in the three continents: Europe, Asia, and North America (Kremer et al., 2012). According to several plastid and nuclear genome studies, the genus Fagus occupies the basal position in the family and has diverged ca. 82 Mya in the late Cretaceous epoch (Kremer et al., 2012; Yang et al., 2018; Zhou et al., 2022) while Castanea and Quercus diverged during the early Paleocene (Zhou et al., 2022). The genus Fagus can be divided in the subgenus Engleriana and Fagus, with the last being further divided into four sections Longipetiolata, Lucida, which includes F. crenata, Fagus, and Grandifolia enclosing F. sylvatica and F. grandifolia, respectively (Jiang et al., 2022). The genus Castanea can be separated into four sections: Eucastanon, comprising C. crenata, C. mollissima, C. sativa, and C. dentata; Balanocastanon containing C. pumila and C. ozarkensis; and Hypocastanon with C. henryi (Lang et al., 2007). Quercus is a very complex genus with more than 500 species divided by two subgenera Cerris and Quercus. For example, Q. suber belongs to the subgenus Cerris, while Q. robur and Q. lobata belong to the subgenus Quercus (Hubert et al., 2014; Zhou et al., 2022).
The economic importance of Fagaceae includes products like cork from the outer bark of Q. suber, and fruits such as chestnuts, mainly from C. sativa, C. crenata, and C. mollissima. The significance of this family has led to the development of several open‐source genetic resources mainly for the three most representative genera Fagus, Quercus, and Castanea: Fagus sylvatica—European beech (Mishra et al., 2018), F. crenata—Japanese beech (Tsukamoto et al., 2020), Q. lobata—Valley oak (Sork et al., 2016), Q. suber—Cork oak (Ramos et al., 2018), Q. robur—English oak (Schmid‐Siegert et al., 2017), C. mollissima—Chinese chestnut (Wang et al., 2020), and C. crenata—Japanese chestnut (Shirasawa et al., 2021) have their genomes sequenced and available to the research community. Several other transcriptomic resources for some other species are also accessible to date.
Although there has been an increasing interest in the epigenetic regulation and its effects in members of Fagaceae (Gugger et al., 2016; Hrivnák et al., 2017; Inácio et al., 2017, 2018, 2022; Michalak et al., 2015; Platt et al., 2015; Ramos et al., 2013; Ribeiro et al., 2009; Rico et al., 2014; Santamaría et al., 2009, 2011; Silva et al., 2020; Vičić et al., 2013; Viejo et al., 2010, 2012) the full identification of the epigenetic regulator toolbox along this family is lacking.
Epigenetic regulators play a key role controlling the expression of genes like those involved in environmental responses and plant development (Lloret et al., 2018). Epigenetic regulators can apply or remove simple chemical residues that result in mitotically and/or meiotically heritable changes in gene expression that do not involve modifications in the DNA sequence (Zhang et al., 2018). The epigenetic modifications such as DNA and histone methylation or acetylation are reversible and can either promote or prevent gene transcription (Albini et al., 2019; Bewick & Schmitz, 2017).
DNA methyltransferases (DNMTs) are the enzymes that catalyze the covalent bond of a methyl group in the carbon 5 of the cytosines (5mC) and can be divided in two groups whether they maintain the DNA methylation state or impose de novo methylation marks (Finnegan & Kovac, 2000; Pavlopoulou & Kossida, 2007). DNA methylation can lead to transcriptional silencing or changes in gene expression, depending on if it happens in the promoters of genes or in the gene bodies (Bewick & Schmitz, 2017; Zhang et al., 2018). In plants, there are four classes of DNMTs that can be classified according to their conserved domains: METHYLTRANSFERASE 1 (MET1), CHROMOMETHYLASEs (CMTs), DNA METHYLTRANSFERASE HOMOLOG 2 (DNMT2), and DOMAINS REARRANGED METHYLTRANSFERASEs (DRMs; Finnegan & Kovac, 2000). All DNMTs have a DNA_Mtase domain besides other specific domains. MET1, an ortholog of the DNMT1 found in animals, is responsible for maintaining CG methylation and is characterized by a Replication Foci Domain (RFD). CMTs have a Bromo‐Adjacent Homology (BAH) and CHRomatin Organization MOdifier (CHROMO) domains and they mediate CHG (CMT3) and CHH (CMT2) modifications, where H means any nucleotide but glycosine (Kumar & Mohapatra, 2021; Schmitz et al., 2019). De novo DNA methylation, performed by DRM2 harboring an UBiquitin Associated‐like domain (UBA), is dependent on the small interfering RNA (siRNA) machinery in a process termed RNA‐directed DNA methylation (RdDM). DRMs can also maintain CHH methylation on euchromatic genomic regions (Zhong et al., 2014). DNMT2 is an enzyme that methylates DNA in protists and Drosophila, while in other eukaryotes it preferably methylates tRNA cytosines (Jeltsch et al., 2017).
DNA demethylases/glycosylases (DDMEs) are enzymes that can actively remove methyl groups through a base excision repair process (Law & Jacobsen, 2010). Demethylation by REPRESSOR OF SILENCING 1 (ROS1) prevents transcriptional gene silencing by maintaining the loci free of methylation, while demethylation by DEMETER (DME) promotes gene expression by establishing a new epigenetic hypomethylated state. Demethylases DEMETER‐LIKE 2 and 3 (DML2 and 3) prevent the accumulation of methylation of genes and their surroundings (Kumar & Mohapatra, 2021).
Like DNA modifications, post translational histone alterations such as histone methylation and acetylation, play a key role in the activation and repression of gene expressions by rendering the associated DNA more or less available for transcription. Histone modifiers act by adding or removing chemical groups to the residues of the N‐terminal tails (Albini et al., 2019). Acetylation, frequently related to increase of gene expression, is mediated by Histone Acetyltransferases (HATs) and can be reversed by Histone Deacetylases (HDACs), leading to transcription repression (Boycheva et al., 2014). HATs are grouped in several families based on sequence homology and activity (Sterner & Berger, 2000): GNAT/MYST superfamily consisting of Gcn5‐related N‐acetyltransferase (GNATs) and MOZ, Ybf2/Sas3, Sas2, and Tip60 (MYSTs); the p300/cAMP‐responsive element‐binding protein‐binding protein family (p300/CBP) and the TATA‐binding protein‐associated factor family (TAFII250). The HDACs are divided in three families: Reduced Potassium Deficiency 3/Histone Deacetylase 1 (RPD3/HDA1), HISTONE DEACETYLASE 2‐type (HD2), and SILENT INFORMATION REGULATOR 2 or sirtuins (SIR2/SRT) based on sequence similarity and cofactor dependency (Albini et al., 2019; Duan et al., 2018). Histone methyltransferases (HMTs) deposit one, two, or three methyl groups on lysines or arginines of histone tails. The most studied alterations are on the lysines of the histones H3 and H4 (Cheng et al., 2020). These enzymes contain a S uppressor of variegation, E nhancer of zeste, and T rithorax (SET) domain responsible for their catalytic activity. In Arabidopsis thaliana HMTs can be organized into five classes: Enhancer of zeste‐like proteins (E[Z]‐like—Class I), A bsent, S mall, or H omeotic (ASH—Class II), Trithorax‐like (ATX—Class III), Trithorax‐related (ATXR—Class IV) and SUppressor of position‐effect VARiegation SU(VAR) (SUV—Class V), according to their conserved domains (Zhou et al., 2020). Each HMT has a specificity to a particular histone residue, and depending on the local and type of modification, a different effect on transcriptional activation or repression takes place: the trimethylation of lysine 4 on histone 3 (H3K4me3) is associated with transcriptional activation, while the mono‐ and di‐methylation of the same histone residue can be associated to both active and inactive loci. Mono‐ and di‐ methylation of H3K9 are usually associated with DNA methylation and transcription silencing, while H3K9me3 is associated with transcriptional activation. A methylation on H3K27 is always associated with gene silencing, while di‐ and tri‐methylation of H3K36 is always associated with transcriptional activation (Cheng et al., 2020). Histone methylation is also reversible by histone demethylases (HDMTs) which have affinity to specific methylated types of residues (Cheng et al., 2020), and can be divided into two types of proteins depending on their conserved domains: lysine‐specific demethylase 1‐like (LSD1) and Jumonji (JmJ) (Albini et al., 2019; Yung et al., 2021; Zhao et al., 2019).
The discovery and characterization of the epigenetic regulators within Fagaceae is a valuable tool to understand their role in the ecological success of this family and how they can be associated with continuous climate change. This characterization may also facilitate further functional studies to disclose how these epigenetic regulators and events influence the development processes like shown in the ones addressed in previous studies (Gugger et al., 2016; Hrivnák et al., 2017; Inácio et al., 2017, 2018; Michalak et al., 2015; Platt et al., 2015; Ramos et al., 2013; Ribeiro et al., 2009; Rico et al., 2014; Santamaría et al., 2009, 2011; Silva et al., 2020; Viejo et al., 2010, 2012).
The aim of this work was to identify and characterize the epigenetic regulators in the species of the three most representative genera of Fagaceae: Fagus, Quercus, and Castanea (Kremer et al., 2012). For this, we took advantage of the resources available online to assemble the transcriptomic data and used them together with the existing assembled genomes. We further characterized the proteins through their predicted conserved domains and studied the phylogenetic relationship between species to help to clarify the evolution of these proteins within this family. This study offers the first wide analysis of epigenetic regulators in the Fagaceae family.
2. MATERIALS AND METHODS
2.1. Accessing genomes and transcriptomes
In this study, species of the Fagaceae family with sequenced genomes were used: Fagus sylvatica L. (Mishra et al., 2018), Fagus crenata Blume (Tsukamoto et al., 2020), Quercus lobata Nee (Sork et al., 2016), Quercus robur L. (Plomion et al., 2018), Quercus suber L. (Ramos et al., 2018), Castanea mollissima Blume (Wang et al., 2020), and Castanea crenata Siebold & Zucc. (Shirasawa et al., 2021). Accession numbers of the genome assemblies used can be found in Table S1.
Freely available RNA‐seq data retrieved from the NCBI Sequence Read Archive (SRA; Leinonen et al., 2011) was used to de novo assemble transcriptomes of the Fagaceae species Fagus grandifolia Ehrh. F. sylvatica, F. crenata, Castanea henryi (Skan) Rehder & E.H. Wilson, and C. crenata. Q. suber transcriptome data were retrieved from the Cork Oak DB (Arias‐Baldrich et al., 2020). The accession numbers from SRA raw‐sequencing data used can be found in Table S2. Read‐quality control was evaluated by FastQC (v. 0.11.7) before and throughout the trimming steps. BBDuk program (from the BBMap package, v. 38.01) was used to trim and filter reads, using the reference resource file supplied with the program. We trimmed the 3′ and 5′ adapters setting a kmer of 35, specifying trimming based on pair overlap detection and both reads to the same length. We also trimmed both ends of the reads for low quality (≥20). After this step, if instability in nucleotide frequencies was still observed in the first few bases, they were removed. Finally, we removed poly‐A tails with more than 10 bp. F. grandifolia reads were analyzed and trimmed on the Galaxy Europe server (https://usegalaxy.eu/), using BBDuk (bbmap v. 38.93) with a 35 kmer size and the remaining parameters were set as default and further trimmed with Trimmomatic (v. 0.38), also with default parameters. Transcriptome de novo assembly was performed using Trinity (v. 2.9.1) in the Galaxy Europe server (https://usegalaxy.eu/), with default settings, or in house using Trinity (v. 2.9.1; Grabherr et al., 2011). We calculated N50 values for each assembly using the R (v. 4.2.0) package CNEr (v. 1.30.0), on RStudio (“Prairie Trillium” Release [8acbd38b, 2022‐04‐19]; Tan et al., 2019; R Core Team, 2022; RStudio Team, 2022). The validation of transcriptome assemblies was done using Benchmarking Universal Single‐Copy Orthologs (BUSCO; Simão et al., 2015), a part of the OmicsBox (v. 2.0; https://www.biobam.com/omicsbox/). We used the transcriptome assembled by Alhinho et al. (2021) for Castanea sativa Mill., the protein library Castanea dentata—Transcriptome Assembly AC454_v3 for Castanea dentata (Marshall) Borkh. Retrieved from Hardwood Genomics (https://hardwoodgenomics.org/), and for Q. robur the OCV3 transcriptome available on the Quercus Portal (https://quercusportal.pierroton.inra.fr/). https://hardwoodgenomics.org/), and for Q. robur the OCV3 transcriptome available on the Quercus Portal (https://quercusportal.pierroton.inra.fr/).
2.2. Identification of DNA (de)methyltransferases and histone modifiers
Protein sequences of A. thaliana DNA methyltransferases and demethylases as well as histone acetyltransferases, deacetylases, methyltransferases, and demethylases were retrieved from The Arabidopsis Information Resource (TAIR) database (www.arabidopsis.org) from January 2021 to March 2022. With the retrieved full sequences as queries, we interrogated the transcriptomic datasets for orthologs using the NCBI tBLASTn online tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and local tBLASTn and BLASTp using BioEdit v.7.2.5 (Hall, 2011). The nomenclature used to describe genes follows the one of A. thaliana: the sequences with higher similarity received the same designation, whereas other less similar received the name “‐like.” Each “‐like” sequence found in a certain genus was also checked in the transcriptomes of other species from the same genus. With the sequences retrieved the genomic location and putative duplications were screened to detect possible paralogs. Paralog protein sequences were aligned by the EMBLE‐EBI MUSCLE alignment tool (Madeira et al., 2022) to retrieve the protein percent identities matrix. When paralogs were detected, the 5000 bp flanking upstream and downstream genomic regions were retrieved and submitted to GIRI Repbase (https://www.girinst.org/) using CENSOR (Kohany et al., 2006) to search for the presence of transposable elements (TEs). Only hits that aligned at least 500 bp or elements that had sequential hits were selected.
2.3. Prediction of protein domains
To check the putative protein structure, we analyzed the presence of specific conserved domains using the Conserved Domain Database (CDD) Batch‐search tool (Marchler‐Bauer et al., 2011; Marchler‐Bauer & Bryant, 2004) and the EBI online tool InterPro 87.0 (Blum et al., 2021).
2.4. Phylogenetic analysis
Phylogenetic and molecular evolutionary analyses were conducted in the MEGA version X (Kumar et al., 2018). For each family of sequences, an alignment was performed by using the MUSCLE algorithm, and trees were inferred by Maximum Likelihood and the Jones–Taylor–Thornton (JTT) matrix‐based model (Jones et al., 1992). The confidence level for each branch of the estimated trees was assessed by the bootstrap method with 1000 replicates. Initial trees for the heuristic search were obtained automatically by applying Neighbor‐Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with superior log likelihood value. Trees were redesigned with the Interactive Tree of Life Tool (iTOL; http://itol.embl.de/; Letunic & Bork, 2021). Incomplete or absent sequences in transcriptomic data were removed from phylogenetic analysis.
3. RESULTS
3.1. Fagus and Castanea transcriptome de novo assembly analysis
To gain knowledge about the epigenetic regulators from the species of Fagus and Castanea genera we de novo assembled the transcriptomes of F. grandifolia, F. sylvatica, F. crenata, C. henryi, and C. crenata by using publicly available data. After the library preparation and assembly, we evaluated their quality (Table S3). Contig number ranged from 514,313 to 111,872 for F. grandifolia and C. henryi, respectively. The average contig length varied between 329 bp for C. crenata and 786 bp for C. henryi and the GC content ranges between 38.9% in F. sylvatica and 42.3% in C. crenata. Comparing all assembled libraries, the lowest N50 was observed for C. crenata (528 bp), and the highest value for F. sylvatica (1771 bp). Transcriptomes assessed by BUSCO showed that the amount of completely assembled genes varied between 98.1% in F. grandifolia and 47.8% in C. crenata, with more duplicates appearing in F. grandifolia and F. sylvatica (>80.0%).
3.2. Sequence identification, characterization, and phylogenetic analysis
To better understand and characterize the epigenetic regulators in the Fagaceae family, we interrogated the genomes and/or the transcriptomes of 11 species of the three most representative genera: Fagus, Quercus, and Castanea. The presence of paralogs was checked in species with available genomes (Tables S4 and S5).
3.2.1. DNA methyltransferases (DNMTs)
We were able to identify MET1, CMT1, CMT2, CMT3, DNMT2, DRM2, and DRM3 proteins in all genera, and all identified proteins shared the conserved catalytic domains with homologs of Arabidopsis thaliana (Figure 1). We identified a duplication in DRM2 in all Quercus and Castanea species, but not in Fagus. Fagaceae DNA methyltransferases were generally longer than A. thaliana orthologs (Table S6), except for CMT2 and DRM2. These differences in length had no effect on the presence of the expected domains except for the DRM proteins where the three UBA domains present in Arabidopsis were never detected in Fagaceae (Figure 1). In all DNA methyltransferases identified, a DNA_methylase (PF00145) domain was present, except for DRM3 where this domain was only present in Fagus species. MET1 presented two DNMT1_Replication Foci domains (DNMT1_RFD—PF12047), and CMTs showed a BAH (PF01426) and a CHROMO domain (PF00385). All DRM2 and DRM3, had a SAM_MeTfrase_DRM (IPR029063) domain of DRMs class proteins, (not shown in Figure 1) and DRM2 displayed one or two UBA (PF00627).
FIGURE 1.

Phylogeny of DNA methyltransferases and their domain organization. The evolutionary history of DNA methyltransferases and their closest paralogs “‐like” was built with: CHROMOMETHYLASES 1–3 (CMT1–3), DNA (CYTOSINE‐5‐)‐METHYLTRANSFERASE 2 (DNMT2), DOMAINS REARRANGED METHYLTRANSFERASE 2 and 3 (DRM2/3), METHYLTRANSFERASE 1 (MET1), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values for 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber. BAH, bromo adjacent homology; CHROMO, chromatin organization modifier; DNMT1‐RFD, DNMT1_Replication foci domain
The phylogenetic analysis of the DNA methyltransferase proteins (Figure 1) clearly showed two main clades: the DRM group and the DNMT2/MET1/CMT group, separating the proteins responsible for de novo DNA methylation (DRM) from the others responsible for the maintenance of methylation. DRMs were grouped by the presence of UBA domains, since no UBA domain was identified in AthDRM3 in Quercus and in Castanea. In the CMT clade, CMT1 and CMT3 grouped together and CMT2 lied apart. All the proteins were clustered by genus supported by high bootstrap values, with Fagus at a basal position, and Castanea and Quercus closer to each other.
3.2.2. DNA demethylases (DDMTs)
We identified the DNA demethylases DME, ROS1, and DML‐like proteins with high similarity to A. thaliana in the three genera. All Fagaceae ROS1 sequences were longer than AthROS1 and DML‐like had almost twice the length of AthDML3 (Table S6).
The DNA demethylase sequences showed the three most characteristic domains (Figure 2): RNA Recognition motif domain (RRM_DME—PF15628), a Permuted version of a single unit of the zf‐CXXC domain (Perm‐CXXC—PF15629) and an Endonuclease III domain belonging to the HhH‐GPD superfamily domain (ENDO3c—PF00730) with its respective iron–sulfur binding subdomain (FES—SM00525) except for CcrDME and FcrDML‐like where the FES subdomain could not be detected (Figure 2). The phylogenetic analysis separated the DML‐like sequences from DME and ROS1, clustering each genus in their own clades.
FIGURE 2.

Phylogeny of DNA demethylases and their domain organization. The evolutionary history of DNA demethylases and their closest paralogs “‐like” was built with DNA demethylases DEMETER (DME), REPRESSOR OF SILENCING 1 (ROS1), DEMETER‐LIKE 3 and “‐like” (DML3/‐like), and inferred by using the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; ENDO3c, endonuclease III; FES, iron‐sulfur binding; perm‐CXXC, permuted version of a single unit of the zinc finger‐CXXC and RRM_DME, RNA recognition motif
3.2.3. Histone acetyltransferases (HATs)
The group of histone acetyltransferases was well represented in the Fagaceae family (Figure 3). We were able to identify six different families of HATs, divided into four distinct groups (G): (G1) GNAT with ELONGATOR COMPLEX PROTEIN 3 (ELP3), GENERAL CONTROL NONDEREPRESSIBLE 5 (GCN5), and HISTONE ACETYLTRANSFERASE OF THE GNAT FAMILY 2 (HAG2); (G2) MYST with HISTONE ACETYLTRANSFERASE OF THE MYST FAMILY 1 (HAM1); (G3) CBP with HISTONE ACETYLTRANSFERASE OF THE CBP FAMILY 1 (HAC1) and (G4) TAFII250 with HISTONE ACETYLTRANSFERASE OF THE TAFII250 FAMILY 1 (HAF1). All genes were present as single copies except for HAC1 of the CBP group with two copies in F. sylvatica, F. crenata, and Q. robur, and three paralogs in C. mollissima, C. crenata, Q. lobata, and Q. suber. There were no major differences in protein size between A. thaliana and the Fagaceae (Table S6), and the sequence domains were found as expected with a few differences like in QsuHAC1‐like2, which is most probably an incomplete sequence (Figure 3).
FIGURE 3.

Phylogeny of histone acetyltransferases and their domain organization. The evolutionary history of histone acetyltransferases was built with: HISTONE ACETYLTRANSFERASE OF THE MYST FAMILY 1 (HAM1), HISTONE ACETYLTRANSFERASE OF THE CBP FAMILY 1 and their closest paralogs “‐like” (HAC1/HAC1‐like1‐2), HISTONE ACETYLTRANSFERASE OF THE TAFII250 FAMILY 1 (HAF1), HISTONE ACETYLTRANSFERASE OF THE GNAT FAMILY 2 (HAG2), ELONGATOR COMPLEX PROTEIN 3 (ELP3), and GENERAL CONTROL NONDEREPRESSIBLE 5 (GCN5), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; Acetyltransf_1, acetyltransferase 1; CHROMO, chromodomain; HAT_KAT11, histone acetyltransferase domain with high similarity to the fungal KAT11 protein; Hat1_N, N‐terminal half of the structure of histone acetyl transferase HAT1; MOZ_SAS, region common to acetyltransferases from MYST family like MOZ and SAS proteins; TBP‐binding, TATA box‐binding protein‐binding; zf‐CCHC, CysCysHisCys type zinc finger knuckle; Zf_Myst, zinc finger domain from MYST class proteins; Zf_PHD_SD, PHD (homeodomain) zinc‐finger domain; zf‐TAZ, transcription adaptor putative zinc finger, and Zf_ZZ, ZZ‐type zinc finger
All GNAT proteins had an acetyltransferase_1 domain (PF00583) homologous to the Gcn5‐related N‐acetyltransferases domain (IPR000182). GCN5 proteins were characterized by also having a Bromodomain (PF00439), ELP3 sequences with the typical Elongator complex Protein‐like (ELP3—IPR006638) domain, and HAG2 proteins had the Hat1_N domain (N‐terminal half of the structure of histone acetyl transferase HAT1—PF10394) which was also present in A. thaliana. All HAM1 proteins had a MOZ_SAS domain (region common to acetyltransferases from MYST family like MOZ and SAS proteins‐PF01853) also called MYST domain (Latrasse et al., 2008) and a Zf_MYST domain (zinc finger domain from MYST class proteins—PF17772). All HAC1 proteins showed the HAT_KAT11 domain (histone acetyltransferase domain with high similarity to fungal KAT11 protein domain‐PF08214) typical for p300/CBP class proteins and the Zf_PHD_SF (PHD [homeodomain] zinc finger domain—PF00628). HAC1 sequences showed two Zf_TAZ domains (Transcription Adaptor putative Zinc finger‐PF02135) at the C‐terminus and at mid‐point positions, while HAC1‐like sequences only had the C‐terminus domain. Regarding the Zf_ZZ domains (ZZ‐type zinc finger domain‐PF00569), all HAC1 proteins had two domains closer to the C‐terminus Zf_TAZ domain, while HAC1‐like sequences only had the one closer to the HAT_KAT11 domain.
All HAF1 proteins found had the same functional domains, a TATA box‐binding protein‐binding domain (TBP‐binding—PF09247), a Ubiquitin domain (PF00240) and a Bromodomain. However, the CysCysHisCys type zinc finger knuckle found in TAFII class proteins (Zf‐CCHC—IPR041670) could not be detected in Q. robur. Interestingly we found a portion with high similarity to a Line‐1 retrotransposable element of Q. suber in the intronic sequence closer to the 5′ end of CcrHAF1, with no direct impact in the conserved domains detected (Figure 3).
The phylogenetic analysis of these proteins clustered them in two major clades with strong bootstrap values, one comprising HAF1 and GCN5 from the TAFII250 and GNAT families respectively, and the other with the remaining GNAT, MYST, and CBP families (Figure 3). In this way the members of the GNAT family did not form one clade: GCN5 was grouped with HAF1 (TAFII250) in the first major clade supported by a remarkably high bootstrap value (99%); while HAG2 is in the second major clade with ELP3 in a sister clade grouped with the other families supported by a medium bootstrap value (53%). The domain differences between ELP3 and the classes MYST and CBP, permitted a second level of separation, whereas HAF1 shared the same Bromodomain with GCN5. HAC1 and HAC1‐like sequences were positioned together, evidencing the differences in number of domains compared with the other HATs, and all the subclades of that group are supported by strong bootstrap values (>80%). The family genera were well individualized in all the proteins, but in HAM1 and ELP3 Arabidopsis sequences showed unusual clustering, supported by low bootstrap values.
3.2.4. Histone deacetylases (HDACs)
The acetylation in the lysine residues of histones H3 and H4 can be reversed by members of the histone deacetylases superfamily. In the Fagaceae we found homologs of eight HDA proteins of the RPD3/HDA group (HDA2, HDA5, HDA6, HDA8, HDA9, HDA14, HDA15, and HDA19), two SRT protein homologs (SRT1 and SRT2) from the Sirtuin group, and one HD2‐type group protein homolog (HDT3). In this superfamily, we found several duplications in the three genera: four genes of HDA14 in Q. robur while only three in Q. suber and Q. lobata; two paralogs of HDA19 and SRT1 in all species, and two SRT2 in C. crenata (Figure 4). All Fagaceae histone deacetylases and their paralogs had sizes similar to A. thaliana proteins (Table S6).
FIGURE 4.

Phylogeny of histone deacetylases and their domain organization. The evolutionary history of histone deacetylases and their closest paralogs “‐like” was built with: RPD3/HDA1 family (HDA1‐15), HDA2‐TYPE FAMILY HISTONES 3 (HDT3), SIRTUIN FAMILY HISTONES 1 and 2 (SRT1/2), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; Hist_deacetyl, histone deacetylase domain; NPL, nucleoplasmine‐like domain; SIR2, sirtuin 2 domain; Zf‐C2H2, C2H2 zinc finger and Zf‐RanBP2‐ new superfamily of C2C2‐type zinc finger motif
Domain analysis showed that all sequences bear their characteristic domains (Figure 4). HDA1 to HDA15 had the typical Histone deacetylase domain (Hist_deacetyl—PF00850), but the C2C2‐type zinc finger motif (Zf‐RanBP2—IPR001876) was only present in HDA15. HDT3 had a Nucleoplasmine‐like domain (NPL—PF17800) and the C2H2‐type zinc finger (Zf‐C2H2—IPR013087) domain while SRT1 and showed the Sirtuin 2 domain (SIR2—IPR026590). When trying to find the N‐terminal part of Ccr_SRT2‐like we found an insertion of a pentatricopeptide repeat‐containing protein At4g20740‐like which was disrupting the normal SRT2 protein.
The phylogenetic analysis of the histone deacetylase proteins first separated the HD2‐type protein HDT3 from the others, supported by a high bootstrap value (100%; Figure 4). The RPD3/HDA1 group is divided in two clades: HDA19, HDA6, and HDA2 joined with sirtuins and another with the remaining HDA. Species of the three genera were generally clustered together in each protein clade except for CcrHDT3‐like and CsaSRT2, which did not group with the other Castanea species.
3.2.5. Histone methyltransferases (HMTs)
Histone methyltransferases are a complex group of proteins with six classes responsible for methylation of a specific residue. In Fagaceae, we identified 37 proteins in total, including duplications (Figures 5, 6, 7, 8, 9, 10) distributed as follows: two Class I proteins (CURLY LEAF [CLF] and SWINGER [SWN]), four Class II proteins (ABSENT, SMALL, OR HOMEOTIC HOMOLOGS 1–3 and RELATED 3 [ASHH1, ASHH2, ASHH3, and ASHR3]), five Class III proteins (TRITHORAX‐LIKE 2–5 and RELATED 3 and 7 [ATX2, ATX3, ATX5, ATXR3, and ATXR7]), two Class IV proteins (ATXR5 and ATXR6), sixteen Class V proteins (SU(VAR) HOMOLOGS 1–9 and RELATED 1–5 [SUVH1, SUVH3, SUVH4, SUVH4‐like, SUVH5, SUVH5‐like1/2/3, SUVH6, SUVH6‐like, SUVH9, SUVR1, SUVR2, SUVR3, SUVR4, and SUVR5]) and eight Class VI proteins (ATXR1‐like, ATXR2, ATXR4, ASHR1, ASHR2, SET10, SET40, and SET41). All protein genes found were present in single copy, except for SUVH4, which was found duplicated in all Fagus and Quercus, and in C. crenata. SUVH5 had four copies in C. mollissima, three in C. crenata and Q. lobata, two in Q. robur and Q. suber and SUVH6 had two copies in C. crenata and C. mollissima. We could not find SUVR4, ATXR4, and SET41 in any Fagus species.
FIGURE 5.

Phylogeny of Class I histone methyltransferases and their domain organization. The evolutionary history of Class I histone methyltransferases and their closest paralogs “‐like” was built CURLY LEAF (CLF) and SWINGER (SWN), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; CXC, C‐X(6)‐C‐X(3)‐C‐X‐C motif; SANT, (switching‐defective protein 3 [Swi3]; adaptor 2 [Ada2]; nuclear receptor co‐repressor [N‐CoR]; transcription factor [TF]IIIB); SET, Su(var)3‐9, Enhancer‐of‐zeste and Trithorax domain
FIGURE 6.

Phylogeny of Class II histone methyltransferases and their domain organization. The evolutionary history of Class II histone methyltransferases and their closest paralogs “‐like” was built with: ABSENT, SMALL OR HOMEOTIC HOMOLOGS 1, 2, and 3 (ASHH1/2/3), ABSENT, SMALL OR HOMEOTIC RELATED 3 (ASHR3), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; AWS, Associated with SET; Post‐SET, C‐terminal associated with SET domain; SET, Su(var)3‐9, Enhancer‐of‐zeste and Trithorax domain; Zf‐CW, CW‐type zinc finger motif; Zf_PHD, PHD‐type zinc finger motif
FIGURE 7.

Phylogeny of Class III histone methyltransferases and their domain organization. The evolutionary history of Class III histone methyltransferases and their closest paralogs “‐like” was built with: TRITHORAX‐LIKE 2, 3, and 5 (ATX2/3/5), TRITHORAX RELATED 3 and 7 (ATXR3/7), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; FYRC, “FY‐rich” domain C‐terminal; FYRN, “FY‐rich” domain N‐terminal; GYF, glycine‐tyrosine‐phenylalanine; post‐SET, C‐terminal associated with SET; PWWP, pro‐Trp‐Trp‐pro motif; SDG2_C, SET DOMAIN GROUP 2 that includes the C terminus post‐SET; SET, Su(var)3‐9, Enhancer‐of‐zeste and Trithorax domain; Zf_PHD, PHD‐type zinc finger motif
FIGURE 8.

Phylogeny of Class IV histone methyltransferases and their domain organization. The evolutionary history of Class IV histone methyltransferases was built with: TRITHORAX RELATED 5 and 6 (ATXR5/6), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber. SET, Su(var)3‐9, Enhancer‐of‐zeste and Trithorax domain; Zf_PHD, PHD‐type zinc finger motif
FIGURE 9.

Phylogeny of Class V histone methyltransferases and their domain organization. The evolutionary history of Class V histone methyltransferases and their closest paralogs “‐like” was built with: SUPPRESSOR OF POSITION‐EFFECT VARIEGATION (SU[VAR]) HOMOLOGS 1, 3, 4, 5, 6, and 9, (SUVH1/3/4/5/9), SU(VAR) RELATED 1, 2, 3, 4, and 5 (SUVR1/2/3/4/5), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; AWS, associated with SET; post‐SET, C‐terminal associated with SET domain; SET, Su(var)3‐9, Enhancer‐of‐zeste, and Trithorax domain; SRA_YGD, SET and ring finger associated, and YDG motif; WIYLD, named after most conserved residues; and Zf_C2H2, C2H2‐type zinc finger motif
FIGURE 10.

Phylogeny of Class VI histone methyltransferases and their domain organization. The evolutionary history of Class VI histone methyltransferases and their closest paralogs “‐like” was built SU(VAR)3‐9, ENHANCER‐OF‐ZESTE AND TRITHORAX 10, 40, and 41 (SET10/40/41), TRITHORAX RELATED 1, 2, and 4 (ATXR1/2/4), ABSENT, SMALL OR HOMEOTIC RELATED 1 and 2 (ASHR1/2), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; SET, Su(var)3–9; Enhancer‐of‐zeste and Trithorax domain; TPR_12, tetratrico peptide repeat region; and zf‐MYND, MYND‐type zinc finger
Taken together, histone methyltransferases were very different in length, and ATXR3 from Class III were the longest proteins (Table S6). FcrATXR3 (2446 amino acids [aa]) had a major difference to AthATXR3 (2335 aa). ASHH2 from Class II is 2084 aa or longer, varying between QroASHH2 (2149 aa) and AthASHH2 more than 648 aa. Class II included the highest range of sequence sizes, from FsyASHH3 and FgrASHH3 with 365 aa to QroASHH2 with 2149 aa. The smallest histone methyltransferase was CmoATXR4 (331 aa) from Class VI.
Despite the differences in length to A. thaliana histone methyltransferases, the HMTs of all classes identified here had their specific domains and showed the typical SET domain (IPR001214), except for SUVH5‐like1, and SUVH5‐like3 (Figure 9). Class I CLF (Figure 5) and SWN had a C‐X(6)‐C‐X(3)‐C‐X‐C motif domain (CXC—IPR026489), and Fagus SWN had additionally a Switching‐defective protein 3 (Swi3), Adaptor 2 (Ada2), Nuclear receptor Co‐Repressor (N‐CoR) and transcription factor (TF)IIIB (SANT—IPR001005) domain, also detected in Arabidopsis. Additionally, all Class II proteins showed an Associated With SET domain (AWS—IPR006560), and except for QsuASHH1, they all had a C‐terminal associated with the SET domain (Post‐SET— IPR003616; Figure 6). Additional specific protein domains such as the CW‐type zinc finger motif (Zf‐CW—PF07496) and the PHD‐type zinc finger motif (Zf_PHD—IPR001965) were also detected in all ASHH2. In Class III proteins (Figure 7), all ATX displayed the SET and Post‐SET domains, and additionally the Pro‐Trp‐Trp‐Pro motif domain (PWWP—PF00855). ATXR 3, instead of the typical Post‐SET, showed a SET DOMAIN GROUP 2 that includes the C terminus Post‐SET domain (SDG2_C—IPR045606). ATX2 presented further specific domains as the “FY‐rich” N‐terminal (FYRN—PF05964), the “FY‐rich” C‐terminal (FYRC—PF05965), and two Zf_PHD. ATX3 and ATX5 showed only three Zf_PHD domains. Moreover, all ATXR7 and AthATXR3 had a Glycine‐tyrosine‐phenylalanine (GYF—IPR025640) domain. All Class IV proteins presented a SET and a Zf_PHD domain (Figure 8). In Class V, all SUVH proteins showed the SET and Ring finger Associated, and YDG motif domain (SRA_YGD—PF02182). Except SUVH5‐like1, which lacked all SET domains, and SUVH5, CdeSUVH1, SUVH9, SUVR2, and SUVR4, that lacked the post‐SET domain, they all had pre‐SET, SET, and post‐SET domains (Figure 9). All SUVR except SUVR2 had pre‐SET, SET, and post‐SET domains. Additionally, all SUVR1, SUVR2, and SUVR4 had a WIYLD domain (named after most conserved residues—PF10440), while SUVR5 showed a Zf_C2H2 domain, and only Castanea SUVR3 displayed an AWS domain. In Class VI, all proteins had SET domains, being the only domain of SET10, SET41, ASHR2, and ATXR4 (Figure 10). SET40 also had a Rubisco LSMT Substrate‐binding domain (RBS—IPR015353). ATXR1 had in addition to the SET domain, a tetratricopeptide repeat region domain (TPR_12—IPR011990), ATXR2 displayed the MYND‐type zinc finger domain (zf‐MYND—PF01753), and ASHR1 had all three domains.
The phylogenetic analysis of histone methyltransferases Class I showed two well supported groups, with the clear distinction between genera (Figure 5), although QsuCLF was separated from the clade of Quercus and Castanea species. In Class II, two major clades separated ASHH2 from the remaining proteins ASHH3, ASHR3, and ASHH1 (Figure 6). All Class II protein sequences respected the genus relationships except QloASHH3, which was grouping with Castanea. Class III sequences were resolved in two main clades, the ATXR and the ATX clades (Figure 7). Class III proteins were grouped within each genus, always showing Fagus as a sister genus of Castanea and Quercus. In Class IV, ATXR5 and ATXR6 formed their own clades (Figure 8) with the genera well resolved. Class V histone methyltransferases were arranged in two main clades (Figure 9): one enclosing SUVR1, SUVR2, and SUVR4 with the similarities between SUVR1 and SUVR2 higher than with SUVR4; the other major clade with the remaining proteins where SUVR3 and SUVR5 were grouped together diverging from all the SUVHs. Also here, the A. thaliana pairs SUVH1/SUVH3 and SUVH5/SUVH6 were closer to each other than with the respective Fagaceae homologs. Class V proteins were generally arranged in genus clades. In Class VI, one main clade comprehended SET10 and SET40 (Figure 10), and the other included ATXR2, ATXR4, ASHR2, ASHR1, SET41, and ATXR1. In ASHR2 the Castanea genus behaves as a sister clade of Quercus and Fagus, which was not in line with the accepted phylogenetic relationship. The Arabidopsis orthologs AthATXR2 and AthSET40 unexpectedly grouped with Quercus and Castanea clade or with the Fagus clade, respectively.
3.2.6. Histone demethylases (HDMTs)
Methylation of histones can be reverted by histone demethylases, which can be divided into two families: JMJ and LSD1/KDM (Figures 11 and 12). In Fagaceae, we found 19 different JMJ proteins (JMJ13, JMJ13‐like, JMJ16, JMJ17, JMJ18, JMJ19, JMJ20, JMJ21, JMJ22, JMJ22‐like, JMJ24, JMJ26, JMJ27, JMJ28, JMJ30, JMJ32, IBM1, ELF6, and REF6), and four LSD1/KDM proteins (FLD, LDL1, LDL2, and LDL3). Except for JMJ13, which was duplicated in Q. lobata, Q. suber, and C. crenata, and JMJ22 with two copies in Q. suber, all other HDMTs protein genes were found as single copies.
FIGURE 11.
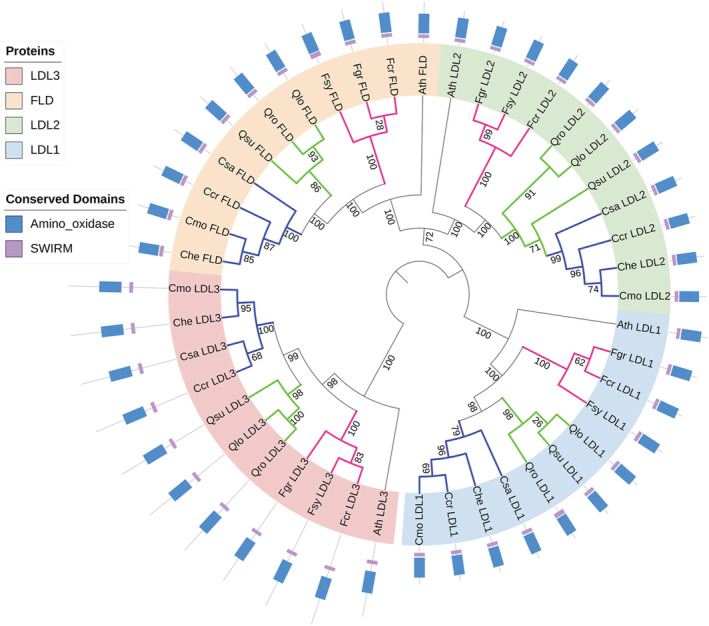
Phylogeny of KDM/LSD1 class histone demethylases and their domain organization. The evolutionary history of KDM/LSD1 class histone demethylases and their closest paralogs “‐like” was built with: LYSINE‐SPECIFIC HISTONE DEMETHYLASE 1 HOMOLOG 1, 2 and 3 (LDL1/2/3) and FLOWERING LOCUS D (FLD), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus; green—Quercus; blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; SWIRM, SWI3, RSC8 and MOIRA domain; and Amino_oxidase, Flavin containing amine oxidoreductase
FIGURE 12.

Phylogeny of JMJ class of histone demethylases and their domain organization. The evolutionary history of JMJ class of histone demethylases and their closest paralogs “‐like” was built with: JUMONJI 13–32 (JMJ13–32), RELATIVE OF EARLY FLOWERING 6 (REF6), EARLY FLOWERING 6 (ELF6) and IMBIBITION‐INDUCIBLE 1 (IBM1), and inferred with the Maximum Likelihood method. The numbers on the branches indicate the bootstrap values from 1000 replicates and the colors of the branches highlight the genera: magenta—Fagus, green—Quercus, blue—Castanea. The protein sequences are represented by a line proportional to amino acid sequence size in which the respective conserved domains are present. Ath, Arabidopsis thaliana; Ccr, C. crenata; Cde, C. dentata; Che, C. henryi; Cmo, C. mollissima; Csa, C. sativa; Fcr, F. crenata; Fgr, F. grandifolia; Fsy, F. sylvatica; Qlo, Q. lobata; Qro, Q. robur; Qsu, Q. suber; ARID, AT‐rich interaction domain; FYRC, “FY‐rich” domain C‐terminal; FYRN, “FY‐rich” domain N‐terminal; JmjC, jumonji family domain with a cupin fold; JmjN, jumonji domain closer to N‐terminal; PLU‐1, PLU‐1‐like domain; PHD, PHD finger; WRC, Trp‐Arg‐Cys motif; Zf_C2H2, zinc finger C2H2‐type; zf‐C5HC2, zinc‐finger C5HC2‐type; and Zf‐RING, zinc‐finger RING type domain
The length of these proteins ranged from around 375 aa in JMJ32 up to around 2100 aa in LDL3 and JMJ27 in all the Fagaceae species (Table S6), the latter showing the major difference between A. thaliana (840 aa) and Castanea (~2086 aa). In most of the proteins, Arabidopsis and Fagaceae proteins had similar lengths.
The Fagaceae histone demethylases KDM/LSD1 class showed the expected domains: SWI 3, R SC8, M OIRA domain (SWIRM—PF04433) and the Flavin containing amine oxidoreductase domain (amino_oxidase—PF01593; Figure 11). All jumonji class proteins had the jumonji family domain with a cupin fold domain (JmjC—PR003347; Figure 12). JMJ20, JMJ30, and JMJ32 only had the JmjC domain. Except for JMJ21, all the Fagaceae proteins shared the same domains and in the same order with the A. thaliana orthologs. However, Fagaceae proteins sometimes presented additional domains. ELF6 and REF6 additionally had a jumonji domain closer to the N‐terminal end (JmjN—PF02375) and four Zf_C2H2 domains. All JMJ13, JMJ13‐like, and JMJ19 also had a zinc‐finger C5HC2‐type domain (zf‐C5HC2—PF02928). Additionally to these latter three domains, JMJ17 displayed an AT‐Rich Interaction Domain (ARID—PF01388), two PLU‐1‐like domains (PLU‐1—PF08429), and two PHD finger domains (PHD—PF00628), while JMJ16 and JMJ18 had a FYRC domain and a FYRN domain. Qsu_JMJ16 missed a large portion of the C′‐terminus region where FYRC and FYRN domains should be located. We were unable to find the missing part because of its assembly into the terminal part of the scaffold 18,358 (Table S5). Additionally to the JmjC domain, all Fagaceae JMJ21 and JMJ22 had an F‐box‐like domain (F‐box—IPR001810). IBM1 and JMJ26 displayed the JmjC domain and zinc‐finger RING type domain (Zf‐RING—IPR001841). JMJ24, JMJ27, and JMJ28 had the JmjC, Zf‐RING, and a Trp‐Arg‐Cys motif domains (WRC—PF08879).
Protein sequences were used to generate two phylogenetic trees: one for the KDM/LSD1 group and another for the JMJ group. The KDM/LSD1 tree (Figure 11) showed the individualization of the LDL3 group from the other LSD1 proteins, with the orthologs clustered by genera except QsuLDL2, which grouped with the Castanea species. The Jumonji class tree (Figure 12) showed all the 17 JMJ proteins separated in distinct clades where the genera were also grouped. Two main groups were individualized: one with JMJ22 and JMJ30 and the proteins harboring the zinc‐finger Ring cladded, including the proteins responsible for the H3K9 demethylation in the same clade; and another group with all JmjN domain‐bearing proteins together with JMJ20, JMJ21, and JMJ32. In this group, JMJ19 and the H3K4 demethylases JMJ16, JMJ17, JMJ18 form their own clade, with the H3K27 demethylases ELF6, REF6, and JMJ13 further away from them.
3.3. Flanking regions in gene duplication events
To assess the similarity of the paralogs found and how recent the duplication events might have occurred, we generated a similarity matrix (Table S7). Similarity values between paralogs range from 37.8% to 99.7% in Q. suber JMJ22 and its paralogs, and in Quercus HDA14 proteins, respectively.
To understand if the duplication events could have been caused by TEs activity, we analyzed the upstream and downstream 5000 bp flanking regions of genes. We selected TEs that aligned in at least 500 bp or that had sequential hits. With this approach, we detected several classes of TEs (Table S8) such as LTR (long terminal repeats), Non‐LTR retroelements, and DNA transposons. Copia‐like and Gypsy‐like LTR retroelements represented 25.0% and 14.3% of the hits respectively, in which SCL‐Bianca and Copia‐31 showed the highest scores (>14,000 and >17,000, respectively). For the Non‐LTR class only LINE elements (long interspersed nuclear elements) were present (39.3%). Finally, DNA transposons represented by the hAT, Helitron, and Harbinger families comprised 17.8% of the elements found. However, only in QsuSRT1 paralogs we were able to detect the LINE1‐39_OS retroelement flanking both 5′ and 3′ ends.
4. DISCUSSION
In this study, we report the Fagaceae family epigenetic regulators toolbox identified by genome‐wide identification of DNA and histone modifiers in seven species belonging to Fagus, Quercus, and Castanea genera. For this, taking advantage of open source genomic and transcriptomic data, we characterized their putative conserved domains, identified gene duplications, and established the phylogenetic relations between protein families in the distinct species.
The assembly genome level and quality proved to be crucial factors to efficiently identify the set of epigenetic regulators of a species. F. sylvatica and Q. lobata are the only species studied which genomes are assembled with chromosome identification (Mishra et al., 2018; Sork et al., 2016). This allowed us to identify and localize the genes in their genomic context. Nevertheless, all genome assemblies permitted us to infer the presence of paralogs, deducing gene duplication events.
At the transcriptome level and based on the resulting percentage of detected genes, a higher number of available reads did not seem to directly influence the quality of our transcriptome assemblies, contrary to what was previously suggested (Raghavan et al., 2022). However, a slight tendency for more complete duplicate (CD) genes can be found as we used 51 SRA libraries in the F. grandifolia assembled transcriptome and the number of CDs was ~85% (Table S3). The reason could be that a redundancy removal was not performed (Ono et al., 2015), while in C. henryi only one SRA library was used, and the number of CDs drops to ~41%.
4.1. Epigenetic regulators in Fagaceae exhibit duplications and are well conserved
In species with available genomes, we identified around 90 epigenetic regulators while in transcriptome data we found only about 80 genes (Figures 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12; Tables S5 and S6). Our transcriptome de novo assemblies evidenced to be very adequate to detect our genes of interest, and the use of both genome and transcriptome libraries proved to be an efficient way to find complete epigenetic regulator sequences, as a combination of data gives more robust results (Bolger et al., 2018).
The phylogenetic and predicted domain analysis suggests that epigenetic regulator proteins are very conserved among Fagaceae and within A. thaliana, indicating that they might have similar functions. This is the case of the histone acetyltransferase GCN5, which is responsible for the acetylation of histones H3 and H4 and has been associated to various developmental processes and resistance to abiotic stresses (Gan et al., 2021). Moreover, high levels of GCN5 were also reported as being associated with dormancy in C. sativa (Santamaría et al., 2011). Nevertheless, there are some differences in the number and domain composition of some proteins when comparing with A. thaliana. This is the case for methyltransferases DRMs (Figure 1) and some HATs (Figure 3), in which both have different numbers in Arabidopsis and Fagaceae (Pandey, 2002; Pavlopoulou & Kossida, 2007; Zhong et al., 2015). Arabidopsis has three DRM genes (DRM1, DRM2, and DRM3) each with specific functions in DNA methylation (Zhong et al., 2015). In Arabidopsis, DRM1 works together with DRM2 to perform de novo DNA methylation (Jullien et al., 2012). In Fagaceae, only homologs for DRM2 and DRM3 were found, and no homolog with similarity to AthDRM1 was detected, although we found two paralogs of DRM2 restricted to Quercus and Castanea. Likewise, A. thaliana has five different HATs (Pandey, 2002). However, we found two to three paralogs of HAC1 proteins in Fagaceae, also restricted to Quercus and Castanea. AtHAC1 has a high affinity to acetylate H3K9 and H3K14 but can also acetylate lysine residues in histone H4 (Earley et al., 2007). This protein is known to be involved in the regulation of flowering time by repressing the FLOWERING LOCUS C in Arabidopsis (Deng et al., 2007). In C. sativa a correlation has been detected between the high level of acetylated H4 and bud burst when compared to bud dormancy (Santamaría et al., 2009), stressing the role of these proteins in important developmental processes in Fagaceae. Both DRM2 and HAC1‐like in Fagaceae have also a different number of domains when compared to A. thaliana. The DRM2 UBA domain is essential for de novo activity and while AtDRM2 exhibits three sub‐units of these domains, the majority of the Fagaceae studied only exhibit one of two UBA subunits, like Nicotiana tabacum (Zhong et al., 2014). In Q. suber DRM2 is essential for the differentiation of cork tissue. Its expression is high in cells derived from the active phellogen (Inácio et al., 2018; Ramos et al., 2013), indicating that the Fagaceae DRM2, although lacking the third UBA subunit, is functional. Such slight differences in the number of predicted domains were found in several proteins of all types of epigenetic regulators. Functional studies would help to unveil if these changes in Fagaceae cause alterations in protein function.
Gene duplications have been detected in all families of epigenetic regulators studied in this work. Histone deacetylases HDA14, HDA19, HDT3, and SRT1 (Figure 4; Tables S5 and S6) showed the highest number of duplication events. The histone deacetylase HDA19 is well known to be involved in plant development, as it directly controls the expression of two flowering repressor genes related to the gibberellin signaling pathway, regulating flowering time in a photoperiod‐dependent way (Ning et al., 2019). In Arabidopsis, four HD2‐type proteins have been described, and they participate in diverse plant processes, such as acetylation of genes related to root and reproductive development (Li et al., 2017; Luo et al., 2022). In Fagaceae, we found duplications of these genes with similarities ranging from >57% in HDT3 up to 99% in HDA14 (Table S7), indicating different duplication events over time. Fagaceae have two HDT3 paralogs in all species, which are present in different chromosomes in all genera. They have similarities around 60% and form well‐defined clades, suggesting an old event of duplication. This is identical to what was observed between SRT1 and SRT1‐like where there is little less than 70% similarity between them (Table S7), and except for Q. robur, are found in different chromosomes. The number of SRTs in Fagaceae is different from A. thaliana where only SRT1 and SRT2 have been described (Zheng, 2020). AtSRT1 has important regulatory functions, such as in stress responses, as it negatively regulates plant tolerance to stress and glycolysis but stimulates mitochondrial respiration (Liu et al., 2017). However, three SRTs have been also described in soybean and in the lower plant Marchantia polymorpha (Zheng, 2020). Moreover, these proteins exist in higher numbers in other organisms, associated with different cellular locations (Szućko, 2016). Contrastingly, HDA14 duplications, restricted to Quercus genera, seem to have occurred more recently as the similarity between sequences is higher (Table S7) and all lie in the same chromosome (Tables S4 and S5) suggesting a proximal duplication event (Qiao et al., 2019).
Although we found homologs in every epigenetic regulator family, some important genes with known functions could not be identified in our data. This is the case of the histone methyltransferase, MEDEA relevant to Arabidopsis embryo development (Simonini et al., 2021), and SUVR4, ATXR4, and SET41 which are absent in Fagus. SUVR4 is a nucleolar histone methyltransferase with preference for monomethylated H3K9 that converts it to a di‐ or tri‐methylated state and is closely related to SUVR1 and SUVR2 (Xu & Jiang, 2020). In Arabidopsis, the SUVR2 seems to lack histone methyltransferase activity although it forms a complex with SUVR1 participating in gene silencing through the RdDM pathway and it was also suggested that the conserved catalytic sites of SUVR2 are dispensable for the function in transcriptional silencing (Han et al., 2014). This stresses the importance of SUVR4 in the tri‐methylation of H3K9, as it is the only known histone methyltransferase able to perform this function in a DNA methylation independent manner, since in Arabidopsis SUVH4/SUVH5/SUVH6 have distinct DNA binding preferences through their SRA domain (Xu & Jiang, 2020). The fact that we were not able to identify it in Fagus (Figure 9), although it was found in Quercus and Castanea suggests that a deletion might have occurred in the beeches after divergence from their closest relative, and the SUVR4 function might have been taken by other proteins, eventually one of the SUVH4 and SUVH5 paralogs, since they also have an affinity to H3K9. Despite the importance of these proteins in controlling gene expression in response to internal and external cues, events of loss and gain of these genes seem to have happened during the evolution of this family.
4.2. TEs could be involved in Fagaceae gene duplication
Phylogenetic analyses allowed us to evidence the diversification of the epigenetic regulators among the Fagaceae species, exposing several cases of duplications such as in DRM2, HAC1, HDA14, HDA19, SRT1, SUVH4, and SUVH5 proteins. Due to less variety of epigenetic regulators in Fagaceae than in Arabidopsis (23 vs. 48 HMTs and 9 vs. 12 HATs; Gao et al., 2021; Zhou et al., 2020), our results suggest that there were less events of duplication in Fagaceae species. This might be related to the two duplication events proposed to have occurred in Arabidopsis (del Pozo & Ramirez‐Parra, 2015) while oaks did not experience a lineage‐specific whole‐genome duplication apart from the triplication event of the ancestral eudicot karyotype (Plomion et al., 2018). An alternative hypothesis is that some Fagaceae proteins might have suffered deletion events which could be related with the presence of high number of TEs that are present in gene rich regions of the Fagaceae genomes (Alves et al., 2012; Mishra et al., 2022; Rocheta et al., 2012). In the human genome, the high abundance of LINE‐1 and Alu repeats favors recombination between non‐homologous loci leading to significant chromosomal rearrangements such as gene duplications and deletions (Chénais, 2022). Non‐LTR and LTR elements are represented in our data (Table S8), which is in overall accordance with what is described for their genomic representativity in these species (Mishra et al., 2022; Ramos et al., 2018; Shirasawa et al., 2021; Wang et al., 2020). Fagaceae genomes are rich in repetitive sequences, namely TEs, with values varying between ~59% and 12% in C. crenata and Q. suber, respectively (Ramos et al., 2018; Shirasawa et al., 2021). They are known to be scattered all over the genome, including in euchromatin regions (Alves et al., 2012; Quesneville, 2020). This genomic disposition may lead to some elements escaping silencing. The di‐methylation of lysine 9 of histone H3 is performed by SUVH4/5/6 proteins associated with methylated DNA, being fundamental to block TE activity (Cheng et al., 2020; Quesneville, 2020). In fact, in Q. suber cork development, QsSUVH4 expression was confirmed in young and traumatic periderms although in very low levels (Inácio et al., 2018), despite the detection of the H3K9me2 mark in cork oak periderms (Ribeiro et al., 2009). The low gene expression level could lead to a poor inactivation likely responsible for the duplication events detected, and for the reallocation of genes observed. In this work, we found a high similarity Line‐1 retrotransposable element of Q. suber in the intronic sequence of CcrHAF1, which could lead to its disruption or silencing (Quesneville, 2020). Moreover, in Arabidopsis some TEs target active gene regions as they recognize the histone marks H3K4me3 and H3K36me3 and tend to select loci responsible for environmental responses (Quesneville, 2020). As such we found evidence of TEs in adjacent areas of the genes (Table S8) although no specific element could be directly associated with the duplications detected and even though retroelements comprised around 80% of the repeats found in the adjacent areas of the paralogs.
5. CONCLUSION
The characterization of protein sequences and domain predictions provide valuable tools to study the evolutionary processes that epigenetic regulators underwent to meet the needs of each species and to cope with the environmental conditions in each ecosystem. The identification, domain prediction, and phylogenetic analysis of multiple Fagaceae epigenetic regulators will facilitate functional studies that will highlight potential additional roles related with gene duplications.
AUTHOR CONTRIBUTIONS
Ângelo Braga, Ana Teresa Alhinho, and Miguel Jesus Nunes Ramos contributed to the transcriptome assemblies. Sofia Alves, Ângelo Braga, Denise Parreira, and Helena Silva performed the bioinformatics analysis. Sofia Alves wrote the manuscript, with contributions from all the authors. Leonor Morais‐Cecílio supervised all analysis. Leonor Morais‐Cecílio and Maria Manuela Ribeiro Costa obtained the funding. All authors have read and agreed to the published version of the manuscript.
FUNDING INFORMATION
This work was supported and co‐financed by the European Regional Development Fund (ERDF), through the Operational Programme for Competitiveness and Internationalization (COMPETE 2020), under Portugal 2020 and by the Fundação para a Ciência e a Tecnologia—FCT I.P. (National Agency for Science and Technology) with the project grant POCI‐01‐0145‐FEDER‐027980/PTDC/ASP‐SIL/27980/2017‐“FlowerCAST”; Centre of Molecular and Environmental Biology (CBMA) was funded by “Contrato‐Programa” UIDB/04050/2020 and LEAF by the project UID/AGR/04129/2020 funded by national funds through the FCT I.P. Sofia Alves and Ana Teresa Alhinho were funded by the FCT grants SFRH/BD/146660/2019 and SFRH/BD/136834/2018, respectively.
Supporting information
Table S1. Genome assemblies accession numbers. Accession numbers of the genome assemblies used in our work. Assembly method and sequencing method was annotated when available in the NCBI assembly database.
Table S2. SRA raw data used for transcriptome library assembly. NCBI Sequence read archive accession numbers and respective information on the strategy, experimental design, and sequencing methods used to obtain the libraries.
Table S3. De novo assembly statistics of Fagaceae transcriptomes and their respective completeness as determined by BUSCO. N50 defines as length of the shortest contig for which longer and equal length contigs cover at least 50% of the assembly. BUSCO (Benchmarking Universal Single‐Copy Orthologous) statistics are Complete‐single‐copy (CSC), Complete Duplicate (CD), Fragmented (Frag), and Missing (Miss).
Table S4. Genomic location of all epigenetic regulators in the current study.
Table S5. Simplified table of the genomic locations of the different epigenetic regulators studied, with their respective paralogs.
Table S6. Protein length of each identified epigenetic regulator. Length units are shown in number of amino acid.
Table S7. Identities between paralogs given by MUSCLE alignment. Numbers are given as percentages.
Table S8. Selection of the results for the 5000 bp adjacent regions of the identified paralogs. Dir‐indicates the orientation of the element regarding the sequence (“d” is for direct and “c” is for complementary); Sim—similarity between the aligned fragments; Pos/Mm:Ts—is the ratio between Positives hits in alignment length (Pos), and the ratio of Mismatches to Transitions in nucleotide alignment (Mm:Ts).
ACKNOWLEDGMENTS
The authors would like to acknowledge Carolina Soares, Rui Figueira, and the PORBIOTA Project, for their contribution to the bioinformatics analysis, and the Galaxy Project for the freely available resources that made several transcriptome assemblies possible.
Alves, S. , Braga, Â. , Parreira, D. , Alhinho, A.T. , Silva, H. , Ramos, M.J.N. et al. (2022) Genome‐wide identification, phylogeny, and gene duplication of the epigenetic regulators in Fagaceae. Physiologia Plantarum, 174(5), e13788. Available from: 10.1111/ppl.13788
Edited by R. Vetukuri
Funding information Fundação para a Ciência e a Tecnologia, Grant/Award Numbers: POCI‐01‐0145‐FEDER‐027980/PTDC/ASP‐SIL/27980/2017, SFRH/BD/136834/2018, SFRH/BD/146660/2019, UID/AGR/04129/2020, UIDB/04050/2020
DATA AVAILABILITY STATEMENT
The data that support the findings were derived from the resources available in the public domain and are available in the Supporting Information of this article.
REFERENCES
- Albini, S. , Zakharova, V. & Ait‐Si‐Ali, S. (2019) Chapter 3—Histone modifications. In: Palacios, D. (Ed.) Epigenetics and regeneration. Cambridge, MA: Academic Press, pp. 47–72. [Google Scholar]
- Alhinho, A.T. , Ramos, M.J.N. , Alves, S. , Rocheta, M. , Morais‐Cecílio, L. , Gomes‐Laranjo, J. et al. (2021) The dynamics of flower development in Castanea sativa Mill. Plants, 10(8), 1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves, S. , Ribeiro, T. , Inácio, V. , Rocheta, M. & Morais‐Cecílio, L. (2012) Genomic organization and dynamics of repetitive DNA sequences in representatives of three Fagaceae genera. Genome, 55(5), 348–359. [DOI] [PubMed] [Google Scholar]
- Arias‐Baldrich, C. , Silva, M.C. , Bergeretti F., Chaves I., Miguel C., Saibo N.J.M., et al (2020) CorkOakDB—the cork oak genome database portal. Database, 2020, baaa114. [DOI] [PMC free article] [PubMed]
- Bewick, A.J. & Schmitz, R.J. (2017) Gene body DNA methylation in plants. Current Opinion in Plant Biology, 36, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum, M. , Chang, H.Y. , Chuguransky, S. , Grego, T. , Kandasaamy, S. , Mitchell, A. et al. (2021) The InterPro protein families and domains database: 20 years on. Nucleic Acids Research, 49(D1), D344–D354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, M.E. , Arsova, B. & Usadel, B. (2018) Plant genome and transcriptome annotations: from misconceptions to simple solutions. Briefings in Bioinformatics, 19(3), 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycheva, I. , Vassileva, V. & Iantcheva, A. (2014) Histone acetyltransferases in plant development and plasticity. Current Genomics, 15(1), 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chénais, B. (2022) Transposable elements and human diseases: mechanisms and implication in the response to environmental pollutants. International Journal of Molecular Sciences, 23(5), 2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, K. , Xu, Y. , Yang, C. , Ouellette, L. , Niu, L. , Zhou, X. et al. (2020) Histone tales: lysine methylation, a protagonist in Arabidopsis development. Journal of Experimental Botany, 71(3), 793–807. [DOI] [PubMed] [Google Scholar]
- del Pozo, J.C. & Ramirez‐Parra, E. (2015) Whole genome duplications in plants: an overview from Arabidopsis. Journal of Experimental Botany, 66(22), 6991–7003. [DOI] [PubMed] [Google Scholar]
- Deng, W. , Liu, C. , Pei, Y. , Deng, X. , Niu, L. & Cao, X. (2007) Involvement of the histone acetyltransferase AtHAC1 in the regulation of flowering time via repression of FLOWERING LOCUS C in Arabidopsis. Plant Physiology, 143(4), 1660–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, C.G. , Zhu, J.K. & Cao, X. (2018) Retrospective and perspective of plant epigenetics in China. Journal of Genetics and Genomics, 45(11), 621–638. [DOI] [PubMed] [Google Scholar]
- Earley, K.W. , Shook, M.S. , Brower‐Toland, B. , Hicks, L. & Pikaard, C.S. (2007) In vitro specificities of Arabidopsis co‐activator histone acetyltransferases: implications for histone hyperacetylation in gene activation. The Plant Journal, 52, 615–626. [DOI] [PubMed] [Google Scholar]
- Finnegan, E.J. & Kovac, K.A. (2000) Plant DNA methyltransferases. Plant Molecular Biology, 43, 189–201. [DOI] [PubMed] [Google Scholar]
- Gan, L. , Wei, Z. , Yang, Z. , Li, F. & Wang, Z. (2021) Updated mechanisms of GCN5—the monkey king of the plant kingdom in plant development and resistance to abiotic stresses. Cell, 10(5), 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, S. , Li, L. , Han, X. , Liu, T. , Jin, P. , Cai, L. et al. (2021) Genome‐wide identification of the histone acetyltransferase gene family in Triticum aestivum . BMC Genomics, 22(1), 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. , Levin, J.Z. , Thompson, D.A. , Amit, I. et al. (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nature Biotechnology, 29(7), 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugger, P.F. , Fitz‐Gibbon, S. , Pellegrini, M. & Sork, V.L. (2016) Species‐wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Molecular Ecology, 25(8), 1665–1680. [DOI] [PubMed] [Google Scholar]
- Hall, T. (2011) BioEdit: an important software for molecular biology. GERF Bulletin of Biosciences, 2(1), 60–61. [Google Scholar]
- Han, Y.F. , Dou, K. , Ma, Z.Y. , Zhang, S.W. , Huang, H.W. , Li, L. et al. (2014) SUVR2 is involved in transcriptional gene silencing by associating with SNF2‐related chromatin‐remodeling proteins in Arabidopsis. Cell Research, 24(12), 1445–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrivnák, M. , Krajmerová, D. , Frýdl, J. & Gömöry, D. (2017) Variation of cytosine methylation patterns in European beech (Fagus sylvatica L.). Tree Genetics & Genomes, 13(6), 117. [Google Scholar]
- Hubert, F. , Grimm, G.W. , Jousselin, E. , Berry, V. , Franc, A. & Kremer, A. (2014) Multiple nuclear genes stabilize the phylogenetic backbone of the genus Quercus . Systematics and Biodiversity, 12(4), 405–423. [Google Scholar]
- Inácio, V. , Barros, P.M. , Costa, A. , Roussado, C. , Gonçalves, E. , Costa, R. et al. (2017) Differential DNA methylation patterns are related to phellogen origin and quality of Quercus suber cork. PLoS One, 12(1), e0169018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inácio, V. , Martins, M.T. , Graça, J. & Morais‐Cecílio, L. (2018) Cork oak young and traumatic periderms show PCD typical chromatin patterns but different chromatin‐modifying genes expression. Frontiers in Plant Science, 9, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inácio, V. , Santos, R. , Prazeres, R. , Graça, J. , Miguel, C.M. & Morais‐Cecílio, L. (2022) Epigenetics at the crossroads of secondary growth regulation. Frontiers in Plant Science, 13, 970342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeltsch, A. , Ehrenhofer‐Murray, A. , Jurkowski, T.P. , Lyko, F. , Reuter, G. , Ankri, S. et al. (2017) Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biology, 14(9), 1108–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, L. , Bao, Q. , He, W. , Fan, D. , Cheng, S. , López‐Pujol, J. et al. (2022) Phylogeny and biogeography of Fagus (Fagaceae) based on 28 nuclear single/low‐copy loci. Journal of Systematics and Evolution, 60(4), 759–772. [Google Scholar]
- Jones, D.T. , Taylor, W.R. & Thornton, J.M. (1992) The rapid generation of mutation data matrices from protein sequences. Bioinformatics, 8(3), 275–282. [DOI] [PubMed] [Google Scholar]
- Jullien, P.E. , Susaki, D. , Yelagandula, R. , Higashiyama, T. & Berger, F. (2012) DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana . Current Biology, 22(19), 1825–1830. [DOI] [PubMed] [Google Scholar]
- Kohany, O. , Gentles, A.J. , Hankus, L. & Jurka, J. (2006) Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics, 7, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer, A. , Abbott, A.G. , Carlson, J.E. , Manos, P.S. , Plomion, C. , Sisco, P. et al. (2012) Genomics of Fagaceae. Tree Genetics & Genomes, 8(3), 583–610. [Google Scholar]
- Kumar, S. & Mohapatra, T. (2021) Dynamics of DNA methylation and its functions in plant growth and development. Frontiers in Plant Science, 12, 596236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. & Tamura, K. (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, P. , Dane, F. , Kubisiak, T.L. & Huang, H. (2007) Molecular evidence for an Asian origin and a unique westward migration of species in the genus Castanea via Europe to North America. Molecular Phylogenetics and Evolution, 43(1), 49–59. [DOI] [PubMed] [Google Scholar]
- Latrasse, D. , Benhamed, M. , Henry, Y. , Domenichini, S. , Kim, W. , Zhou, D.‐X. et al. (2008) The MYST histone acetyltransferases are essential for gametophyte development in Arabidopsis. BMC Plant Biology, 8(1), 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, J.A. & Jacobsen, S.E. (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics, 11(3), 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen, R. , Sugawara, H. & Shumway, M. (2011) The sequence read archive. Nucleic Acids Research, 39 (Database), D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. & Bork, P. (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Research, 49(W1), W293–W296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Torres‐Garcia, J. , Latrasse, D. , Benhamed, M. , Schilderink, S. , Zhou, W. et al. (2017) Plant‐specific histone deacetylases HDT1/2 regulate GIBBERELLIN 2‐OXIDASE2 expression to control Arabidopsis root meristem cell number. The Plant Cell, 29(9), 2183–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Wei, W. , Zhu, W. , Su, L. , Xiong, Z. , Zhou, M. et al. (2017) Histone deacetylase AtSRT1 links metabolic flux and stress response in Arabidopsis. Molecular Plant, 10(12), 1510–1522. [DOI] [PubMed] [Google Scholar]
- Lloret, A. , Badenes, M.L. & Ríos, G. (2018) Modulation of dormancy and growth responses in reproductive buds of temperate trees. Frontiers in Plant Science, 9, 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y. , Shi, D.Q. , Jia, P.F. , Bao, Y. , Li, H.J. & Yang, W.C. (2022) Nucleolar histone deacetylases HDT1, HDT2, and HDT3 regulate plant reproductive development. Journal of Genetics and Genomics, 49(1), 30–39. [DOI] [PubMed] [Google Scholar]
- Madeira, F. , Pearce, M. , Tivey, A.R.N. , Basutkar, P. , Lee, J. , Edbali, O. et al. (2022) Search and sequence analysis tools services from EMBL‐EBI in 2022. Nucleic Acids Research, 50(W1), W276–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler‐Bauer, A. & Bryant, S.H. (2004) CD‐search: protein domain annotations on the fly. Nucleic Acids Research, 32 (Web Server), W327–W331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler‐Bauer, A. , Lu, S. , Anderson, J.B. , Chitsaz, F. , Derbyshire, M.K. , DeWeese‐Scott, C. et al. (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Research, 39 (Database), D225–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak, M. , Plitta‐Michalak, B.P. , Naskręt‐Barciszewska, M. , Barciszewski, J. , Bujarska‐Borkowska, B. & Chmielarz, P. (2015) Global 5‐methylcytosine alterations in DNA during ageing of Quercus robur seeds. Annals of Botany, 116(3), 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, B. , Gupta, D.K. , Pfenninger, M. , Hickler, T. , Langer, E. , Nam, B. et al. (2018) A reference genome of the European beech (Fagus sylvatica L.). GigaScience, 7(6), giy063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, B. , Ulaszewski, B. , Meger, J. , Aury, J.‐M. , Bodénès, C. , Lesur‐Kupin, I. et al. (2022) A chromosome‐level genome assembly of the European beech (Fagus sylvatica) reveals anomalies for organelle DNA integration, repeat content and distribution of SNPs. Frontiers in Genetics, 12, 2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning, Y. , Chen, Q. , Lin, R. , Li, Y. , Li, L. , Chen, S. et al. (2019) The HDA19 histone deacetylase complex is involved in the regulation of flowering time in a photoperiod‐dependent manner. The Plant Journal, 98(3), 448–464. [DOI] [PubMed] [Google Scholar]
- Ono, H. , Ishii, K. , Kozaki, T. , Ogiwara, I. , Kanekatsu, M. & Yamada, T. (2015) Removal of redundant contigs from de novo RNA‐Seq assemblies via homology search improves accurate detection of differentially expressed genes. BMC Genomics, 16(1), 1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey, R. (2002) Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Research, 30(23), 5036–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlopoulou, A. & Kossida, S. (2007) Plant cytosine‐5 DNA methyltransferases: structure, function, and molecular evolution. Genomics, 90(4), 530–541. [DOI] [PubMed] [Google Scholar]
- Platt, A. , Gugger, P.F. , Pellegrini, M. & Sork, V.L. (2015) Genome‐wide signature of local adaptation linked to variable CpG methylation in oak populations. Molecular Ecology, 24(15), 3823–3830. [DOI] [PubMed] [Google Scholar]
- Plomion, C. , Aury, J.M. , Amselem, J. , Leroy, T. , Murat, F. , Duplessis, S. et al. (2018) Oak genome reveals facets of long lifespan. Nature Plants, 4(7), 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, X. , Li, Q. , Yin, H. , Qi, K. , Li, L. , Wang, R. et al. (2019) Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biology, 20(1), 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesneville, H. (2020) Twenty years of transposable element analysis in the Arabidopsis thaliana genome. Mobile DNA, 11(1), 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2022) R: a language and environment for statistical computing. Vienna, Austria: R Core Team. https://www.R-project.org/ [Google Scholar]
- Raghavan, V. , Kraft, L. , Mesny, F. & Rigerte, L. (2022) A simple guide to de novo transcriptome assembly and annotation. Briefings in Bioinformatics, 23(2), bbab563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, A.M. , Usié, A. , Barbosa, P. , Barros, P.M. , Capote, T. , Chaves, I. et al. (2018) The draft genome sequence of cork oak. Scientific Data, 5(1), 180069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, M. , Rocheta, M. , Carvalho, L. , Inácio, V. , Graça, J. & Morais‐Cecilio, L. (2013) Expression of DNA methyltransferases is involved in Quercus suber cork quality. Tree Genetics & Genomes, 9(6), 1481–1492. [Google Scholar]
- Ribeiro, T. , Viegas, W. & Morais‐Cecílio, L. (2009) Epigenetic marks in the mature pollen of Quercus suber L. (Fagaceae). Sexual Plant Reproduction, 22(1), 1–7. [DOI] [PubMed] [Google Scholar]
- Rico, L. , Ogaya, R. , Barbeta, A. & Peñuelas, J. (2014) Changes in DNA methylation fingerprint of Quercus ilex trees in response to experimental field drought simulating projected climate change. Plant Biology, 16(2), 419–427. [DOI] [PubMed] [Google Scholar]
- Rocheta, M. , Carvalho, L. , Viegas, W. & Morais‐Cecílio, L. (2012) Corky, a gypsy‐like retrotransposon is differentially transcribed in Quercus suber tissues. BMC Research Notes, 5(1), 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, R. (2004) Temperate ecosystems ‐ Fagaceae. In: Encyclopedia of forest sciences. NX Amsterdam, The Netherlands: Elsevier, pp. 1419–1427. [Google Scholar]
- RStudio Team . (2022) RStudio: integrated development for R. Boston, MA: RStudio, PBC. http://www.rstudio.com/ [Google Scholar]
- Santamaría, M.E. , Hasbún, R. , Valera, M.J. , Meijón, M. , Valledor, L. , Rodríguez, J.L. et al. (2009) Acetylated H4 histone and genomic DNA methylation patterns during bud set and bud burst in Castanea sativa . Journal of Plant Physiology, 166(13), 1360–1369. [DOI] [PubMed] [Google Scholar]
- Santamaría, M.E. , Rodríguez, R. , Cañal, M.J. & Toorop, P.E. (2011) Transcriptome analysis of chestnut (Castanea sativa) tree buds suggests a putative role for epigenetic control of bud dormancy. Annals of Botany, 108(3), 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid‐Siegert, E., Sarkar, N., Iseli, C., Calderon, S., Gouhier‐Darimont, C., Chrast, J. et al. (2017) Low number of fixed somatic mutations in a long‐lived oak tree. Nature Plants 3, 926–929. [DOI] [PubMed] [Google Scholar]
- Schmitz, R.J. , Lewis, Z.A. & Goll, M.G. (2019) DNA methylation: shared and divergent features across eukaryotes. Trends in Genetics, 35(11), 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasawa, K. , Nishio, S. , Terakami, S. , Botta, R. , Marinoni, D.T. & Isobe, S. (2021) Chromosome‐level genome assembly of Japanese chestnut (Castanea crenata Sieb. et Zucc.) reveals conserved chromosomal segments in woody rosids. DNA Research, 28(5), dsab016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, H.G. , Sobral, R.S. , Magalhães, A.P. , Morais‐Cecílio, L. & Costa, M.M.R. (2020) Genome‐wide identification of epigenetic regulators in Quercus suber L. International Journal of Molecular Sciences, 21(11), 3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. & Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31(19), 3210–3212. [DOI] [PubMed] [Google Scholar]
- Simonini, S. , Bemer, M. , Bencivenga, S. , Gagliardini, V. , Pires, N.D. , Desvoyes, B. et al. (2021) The Polycomb group protein MEDEA controls cell proliferation and embryonic patterning in Arabidopsis. Developmental Cell, 56(13), 1945–1960.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sork, V.L. , Fitz‐Gibbon, S.T. , Puiu, D. , Crepeau, M. , Gugger, P.F. , Sherman, R. et al. (2016) First draft assembly and annotation of the genome of a California endemic oak Quercus lobata Née (Fagaceae). G3: Genes, Genomes, Genetics, 6(11), 3485–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner, D.E. & Berger, S.L. (2000) Acetylation of histones and transcription‐related factors. Microbiology and Molecular Biology Reviews, 64(2), 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szućko, I. (2016) Sirtuins: not only animal proteins. Acta Physiologiae Plantarum, 38(10), 237. [Google Scholar]
- Tan, G. , Polychronopoulos, D. & Lenhard, B. (2019) CNEr: a toolkit for exploring extreme noncoding conservation. PLoS Computational Biology, 15(8), e1006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto, M. , Akada, S. , Matsuda, S. , Jouyu, H. , Kisanuki, H. , Tomaru, N. et al. (2020) Assessments of fine‐scale spatial patterns of SNPs in an old‐growth beech forest. Heredity, 125(4), 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vičić, V. , Barišić, D. , Horvat, T. , Biruš, I. & Zoldos, V. (2013) Epigenetic characterization of chromatin in cycling cells of pedunculate oak, Quercus robur L. Tree Genetics & Genomes, 9(5), 1247–1256. [Google Scholar]
- Viejo, M. , Rodríguez, R. , Valledor, L. , Pérez, M. , Cañal, M.J. & Hasbún, R. (2010) DNA methylation during sexual embryogenesis and implications on the induction of somatic embryogenesis in Castanea sativa Miller. Sexual Plant Reproduction, 23(4), 315–323. [DOI] [PubMed] [Google Scholar]
- Viejo, M. , Santamaría, M.E. , Rodríguez, J.L. , Valledor, L. , Meijón, M. , Pérez, M. et al. (2012) Epigenetics, the role of DNA methylation in tree development. In: Loyola‐Vargas, V.M. & Ochoa‐Alejo, N. (Eds.) Methods in molecular biology. Totowa, NJ: Humana Press, pp. 277–301. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Tian, S. , Sun, X. , Cheng, X. , Duan, N. , Tao, J. et al. (2020) Construction of pseudomolecules for the Chinese chestnut (Castanea mollissima) genome. G3: Genes, Genomes, Genetics, 10(10), 3565–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. & Jiang, H. (2020) Writing and reading histone H3 lysine 9 methylation in Arabidopsis. Frontiers in Plant Science, 11, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Zhu, J. , Feng, L. , Zhou, T. , Bai, G. , Yang, J. et al. (2018) Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: highlighting the effect of codon composition bias in phylogenetic inference. Frontiers in Plant Science, 9, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung, W. , Li, M. , Sze, C. , Wang, Q. & Lam, H. (2021) Histone modifications and chromatin remodelling in plants in response to salt stress. Physiologia Plantarum, 173(4), 1495–1513. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Lang, Z. & Zhu, J.K. (2018) Dynamics and function of DNA methylation in plants. Nature Reviews Molecular Cell Biology, 19(8), 489–506. [DOI] [PubMed] [Google Scholar]
- Zhao, T. , Zhan, Z. & Jiang, D. (2019) Histone modifications and their regulatory roles in plant development and environmental memory. Journal of Genetics and Genomics, 46(10), 467–476. [DOI] [PubMed] [Google Scholar]
- Zheng, W. (2020) Review: the plant sirtuins. Plant Science, 293, 110434. [DOI] [PubMed] [Google Scholar]
- Zhong, X. , Du, J. , Hale, C.J. , Gallego‐Bartolome, J. , Feng, S. , Vashisht, A.A. et al. (2014) Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell, 157(5), 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, X. , Hale, C.J. , Minh, N. , Israel, A. , Martin, G. , Jonathan, H. et al. (2015) Domains rearranged methyltransferase3 controls DNA methylation and regulates RNA polymerase V transcript abundance in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America, 112(3), 911–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, B.‐F. , Yuan, S. , Crowl, A.A. , Liang, Y.‐Y. , Shi, Y. , Chen, X.‐Y. et al. (2022) Phylogenomic analyses highlight innovation and introgression in the continental radiations of Fagaceae across the Northern Hemisphere. Nature Communications, 13(1), 1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Liu, Y. , Liang, Y. , Zhou, D. , Li, S. , Lin, S. et al. (2020) The function of histone lysine methylation related SET domain group proteins in plants. Protein Science, 29(5), 1120–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genome assemblies accession numbers. Accession numbers of the genome assemblies used in our work. Assembly method and sequencing method was annotated when available in the NCBI assembly database.
Table S2. SRA raw data used for transcriptome library assembly. NCBI Sequence read archive accession numbers and respective information on the strategy, experimental design, and sequencing methods used to obtain the libraries.
Table S3. De novo assembly statistics of Fagaceae transcriptomes and their respective completeness as determined by BUSCO. N50 defines as length of the shortest contig for which longer and equal length contigs cover at least 50% of the assembly. BUSCO (Benchmarking Universal Single‐Copy Orthologous) statistics are Complete‐single‐copy (CSC), Complete Duplicate (CD), Fragmented (Frag), and Missing (Miss).
Table S4. Genomic location of all epigenetic regulators in the current study.
Table S5. Simplified table of the genomic locations of the different epigenetic regulators studied, with their respective paralogs.
Table S6. Protein length of each identified epigenetic regulator. Length units are shown in number of amino acid.
Table S7. Identities between paralogs given by MUSCLE alignment. Numbers are given as percentages.
Table S8. Selection of the results for the 5000 bp adjacent regions of the identified paralogs. Dir‐indicates the orientation of the element regarding the sequence (“d” is for direct and “c” is for complementary); Sim—similarity between the aligned fragments; Pos/Mm:Ts—is the ratio between Positives hits in alignment length (Pos), and the ratio of Mismatches to Transitions in nucleotide alignment (Mm:Ts).
Data Availability Statement
The data that support the findings were derived from the resources available in the public domain and are available in the Supporting Information of this article.