

Summary
Crop domestication usually leads to the narrowing genetic diversity. However, human selection mainly focuses on visible traits, such as yield and plant morphology, with most metabolic changes being invisible to the naked eye. Buckwheat accumulates abundant bioactive substances, making it a dual‐purpose crop with excellent nutritional and medical value. Therefore, examining the wiring of these invisible metabolites during domestication is of major importance. The comprehensive profiling of 200 Tartary buckwheat accessions exhibits 540 metabolites modified as a consequence of human selection. Metabolic genome‐wide association study illustrates 384 mGWAS signals for 336 metabolites are under selection. Further analysis showed that an R2R3‐MYB transcription factor FtMYB43 positively regulates the synthesis of procyanidin. Glycoside hydrolase gene FtSAGH1 is characterized as responsible for the release of active salicylic acid, the precursor of aspirin and indispensably in plant defence. UDP‐glucosyltransferase gene FtUGT74L2 is characterized as involved in the glycosylation of emodin, a major medicinal component specific in Polygonaceae. The lower expression of FtSAGH1 and FtUGT74L2 were associated with the reduction of salicylic acid and soluble EmG owing to domestication. This first large‐scale metabolome profiling in Tartary buckwheat will facilitate genetic improvement of medicinal properties and disease resistance in Tartary buckwheat.
Keywords: buckwheat, metabolite variation, domestication, mGWAS, traditional medicine
Introduction
Tartary buckwheat (Fagopyrum tataricum Gaertn.) is one of the cultivated species of the Fagopyrum (buckwheat) genus. Buckwheat has been considered to originate in the Tibetan plateau or nearby mountains of Yunnan, southwest China. Wild Tartary buckwheat, F. tartaricum ssp. potanini, is more widespread on the Tibetan plateau (Campbell, 1976; Ohnishi, 1998; Tsuji and Ohnishi, 2001; Weisskopf and Fuller, 2014). Moreover, cultivated Tartary buckwheat is distributed in many high‐altitude areas, such as southwestern China, Nepal, and Bhutan. It is used as a staple food and an important raw material for functional food production after selection for annual habit, non‐shattering inflorescence, low seed dormancy, and increased seed size (Fuller and Allaby, 2009; Hunt et al., 2018). Besides the well‐balanced amino acid composition and rich content of dietary fibre, mineral elements and vitamins, Tartary buckwheat contains diverse bioactive compounds, such as flavonoids, anthraquinones and phenolic acids, which endues this gluten‐free crop with medical values (Kreft et al., 2020; Zhu, 2016). Rutin is the distinctive flavonoids of Tartary buckwheat and has a pharmaceutical function for cardiovascular disorders such as hypertension, hyperlipidemia and myocardial infarction (Sharma et al., 2013). Emodin is a specific natural occurring anthraquinone derivative in many Chinese medicinal herb plants of Polygonaceae displaying diuretic, anti‐cardiovascular and anti‐cancer bioactive effects (Dong et al., 2016; Li et al., 2020a; Peng et al., 2013). Given the nutritional and medical values, Tartary buckwheat is often considered as an ideal functional food with diverse health benefits including pain relief, neuroprotection, anti‐oxidation, anti‐microbial, anti‐inflammatory, anti‐diabetic, anti‐cancer, anti‐hypertension and cholesterol level reduction (Jing et al., 2016; Joshi et al., 2020).
In many mountainous areas where other major crops fail to perform optimally or even survive, Tartary buckwheat has been planted as a staple food, owing to its properties of short growth period, high ecological adaptability and tolerance in low‐nutrient conditions (Kumari and Chaudhary, 2020). Some bioactive substances with pharmaceutical effects also help buckwheat resistant to the harsh environment. Fagopyritols are structurally similar with putative insulin mediator D‐chiro‐inositol galactosamine derivative and may be used to treat with non‐insulin dependent diabetes mellitus (Cid et al., 2004). These compounds compose almost 50% of the total soluble carbohydrate in buckwheat embryos and provide buckwheat with great ability to survive in high altitude regions with low rainfall (Jing et al., 2016). Salicylic acid (SA) is the precursor and the active ingredient of aspirin (acetylsalicylic acid), the most widely consumed analgesic, antipyretic and anti‐inflammatory drug around the world for centuries (Vlot et al., 2009). Its extensive signalling functions as a plant phytohormone inducing plant defence against biotrophic pathogens invasions, especially a soil‐borne plant pathogen Rhizoctonia solani causing a universal crop disease in major cereal crops, legumes, vegetables, as well as buckwheat (Kouzai et al., 2018; Li et al., 2019b, 2020b; Oladzad et al., 2019; Zrenner et al., 2020). Though the structures of these metabolites are different, chemical modifications commonly occur in plant cells, such as glycosylation (Maruri‐López et al., 2019; Peng et al., 2013; Yin et al., 2020). As carbohydrates could access various chemical space, the glycosylation of metabolites could influence their solubility as well as stability, thus help them to perform different functions in various cell components (Singh et al., 2012). However, the biosynthetic pathways of these metabolites, especially later glycosylation reactions, in Tartary buckwheat remain unclear to date (Maruri‐López et al., 2019; Yin et al., 2020).
In the process of crop domestication, human selection mainly focuses on the visible agronomic traits, such as yield, seed shattering, erect plant architecture, grain size and quality and hull colour. Early selection however largely ignored the changes of many invisible traits, for example, the content of many bioactive compounds, resulted from the decrease of genetic diversity during domestication. These compounds protect plants from biotic and abiotic stress and have potential benefits to human health as pharmaceuticals. The tomato domestication resulted in reduced salt tolerance and the accumulation of steroidal glycoalkaloids (Zhu et al., 2018). The selection of non‐bitter cucumber led to the loss of pharmaceutical properties (Shang et al., 2014). Tartary buckwheat is undergoing an early stage of domestication. Compliant to human demand, yield has been improved and the growth period has been shortened, while our previous study found that the artificial selection resulted in decreased content of some flavonoids (Zhang et al., 2021). By the whole‐genome resequencing, Zhang et al. (2021) divided 510 accessions into the Himalayan wild (HW) group, the Southwestern Landraces (SL) and the Northern Landraces (NL) of China. They also identified numerous domestication sweeps in the SL and NL groups compared to the HW group. However, many domestication sweeps did not overlap with GWAS signals corresponding to agronomic traits. We speculate that these may, however, play important roles in the shaping of bioactive compounds during Tartary buckwheat domestication.
In order to investigate this hypothesis, we conducted a comprehensive metabolic profiling and a subsequent mGWAS of 200 accessions in Tartary buckwheat (Figure S1). A total of 540 differential metabolites were detected among HW, SL and NL groups, and 1253 mGWAS signals were found. Among them, 384 mGWAS signals corresponding to 336 metabolites were identified to be subjected to domestication. By enzymatic validation and genetic transformation, the potential mechanism(s) responsible for metabolome changes during Tartary buckwheat domestication were also revealed. As such, this study will enable breeding programs to enhance levels of buckwheat bioactives, enhancing the appeal of this much grown species as a source for improving human diet.
Results
Metabolic profiling of Tartary buckwheat grains
To assess the natural variation of the metabolome in Tartary buckwheat grains, we collected grain samples from a diverse global collection of 200 Tartary buckwheat accessions based on their passport information, morphological traits and phylogenetic relationships. These accessions include 14 HW accessions, 80 SL accessions and 106 NL accessions (Figure S2A and Table S1). We next determined the relative content of 1092 distinct metabolic features (Tables S2 and S3) in seeds of Tartary buckwheat using a broadly targeted liquid chromatography–tandem mass spectrometry (LC–MS/MS)‐based metabolic profiling method (Chen et al., 2013). Of these metabolites, the chemical structures of 567 were annotated, including metabolites belonging to the flavonoids, phenolic acids, amino acids, lipids, organic acids and nucleotides and derivatives (Figure S2B and Table S2). These metabolites exhibited normally distributed coefficients of variation (CV) (Figure S2C and Table S2), among the 200 accessions. Moreover, the phenotypic variation of HW (average CV is 0.42) was higher than NL (average CV is 0.40) and SL (average CV is 0.34), which is consistent with genetic diversity levels of three groups.
The relationships among the metabolite contents were evaluated using Spearman's rank correlation (Figure S3 and Table S4). Rutin and (epi)‐catechins, which represent the main bioactive phenolic compound in buckwheat (Martín‐García et al., 2019), were found to be significantly correlated with multiple compounds (Figure S4A,B). Most of the annotated correlated metabolites are their precursors or condensed or structurally similar compounds with a similar result being found for fagopyritols (Figure S4C), which represent the major soluble carbohydrate in buckwheat and are necessary for desiccation tolerance during buckwheat seed maturation (Horbowicz et al., 1998). Given that the synthetic pathways with regard to these metabolites are not completely clear, the manner in which they interact is likely of interest.
Genetic basis of metabolism in Tartary buckwheat grains
In order to dissect the genetic basis underlying the natural variation of metabolites in Tartary buckwheat, the sequences from a diverse global collection of 200 Tartary buckwheat accessions were used in a GWAS (Zhang et al., 2021). Following imputation, single‐nucleotide polymorphisms (SNP) with minor allele frequencies (MAFs) > 0.05 or missing rates of <10% were selected, resulting in a total of 1 004 824 SNPs for further analysis. We then performed mGWAS for 567 known metabolites using both the Efficient Mixed‐Model Association eXpedited program (EMMAx) and the factored spectrally transformed linear mixed models (FaST‐LMM). Among 398 metabolite features, a total of 1253 lead SNPs were detected, with 291 SNPs corresponding to flavonoids and 171 SNPs corresponding to phenolic acids (Table S5). 67.8% of the metabolite features (270 out of 398) had more than one significant association, with an average of 4.16 associations per metabolite. 128 metabolites were controlled by only one lead SNP, such as mws0054 (catechin), mws1434 (isovitexin) and mws0045 (quercitrin).
Following our mGWAS analysis, we found that procyanidin B1, a flavonoid dimer formed from catechin and epicatechin, was significantly as sociated with the lead SNP 13164411 on Chr 1, which located in the −4236 bp upstream FtPinG0100324300.01 (Figures 1a and S5, and Table S5). G‐hap exhibited a higher content of procyanidins (Figure S6 and Table S7). Further study established that a significant positive correlation with the procyanidin B1 content between the gene expression level of FtPinG0100324300.01 (Figure 1b). FtPinG0100324300.01 was in linkage disequilibrium (LD) with SNP 13164441 (Figure 1c,d and Table S6). That suggested FtPinG0100324300.01 may controlled the accumulation of procyanidin B1 in Tatary buckwheat. Phylogenetic analysis showed, FtPinG0100324300.01 may encoding a R2R3‐MYB transcription factor named FtMYB43 (Figure 1e). Auto‐activation tests revealed that FtMYB43 exhibited transcriptional activation activity (Figure 1f). Overexpression in hairy roots exhibited higher expression of the key genes (FtDFR, FtANS and FtFLS) responsible for procyanidin synthesis than the corresponding negative control (Figure 1g). Ectopic expression of FtMYB43 in Arabidopsis showed that not only the expression of AtANS, AtDFR, and AtTT10, but also the total procyanidins content increased (Figure 1h,i), suggesting FtMYB43 may regulate procyanidin synthesis by up‐regulating flavonoid synthesis. As homologues of FtMYB43 have already been confirmed as participating in the regulation of anthocyanin or rutin synthesis (Huang et al., 2019; Li et al., 2019a; Wang et al., 2019), this result illustrates that our mGWAS results are reliable, which enables us to effectively find genes responsible for metabolites variations in Tartary buckwheat.
Figure 1.
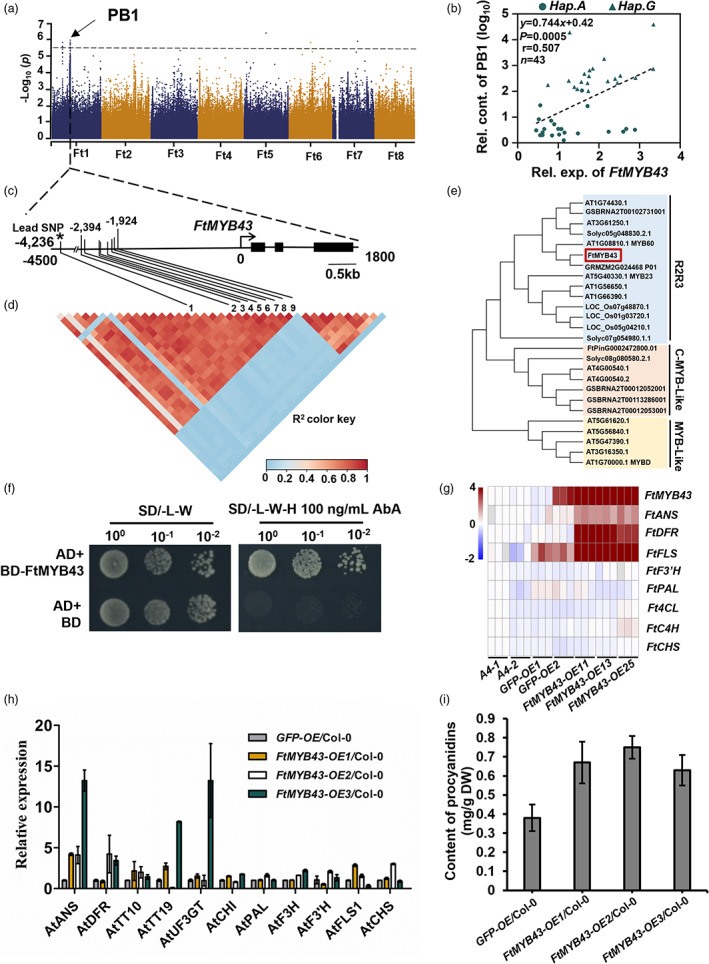
Genetic basis of metabolic profiling in Tartary buckwheat grains. (a) Manhattan plot displays the GWAS result of procyanidin B1 content. The dashed line indicates the threshold −log10 P = 5.68. (b) Correlation between the relative contents of procyanidin B1 and the transcription level of FtMYB43 in 37 buckwheat varieties, including 20 A‐hap and 23 G‐hap. P value was calculated using Student's t‐test. (c) Gene model of FtMYB43. Filled black boxes represent coding sequence. The black vertical lines mark the polymorphic sites identified by high‐throughput sequencing, and the star (*) represents the lead SNP Ft1: 13164411 located in located in the −4236 bp upstream FtMYB43. (d) A representation of pairwise r 2 value (a measure of LD) among all polymorphic sites in approximately 26.5 kb region around the lead SNP Ft1: 13164411. The colour gradation of each box corresponds to the r 2 value according to the legend and the number “1” represents the lead SNP Ft1: 13164411, “2–9” represent the SNPs in the promoter region of FtMYB43. (e) Phylogenetic analysis of the candidate gene FtMYB43. (f) Analysis of transcriptional activation activity of FtMYB43. (g) Expression pattern of procyanidin biosynthesis genes in FtMYB43 overexpression hairy root evaluated by qRT‐PCR. The buckwheat hairy roots were infected with Agrobacterium rhizogenes strains A4 containing different recombined plasmid. The hairy roots infected with A4 or GFP‐OE (35S: GFP) were used as negative control. (h) Expression pattern of procyanidin biosynthesis genes in FtMYB43 ectopic expression in Arabidopsis evaluated by qRT‐PCR. The Col‐0 (wild type) and ectopic expression of GFP (35S: GFP) were used as negative control. (i) Procyanidin content in ectopic expression of FtMYB43 in Arabidopsis determined by DCMCA‐HCl. The Col‐0 (wild type) and ectopic expression of GFP (35S: GFP) were used as negative control. All experiments in (g), (h) and (i) were performed as three biologically independent experiments and the data in (e), (h) and (i) are presented as mean ± SD. Significant differences are indicated by asterisks: **, P < 0.01, ***, P < 0.001, Student's t test.
Metabolome alteration during Tartary buckwheat domestication
The population structure of Tartary buckwheat germplasm collection has been well characterized (Zhang et al., 2021). However, little is known concerning how these distinctions affect metabolite profiles. There are 245 differentially abundant metabolites in NL and 265 differentially abundant metabolites in SL compared to HW, of which 98 metabolites exhibited similar dynamic trends (Figure 2a and Table S8). For example, fagopyritol A1 (FPA1) and B2 (FPB2) both accumulated in SL and NL, while emodin‐8‐O‐β‐d‐glucoside (EmG) and SA both decreased in SL and NL (Figure 2b). 328 metabolites were present at significantly different abundance between NL and SL. Among these metabolites, quercetin‐3,7‐O‐β‐d‐diglucoside (QDG), epiafzelechin (EAF), catechin (Cat) and catechin‐7‐O‐glucoside (C7G) were higher in SL compared to NL (Figure 2b), suggesting metabolome alteration in NL and SL is probably a result of domestication. These subspecies‐specific metabolites likely reflect the subspecies differentiation of Tartary buckwheat, especially between the three major subspecies.
Figure 2.
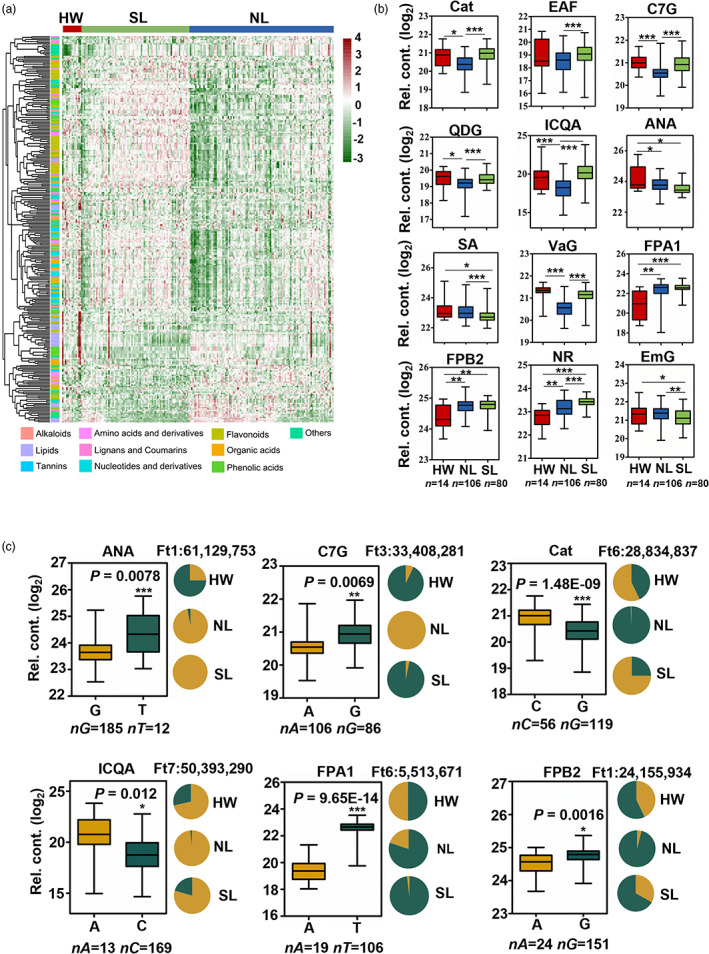
Comparison of metabolic profiles in 200 Tartary buckwheat accessions. (a) Heatmap visualization of relative different metabolites among HW, NL and SL accessions. The contents of each metabolite were normalized before performing linkage hierarchical clustering. Red indicates high abundance and green indicates low content. (b) Box plot of some important metabolites in Tartary buckwheat with different variations among HW, NL and SL groups. Including flavanols (Cat, Catechin; EAF, epiafzelechin; QDG, quercetin‐3,7‐O‐β‐d‐diglucoside; C7G, (+)‐catechin‐7‐O‐glucoside), phenolic acids (ICQA, isochlorogenic acid A; ANA, anchoic acid; SA, salicylic acid; VaG, 1′‐O‐vanilloyl‐β‐d‐glucoside), saccharides and alcohols (FPA1, fagopyritol A1; FPB2, fagopyritol B2), vitamin (NR, nicotinate d‐ribonucleoside) and anthraquinone (EmG, emodin‐8‐O‐β‐d‐glucoside). nHW = 14, nNL = 106 and nSL = 80 for all plots. (c) Frequencies of the high/low‐level trait‐related allele in three groups. Box plot indicates the relative contents of the corresponding metabolites. The metabolic data was log2 transformed. Data in (b) and (c) are presented as mean ± SD. Significant differences are indicated by asterisks: *, P < 0.05, **, P < 0.01, ***, P < 0.001, Student's t test.
We have previously demonstrated two genetically and geographically distinct groups NL and SL as being subjected to independent domestication process, with 150 domestication sweeps between HW and SL groups, and 156 domestication sweeps between HW and NL groups, respectively (Zhang et al., 2021). However, the function of numerous domestication sweeps remains ambiguous. By comparing mGWAS signals with domestication sweeps, we found a total of 384 lead SNPs for 336 metabolites were located in domestication sweeps, with 204 signals for 172 metabolites in HW vs NL (Table S9), and 217 signals for 182 metabolites in HW vs SL (Table S10). However, only 37 mGWAS signals for 29 metabolites were mapped to both groups' domestication sweeps, suggesting that the abundance of most metabolite is subject to independent domestication process.
Some buckwheat bioactive compounds were subjected to domestication. For example, Cat and its derivative C7G were both present at lower levels in NL compared to HW (Figure 2b). The mGWAS signals for C7G on Chr 3 and Cat on Chr 6 overlapped with domestication sweeps of NL vs HW (Figures S5, S7, S8I and S9A). The allele A of the lead SNP 33408281 on Chr 3 for lower C7G content increased from HW to NL (Figure 2c). FtPinG0303261500.01, encoding a NAC transcription factor, was located 21 727 bp downstream of SNP 33408281 on Chr 3 (Figures S5 and S9, and Table S11). Although FtPinG0303261500.01 is not the highest expression gene in this interval, the expression of this NAC gene varies between the two haplotypes (Figure S9D,F). These results suggest this NAC transcription factor might be involved in C7G decrease during Tartary buckwheat domestication. FPA1 accumulated in NL and SL compared to HW (Figure 2b). Intriguingly, the mGWAS signals for FPA1 on Chr 6 overlapped with the domestication sweep of SL vs HW (Figures S5, S7 and S9), and the allele for higher content increased from HW to SL (Figure 2c). FtPinG0606262800.01, encoding a Phenylalanine Ammonia‐Lyase (PAL) was located 2727 bp downstream of SNP 14120713 (Figures S5 and S10A–D, and Table S12), and the expression of FtPinG0606262800.01 varies between the two haplotypes (Figure S10E), suggesting FtPAL was involved in FPA1 change during Tartary buckwheat domestication. We additionally observed the similar domestication phenomenon in other metabolites, such as anchoic acid (ANA), isochlorogenic acid A (ICQA) and FPB2 (Figures 2b,c, S7 and S8). These results suggest that not only the agronomic traits changed, but also metabolites contents were affected during Tartary buckwheat domestication. Candidate genes identified by GWAS may help to reveal biosynthesis mechanism of these bioactive compounds.
Decrease of SA level results in diminished disease resistance
Salicylic acid initially accumulates in locally infected tissues and then spreads throughout the plant thereby inducing systemic acquired resistance (Maruri‐López et al., 2019). That said, free SA is poorly soluble in water. It can be subsequently glycosylated into inactive and soluble salicylic acid 2‐O‐β‐glucoside (SAG) stored in vacuoles until SA triggered responses are activated (Maruri‐López et al., 2019). Within our metabolomics data, SA was found to be decreased in SL compared to HW, suggesting a negative selection of SA during Tartary buckwheat domestication (Figure 2b). mGWAS identified five major signals associated with SA (Figure S4 and Table S5), and one (SNP 14120713 on Chr 7) of them, located in the 3180 bp downstream FtPinG0707407000.01 overlapped with a domestication sweep between HW and SL (Figures 3a,b and S7). FtPinG0707407000.01, encoding a glycoside hydrolase, named Salicylic Acid Glycoside Hydrolase 1 (FtSAGH1), was within the LD interval spanned by SNP 14120713 (Figure 3c,d and Table S13). The allele (T) for lower SA content increased from 60.71% in HW to 99.38% in SL (Figure 3e), suggesting that the closely linked gene FtSAGH1 likely determined the SA domestication. Further investigation revealed that the expression of the FtSAGH1 gene was positively associated with SA content (Figure 3f). Meanwhile, the expression of FtSAGH1 being prominent in the buckwheat stem, leaf and mature seed (Figure S11). Following the expression of FtSAGH1 in E. coli BL21, its function of hydrolyzing SAG to SA was confirmed in vitro enzyme assays (Figures 3g, S12 and S13). We also noticed that there were two other glycoside hydrolase encoded genes (FtPinG0707406600.01 and FtPinG0707407400.01) in the candidate region of SNP Ft7: 14120713 (Table S13). We also test the ability of hydrolyzing SAG to SA in vitro enzyme assays, but we did not detect the SA in the same and suitable conditions (Figure S13B). In addition, overexpression of FtSAGH1, in hairy roots, resulted in elevated SA content coupled to diminished SAG content (Figure 3h and Table S14). In order to study whether SA content is related to Tartary buckwheat disease resistance, Rhizoctonia solani AG‐4 HGI (Li et al., 2020b), which causes severe canker disease of buckwheat was used to infect hypocotyls of Tartary buckwheat (Figure S14). The disease resistance of Tartary buckwheat was higher within the SL with comparison to the HW genotypes (Figure 3i and Table S15), negatively correlating with the SA content (Figure 2b). Furthermore, the resistance of pathogen was enhanced in FtSAGH1 overexpression in Arabidopsis (Figure 3j,k and Table S16). The above results suggest that FtSAGH1 responsible for SAG hydrolyzation to SA, which was involved in SA content and subsequent disease resistance domestication in Tartary buckwheat.
Figure 3.
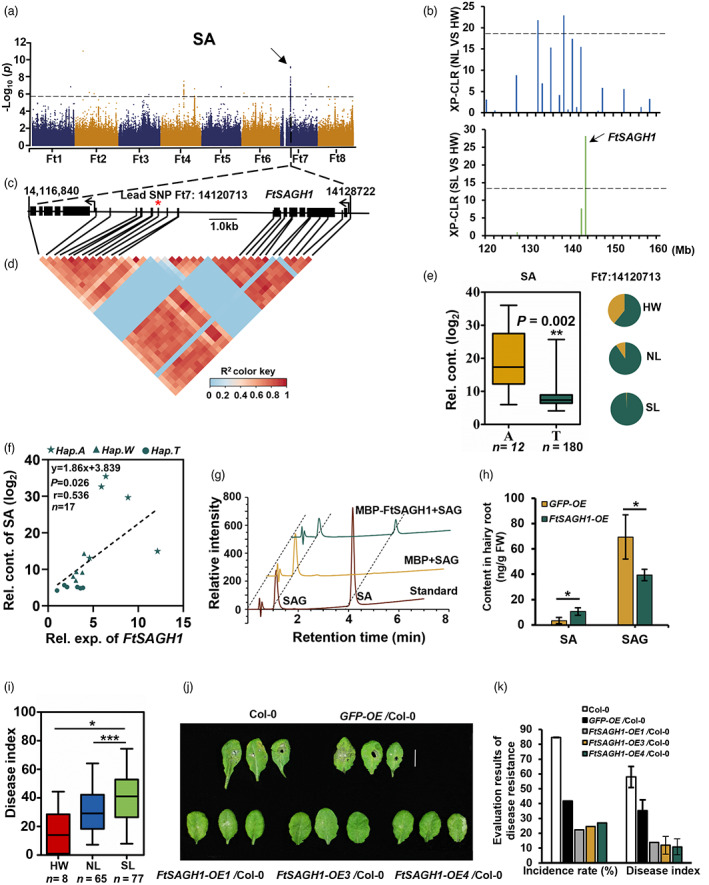
Decrease of SA induces disease resistance changed during Tartary buckwheat domestication. (a) Manhattan plot and QQ plot display the GWAS results of salicylic acid (SA) content. Metabolite content was genetically associated with the sweep harbouring FtSAGH1. The dashed line indicates the threshold −log10 P = 5.68. (b) The cross population composite likelihood ratio (XP‐CLR) between NL and HW (upper), and between SL and HW (lower) on chromosome 7. The black dashed horizontal lines indicate top 10% threshold for entire chromosome 7 (threshod with 18 in NL/HW and 14 in SL/HW). The red arrows indicate the position of FtSAGH1 in the sweeps. (c) Gene model of FtSAGH1 and neighbouring gene FtPinG0707406600.01. Filled black boxes represent coding sequence. The black vertical lines mark the polymorphic sites identified by high‐throughput sequencing, and the star (*) represents the lead SNP Ft7: 14120713, located in the 3180 bp downstream FtSAGH1. (d) A representation of pairwise r 2 value (a measure of LD) among all polymorphic sites in approximately 11.9 kb region around the lead SNP. The colour gradation of each box corresponds to the r 2 value according to the legend. (e) Frequencies of the high/low‐level trait‐related allele in three groups. Box plot indicates the relative contents of SA, plotted as a function of genotypes at SNP Ft7: 14120713. The SA contents were log2 transformed, nHap.A = 12, nHap.T = 180. (f) Correlation between the relative contents of SA and the transcription level of FtSAGH1 in 17 buckwheat varieties, including 5 A‐hap, 6 W(A/T)‐hap and 6 T‐hap. P value was calculated using Student's t‐test. (g) FtSAGH1 enzymatic assay in vitro. The different reaction curves by LC–MS were showed. Protein extract from E. coli containing the pMAL‐C5X empty vector was used as a negative control. (h) SA and SAG (salicylic acid‐2‐O‐β‐glucoside) content in FtSAGH1 overexpression hairy roots mixed from three independent lines and five independent experiments were performed. The hairy roots GFP ‐ OE (infected by A4 containing 35S: GFP) were used as negative control. (i) Box plot of disease index infected by Rhizoctionia solani in different variations among HW, NL and SL groups. nHW = 8, nNL = 65, nSL = 77. (j) Phenotype of disease resistance to Rhizoctonia solani in ectopic expression of FtSAGH1 in Arabidopsis. (k) Evaluation of disease resistance to Rhizoctonia solani in (j). Three biological replicates were performed each lines except FtSAGH1‐OE3/Col‐0. Data in (e), (f), (h), (i) and (k) are presented as mean ± SD. Significant differences are indicated by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001, Student's t‐test.
Decrease of emodin during domestication and identification of the biosynthetic enzyme FtUGT74L2
Emodin is a bitter tasting compound widely present in Polygonaceae plants being the main functional ingredient of many herbs of the Polygonaceae (Dong et al., 2016; Li et al., 2020a). Glycosylation of emodin can increase both its solubility and bioavailability (Xie et al., 2014; Yin et al., 2020). From the metabolomics analysis, EmG was decreased from HW to SL (Figure 2b), suggesting a negative selection for glycosylated emodin during Tartary buckwheat domestication. One major signal SNP 7097024 (A/G) on Chr 4 overlapped with a clear domestication sweep between HW and SL (Figures 4a,b and S5). The allele (G) for lower EmG content increases in proportion from 53.57% in HW to 79.11% in SL (Figure 4e). The SNP Ft4: 7097024 was located in the −3336 bp upstream FtPinG0403865200.01, encoding a UDP‐glucosyltransferase, named UDP‐glycosyltransferase 74‐like 2, FtUGT74L2, suggesting this gene might catalyse the key step of emodin glycosylation (Figures 4c,d and S15, and Table S17). Further study revealed that FtUGT74L2 was expressed in almost all tissues, and the expression of FtUGT74L2 gene was positively associated with EmG content (Figures 4F and S16). Following the enzymatical assay of FtUGT74L2, we could demonstrate that it converted emodin to EmG (Figures 4g and S17), providing a further validation of its function. Overexpression in hairy roots further confirmed this result (Figure 4h and Table S18). We thus speculate that FtUGT74L2 glycosylates emodin in vivo and that the activity of this reaction is reduced during domestication in Tartary buckwheat.
Figure 4.
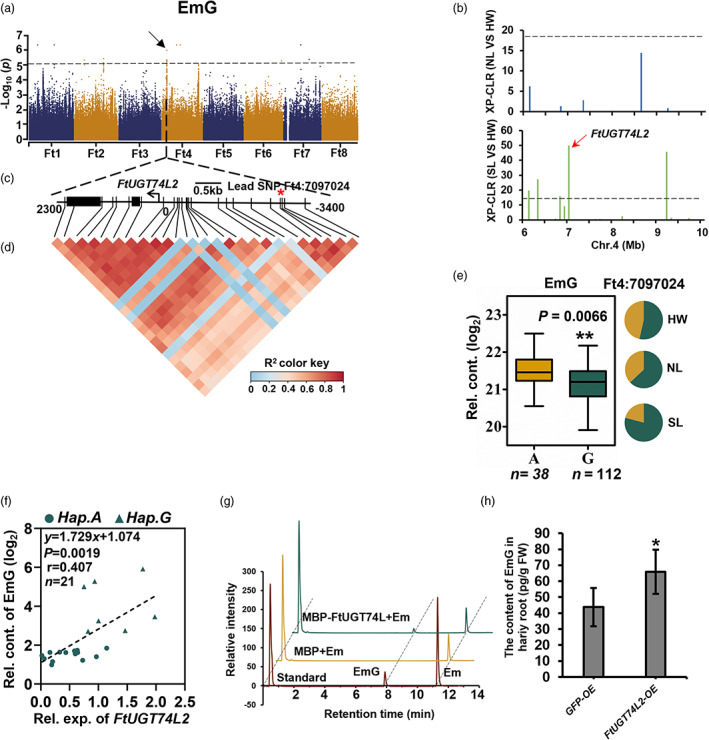
Decrease of emodin during domestication and identification of the biosynthetic enzyme FtUGT74L2. (a) Manhattan plot and QQ plot display the GWAS results of EmG content. The dashed line indicates the threshold −log10 P = 5. (b) The cross population composite likelihood ratio (XP‐CLR) between NL and HW (upper), and between SL and HW (lower) on chromosome 4. The black dashed horizontal lines indicate top 10% threshold for entire chromosome 4 (threshod with 18 in NL/HW and 14 in SL/HW). The red arrows indicate the position of FtUGT74L2 in the sweeps. (c) Gene model of FtUGT74L2. Filled black boxes represent coding sequence. The black vertical lines mark the polymorphic sites identified by high‐throughput sequencing, and the star (*) represents the lead SNP Ft4:7097024, located in the −3336 bp upstream FtUGT74L2. (d) A representation of pairwise r 2 value (a measure of LD) among all polymorphic sites in approximately 5.6 kb region around the lead SNP. The colour gradation of each box corresponds to the r 2 value according to the legend. (e) Frequencies of the high/low‐level trait‐related allele in three groups. Box plot indicates the relative content of EmG, plotted as a function of genotypes at SNP Ft4:7097024. The EmG content was log2 transformed, nHap.A = 38, nHap.G = 112. (f) Correlation between the relative contents of EmG and the transcription level of FtUGT74L2 in 21 buckwheat varieties, including 7 A‐hap and 14 G‐hap. P value was calculated using Student's t‐test. (g) FtUGT74L2 enzymatic assay in vitro. The different reaction curves by LC–MS were showed. Protein extract from E. coli containing the pMAL‐C5X empty vector was used as a negative control. (h) EmG content in FtUGT74L2 overexpression hairy roots mixed from three independent lines and three independent experiments were performed. The hairy roots of infected by A4 containing 35S: GFP(GFP‐OE) were used as negative control. Data in (e), (f) and (h) are presented as mean ± SD. Significant differences are indicated by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001, Student's t‐test.
Discussion
Plants accumulate a wide spectrum of primary and specialized metabolites. These classes of metabolites are known to play vital roles in general plant growth and environmental adaptation, as well as providing indispensable resources of energy, nutrition and pharmaceutically relevant molecules to humans (De Luca et al., 2012; Saito and Matsuda, 2010). Within the past decades, widely targeted metabolomics studies have not only comprehensively defined the natural variation in metabolite abundance in rice, maize and wheat (Chen et al., 2014, 2016, 2020a; Wen et al., 2014), but also revealed the metabolite changes that have occurred on the domestication of wheat, tomato and qingke, respectively (Beleggia et al., 2016; Gao et al., 2019; Giovannoni, 2018; Zeng et al., 2020). Despite the relevance of these studies is analysing high yield crops, they have exclusively been used to examine food crops (Fernie and Yan, 2019). Changes in the levels of pharmaceutically relevant molecules and their genetic basis in buckwheat, a dual‐purpose crop, during domestication have, as yet, not been reported. In the current metabolomics analysis, we identified a wide variety of bioactive compounds, including catechins, fagopyritols, salicylic acid and emodin, whose levels changed during Tartary buckwheat domestication, with multiple metabolism associated genes being seen to follow similar pattern. The combined results of these studies thereby explained the effect of domestication on specialized metabolite accumulation, and thereby provide a defined knowledge bases for the breeding of Tartary buckwheat with high content of bioactive substances.
We additionally verified some candidate genes responsible for changes in abundance of metabolites in Tartary buckwheat. Among them, MYB transcription factors are one of the most important and consequently well studied transcription factor families annotated in plants, including in Tartary buckwheat (Dong et al., 2020; Huang et al., 2019). In our mGWAS analysis, we identified that FtMYB43 appeared to be involved in regulation of procyanidin B1 content. Procyanidin B1 is a flavonoid dimer formed from catechin and epicatechin. C7G is the glycosylated form of catechin, which is another important flavonoid derived from phenylalanine in buckwheat (Joshi et al., 2020). Given that a NAC transcription factor was involved in regulation of glycosyltransferase gene expression, we speculated that NAC transcription factor associated with C7G content may be involved in the glycosylation of catechin (Lee et al., 2017). Fagopyritols are natural D‐chiro‐inositol derivatives possibly be used to treated non‐insulin dependent diabetes mellitus (Cid et al., 2004). These compounds are mainly soluble carbohydrates in buckwheat embryos and participate in buckwheat desiccation tolerance (Jing et al., 2016). As fagopyritols biosynthesis pathway is not clear, the mechanism of PAL regulating FPA1 contents needs to be further studied. Emodin is a common bioactive molecule of Polygonaceae plants (Dong et al., 2016; Li et al., 2020a), whereas SA plays an important role in plant disease resistance (Ma et al., 2019; Zhang and Li, 2019). The glycosylation of these metabolites could both enhance their solubility and bioavailability (Ding and Ding, 2020; Maruri‐López et al., 2019). However, only a few glycosyltransferases or glycosyl hydrolases have been identified that catalyse such (de)glycosylation reactions in plants (Dean and Delaney, 2008; Ding and Ding, 2020; Maruri‐López et al., 2019; Yao et al., 2007; Yin et al., 2020; Zhang and Li, 2019). In our current study, we found that FtUGT74L2 can glycosylate emodin to EmG, and FtSAGH1 can hydrolyze SAG to SA. As these (de)glycosylation processes can make alter the function of bioactive substances, these results may enrich the chemical modification and regulation mechanism of metabolites in plants, and help to breeding buckwheat varieties with higher content and higher stability of bioactive substances to better fit human's demands and climate changes.
With the increasing population and the changing climate, researchers have been trying to look for ways to increase the yield of crops. The plant metabolome is usually regarded as a bridge between genome and phenotype. As such the balance between plant metabolites biosynthesis and yield is essential in plant precisely regulate growth and stress resistance. It is generally believed that the biosynthesis of secondary metabolites in plants is at the expense of slow growth (Chen et al., 2016). For example, reduction of trehalose‐6‐phosphate, a signal molecule regulating sucrose use and allocation, through over‐expression of the trehalose‐6‐phosphate phosphatase could increase maize yield in both water‐deficit and well‐watered treatment fields (Nuccio et al., 2015). Moreover, the reduction of brassinosteroid, a hormone regulating plant architecture, through over‐expression of GmMYB14 could increase the yield of soybean under high planting densities (Chen et al., 2021). While some metabolites were found to positively correlated with crop yield. For example, mutation of a sorghum 3‐deoxyd‐arabino‐heptulosonate‐7‐phosphate synthase gene catalysing a key step in the shikimate biosynthesis pathway exhibit decreased grain yield (Chen et al., 2020b). Similarly, auxin synthesis related metabolites were found positively associated with grain numbers per spike (Shi et al., 2020).
These results show that these secondary metabolites play a comprehensive role in crop yield regulation. Through correlation analysis, we found the contents of SA, EmG, C7G and procyanidin A3 were negatively correlated with grain yield of Tartary buckwheat (Figure S18). We scanned the signification loci of mGWAS for the above four metabolites in that of the pGWAS for grain yield, unfortunately, we did not find the same loci. Thus, whether these metabolite variationsare responsible for the agronomic traits change requires further analysis. We subsequently performed pGWAS of some domestication associated traits, including plant height and whole growth period, as illustrated in Table S1. Further analysis found four lead SNPs overlapped with mGWAS signals (Figure S19). Among them, one of the significant loci of whole growth period was overlapped with that of 1‐hydroxyterpinin mono‐glucoside, a metabolite belonging to lignan. Three of the significant loci of seed diameter were overlapped with that of 3,4‐Dicaffeoylquinic acid. We postulate that these loci could be used to develop potential molecular markers for domestication associated traits. As Tartary buckwheat has both considerable edible and medicinal value, maintaining the levels of these pharmacological‐relevant metabolites while increasing yield is the most important problem currently facing Tartary buckwheat breeding. These associations between pharmacological‐relevant metabolites and grain yield could not only contribute to the improvement of buckwheat breeding, but also provide a theoretical basis for predicting complex agronomic traits through metabolite content, so as to accelerate the buckwheat breeding process.
Given that agronomic traits were generally regulated by specialized metabolites, the basis behind domestication was the selection of the specialized metabolite composition (Ku et al., 2020). At present, it has been found that domestication could result in multiple metabolite changes in plants (Beleggia et al., 2016; Chen et al., 2020a; Giovannoni, 2018). These changes may be directly caused by one gene or indirectly caused by the linkage of genes within the corresponding domestication sweeps (Giovannoni, 2018). As plant defence responses often occurs at the cost of growth and reproduction (Karasov et al., 2017), crop domestication with high yield usually leads to the disappearance of resistant traits (Tian et al., 2018), In addition, many resistant related specialized metabolites have bitter taste, while consumers tend to prefer less bitter crops, resulting in the decreased content of resistant related substance. For instance, in tomato domestication, people preferred to choose less bitter fruit, resulting in decreased content of bitter steroidal glycoalkaloid (Giovannoni, 2018). Similar results were reported in legumes (loss of alkaloids and saponins during domestication) and in cucumber (loss of cucurbitacins during domestication) (Ku et al., 2020; Shang et al., 2014). Thus, domesticated crops usually have better yield, taste and appearance, however as a cost, the content of specialized metabolites as well as the resistance to biotic stress are reduced (Ku et al., 2020). However, as evidenced by a recent review there is little metabolic conservation with respect to domestication with the majority of major crops displaying different changes in metabolite abundance across major crop species (Alseekh et al., 2021). Moreover, we have previous confirmed, in Tartary buckwheat, that in order to fit human demand and growth environment, domestication of grain weight and growth period have taken predominance (Zhang et al., 2021). In our mGWAS, we found that the content of SA was decreased in the SL cultivated group with respect to the Himalayan wild accessions HW. Since SA is an important disease resistant signalling molecule which exhibits a sharp aroma (Zhang and Li, 2019), we speculate that the change of SA content caused the disease resistance ability modified during Tartary buckwheat domestication. We also found the content of glycosylated emodin decreased during Tartary buckwheat domestication, indicating humans instinctively selected for less bitter crops, progressively selecting plants with tastier crops and consequently, reducing the levels of the more bitter medicinal ingredients glycosylated emodin.
Conclusion
Our mGWAS analysis revealed a spectrum of specialized metabolites, including disease resistant molecules and bioactive compounds, changed during domestication of Tartary buckwheat, and a variety of genes eventually responsible for metabolites changes. These results will further enrich the regulation mechanism of specialized metabolism of Tartary buckwheat and provide a theoretical basis for breeding Tartary Buckwheat varieties with high active ingredients and better agronomic properties.
Experimental procedures
Plant material and phenotyping
A diverse worldwide collection of 200 Tartary buckwheat accessions based on the maximization of the allelic diversity/richness, phylogenetic relationships, passport information and morphological traits according to Core Hunter software (Thachuk et al., 2009). These 200 accessions include 14 HW accessions, 80 SL accessions and 106 NL accessions (Zhang et al., 2021). The nucleotide diversity values (π) of the 200 accessions and 510 accessions were calculated using the TASSEL5. The detailed information about these accessions, including variety name, place of origin, subpopulation identity and morphological traits was listed in Table S1.
For metabolic profiling, these 200 Tartary buckwheat accessions were repeatedly sown in two plots in April and harvested in July of 2019 in Liangshan (27°59′N, 102°50′ E, Sichuan, China). In detail, the time of sowing and the altitudes of the planting plots were different. For repeat 1, all accessions were sown in a plot at an altitude of 2063 m, located in Liangshan in early 8 April, 2019. After 10 days, repeat 2 were sown at an altitude of 2550 m in another plot that is approximately 600 m away from repeat 1. The horizontal and vertical distance between the two plots is approximately 300 and 487 m, respectively (Figure S1). The two plots were both plots 0.2 ha in size and had the same soil conditions and management. 20 plants per accession as one biological replication were planted with density of 35 cm between plants in a row and 45 cm between rows. The whole of accessions in each plot was arranged uninterruptedly by serial number listed in Table S1. Field management, including irrigation, fertilization, weeding, and pest control, followed the standard agricultural practices in buckwheat production. About 50 mature grains were harvested from five plants per accession with representative phenotype randomly selected from repeat 1 and then pooled for metabolic profiling. The other biological replicate was pooled from the repeat 2 in the same way.
For phenotyping, the 200 Tartary buckwheat accessions were grown in Liangshan in 2019. Three individual plants from each accession in each replication were used for measurements of plant height. The whole growth period was defined as the growing days from sowing to maturity. Mature seeds were harvested from the selected six plants for measurements of 1000‐grain weight, grain yield per plant, seed length, seed width, pericarp colour and seed diameter and seed morphological.
Metabolite profiling and metabolomics
A liquid chromatography‐electrospray ionization‐tandem mass spectrometry (LC–MS/MS) system was used for the relative quantification of widely targeted metabolites in dried Tartary buckwheat seed samples (Chen et al., 2013). The lyophilized seeds were grounded using a mixer mill (MM400, Retsch) with a zirconia bead for 1.5 min at 30 Hz. The powder of each accession was stored at −80 °C prior to extraction. The 100 mg powder was weighed and extracted with 1.2 mL 70% (v/v) aqueous methanol. The mixture was further vortexed, every 10 min, three times before being kept at 4 °C overnight. Following centrifugation at 10 000 g for 10 min, all the supernatants were combined (CNWBON Carbon‐GCB SPE Cartridge, 250 mg, 3 mL; ANPEL, Shanghai, China, http://www.anpel.com.cn) and filtrated (SCAA‐104, 0.22 μm pore size; ANPEL, Shanghai, China, http://www.anpel.com.cn) before HPLC‐MS/MS analysis.
The sample extracts were analysed using an UPLC‐ESI‐MS/MS system (UPLC, Shim‐pack UFLC Shimadzu CBM30A system, www.shimadzu.com.cn; MS, Applied Biosystems 6500 QTRAP, www.appliedbiosystems.com.cn). The analytical conditions were as follows, UPLC: column, Agilent SB‐C18 (1.8 μm, 2.1 mm × 100 mm; Agilent Technologies, Santa Clara, California, USA); solvent A, pure water with 0.1% formic acid, and solvent B, acetonitrile. The gradient program was as follows, the starting conditions of 95% A, 5% B; within 9 min, a linear gradient to 5% A, 95% B was programmed, and a composition of 5% A, 95% B was kept for 1 min; a composition of 95% A, 5% B was adjusted within 1.10 min and kept for 2.9 min. The column oven was set to 40 °C; The injection volume was 2 μL. The effluent was alternatively connected to an ESI‐triple quadrupole–linear ion trap (QTRAP)‐MS.
Linear ion trap (LIT) and triple quadrupole (QQQ) scans were acquired on a QTRAP mass spectrometer using an API 6500 QTRAP UPLC/MS/MS System, equipped with an ESI Turbo Ionspray interface operated in positive and negative ion mode and controlled by Analyst 1.6.3 software (ABSciex, Redwood, California, USA). The ESI source operation parameters were as follows: ion source, turbo spray; source temperature 550 °C; ion spray voltage (IS) 5500 V (positive ion mode)/−4500 V (negative ion mode); ion source gas I (GSI), gas II (GSII), curtain gas (CUR) was set at 50, 60, and 30.0 psi, respectively; the collision gas (CAD) was high. Instrument tuning and mass calibration were performed with 10 and 100 μm polypropylene glycol solutions in QQQ and LIT modes, respectively. The QQQ scans were acquired as multiple reaction monitoring (MRM) experiments with collision gas (nitrogen) set to 5 psi. The declustering potential (DP) and collision energy (CE) for individual MRM transitions were done with further DP and CE optimization. A specific set of MRM transitions were monitored for each period according to the metabolites eluted within this period (Chen et al., 2014).
Metabolite annotation
Qualitative analysis of the metabolite data was performed base on the Metware Database (Metware Biotechnology Co., Ltd. Wuhan, China). A home‐made software reference to MetDNA of Zhu lab and Master view from AB Sciex were used to give the matching score of metabolites with the Metware Database. The quantitative analysis of metabolites used multiple reaction monitoring (MRM). In the MRM mode, the precursor ions were fragmented after the induced ionization of the collision chamber to form many fragment ions, then selected a characteristic fragment ion by triple quadrupole filtering to eliminate the interference of non‐target ions. After obtaining the metabolite mass spectrometry data of different samples, the peak area of all substance mass peaks was integrated, and the peaks of the same metabolite in different samples were integrated and corrected (Fraga, 2013).
Genome‐wide association analyses
A total of 1 004 824 SNPs (MAF >0.05 and Missing rate < 10%) for 200 accessions were used to perform the mGWAS. Efficient Mixed‐Model Association eXpedited program (EMMAx) and factored spectrally transformed linear mixed models (FaST‐LMM) were used to conduct all associations (Kang et al., 2010; Lippert et al., 2011). The effective number of independent markers (N) was calculated as 488 832 using the GEC tool (Li et al., 2012). and the suggestive P value (1/N) was set as the threshold. Linkage disequilibrium (LD) was estimated using standardized disequilibrium coefficients (D′) and squared allele‐frequency correlations (r 2) for pairs of SNP loci according to the TASSEL5 program. LD plots were generated in R software (version 4.0.4) under default parameters, wherein the r 2 values were indicated as percentages.
Identification of different metabolites
Identification of different metabolites between different groups was determined by partial least squares‐discriminate analysis (PLS‐DA) with variable importance for the projection (VIP) values >1, followed by ANOVA (P ≤ 0.05).
Phylogenetic and population structure analyses
A subset of 1 004 824 SNPs was used for phylogenetic and population structure analysis. Vcf files were converted to hapmap format with custom perl scripts and to PLINK format file by PLINK v1.90 (http://pngu.mgh.harvard.edu/purcell/plink/). Under the p‐distances model with bootstrapping (100), a neighbour‐joining tree of all samples was constructed with TreeBest 1.9.2.
Expression of candidate genes
Total RNA from Tartary buckwheat seedlings were extracted by the RNApre Pure Plant Plus kit (DP441; Tiangen, Beijing, China) according to the manufacturer's instructions. The first‐strand cDNA was synthesized using HiScript III RT SuperMix for qPCR (R323‐01; Vazyme, Nanjing, China) according to the manufacturer's protocol. Real‐time PCR was performed by using ChamQ Universal SYBR qPCR Master Mix (Q711; Vazyme). The expression measurements were obtained using the relative quantification method. Primers used in this study were shown in Table S19. For each construct, at least three independent over‐expression plants were selected for metabolites analysis.
Transgenic hairy roots initiation
The CDS of FtMYB43 was constructed into pCAMBIA 1307 vector (35S:FtMYB43‐cmyc), while FtSAGH1 and FtUGT74L2 were inserted into pCAMBIA 1302 vector (35S: FtSAGH1‐GFP and 35S: FtUGT74L2‐GFP). Then the vectors were transformed into Tartary buckwheat cv. Pinku by Agrobacterium rhizogenes A4 as previously described (Zhou et al., 2012). In detail, the methods were conducted as the following procedures: (i) Preparation of Pinku sterile seedlings, (ii) A. rhizogenes culture, (iii) infection and cocultivation of explants to produce hairy root cultures, (iv) selective culture and (v) subculturing the transgenic hairy roots. Dried hairy roots were used for measurement of corresponding metabolites. For transgenic Arabidopsis, FtMYB43 and FtSAGH1 were constructed into pCAMBIA1302 vector were transformed into Arabidopsis by Agrobacterium rhizogenes GV3101‐mediated transformation using the floral dip method (Zhang et al., 2006). The PCR and qRT‐PCR were used to test positively transgenic lines (Figures S20–S22).
Recombinant protein purification and enzyme assay
The full‐length cDNA of candidate gene was amplified and constructed into the pMAL‐C5X expression vector. Recombinant proteins were expressed in E.coli BL21 (DE3) chemically competent cells (CD601‐02; TransGen, Beijing, China) following induction by addition of 0.2 mm isopropyl‐d‐1‐thiogalactopyranoside (IPTG) and growing continually for 12 h at 16 °C. Cells were harvested pellets were re‐suspended and lysed by sonication. The crude extract was collected by centrifugation at 12 000 g for 10 min at 4 °C, and the supernatant was stored at −80 °C. Purification of the MBP‐tagged proteins was performed using amylase resin (New England Biolabs, Ipswich, Massachusetts, USA) following the manufacturer's instructions. Immunoblotting of MBP‐FtSAGH1 and MBP‐FtUGT74L2 were performed with anti‐MBP (1:2000; CW0299, CWBIO, Taizhou, Jiangsu, China) and anti‐mouse IgG (1:8000; CW0102, CWBIO) antibodies.
For FtSAGH1 activity assay, 0.5 μg purified protein was added to the reaction buffer (100 mm Tris–HCl, 14 mm β‐mercaptoethanol, 1 mm salicylic acid 2‐O‐β‐glucoside (ZC‐25543; SHANGHAI ZZBIO, Shanghai, China), 1 mm ATP, pH 8.0) and incubated at 37 °C for 30 min. For FtUGT74L2 activity assay, 0.5 μg purified protein was added to the reaction buffer (100 mm Tris–HCl, 14 mm β‐mercaptoethanol, 1 mm emodin (ZC‐48749; SHANGHAI ZZBIO), 9 mm UDP‐glucoside, pH 8.0) and incubated at 37 °C for 30 min. After terminated by freeze dryer at −40 °C, the dried reaction products were re‐dissolved in 80% methanol for analysed by LC–MS (Agilent G6500 Series HPLC‐QTOF). The standards of salicylic acid (S5922; Sigma Aldrich, Taufkirchen, Germany) and emodin‐8‐O‐β‐d‐glucoside (ZPG‐83849; SHANGHAI ZZBIO) were used to determine the product.
Yeast auto‐activation test
LiAc yeast transformation was used as described in the Matchmarker Gold Yeast Two‐Hybrid System User Manual (Clontech, San Jose, California, USA). The FtMYB43 were constructed into pGBKT7 and co‐transformed with pGADT7 into yeast strain Y2HGold. Then, the transformants were selected on SD/−L‐W. Auto‐activation test on SD/−L‐W‐H with 100 ng/mL Aureobasidin A (AbA,630466; Clontech).
Plant disease resistance test
Rhizoctonia solani AG‐4 HGI was used to test the disease resistance of Tartary buckwheat hypocotyl and transgenic Arabidopsis leaves (Li et al., 2020b). For infection of Tartary buckwheat hypocotyl, R. solani was pre‐cultured in PDB (potato dextrose broth) medium at 28 °C for 3 days. After dilution for 50 times, pathogen medium was used to infect Tartary buckwheat hypocotyls which germinated for 4 days. For infection of Arabidopsis leaves, R. solani was pre‐cultured in PDA (potato dextrose agar) plate for 5 days and placed the pathogen cake on detached leaves of 2‐weeks old. The disease index and incidence rate were conducted after infection for 16 h as previously described (Park et al., 2008).
Statistical analysis
Metabolite relative contents were log2‐transformed to fit the normal distribution. The coefficient of variation values was calculated independently for each metabolite. The hierarchical clustering analysis (HCA) and the heatmaps were obtained through R software (version 4.0.4), and the network construction was achieved using Cytoscape (version 3.7.1) on the basis of Spearman's correlation coefficients (P < 0.001). Data for gene relative expression level, metabolite relative content, disease index were presented as mean ± SD. Statistical differences were determined by two‐tailed Student's t‐test using GraphPad Prism software (version 9.2) and significance values was defined as of P < 0.05.
Conflict of interest
The authors declare no conflicts of interest.
Author contributions
M.Z. designed and managed the project. M.Z., D.J., V.M., M.I.G. X.Z. and M.L. organized the funding for this research. Y.C. performed the annotation of the metabolites. H.Z., Y.H., K.Z., M.H., B.G. and M.I.G. performed the data analysis and/or figure design. H.Z., S.L., F.H., D.Y. and Y.F. performed the most of experiments. Y.H., H.Z., N.G.‐G., C.H. and M.Z. wrote the manuscript. Y.H., H.Z., A.R.F., N.G.‐G., C.H., D.J., V.M. and M.Z. finalized the manuscript. All authors read and approved the paper.
Supporting information
Figure S1 Workflow of this study.
Figure S2 Characters of metabolome in three groups of Tartary buckwheat.
Figure S3 Correlation analyses of metabolites.
Figure S4 Networks for important substances in Tartary buckwheat.
Figure S5 Mul‐Manhattan plots and Quantile–Quantile (Q–Q) plots of the two biological replicates' main metabolites based on the FaST‐LMM and EMMAx model.
Figure S6 FtMYB43 transcription factor is involved in procyanidins biosynthesis in Tartary buckwheat.
Figure S7 Selective signals in domestication of NL (blue) and in SL (green) on each chromosome.
Figure S8 Manhattan plots and Quantile–Quantile (Q–Q) plots of the domesticated metabolites on the basis of a different model.
Figure S9 FtNAC is involved in regulating C7G biosynthesis.
Figure S10 FtPAL plays an important role in FPA1 biosynthesis.
Figure S11 Identification of FtSAGH1 tissue‐specific expression by qRT‐PCR
Figure S12 Western blotting with MBP antibody detected the expression of purified recombinant MBP‐FtSAGH1 and MBP‐FtUGT74L2.
Figure S13 LC–MS/MS fragmentation of salicylic acid and salicylic acid 2‐O‐β‐glucoside from enzymatic assays and other two candidate genes enzymatic assay in vitro.
Figure S14 Evaluation of disease resistance to Rhizoctonia solani in different buckwheat accessions.
Figure S15 The analysis of the proposed function of FtUGT74L2.
Figure S16 Identification of FtUGT74L2 tissue‐specific expression by qRT‐PCR.
Figure S17 Chemical structures and LC–MS/MS fragmentation of emodin and emodin‐8‐O‐β‐d‐glucoside from enzymatic assays.
Figure S18 Correlation analyses of agronomic traits and partial metabolites.
Figure S19 Manhattan plots display the mGWAS of 1‐Hydroxyterpinin mono‐glucoside and pGWAS of whole growth period (left), and mGWAS of 3,4‐Dicaffeoylquinic acid and pGWAS of seed diameter (right). The dashed line indicates the threshold ‐log10P=5. The same color arrows indicate the same loci.
Figure S20 All related transgenic hairy roots were verified by PCR.
Figure S21 Ectopic expression of FtSAGH1 and FtMYB43 in Arabidopsis were verified by PCR.
Figure S22 All related transgenic hairy roots were verified by qRT‐PCR.
Table S1 The list of 200 buckwheat accessions used in this study.
Table S2 Detailed information for metabolites detected in the current study.
Table S3 The relative contents of metabolites.
Table S4 Correlation between the detected metabolites.
Table S5 Significant mGWAS signals for all annotated metabolites.
Table S6 Identification of candidate genes associated with Ft1:13164441 on Chr.1 for procyanidin B1 content.
Table S7 The relationship bewteen the relative expression of FtMYB43 and the relative content of Procyanidins.
Table S8 The significantly different metabolites between different groups.
Table S9 Overlapping of the significant mGWAS loci for the metabolites with HW and NL domestication regions based on XP‐CLR analysis.
Table S10 Overlapping of the significant mGWAS loci for the metabolites with HW and SL domestication regions based on XP‐CLR analysis.
Table S11 Identification of candidate genes associated with Ft3:33408281 on Chr.3 for catechin‐7‐O‐glucoside content.
Table S12 Identification of candidate genes associated with Ft6:5513671 on Chr.6 for fagopyritol A1 content.
Table S13 Identification of candidate genes associated with Ft7:14120713 on Chr.7 for salicylic acid content.
Table S14 LC–MS detected the content of SAG/SA in hairy roots.
Table S15 Evaluation results of disease resistance in different groups.
Table S16 Evaluation results of disease resistance in transgenic Arabidopsis.
Table S17 Identification of candidate genes associated with Ft4:7097024 on Chr.4 for emodin‐8‐O‐β‐d‐glucoside content.
Table S18 LC–MS detected the content of EmG in hairy roots.
Table S19 Primers used in this study.
Acknowledgements
The authors appreciate the critical reading and helpful comments on the manuscript made by Tao Lin from China Agricultural University, China. The authors thank Yanrong Hao, Qian Zuo and Xiang Lu for their efforts on the preparation of plant samples and molecular experiments. This work was supported by the National Key R&D Program of China (2019YFD1000700/2019YFD1000701), the National Natural Science Foundation of China (31871536, 31801427, 31901511), the European Union Horizon 2020 project ECOBREED (771367), the Planta SYST (SGA‐CSA No. 739582 under FPA No.664620), and the China National Postdoctoral Program for Innovative Talents (BX20200377).
References
- Alseekh, S. , Scossa, F. , Wen, W. , Luo, J. , Yan, J. , Beleggia, R. , Klee, H.J. et al. (2021) Domestication of crop metabolomes: desired and unintended consequences. Trends Plant Sci. 26, 650–661. [DOI] [PubMed] [Google Scholar]
- Beleggia, R. , Rau, D. , Laidò, G. , Platani, C. , Nigro, F. , Fragasso, M. , De Vita, P. et al. (2016) Evolutionary metabolomics reveals domestication‐associated changes in tetraploid wheat kernels. Mol. Biol. Evol. 33, 1740–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, C.G. (1976) Buckwheat: Fagopyrum (Polygonaceae). In Evolution of Crop Plants ( Simmonds, N.W. , ed), pp. 235–237. London and New York: Longman. [Google Scholar]
- Chen, W. , Gong, L. , Guo, Z. , Wang, W. , Zhang, H. , Liu, X. , Yu, S. et al. (2013) A novel integrated method for large‐scale detection, identification, and quantification of widely targeted metabolites: application in the study of rice metabolomics. Mol. Plant, 6, 1769–1780. [DOI] [PubMed] [Google Scholar]
- Chen, W. , Gao, Y. , Xie, W. , Gong, L. , Lu, K. , Wang, W. , Li, Y. et al. (2014) Genome‐wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 46, 714–721. [DOI] [PubMed] [Google Scholar]
- Chen, W. , Wang, W. , Peng, M. , Gong, L. , Gao, Y. , Wan, J. , Wang, S. et al. (2016) Comparative and parallel genome‐wide association studies for metabolic and agronomic traits in cereals. Nat. Commun. 7, 12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Hu, X. , Shi, T. , Yin, H. , Sun, D. , Hao, Y. , Xia, X. et al. (2020a) Metabolite‐based genome‐wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnol. J. 18, 1722–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Zhu, M. , Liu, R. , Zhang, M. , Lv, Y. , Liu, Y. , Xiao, X. et al. (2020b) BIOMASS YIELD 1 regulates sorghum biomass and grain yield via the shikimate pathway. J. Exp. Bot. 71, 5506–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Yang, H. , Fang, Y. , Guo, W. , Chen, H. , Zhang, X. , Dai, W. et al. (2021) Overexpression of GmMYB14 improves high‐density yield and drought tolerance of soybean through regulating plant architecture mediated by the brassinosteroid pathway. Plant Biotechnol. J. 19, 702–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid, M.B. , Alfonso, F. and Martín‐Lomas, M. (2004) Synthesis of fagopyritols A1 and B1 from D‐chiro‐inositol. Carbohydr. Res. 339, 2303–2307. [DOI] [PubMed] [Google Scholar]
- De Luca, V. , Salim, V. , Atsumi, S.M. and Yu, F. (2012) Mining the biodiversity of plants: a revolution in the making. Science, 336, 1658–1661. [DOI] [PubMed] [Google Scholar]
- Dean, J.V. and Delaney, S.P. (2008) Metabolism of salicylic acid in wild‐type, ugt74f1 and ugt74f2 glucosyltransferase mutants of Arabidopsis thaliana . Physiol. Plant. 132, 417–425. [DOI] [PubMed] [Google Scholar]
- Ding, P. and Ding, Y. (2020) Stories of salicylic acid: a plant defense hormone. Trends Plant Sci. 25, 549–565. [DOI] [PubMed] [Google Scholar]
- Dong, X. , Fu, J. , Yin, X. , Cao, S. , Li, X. , Lin, L. and Ni, J. (2016) Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother. Res. 30, 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, Q. , Zhao, H. , Huang, Y. , Chen, Y. , Wan, M. , Zeng, Z. , Yao, P. et al. (2020) FtMYB18 acts as a negative regulator of anthocyanin/proanthocyanidin biosynthesis in Tartary buckwheat. Plant Mol. Biol. 104, 309–325. [DOI] [PubMed] [Google Scholar]
- Fernie, A.R. and Yan, J. (2019) De Novo domestication: an alternative route toward new crops for the future. Mol. Plant, 12, 615–631. [DOI] [PubMed] [Google Scholar]
- Fraga, B.M. (2013) Natural sesquiterpenoids. Nat. Prod. Rep. 30, 1226–1264. [DOI] [PubMed] [Google Scholar]
- Fuller, D.Q. and Allaby, R. (2009) Seed dispersal and crop domestication: shattering, germination and seasonality in evolution under cultivation. Ann. Plant Rev. 38, 238–295. [Google Scholar]
- Gao, L. , Gonda, I. , Sun, H. , Ma, Q. , Bao, K. , Tieman, D.M. , Burzynski‐Chang, E.A. et al. (2019) The tomato pan‐genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 51, 1044–1051. [DOI] [PubMed] [Google Scholar]
- Giovannoni, J. (2018) Tomato multiomics reveals consequences of crop domestication and improvement. Cell, 172, 6–8. [DOI] [PubMed] [Google Scholar]
- Horbowicz, M. , Brenac, P. and Obendorf, R.L. (1998) Fagopyritol B1, O‐alpha‐D‐galactopyranosyl‐(1→2)‐D‐chiro‐inositol, a galactosyl cyclitol in maturing buckwheat seeds associated with desiccation tolerance. Planta, 205, 1–11. [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Wu, Q. , Wang, S. , Shi, J. , Dong, Q. , Yao, P. , Shi, G. et al. (2019) FtMYB8 from Tartary buckwheat inhibits both anthocyanin/proanthocyanidin accumulation and marginal trichome initiation. BMC Plant Biol. 19, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt, H.V. , Shang, X. and Jones, M.K. (2018) Buckwheat: a crop from outside the major Chinese domestication centres? A review of the archaeobotanical, palynological and genetic evidence. Veg. Hist. Archaeobot. 27, 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing, R. , Li, H.Q. , Hu, C.L. , Jiang, Y.P. , Qin, L.P. and Zheng, C.J. (2016) Phytochemical and pharmacological profiles of three Fagopyrum Buckwheats. Int. J. Mol. Sci., 17, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi, D.C. , Zhang, K. , Wang, C. , Chandora, R. , Khurshid, M. , Li, J. , He, M. et al. (2020) Strategic enhancement of genetic gain for nutraceutical development in buckwheat: a genomics‐driven perspective. Biotechnol. Adv. 39, 107479. [DOI] [PubMed] [Google Scholar]
- Kang, H.M. , Sul, J.H. , Service, S.K. , Zaitlen, N.A. , Kong, S.Y. , Freimer, N.B. , Sabatti, C. et al. (2010) Variance component model to account for sample structure in genome‐wide association studies. Nat. Genet. 42, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasov, T.L. , Chae, E. , Herman, J.J. and Bergelson, J. (2017) Mechanisms to mitigate the trade‐off between growth and defense. Plant Cell, 29, 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzai, Y. , Kimura, M. , Watanabe, M. , Kusunoki, K. , Osaka, D. , Suzuki, T. , Matsui, H. et al. (2018) Salicylic acid‐dependent immunity contributes to resistance against Rhizoctonia solani, a necrotrophic fungal agent of sheath blight, in rice and Brachypodium distachyon . New Phytol. 217, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreft, I. , Zhou, M. , Golob, A. , Germ, M. , Likar, M. , Dziedzic, K. and Luthar, Z. (2020) Breeding buckwheat for nutritional quality. Breed. Sci. 70, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku, Y.S. , Contador, C.A. , Ng, M.S. , Yu, J. , Chung, G. and Lam, H.M. (2020) The effects of domestication on secondary metabolite composition in Legumes . Front. Genet. 11, 581357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari, A. and Chaudhary, H.K. (2020) Nutraceutical crop buckwheat: a concealed wealth in the lap of Himalayas. Crit. Rev. Biotechnol. 40, 539–554. [DOI] [PubMed] [Google Scholar]
- Lee, D.K. , Chung, P.J. , Jeong, J.S. , Jang, G. , Bang, S.W. , Jung, H. , Kim, Y.S. et al. (2017) The rice OsNAC6 transcription factor orchestrates multiple molecular mechanisms involving root structural adaptions and nicotianamine biosynthesis for drought tolerance. Plant Biotechnol. J. 15, 754–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M.X. , Yeung, J.M. , Cherny, S.S. and Sham, P.C. (2012) Evaluating the effective numbers of independent tests and significant p‐value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Zhang, K. , Meng, Y. , Li, Q. , Ding, M. and Zhou, M. (2019a) FtMYB16 interacts with Ftimportin‐α1 to regulate rutin biosynthesis in Tartary buckwheat. Plant Biotechnol. J. 17, 1479–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. , Lin, B. , Wang, H. , Li, X. , Yang, F. , Ding, X. , Yan, J. et al. (2019b) Natural variation in ZmFBL41 confers banded leaf and sheath blight resistance in maize. Nat. Genet. 51, 1540–1548. [DOI] [PubMed] [Google Scholar]
- Li, Q. , Gao, J. , Pang, X. , Chen, A. and Wang, Y. (2020a) Molecular mechanisms of action of emodin: as an anti‐cardiovascular disease drug. Front. Pharmacol. 11, 559607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Zhang, K. , Khurshid, M. , Fan, Y. , Xu, B. and Zhou, M. (2020b) First report of Rhizoctonia solani AG‐4 HGI causing stem canker on Fagopyrum tataricum (Tartary buckwheat) in China. Plant Dis. 104, 3263. [DOI] [PubMed] [Google Scholar]
- Lippert, C. , Listgarten, J. , Liu, Y. , Kadie, C.M. , Davidson, R.I. and Heckerman, D. (2011) FaST linear mixed models for genome‐wide association studies. Nat. Methods, 8, 833–835. [DOI] [PubMed] [Google Scholar]
- Ma, Y. , Liu, M. , Stiller, J. and Liu, C. (2019) A pan‐transcriptome analysis shows that disease resistance genes have undergone more selection pressure during barley domestication. BMC Genomics, 20, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín‐García, B. , Pasini, F. , Verardo, V. , Gómez‐Caravaca, A.M. , Marconi, E. and Caboni, M.F. (2019) Distribution of free and bound phenolic compounds in buckwheat milling fractions. Foods, 8, 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruri‐López, I. , Aviles‐Baltazar, N.Y. , Buchala, A. and Serrano, M. (2019) Intra and extracellular journey of the phytohormone salicylic acid. Front. Plant Sci. 10, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuccio, M.L. , Wu, J. , Mowers, R. , Zhou, H.P. , Meghji, M. , Primavesi, L.F. , Paul, M.J. et al. (2015) Expression of trehalose‐6‐phosphate phosphatase in maize ears improves yield in well‐watered and drought conditions. Nat. Biotechnol. 33, 862–869. [DOI] [PubMed] [Google Scholar]
- Ohnishi, O. (1998) Search for the wild ancestor of buckwheat III. The wild ancestor of cultivated common buckwheat, and of Tartary buckwheat. Econ. Bot. 52, 123–133. [Google Scholar]
- Oladzad, A. , Zitnick‐Anderson, K. , Jain, S. , Simons, K. , Osorno, J.M. , McClean, P.E. and Pasche, J.S. (2019) Genotypes and genomic regions associated with Rhizoctonia solani resistance in common bean. Front. Plant Sci. 10, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, D.S. , Sayler, R.J. , Hong, Y.G. , Nam, M.H. and Yang, Y. (2008) A method for inoculation and evaluation of rice sheath blight disease. Plant Dis. 92, 25–29. [DOI] [PubMed] [Google Scholar]
- Peng, L.X. , Wang, J.B. , Hu, L.X. , Zhao, J.L. , Xiang, D.B. , Zou, L. and Zhao, G. (2013) Rapid and simple method for the determination of emodin in Tartary buckwheat (Fagopyrum tataricum) by high‐performance liquid chromatography coupled to a diode array detector. J. Agric. Food Chem. 61, 854–857. [DOI] [PubMed] [Google Scholar]
- Saito, K. and Matsuda, F. (2010) Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 61, 463–489. [DOI] [PubMed] [Google Scholar]
- Shang, Y. , Ma, Y. , Zhou, Y. , Zhang, H. , Duan, L. , Chen, H. , Zeng, J. et al. (2014) Biosynthesis, regulation, and domestication of bitterness in cucumber. Science, 346, 1084–1088. [DOI] [PubMed] [Google Scholar]
- Sharma, S. , Ali, A. , Ali, J. , Sahni, J.K. and Baboota, S. (2013) Rutin: therapeutic potential and recent advances in drug delivery. Expert Opin. Investig. Drugs, 22, 1063–1079. [DOI] [PubMed] [Google Scholar]
- Shi, T. , Zhu, A. , Jia, J. , Hu, X. , Chen, J. , Liu, W. , Ren, X. et al. (2020) Metabolomics analysis and metabolite‐agronomic trait associations using kernels of wheat (Triticum aestivum) recombinant inbred lines. Plant J. 103, 279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, S. , Phillips, G.N., Jr. and Thorson, J.S. (2012) The structural biology of enzymes involved in natural product glycosylation. Nat. Prod. Rep. 29, 1201–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thachuk, C. , Crossa, J. , Franco, J. , Dreisigacker, S. , Warburton, M. and Davenport, G.F. (2009) Core hunter: an algorithm for sampling genetic resources based on multiple genetic measures. BMC Bioinformatics, 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, L. , Shi, S. , Nasir, F. , Chang, C. , Li, W. , Tran, L.P. and Tian, C. (2018) Comparative analysis of the root transcriptomes of cultivated and wild rice varieties in response to Magnaporthe oryzae infection revealed both common and species‐specific pathogen responses. Rice, 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji, K. and Ohnishi, O. (2001) Phylogenetic relationships among wild and cultivated Tartary buckwheat(Fagopyrum tataricum Gaert) populations revealed by AFLP analyses. Genes Genet. Syst. 76, 47–52. [DOI] [PubMed] [Google Scholar]
- Vlot, A.C. , Dempsey, D.A. and Klessig, D.F. (2009) Salicylic acid, a multifaceted hormone to combat disease. Annu. Rev. Phytopathol. 47, 177–206. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Lu, W. , Ran, L. , Dou, L. , Yao, S. , Hu, J. , Fan, D. et al. (2019) R2R3‐MYB transcription factor MYB6 promotes anthocyanin and proanthocyanidin biosynthesis but inhibits secondary cell wall formation in Populus tomentosa . Plant J. 99, 733–751. [DOI] [PubMed] [Google Scholar]
- Weisskopf, A. and Fuller, D.Q. (2014) Buckwheat: origins and development. In Encyclopedia of Global Archaeology ( Smith, C. , ed), pp. 1025–1028. New York, NY: Springer New York. [Google Scholar]
- Wen, W. , Li, D. , Li, X. , Gao, Y. , Li, W. , Li, H. , Liu, J. et al. (2014) Metabolome‐based genome‐wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 5, 3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, K. , Chen, R. , Li, J. , Wang, R. , Chen, D. , Dou, X. and Dai, J. (2014) Exploring the catalytic promiscuity of a new glycosyltransferase from Carthamus tinctorius . Org. Lett. 16, 4874–4877. [DOI] [PubMed] [Google Scholar]
- Yao, J. , Huot, B. , Foune, C. , Doddapaneni, H. and Enyedi, A. (2007) Expression of a beta‐glucosidase gene results in increased accumulation of salicylic acid in transgenic Nicotiana tabacum cv. Xanthi‐nc NN genotype. Plant Cell Rep. 26, 291–301. [DOI] [PubMed] [Google Scholar]
- Yin, Q. , Han, X. , Han, Z. , Chen, Q. , Shi, Y. , Gao, H. , Zhang, T. et al. (2020) Genome‐wide analyses reveals a glucosyltransferase involved in rutin and emodin glucoside biosynthesis in Tartary buckwheat. Food Chem. 318, 126478. [DOI] [PubMed] [Google Scholar]
- Zeng, X. , Yuan, H. , Dong, X. , Peng, M. , Jing, X. , Xu, Q. , Tang, T. et al. (2020) Genome‐wide dissection of co‐selected UV‐B responsive pathways in the UV‐B adaptation of qingke. Mol. Plant 13, 112–127. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. and Li, X. (2019) Salicylic acid: biosynthesis, perception, and contributions to plant immunity. Curr. Opin. Plant Biol. 50, 29–36. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Henriques, R. , Lin, S.S. , Niu, Q.W. and Chua, N.H. (2006) Agrobacterium‐mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 1, 641–646. [DOI] [PubMed] [Google Scholar]
- Zhang, K. , He, M. , Fan, Y. , Zhao, H. , Gao, B. , Yang, K. , Li, F. et al. (2021) Resequencing of global Tartary buckwheat accessions reveals multiple domestication events and key loci associated with agronomic traits. Genome Biol. 22, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, M.L. , Zhu, X.M. , Shao, J.R. , Wu, Y.M. and Tang, Y.X. (2012) An protocol for genetic transformation of Catharanthus roseus by Agrobacterium rhizogenes A4. Appl. Biochem. Biotechnol. 166, 1674–1684. [DOI] [PubMed] [Google Scholar]
- Zhu, F. (2016) Chemical composition and health effects of Tartary buckwheat. Food Chem. 203, 231–245. [DOI] [PubMed] [Google Scholar]
- Zhu, G. , Wang, S. , Huang, Z. , Zhang, S. , Liao, Q. , Zhang, C. , Lin, T. et al. (2018) Rewiring of the fruit metabolome in tomato breeding. Cell, 172, 249–261.e212. [DOI] [PubMed] [Google Scholar]
- Zrenner, R. , Genzel, F. , Verwaaijen, B. , Wibberg, D. and Grosch, R. (2020) Necrotrophic lifestyle of Rhizoctonia solani AG3‐PT during interaction with its host plant potato as revealed by transcriptome analysis. Sci. Rep. 10, 12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Workflow of this study.
Figure S2 Characters of metabolome in three groups of Tartary buckwheat.
Figure S3 Correlation analyses of metabolites.
Figure S4 Networks for important substances in Tartary buckwheat.
Figure S5 Mul‐Manhattan plots and Quantile–Quantile (Q–Q) plots of the two biological replicates' main metabolites based on the FaST‐LMM and EMMAx model.
Figure S6 FtMYB43 transcription factor is involved in procyanidins biosynthesis in Tartary buckwheat.
Figure S7 Selective signals in domestication of NL (blue) and in SL (green) on each chromosome.
Figure S8 Manhattan plots and Quantile–Quantile (Q–Q) plots of the domesticated metabolites on the basis of a different model.
Figure S9 FtNAC is involved in regulating C7G biosynthesis.
Figure S10 FtPAL plays an important role in FPA1 biosynthesis.
Figure S11 Identification of FtSAGH1 tissue‐specific expression by qRT‐PCR
Figure S12 Western blotting with MBP antibody detected the expression of purified recombinant MBP‐FtSAGH1 and MBP‐FtUGT74L2.
Figure S13 LC–MS/MS fragmentation of salicylic acid and salicylic acid 2‐O‐β‐glucoside from enzymatic assays and other two candidate genes enzymatic assay in vitro.
Figure S14 Evaluation of disease resistance to Rhizoctonia solani in different buckwheat accessions.
Figure S15 The analysis of the proposed function of FtUGT74L2.
Figure S16 Identification of FtUGT74L2 tissue‐specific expression by qRT‐PCR.
Figure S17 Chemical structures and LC–MS/MS fragmentation of emodin and emodin‐8‐O‐β‐d‐glucoside from enzymatic assays.
Figure S18 Correlation analyses of agronomic traits and partial metabolites.
Figure S19 Manhattan plots display the mGWAS of 1‐Hydroxyterpinin mono‐glucoside and pGWAS of whole growth period (left), and mGWAS of 3,4‐Dicaffeoylquinic acid and pGWAS of seed diameter (right). The dashed line indicates the threshold ‐log10P=5. The same color arrows indicate the same loci.
Figure S20 All related transgenic hairy roots were verified by PCR.
Figure S21 Ectopic expression of FtSAGH1 and FtMYB43 in Arabidopsis were verified by PCR.
Figure S22 All related transgenic hairy roots were verified by qRT‐PCR.
Table S1 The list of 200 buckwheat accessions used in this study.
Table S2 Detailed information for metabolites detected in the current study.
Table S3 The relative contents of metabolites.
Table S4 Correlation between the detected metabolites.
Table S5 Significant mGWAS signals for all annotated metabolites.
Table S6 Identification of candidate genes associated with Ft1:13164441 on Chr.1 for procyanidin B1 content.
Table S7 The relationship bewteen the relative expression of FtMYB43 and the relative content of Procyanidins.
Table S8 The significantly different metabolites between different groups.
Table S9 Overlapping of the significant mGWAS loci for the metabolites with HW and NL domestication regions based on XP‐CLR analysis.
Table S10 Overlapping of the significant mGWAS loci for the metabolites with HW and SL domestication regions based on XP‐CLR analysis.
Table S11 Identification of candidate genes associated with Ft3:33408281 on Chr.3 for catechin‐7‐O‐glucoside content.
Table S12 Identification of candidate genes associated with Ft6:5513671 on Chr.6 for fagopyritol A1 content.
Table S13 Identification of candidate genes associated with Ft7:14120713 on Chr.7 for salicylic acid content.
Table S14 LC–MS detected the content of SAG/SA in hairy roots.
Table S15 Evaluation results of disease resistance in different groups.
Table S16 Evaluation results of disease resistance in transgenic Arabidopsis.
Table S17 Identification of candidate genes associated with Ft4:7097024 on Chr.4 for emodin‐8‐O‐β‐d‐glucoside content.
Table S18 LC–MS detected the content of EmG in hairy roots.
Table S19 Primers used in this study.