

Abstract
The primary mode of metabolism of nicotine is via the formation of cotinine by the enzyme CYP2A6. Cotinine undergoes further CYP2A6-mediated metabolism by hydroxylation to 3-hydroxycotinine and norcotinine, but can also form cotinine-N-glucuronide and cotinine-N-oxide (COX). The goal of this study was to investigate the enzymes that catalyze COX formation and determine whether genetic variation in these enzymes may affect this pathway. Specific inhibitors of major hepatic cytochrome P450 (P450) enzymes were used in cotinine-N-oxidation reactions using pooled human liver microsomes (HLMs). COX formation was monitored by ultrahigh-pressure liquid chromatography–tandem mass spectrometry and enzyme kinetic analysis was performed using microsomes from P450-overexpressing human embryonic kidney 293 (HEK293) cell lines. Genotype-phenotype analysis was performed in a panel of 113 human liver specimens. Inhibition of COX formation was only observed in HLMs when using inhibitors of CYP2A6, CYP2B6, CYP2C19, CYP2E1, and CYP3A4. Microsomes from cells overexpressing CYP2A6 or CYP2C19 exhibited similar N-oxidation activity against cotinine, with maximum reaction rate over Michaelis constant values (intrinsic clearance) of 4.4 and 4.2 nL/min/mg, respectively. CYP2B6-, CYP2E1-, and CYP3A4-overexpressing microsomes were also active in COX formation. Significant associations (P < 0.05) were observed between COX formation and genetic variants in CYP2C19 (*2 and *17 alleles) in HLMs. These results demonstrate that genetic variants in CYP2C19 are associated with decreased COX formation, potentially affecting the relative levels of cotinine in the plasma or urine of smokers and ultimately affecting recommended smoking cessation therapies.
SIGNIFICANCE STATEMENT
This study is the first to elucidate the enzymes responsible for cotinine-N-oxide formation and genetic variants that affect this biological pathway. Genetic variants in CYP2C19 have the potential to modify nicotine metabolic ratio in smokers and could affect pharmacotherapeutic decisions for smoking cessation treatments.
Introduction
The major mode of metabolism of nicotine in smokers (∼70%) is via the formation of cotinine (COT) by the CYP2A6 enzyme. COT is slowly eliminated from plasma with a half-life of 10–27 hours (Jarvis et al., 1988), undergoing metabolism by hydroxylation to 3-hydroxy-COT (3HC) (Nakajima et al., 1996a) and N-(hydroxyl-methyl) norcotinine (Brown et al., 2005), glucuronidation to cotinine-N-glucuronide (COT-Gluc) (Chen et al., 2007), and oxidation to COT-N-oxide (COX), accounting for an average of 63%, 4%, 24%, and 9% of cotinine metabolites, respectively, in the urine of Caucasian smokers (Fig. 1) (Rangiah et al., 2011). While extensive studies have been performed examining the enzymes responsible for the formation of COT (Nakajima et al., 1996b), N-(hydroxymethyl)-norcotinine (Brown et al., 2005), and COT-Gluc (Chen et al., 2010), no studies have identified the enzymes responsible for COX formation (Yamanaka et al., 2004; Yildiz, 2004).
Fig. 1.

Schematic of the cotinine metabolism pathway. (1) Nicotine, (2) COT, (3) 3HC, (4) COX, (5) COT-glucuronide, and (6) norcotinine. Percentages [taken from (Rangiah et al., 2011)] indicate the levels of each metabolite as a percentage of total cotinine metabolites in the urine from smokers.
A potentially important role for COX may be as a factor that affects the nicotine metabolic ratio (NMR), which describes the ratio of plasma 3HC to COT in individual smokers (Dempsey et al., 2004), as a biomarker of nicotine addiction. This ratio is highly correlated with nicotine clearance in humans and has been generally accepted as an in vivo marker for CYP2A6 activity (Dempsey et al., 2004). The NMR is widely used to assess nicotine dependence and smoking behavior (Falcone et al., 2011), and to aid clinicians in determining the most efficacious pharmacotherapy for smoking cessation (Lerman et al., 2006, 2015; Malaiyandi et al., 2006; Chen et al., 2018). It has been shown that the NMR is highly variable among different demographic groups and that these differences could be affecting smoking cessation rates at the population level (Baurley et al., 2016; Fix et al., 2017). It has also been widely reported that nicotine metabolism varies among patients and that some of this variation can be explained by genetic variation in genes involved in the nicotine metabolism pathway (Hukkanen et al., 2005; Chenoweth et al., 2014). While alterations in the levels of 3HC-Gluc did not affect the NMR in African American subjects regardless of uridine 5′-diphospho-glucuronosyltransferase (UGT) 2B17 genotype (Zhu et al., 2013), it was reported that the NMR is influenced by the levels of COT-Gluc formation in both Caucasian and African American smokers (Berg et al., 2010; Jacobson and Ferguson, 2014; Murphy et al., 2014, 2017), and variability in NMR due to differences in COT-Gluc formation were suggested to be of particular importance in populations with a high prevalence of functional UGT2B10 genetic variants (e.g., African Americans) (Murphy, 2017).
The presence of COX was first reported by Dagne et al. in male rhesus monkey urine in 1972 (Dagne and Castagnoli, 1972). Recent evidence has suggested that urinary COX is an effective biomarker of the effects of environmental or secondhand tobacco smoking on skin autofluorescence, a tool used to predict diabetes-related cardiovascular complications (Van Waateringe et al., 2017). COX has also been used as a biomarker for active nicotine consumption in athletes (Marclay et al., 2011) and as a biomarker of nicotine exposure in breast milk (Pellegrini et al., 2007). Studies examining the potential role of the flavin monooxygenase enzymes demonstrated that none of these enzymes exhibited COX formation activity (Gorrod and Peyton, 1999; Tsai and Gorrod, 1999). Additional studies using cytochrome P450 (P450) inhibitors demonstrated that COX is formed from COT in hamster and guinea pig liver microsomes, potentially by P450 enzymes (Jenner et al., 1971). This was validated in further studies where phenobarbital (a strong P450 inducer) pretreatment in rat liver increased COX production eightfold, an effect that was negated when cotreated with the P450 inhibitor metyrapone (Foth et al., 1992). The goal of this study was to identify the human P450 enzymes responsible for COX formation and to determine whether genetic variations in these enzymes affect the levels of COX formation in a panel of human liver specimens.
Materials and Methods
Chemicals and Materials
Dulbecco’s Modified Eagles Medium, Dulbecco’s phosphate-buffered saline, fetal bovine serum, and geneticin (G418) were purchased from Gibco (Grand Island, New York). Anti–V5-horseradish peroxide (HRP) antibody was obtained from Invitrogen (Carlsbad, California) while the anti–calnexin-HRP antibody was purchased from Abcam (Cambridge, UK). The BCA protein assays used for total protein quantification were purchased from Pierce (Rockford, Illinois) and the NADPH regeneration system was purchased from Corning (Corning, New York). The following chemicals used for in vitro N-oxidation activity assays were purchased from Sigma Aldrich (St. Louis, Missouri): phenacetin, bupropion, amodiaquine, diclofenac, omeprazole, dextromethorphan, chlorzoxazone, midazolam, furafylline, clopidogrel, montelukast, sulfaphenazole, tranylcypromine, quinidine, clomethiazole, ketoconazole, and cotinine. Pooled human liver microsomes (HLMs) were purchased from Sekisui XenoTech (Kansas City, Kansas) while pooled human liver RNA was purchased from Biochain (Newark, CA). SuperScript VILO synthesis kit was purchased from Thermo Fisher Scientific (Waltman, MA) and TaqMan probes were purchased from AB Applied Biosystems (Foster City, CA). High-performance liquid chromatography-grade ammonium acetate and acetonitrile were purchased from Fisher Scientific (Pittsburgh, Pennsylvania) while the ACQUITY UPLC BEH-HILIC (1.7µm 2.1 × 100 mm) column was purchased from Waters (Milford, Massachusetts). Cotinine, COX, 3HC, and the internal standards D3-COX and D3-3HC were purchased from Toronto Research Chemicals (Ontario, Canada).
Biological Specimens
Normal human liver specimens and matching genomic DNA samples from 113 subjects were provided by the Tissue Procurement Facility at the H. Lee Moffitt Cancer Center (Tampa, FL) as previously described (Chen et al., 2012). All subjects were Caucasian, 42% (n = 48) were female, and the mean age of the subjects was 64 years. Microsomes were prepared as described previously and were stored at −80°C (Coughtrie et al., 1987; Yokota et al., 1989). All protocols involving tissue specimens were approved by the institutional review board at the H. Lee Moffitt Cancer Center and in accordance with assurances filed with and approved by the United States Department of Health and Human Services.
P450 Inhibition Assays in HLMs
Inhibition reactions (final volume = 25 µL) contained 25 µg of total pooled HLM protein, 50 mM potassium phosphate, NADPH-regenerating system (1.55 mmol/L NADP+, 3.3 mmol/L glucose-6-phosphate, 3.3 mmol/L MgCl2, and 0.5 U of 40 U/mL glucose-6-phosphate dehydrogenase), and 500 μM COT. Specific P450 inhibitors were added at concentrations of 1 or 10 µM: furafylline [CYP1A2 (Sesardic et al., 1990)], tranylcypromine [CYP2A6 and CYP2C19 (Draper et al., 1997; Taavitsainen et al., 2001)], clopidogrel [CYP2B6 (Richter et al., 2004)], montelukast [CYP2C8 (Walsky et al., 2005)], sulfaphenazole [CYP2C9 (Miners et al., 1988)], quinidine [CYP2D6 (von Bahr et al., 1985)], clomethiazole [CYP2E1 (Stresser et al., 2016)], or ketoconazole [CYP3A4 (Maurice et al., 1992)] (see Supplemental Table 1). All reactions were incubated for 30 minutes at 37°C, with reactions terminated by the addition of 25 μL ice-cold acetonitrile containing 0.001 ppm internal standard (D3-COX). Supernatants were collected after centrifugation at 16,100g for 10 minutes at 4°C for subsequent ultrahigh-pressure liquid chromatography–tandem mass spectrometry (UHPLC-MS/MS) analysis. Pooled HLMs without any inhibitor was used as the reference reaction, and all assays were performed in triplicate.
Enzyme Kinetic Assays
Human embryonic kidney 293 (HEK293) cells individually overexpressing V5-tagged CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 were previously described and used in the present kinetic analyses (Peterson et al., 2017a). Microsomal membrane fractions of P450-overexpressing cell lines were prepared by differential centrifugation as previously described (Dellinger et al., 2006; Peterson et al., 2017b). For the determination of relative P450 quantification of each microsomal preparation, equal amounts of microsomal protein (20 µg) were loaded on 10% SDS-polyacrylamide gels, with P450 protein quantity determined by western blot analysis using the anti–V5-HRP antibody at a 1:2,500 dilution. As a loading control for microsomal fractions, the anti–calnexin-HRP antibody was used at a 1:5,000 dilution for all western blots. Image J software was used to perform densitometry analysis, and the relative expression of each P450-containing microsomal preparation was used for normalization in N-oxidation activity assays.
N-oxidation reactions (final volume = 12.5 µL) contained 50 µg of total microsomal protein, 50 mM potassium phosphate, an NADPH-regenerating system (1.55 mmol/L NADP+, 3.3 mmol/L glucose-6-phosphate, 3.3 mmol/L MgCl2, 0.5 of 40 U/mL glucose-6-phosphate dehydrogenase) and varying concentrations of COT (0.05–1,000 μM). All incubations were performed for 30 minutes at 37°C, with reactions terminated by the addition of ice-cold acetonitrile to a final volume of 125 µL. Supernatants were collected after centrifugation at 16,100g for 10 minutes at 4°C and 0.001 ppm internal standard (D3-COX) was added prior to subsequent UHPLC-MS/MS analysis. Pooled commercial HLMs and untransfected parent HEK293 cell microsomes (50 µg) were used as the source for positive and negative assay controls, respectively.
UHPLC-MS/MS Analysis
UHPLC-MS/MS was performed on a UPLC-BEH-HILIC column (2.1 × 100 mm, 1.7 μm) with a mobile phase that consisted of 5 mmol/L sodium acetate (pH 6.7) in either 50% acetonitrile (v/v) (buffer A) or 90% acetonitrile (buffer B). A gradient elution was used as follows: 20% buffer A for 1.5 minutes, a linear gradient to 100% buffer A from 1.5–2.5 minutes, maintenance of 100% buffer A for 3 minutes, and a re-equilibrium step to the initial 20% buffer A conditions from 5.5–7 minutes (flow rate 0.4 mL·min−1). The injection volume was 5 µL and the column temperature was 30°C. Metabolite detection was performed in a Waters ACQUITY XEVO TQD instrument in MRM ESI+ mode. A multiple reaction monitoring method was performed using the following mass-to-charge ratio (m/z) transitions: 177.103→98.0, 193.1→96.0, and 196.1→96.0 to monitor COT, COX, and D3-COX, respectively. The desolvation temperature was 500°C, with 800 L/h of nitrogen gas. The collision energy was optimized at 27 V, 21 V, and 21 V for COT, COX, and D3-COX, respectively. A cone voltage of 30 V and 0.5-second dwell time resulted in high-sensitivity detection of COX and D3-COX. Peak retention times observed in the enzymatic incubations were compared with the retention time of the respective D3 internal standard.
The specificity of all UHPLC-MS/MS methods were validated using positive and negative control samples as well as by comparing peaks with those observed for purchased standards and isotope-labeled internal standards. Linearity was validated by r2 > 0.997 for all standard curves. Since isotope-labeled internal standards were used in all UHPLC-MS/MS analysis, matrix effects on analyte quantification were minimal. For quantification of COX and 3HC, HLM assays were directly spiked with COX or 3HC standard (0.001 ppm each) and then analyzed by UHPLC-MS/MS without sample extraction. Therefore, recovery or dilution effects were not a concern. Analyte quantification was based on the ratio of signal peak area versus the peak area of the internal standard.
Stability studies were performed by quantifying COX levels postextraction in HLM assays by UHPLC-MS/MS, and then repeating the quantificationafter incubating the extracted COX for 24 hours at 8°C. This was the longest time required to run any given sample in our UHPLC-MS/MS analysis. The stability of COX varied by no more than 10%.
Assay precision and accuracy was validated by repeated sample quantification. This was performed by preparing 5 individual samples of reaction matrix containing 1 ppm of COX standard, processing them for UHPLC-MS/MS, and immediately measuring COX concentrations for each sample, as compared to individually prepared standard curves also measured by UHPLC-MS/MS. The mean recovery for the 5 samples was 88.3% and the CV for these samples was 3.1%, indicating high accuracy and precision. The lower limit of detection for this experimental system ranged from 0.049 to 0.98 µM for cotinine, COX, and 3HC, with quantification ranging from 0.049 to 10 µM for cotinine and COX and from 0.098 to 10 µM for 3HC (see UHPLC-MS/MS performance parameters in Supplemental Table 2).
COT N-Oxidation Assays in HLMs
Microsomes from the 113 individual human liver specimens obtained from the H. Lee Moffitt Cancer Center were used for these studies. Assays were performed as described above using 25 µg of total microsomal protein and 500 µM of cotinine as substrate. D3-3HC and D3-COX (0.001 ppm each) were added to the reaction immediately prior to UHPLC-MS/MS analysis. In addition to the detection of COX, D3-COX, and COT, 3HC and D3-3HC formation were also monitored in the same reaction. The same UHPLC-MS/MS method described above was utilized, with the addition of the following MS/MS mass transitions: m/z 193.1→80.0 and 196.1→80.0, to monitor 3HC and D3-3HC, respectively. The cone voltage and collision energy were optimized at 20 V for both 3HC and D3-3HC. Metabolite retention times observed in the enzymatic incubations were compared with retention times of their corresponding D3 internal standard metabolites. Tissue activity assays were performed in triplicate.
Genotyping of CYP2A6, CYP2B6, and CYP2C19 in Human Liver Specimens
The potential impact of genetic variation in CYP2A6, CYP2B6, and CYP2C19 on COT-N-oxidation activity was examined by genotyping the same 113 liver specimens used for microsomal activity assays described above for allelic variants that have been associated with altered enzyme expression and/or function and that have a minor allele frequency (MAF) of > 0.10 in Caucasians [CYP2A6: *2, *9, and *14; CYP2B6: *2, *5, *9, Int1, and Int2; and CYP2C19: *2 and *17; (Fernandez-Salguero et al., 1995; Pianezza et al., 1998; Gervot et al., 1999; Pitarque et al., 2001; Desta et al., 2002; Hulot et al., 2006; Sim et al., 2006; Binnington et al., 2012; Bloom et al., 2013a; Bloom et al., 2013b; Ahmad et al., 2017; Bloom et al., 2019; Wang et al., 2019)]. High-prevalence functional alleles (MAF > 0.05) are not observed for CYP3A4 (Eiselt et al., 2001; Zhou et al., 2017). While high-prevalence alleles are observed for CYP2E1, their functional role has not been clearly established (Hu et al., 1997; Zhou et al., 2017). The nonfunctional CYP2A6*4 and CYP2A6*7 alleles were not analyzed in this group since all liver donors were from Caucasian subjects, who have a low MAF (< 0.03) for these alleles (López-Flores et al., 2017). Genomic DNA from the 113 liver specimens was genotyped using TaqMan probes following manufacturer’s suggested protocols. Dilutions of 5 ng DNA/µL were used to perform all genotyping analysis. The following alleles were examined in the liver specimens using Taqman probes: CYP2A6 [*2 (rs1801272; C__27861808), *9 (rs28399433; C__30634332_10) and *14 (rs rs28399435; C__30634234_10)], CYP2B6 [*2 (rs8192709; C__2818162_20), *5 (rs3211371; C__30634242_40), *9 (rs3745274; C__7817765_60), Int1 (rs4803419; C__7817764_10) and Int 2 (rs8109525; C__2818157_10)], and CYP2C19 [*2 (rs4244285; C__25986767_70) and *17 (rs12248560; C__4625986767_70)]. All genotyping reactions were performed in quadruplicate. As the CYP2B6 intron variants 1 and 2 (Int 1 and Int 2) exhibited high linkage disequilibrium in this population (R2= 0.773), they were considered as a single variant (termed ‘Int’) for CYP2B6 genotyping analysis.
Statistical Analyses
Kinetic parameters were determined from the Michaelis-Menten equation using GraphPad Prism version 6.01 (GraphPad Software, San Diego). Relative maximum reaction rates (Vmax) were calculated as:
with values normalized to the relative expression of each P450-overexpressed HEK293 cell microsomal protein preparation as determined by western blot analysis using the anti-V5 antibody and Image J software as described above.
All reported values represent the results [e.g., mean ± S.D.] of three independent experiments. The inhibition of each P450 enzyme in HLMs was compared with the reaction without any specific inhibitor using a two-tailed Student’s t test. One-way ANOVA followed by a Kruskal-Wallis H test was used to analyze different metabolism phenotype groups within the panel of liver specimens; P values from ANOVA are reported unless indicated otherwise. A P value of less than 0.05 was considered the threshold for statistical significance.
Results
Inhibition Assays of COX Formation in HLMs
The effects of enzyme-specific P450 inhibitors in HLM-mediated conversion of COT to COX was examined using UHPLC-MS/MS. As shown in Fig. 2A, a COX peak was observed at a retention time of 1.80 minutes (middle panel), while the COT substrate peak was observed at 1.06 minutes (top panel). The retention time of the COX peak was identical to those of D3-labeled COX standard (bottom panel). In incubations using 10 µM specific inhibitors for enzymes CYP2B6, CYP2E1, and CYP3A4, significant decreases in COX formation of 21% (P = 0.033), 16% (P = 0.026), and 33% (P = 0.013), respectively, were observed compared with control assays without inhibitor. No significant inhibition was observed when using these inhibitors at 1 µM (Fig. 2B). When tranylcypromine was used to inhibit both CYP2A6 and CYP2C19 in HLMs, significant decreases in COX formation of 15% (P = 0.004) and 54% (P = 0.002) were observed using 1 µM and 10 µM, respectively. No significant decrease in COX formation was observed when using inhibitors for CYP1A2, CYP2C8, CYP2C9, or CYP2D6 at both 1 µM and 10 µM.
Fig. 2.
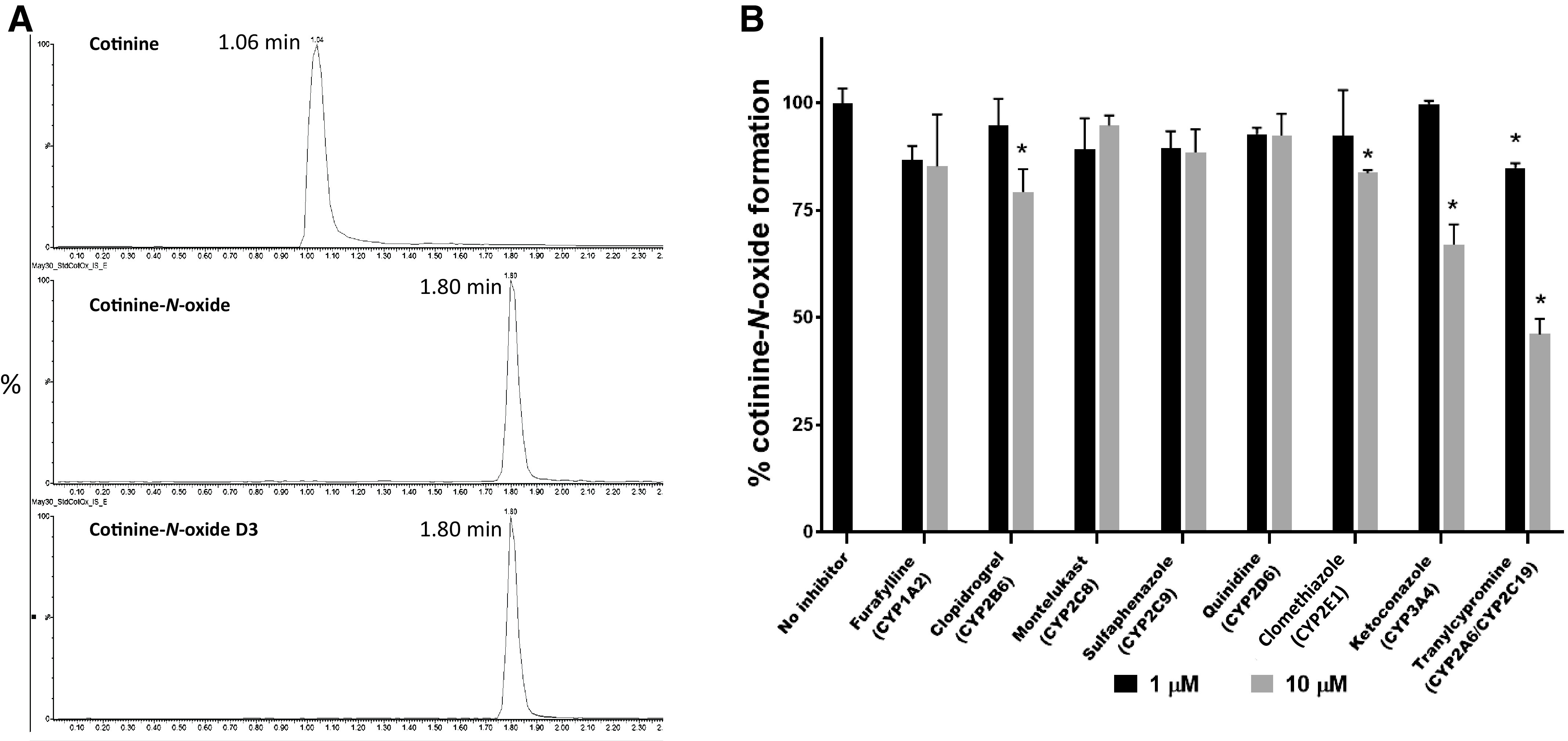
COX detection by MS/MS and inhibition in pooled HLMs. (A) Representative UHPLC-MS/MS traces of COX formation. Top panel, COT (retention time = 1.06 minutes, mass transition m/z: 177.103 > 98); middle panel, COX (retention time = 1.80 minutes, mass transition m/z: 193.1 > 96); bottom panel, deuterium (D3)-labeled COX (retention time = 1.80 minutes, mass transition m/z: 196.1 > 96). (B) Specific inhibition of P450-mediated COT metabolism in pooled HLMs. Data represent means of triplicate independent assays. *P < 0.05 as compared with incubations without inhibitor.
Kinetic Analysis of COT-N-Oxide Formation
The P450 enzymes that showed significant inhibition for COX formation in HLMs when using enzyme-specific inhibitors (CYP2A6, CYP2B6, CYP2C19, CYP2E1, and 3A4) were further analyzed for kinetic parameters of COX formation using microsomes from HEK293 cells overexpressing these V5-tagged P450 enzymes (Peterson et al., 2017a), with the relative Vmax values of COX formation calculated based on the relative expression of each P450 enzyme, measured by densitometry analysis of the V5 antibody signal in western blots (Supplemental Fig. 1). The chosen incubation times and protein concentrations were within the linear range of COT-N-oxidation velocity curves for each P450 enzyme tested (data not shown). Representative kinetic plots for COX formation in microsomes from P450-overexpressing cell lines and in HLMs are shown in Fig. 3. The highest COX formation activities were observed for microsomes from the CYP2A6- and CYP2C19-overexpressing cell lines (Vmax/KM = 44 and 42 nL•min-1•mg-1, respectively; Table 1). CYP2A6 and CYP2C19 also exhibited similar KM values of 390 µM and 405 µM, respectively; CYP2B6 exhibited a somewhat higher KM of 810 µM. The average KM values observed for these three enzymes for COX formation was very similar to the KM observed for HLMs (KM = 550 µM). In contrast, low affinities for COX formation were observed for CYP2E1 and CYP3A4, with KM values > 10 mM.
Fig. 3.

Kinetic analysis of COX formation in P450-overexpressing cell lines. Michaelis-Menten curves for microsomes of HEK293 cells overexpressing CYP2A6 (A), CYP2B6 (B), CYP2C19 (C), CYP2E1 (D), and CYP3A4 (E) and HLMs (F). Each curve is representative of one of three different experiments. For cell line microsomes, the rate of COX formation was adjusted per milligram of microsomal protein normalized based on V5 expression (determined using a V5 antibody) and on calnexin expression as determined by western blot as described in Materials and Methods.
TABLE 1.
Kinetic analysis of cotinine-N-oxide formation by P450 enzymes.
Data are expressed as the mean ± S.D. of three independent experiments. Cotinine concentrations of 0.005–10 mM were used for kinetic analysis.
| P450 Enzyme | KM | Vmax | Vmax/KM |
|---|---|---|---|
| (uM) | (pmol • min-1 • mg-1)a | (nL • min-1 • mg-1)a | |
| 2A6 | 390 ± 87 | 17 ± 1.5 | 44 ± 7.6 |
| 2B6 | 810 ± 24 | 16 ± 0.01 | 22 ± 6.9 |
| 2C19 | 405 ± 19 | 15 ± 1.6 | 42 ± 19 |
| 2E1 | >10 mM | ND | ND |
| 3A4 | >10 mM | ND | ND |
| HLMb | 550 ± 49 | 587 ± 26 | 1070 ± 8.4 |
ND, not determined.
aThe Vmax was calculated per total microsomal protein levels after normalization based on microsomal P450 expression levels as determined by western blot analysis.
bPooled human liver microsomes from 20 individuals.
COX Formation Versus P450 Genotypes in Human Liver Specimens
In vitro COX formation was measured in a panel of 113 HLMs by UHPLC-MS/MS as described in the Materials and Methods. The mean rate of COX formation observed for these specimens was 11.8 ± 6.4 pmol•min−1•mg microsomal protein−1, ranging from 0.71 to 31.2 pmol•min−1•mg microsomal protein−1 (Table 2). As a measure of CYP2A6 activity, levels of 3HC formation were also quantified in the same specimens. The mean rate of 3HC formation in the same HLM specimens was 28.1 ± 14.9 pmol•min−1•mg microsomal protein−1 of 3HC with a range of 3.4 to 101 pmol•min−1•mg microsomal protein−1.
TABLE 2.
Rate of COX and 3HC production in HLM specimens.
n = 113 HLM specimens.
| Mean ± S.D.(pmol • min−1• mg−1) | Range(pmol • min−1• mg−1) | |
|---|---|---|
| COX | 11.8 ± 6.4 | 0.71–31.2 |
| 3HC | 28.1 ± 14.9 | 3.4–101 |
Informative genotyping data were obtained for 113, 109, and 102 subjects tested for polymorphisms in CYP2C19, CYP2A6, and CYP2B6, respectively, with informative genotypes obtained for each of the 113 subjects for all CYP2C19 single nucleotide polymorphisms (SNPs) examined, each of the 109 subjects for all CYP2A6 SNPs examined, and each of the 102 subjects for all CYP2B6 SNPs examined. The MAF observed for each allele in the genotyped specimens is similar to those reported for the Northern Europeans from Utah population in the 1000 Genomes Project (Supplemental Table 3; http://uswest.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000255974;r=19:40843541–40850447).
The impact of CYP2C19 variants on COX formation was assessed with CYP2C19 genotype categorized according to the recommendations of the Clinical Pharmacogenetics Implementation Consortium (Scott et al., 2013), with ultrarapid metabolizers of 2C19 (UM2C19) containing the (*17/*17) and (*17/*1) genotypes, extensive metabolizers of CYP2C19 (EM2C19) consisting of the (*1/*1) wild-type genotype, intermediate metabolizers of 2C19 (IM2C19) containing the (*2/*17) or (*2/*1) genotypes, and poor metabolizers of 2C19 (PM2C19) consisting of the (*2/*2) genotype. The rate (mean ± S.D., expressed as pmol•min−1•mg−1 microsomal protein) of COX formation in the different metabolizing groups were as follows: UM2C19 (12.6 ± 5.3; n = 26), EM2C19 (11.9 ± 6.5; n = 62), IM2C19 (10.7 ± 5.4; n = 21), and PM2C19 (8.4 ± 4.0; n = 4; Fig. 4A). While the decreases observed in the different CYP2C19 metabolizing groups were not significant (ptrend = 0.26), the difference approached significance (p = 0.089) when comparing the combined UM2C19 + EM2C19 groups (11.9 ± 6.4 pmol•min−1•mg−1 microsomal protein) versus the combined IM2C19 + PM2C19 groups (10.1 ± 5.3 pmol•min−1•mg−1 microsomal protein; Fig. 4B). To normalize the specimens based on differences in sample quality and overall enzymatic activity, the COX/3HC ratio was also used as a phenotypic measure. A significant (Kruskal-Wallis test P = 0.026) trend toward decreased mean COX/3HC ratio was observed when comparing the UM2C19 (0.55 ± 0.11), EM2C19 (0.44 ± 0.19), IM2C19 (0.36 ± 0.12), and PM2C19 (0.32 ± 0.050) groups (Fig. 4C). Significant differences in COX/3HC ratio were also observed when comparing the UM2C19 group with the IM2C19 (P = 0.0032) and PM2C19 (P = 0.0058) groups, respectively (Fig. 4C). Furthermore, a significant difference in COX/3HC was observed when comparing the combined UM2C19 + EM2C19 groups versus the combined IM2C19 + PM2C19 groups (P = 0.017; Fig. 4D). While not statistically significant, similar trends were observed for CYP2C19 genotype groups when the analysis was performed only for subjects in the CYP2A6 EM group (n = 89, described below; P = 0.086; Supplemental Fig. 2A) or only in the CYP2B6 EM group (n = 44, described below; P = 0.33; Supplemental Fig. 2B).
Fig. 4.

CYP2C19 genotype-phenotype analysis in human liver specimens. (A) COX formation in specimens stratified by CYP2C19 genotypes (UM2C19, EM2C19, IM2C19, and PM2C19; n = 113). (B) COX formation in specimens stratified by combined CYP2C19 genotypes (UM2C19/EM2C19 versus IM2C19/PM2C19). (C) The ratio of COX/3HC in specimens stratified by CYP2C19 genotypes (UM2C19, EM2C19, IM2C19 and PM2C19). (D) The ratio of COX/3HC in specimens stratified by combined CYP2C19 genotypes (UM2C19/EM2C19 versus IM2C19/PM2C19). Genotype classifications are described in Materials and Methods. KW test, Kruskal-Wallis test. *P = 0.017, **P < 0.01.
To minimize CYP2C19 genotype-mediated effects, the effect of CYP2A6 genotype was analyzed using specimens that were categorized as EM for CYP2C19 (n = 62). CYP2A6 genotype was categorized according to previously reported EM and IM CYP2A6 groups as follows: the EM2A6 group included specimens exhibiting the (*1/*1), (*1/*2), (*1/*9), or (*1/*14) genotypes, while the IM2A6 group included the (*2/*2), (*9/*9), (*2/*9), (*2/*14), and (*9/*14) genotypes [no subjects were (*14/*14)] (Benowitz et al., 2006). Slow metabolizers for CYP2A6 were not screened in this study since the low-prevalence (*4 or *7 alleles were not examined in this population). No significant differences (P = 0.70) in the rate (mean ± S.D. expressed as pmol•min−1•mg microsomal protein−1) of COX formation were observed between the EM2A6 (12.2 ± 6.6) and IM2A6 groups (10.6 ± 6.4; Supplemental Fig. 2C).
Using specimens that exhibited only EM genotypes for both CYP2C19 and CYP2A6 (n = 50), CYP2B6 genotype groups were stratified as follows: the EM2B6 group included specimens that exhibited the (*1/*1), (*1/*2), or (*1/*5) genotypes; the IM2B6 group included HLMs exhibiting the (*1/*9), (*1/Int), (*2/*2), (*2/*5), (*2/*9), (*2/Int), (*5/*5), (*5/*9), or (*5/Int) genotypes; and the PM2B6 group included specimens with the (*9/*9), (*9/Int), or (Int/Int) genotypes. No significant differences in the rate (mean ± S.D. expressed as pmol•min−1•mg−1 microsomal protein) of COX formation were observed in the different CYP2B6 metabolism phenotype groups [EM2B6 (12.48 ± 5.32) versus IM2B6 (10.13 ± 6.28) versus PM2B6 (9.45 ± 7.41); p = 0.14; results not shown]. The COX/3HC ratios of 0.46 (n = 7) for the EM2B6 group, 0.50 (n = 20) for the IM2B6 group and 0.40 (n = 23) for the PM2B6 group were also not significantly different (P = 0.51; Supplemental Fig. 2D). In addition, no significant differences in COX formation or COX/3HC ratio were observed when comparing the PM2B6 versus EM2B6 + IM2B6 groups (results not shown).
Discussion
The involvement of CYP2A6 in the formation of COT from nicotine (Nakajima et al., 1996b), and 3HC from COT is well-established (Nakajima et al., 1996a). Studies examining the enzymology of cotinine metabolite formation have also been previously performed for COT-glucuronide and norcotinine (Brown et al., 2005; G. Chen et al., 2007). The present study is the first to examine the major hepatic enzymes in the formation of COX, which accounts for up to 9% of total cotinine metabolites in the urine of smokers (Rangiah et al., 2011) This pathway may be particularly important for COT excretion in individuals with functionally deficient CYP2A6 and/or UGT2B10 genotypes where urinary COT levels may be altered, an effect similar to that observed for nicotine-N’-oxide formation in subjects with altered CYP2A6 activities (Yamanaka et al., 2004; Perez-Paramo et al., 2019). In the present studies, experiments using HLMs demonstrated that up to 54% of COX formation was inhibited using tranylcypromine, an inhibitor of both CYP2A6 and CYP2C19, while COX formation decreased by 21% in HLMs using clopidogrel, a specific inhibitor of CYP2B6. Up to 33% of COX formation was inhibited using ketoconazole, an inhibitor of CYP3A4, suggesting that this enzyme may also play an important role in COX formation. However, while microsomes from CYP3A4-overexpressing cells exhibited a high Vmax [a pattern similar to that observed previously for this enzyme for other substrates (Sevrioukova & Poulos, 2013)], the substrate affinity of this enzyme for COT was low. It is possible that CYP3A4 may play more of a role in COX formation when cotinine levels are very high within a smoker. Other than CYP2E1, which showed low substrate affinity in CYP2E1 overexpressing cell microsomes and marginal activity in inhibition studies, no other hepatic P450 enzyme screened in this study (CYP1A2, CYP2C8, CYP2C9, and CYP2D6) exhibited detectable COX formation activity. Kinetic analysis using microsomes from P450-overexpressing cell lines demonstrated similar KM values for CYP2A6 (390 µM) and CYP2C19 (405 µM) for COX formation; a nearly two-fold KM was observed for microsomes from CYP2B6-overexpressing cells (810 µM). The KM values of these three P450 enzymes were similar to that observed for pooled HLMs (KM = 550 µM), suggesting that potentially all three of these P450 enzymes could contribute to COX formation in smokers.
This is the first study demonstrating the importance of CYP2C19 in nicotine metabolism. The intrinsic clearance of COX formation by microsomes from CYP2C19-overexpressing cells in the present study was very similar to that observed for CYP2A6 (4.2 versus 4.4 nL•mL−1•mg microsomal protein−1, normalized to P450 enzyme expression as determined by western blotting). Previous studies have suggested that CYP2C19 protein expression is at least three times that of CYP2A6 in human livers (Shimada et al., 1994; Achour et al., 2014), suggesting that CYP2C19 may play a more important role than CYP2A6 in COX formation.
CYP2B6 has been previously reported to be involved in nicotine metabolism, including in the formation of COT from nicotine, where it exhibits a KM (550 µM) (Yamanaka et al., 2005) approximately 5.8x higher than that observed for CYP2A6 (95 µM) (Yamanaka et al., 2005). CYP2B6 was also suggested to be involved in the formation of (S)-nornicotine-iminium and (S)-nornicotine, with KM values ranging from 184 to 269 µM (Bloom et al., 2019). This contrasts with the higher KM observed for CYP2B6 for COX formation (810 µM). CYP2B6 was shown in previous studies to exhibit lower levels of hepatic expression than CYP2C19 and to be expressed at levels similar to those observed for CYP2A6 (Shimada et al., 1994; Achour et al., 2014). Together these data suggest that, of the three enzymes, CYP2B6 may be playing the least significant role in COX formation.
Most interestingly, COX formation in HLMs was significantly associated with CYP2C19 genotypes in the present study. CYP2C19 variants significantly modified COX formation by up to 35% when comparing the UM2C19 group and PM2C19 groups, and 25% when comparing the UM2C19 and IM2C19 groups. These results are consistent with those observed previously for CYP2C19 genotypes on drug metabolism, including agents like clopidogrel, omeprazole, and voriconazole, which show increased enzyme efficacy by up to 35%–40% in the UM2C19/EM2C19 genotype groups when compared with the PM2C19 genotype group (Li-Wan-Po et al., 2010).
While CYP2A6 showed similar activity as CYP2C19 in the kinetics analysis, CYP2A6 genetic variants were not significantly associated with an altered COX formation phenotype. This pattern is consistent with the lower level of expression of CYP2A6 as compared with CYP2C19 in human livers (Shimada et al., 1994; Achour et al., 2014). However, since CYP2A6 is the enzyme responsible for 3HC conversion (Nakajima et al., 1996a), the COX/3HC ratio could not be used in this case to correct for differences in enzyme quality between specimens. Additionally, CYP2A6 alleles that would correspond with poor metabolizer subjects (carriers of the CYP2A6 *4 or *7 alleles) were not included in this study due to low allelic frequency in the studied population.
No differences in the levels of observed COX formation or as COX expressed as a ratio with 3HC were observed for CYP2B6 genotypes in the present study. Previous studies have suggested that CYP2B6 may be more important in nicotine metabolism when CYP2A6 function is impaired (Dicke et al., 2005; Al Koudsi and Tyndale, 2010). Additionally, an effect by variant CYP2B6 genotypes on COX formation could not be adequately tested in subjects with CYP2A6-deficient genotypes given the small sample number of specimens from subjects who were IM2A6 (n = 5) and the fact that poor metabolizers of CYP2A6 were not assessed in this population. Further analysis of a large sample size of intermediate and poor CYP2A6 metabolizer subjects will be required to better determine the extent of the influence of CYP2B6 and CYP2A6 on COX formation.
In summary, this is the first study to report on the major hepatic enzymes important in COX formation, with CYP2C19 playing an important role. This study also demonstrates CYP2C19-mediated genetic effects on COX formation in a panel of human liver specimens, an effect that could potentially modify the NMR in smokers and could affect pharmacotherapeutic decisions for smoking cessation treatment. Further population-based studies involving genotype-phenotype associations should be performed to better assess the effect of CYP2C19 on COX formation and the NMR.
Acknowledgments
The authors thank Dr. Senthil Natesan in the Department of Pharmaceutical Sciences at the Washington State University College of Pharmacy and Pharmaceutical Sciences for his helpful suggestions and discussion. The authors also thank the Mass Spectrometry Core facility at Washington State University Spokane for their help with UHPLC/MS.
Abbreviations
- COT
cotinine
- COT-Gluc
cotinine-N- glucuronide
- COX
cotinine-N-oxide
- EM
extensive metabolizer
- 3HC
3-hydroxy cotinine
- HEK293
human embryonic kidney 293
- HLM
human liver microsome
- HRP
horseradish peroxidase
- IM
intermediate metabolizer
- KM
Michaelis constant
- m/z
mass-to-charge ratio
- MAF
minor allele frequency
- NMR
nicotine metabolic ratio
- P450
cytochrome P450
- PM
poor metabolizer
- SNP
single nucleotide polymorphism
- UGT
uridine 5′-diphospho-glucuronosyltransferase
- UM
ultrarapid metabolizers
- UHPLC-MS/MS
ultrahigh-pressure liquid chromatography–tandem mass spectrometry
- Vmax
maximum reaction rate
Authorship Contributions
Participated in research design: Perez-Paramo, Watson, Chen, Lazarus.
Conducted experiments: Perez-Paramo.
Performed data analysis: Perez-Paramo, Watson, Chen, Lazarus.
Wrote or contributed to the writing of the manuscript: Perez-Paramo, Watson, Lazarus.
Footnotes
This work was supported by National Institutes of Health National Institutes of Environmental Health Sciences [Grant R01-ES025460] (to P.L.), the Fulbright-Garcia Robles Program, and a CONACyT dissertation grant (to Y.X.P.-P.), and the Health Sciences and Services Authority of Spokane County [Grant WSU002292] (to WSU College of Pharmacy and Pharmaceutical Sciences).
No author has an actual or perceived conflict of interest with the contents of this article.
1Current affiliation: Genentech, Inc., South San Francisco, California.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Achour B, Barber J, Rostami-Hodjegan A (2014) Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: a meta-analysis. Drug Metab Dispos 42:1349–1356 10.1124/dmd.114.058834. [DOI] [PubMed] [Google Scholar]
- Ahmad T, Sabet S, Primerano DA, Richards-Waugh LL, Rankin GO (2017) Tell-Tale SNPs: The Role of CYP2B6 in Methadone Fatalities. J Anal Toxicol 41:325–333 10.1093/JAT/BKW135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Koudsi N, Tyndale RF (2010) Hepatic CYP2B6 is altered by genetic, physiologic, and environmental factors but plays little role in nicotine metabolism. Xenobiotica 40:381–392 10.3109/00498251003713958. [DOI] [PubMed] [Google Scholar]
- Baurley JW, Edlund CK, Pardamean CI, Conti DV, Krasnow R, Javitz HS, Hops H, Swan GE, Benowitz NL, Bergen AW (2016) Genome-Wide Association of the Laboratory-Based Nicotine Metabolite Ratio in Three Ancestries. Nicotine Tob Res 18:1837–1844 10.1093/ntr/ntw117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL, Swan GE, Jacob P 3rd, Lessov-Schlaggar CN, Tyndale RF (2006) CYP2A6 genotype and the metabolism and disposition kinetics of nicotine. Clin Pharmacol Ther 80:457–467 10.1016/j.clpt.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Berg JZ, von Weymarn LB, Thompson EA, Wickham KM, Weisensel NA, Hatsukami DK, Murphy SE (2010) UGT2B10 genotype influences nicotine glucuronidation, oxidation, and consumption. Cancer Epidemiol Biomarkers Prev 19:1423–1431 10.1158/1055-9965.EPI-09-0959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnington MJ, Zhu AZX, Renner CC, Lanier AP, Hatsukami DK, Benowitz NL, Tyndale RF (2012) CYP2A6 and CYP2B6 genetic variation and its association with nicotine metabolism in South Western Alaska Native people. Pharmacogenet Genomics 22:429–440 10.1097/FPC.0b013e3283527c1c.CYP2A6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom AJ, Harari O, Martinez M, Zhang X, McDonald SA, Murphy SE, Goate A (2013a) A compensatory effect upon splicing results in normal function of the CYP2A6*14 allele. Pharmacogenet Genomics 23:107–116 DOI: 10.1016/j.biotechadv.2011.08.021.Secreted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom AJ, Martinez M, Chen LS, Bierut LJ, Murphy SE, Goate A (2013b) CYP2B6 non-coding variation associated with smoking cessation is also associated with differences in allelic expression, splicing, and nicotine metabolism independent of common amino-acid changes. PLoS One 8:e79700 DOI: 10.1371/journal.pone.0079700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom AJ, Wang PF, Kharasch ED (2019) Nicotine oxidation by genetic variants of CYP2B6 and in human brain microsomes. Pharmacol Res Perspect 7:e00468 DOI: 10.1002/prp2.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, von Weymarn LB, Murphy SE (2005) Identification of N-(hydroxymethyl) norcotinine as a major product of cytochrome P450 2A6, but not cytochrome P450 2A13-catalyzed cotinine metabolism. Chem Res Toxicol 18:1792–1798 DOI: 10.1021/tx0501381.16359169 [DOI] [PubMed] [Google Scholar]
- Chen G, Blevins-Primeau AS, Dellinger RW, Muscat JE, Lazarus P (2007) Glucuronidation of nicotine and cotinine by UGT2B10: loss of function by the UGT2B10 Codon 67 (Asp>Tyr) polymorphism. Cancer Res 67:9024–9029 10.1158/0008-5472.CAN-07-2245. [DOI] [PubMed] [Google Scholar]
- Chen G, Giambrone NE, Lazarus P (2012) Glucuronidation of trans-3′-hydroxycotinine by UGT2B17 and UGT2B10. Pharmacogenet Genomics 22:183–190 10.1097/FPC.0b013e32834ff3a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Giambrone NE Jr, Dluzen DF, Muscat JE, Berg A, Gallagher CJ, Lazarus P (2010) Glucuronidation genotypes and nicotine metabolic phenotypes: Importance of UGT2B10 and UGT2B17 knock-out polymorphisms. Cancer Res 70:7543–7552 10.1158/0008-5472.CAN-09-4582.Glucuronidation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L-S, Horton A, Bierut L (2018) Pathways to precision medicine in smoking cessation treatments. Neurosci Lett 669:83–92 10.1016/j.neulet.2016.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenoweth MJ, Novalen M, Hawk LW Jr, Schnoll RA, George TP, Cinciripini PM, Lerman C, Tyndale RF (2014) Known and novel sources of variability in the nicotine metabolite ratio in a large sample of treatment-seeking smokers. Cancer Epidemiol Biomarkers Prev 23:1773–1782 10.1158/1055-9965.EPI-14-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughtrie MWH, Burchell B, Bend JR (1987) Purification and properties of rat kidney UDP-glucuronosyltransferase. Biochem Pharmacol 36:245–251 10.1016/0006-2952(87)90696-4. [DOI] [PubMed] [Google Scholar]
- Dagne E, Castagnoli N Jr (1972) Cotinine N-oxide, a new metabolite of nicotine. J Med Chem 15:840–841 10.1021/jm00278a011. [DOI] [PubMed] [Google Scholar]
- Dellinger RW, Fang JL, Chen G, Weinberg R, Lazarus P (2006) Importance of UDP-glucuronosyltransferase 1A10 (UGT1A10) in the detoxification of polycyclic aromatic hydrocarbons: decreased glucuronidative activity of the UGT1A10139Lys isoform. Drug Metab Dispos 34:943–949 10.1124/dmd.105.009100. [DOI] [PubMed] [Google Scholar]
- Dempsey D, Tutka P, Jacob P 3rd, Allen F, Schoedel K, Tyndale RF, Benowitz NL (2004) Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther 76:64–72 10.1016/j.clpt.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Desta Z, Zhao X, Shin JG, Flockhart DA (2002) Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 41:913–958 10.2165/00003088-200241120-00002. [DOI] [PubMed] [Google Scholar]
- Dicke KE, Skrlin SM, Murphy SE (2005) Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-butanone metabolism by cytochrome P450 2B6. Drug Metab Dispos 33:1760–1764 10.1124/dmd.105.006718.nornicotine. [DOI] [PubMed] [Google Scholar]
- Draper AJ, Madan A, Parkinson A (1997) Inhibition of coumarin 7-hydroxylase activity in human liver microsomes. Arch Biochem Biophys 341:47–61 10.1006/abbi.1997.9964. [DOI] [PubMed] [Google Scholar]
- Eiselt R, Domanski TL, Zibat A, Mueller R, Presecan-Siedel E, Hustert E, Zanger UM, Brockmoller J, Klenk HP, Meyer UA, et al. (2001) Identification and functional characterization of eight CYP3A4 protein variants. Pharmacogenetics 11:447–458 10.1097/00008571-200107000-00008. [DOI] [PubMed] [Google Scholar]
- Falcone M, Jepson C, Benowitz N, Bergen AW, Pinto A, Wileyto EP, Baldwin D, Tyndale RF, Lerman C, Ray R (2011) Association of the nicotine metabolite ratio and CHRNA5/CHRNA3 polymorphisms with smoking rate among treatment-seeking smokers. Nicotine Tob Res 13:498–503 10.1093/ntr/ntr012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Salguero P, Hoffman SMG, Cholerton S, Mohrenweiser H, Raunio H, Rautio A, Pelkonen O, Huang JD, Evans WE, Idle JR, et al. (1995) A genetic polymorphism in coumarin 7-hydroxylation: sequence of the human CYP2A genes and identification of variant CYP2A6 alleles. Am J Hum Genet 57:651–660. [PMC free article] [PubMed] [Google Scholar]
- Fix BV, O’Connor RJ, Benowitz N, Heckman BW, Cummings KM, Fong GT, Thrasher JF (2017) Nicotine Metabolite Ratio (NMR) Prospectively Predicts Smoking Relapse: Longitudinal Findings From ITC Surveys in Five Countries. Nicotine Tob Res 19:1040–1047 10.1093/ntr/ntx083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foth H, Aubrecht J, Höhne M, Walther UI, Kahl GF (1992) Increased cotinine elimination and cotinine-N-oxide formation by phenobarbital induction in rat and mouse. Clin Investig 70:175–181. [DOI] [PubMed] [Google Scholar]
- Gervot L, Rochat B, Gautier JC, Bohnenstengel F, Kroemer H, de Berardinis V, Martin H, Beaune P, de Waziers I (1999) Human CYP2B6: expression, inducibility and catalytic activities. Pharmacogenetics 9:295–306. [PubMed] [Google Scholar]
- Gorrod JW, Peyton JI (1999) Analytical Determination of Nicotine and Related Compounds and their Metabolites, in Analytical Determination of Nicotine and Related Compounds and their Metabolites, Elsevier, Amsterdam, Netherlands DOI: 10.1016/b978-0-444-50095-3.x5000-1. [DOI] [Google Scholar]
- Hu Y, Oscarson M, Johansson I, Yue QY, Dahl ML, Tabone M, Arincò S, Albano E, and Ingelman-Sundberg M (1997) Genetic polymorphism of human CYP2E1: characterization of two variant alleles. Mol Pharmacol 51:370–376. [PubMed] [Google Scholar]
- Hukkanen J, Jacob P 3rd, Benowitz NL (2005) Metabolism and disposition kinetics of nicotine. Pharmacol Rev 57:79–115 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- Hulot JS, Bura A, Villard E, Azizi M, Remones V, Goyenvalle C, Aiach M, Lechat P, Gaussem P (2006) Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood 108:2244–2247 10.1182/blood-2006-04-013052. [DOI] [PubMed] [Google Scholar]
- Jacobson GA, Ferguson SG (2014) Relationship between cotinine and trans-3′-hydroxycotinine glucuronidation and the nicotine metabolite ratio in Caucasian smokers. Biomarkers 19:679–683 10.3109/1354750X.2014.966254. [DOI] [PubMed] [Google Scholar]
- Jarvis MJ, Russell MAH, Benowitz NL, Feyerabend C (1988) Elimination of cotinine from body fluids: implications for noninvasive measurement of tobacco smoke exposure. Am J Public Health 78:696–698 10.2105/AJPH.78.6.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P, Gorrod JW, Beckett AH (1971) Comparative C- and N-oxidation of (plus)- and (minus)-nicotine by various species. Xenobiotica 1:497–498 10.3109/00498257109041515. [DOI] [PubMed] [Google Scholar]
- Lerman C, Schnoll RA, Hawk LW Jr, Cinciripini P, George TP, Wileyto EP, Swan GE, Benowitz NL, Heitjan DF, Tyndale RF; PGRN-PNAT Research Group (2015) Use of the nicotine metabolite ratio as a genetically informed biomarker of response to nicotine patch or varenicline for smoking cessation: a randomised, double-blind placebo-controlled trial. Lancet Respir Med 3:131–138 10.1016/S2213-2600(14)70294-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman C, Tyndale R, Patterson F, Wileyto EP, Shields PG, Pinto A, Benowitz N (2006) Nicotine metabolite ratio predicts efficacy of transdermal nicotine for smoking cessation. Clin Pharmacol Ther 79:600–608 10.1016/j.clpt.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Li-Wan-Po A, Girard T, Farndon P, Cooley C, Lithgow J (2010) Pharmacogenetics of CYP2C19: functional and clinical implications of a new variant CYP2C19*17. Br J Clin Pharmacol 69:222–230 10.1111/j.1365-2125.2009.03578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Flores LA, Pérez-Rubio G, Falfán-Valencia R (2017) Distribution of polymorphic variants of CYP2A6 and their involvement in nicotine addiction. EXCLI J 16:174–196 10.17179/excli2016-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaiyandi V, Lerman C, Benowitz NL, Jepson C, Patterson F, Tyndale RF (2006) Impact of CYP2A6 genotype on pretreatment smoking behaviour and nicotine levels from and usage of nicotine replacement therapy. Mol Psychiatry 11:400–409 10.1038/sj.mp.4001794. [DOI] [PubMed] [Google Scholar]
- Marclay F, Grata E, Perrenoud L, Saugy M (2011) A one-year monitoring of nicotine use in sport: frontier between potential performance enhancement and addiction issues. Forensic Sci Int 213:73–84 10.1016/j.forsciint.2011.05.026. [DOI] [PubMed] [Google Scholar]
- Maurice M, Pichard L, Daujat M, Fabre I, Joyeux H, Domergue J, Maurel P (1992) Effects of imidazole derivatives on cytochromes P450 from human hepatocytes in primary culture. FASEB J 6:752–758 10.1096/FASEBJ.6.2.1371482. [DOI] [PubMed] [Google Scholar]
- Miners JO, Smith KJ, Robson RA, McManus ME, Veronese ME, Birkett DJ (1988) Tolbutamide hydroxylation by human liver microsomes. Kinetic characterisation and relationship to other cytochrome P-450 dependent xenobiotic oxidations. Biochem Pharmacol 37:1137–1144 10.1016/0006-2952(88)90522-9. [DOI] [PubMed] [Google Scholar]
- Murphy SE (2017) Nicotine Metabolism and Smoking: Ethnic Differences in the Role of P450 2A6. Chem Res Toxicol 30:410–419 10.1021/acs.chemrestox.6b00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SE, Park SSL, Thompson EF, Wilkens LR, Patel Y, Stram DO, Le Marchand L (2014) Nicotine N-glucuronidation relative to N-oxidation and C-oxidation and UGT2B10 genotype in five ethnic/racial groups. Carcinogenesis 35:2526–2533 10.1093/carcin/bgu191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SE, Sipe CJ, Choi K, Raddatz LM, Koopmeiners JS, Donny EC, Hatsukami DK (2017) Low cotinine glucuronidation results in higher serum and saliva cotinine in African American compared to White smokers. Cancer Epidemiol Biomarkers Prev 26:1093–1099 10.1158/1055-9965.EPI-16-0920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M, Yamamoto T, Nunoya K, Yokoi T, Nagashima K, Inoue K, Funae Y, Shimada N, Kamataki T, Kuroiwa Y (1996a) Characterization of CYP2A6 involved in 3′-hydroxylation of cotinine in human liver microsomes. J Pharmacol Exp Ther 277:1010–1015. [PubMed] [Google Scholar]
- Nakajima M, Yamamoto T, Nunoya K, Yokoi T, Nagashima K, Inoue K, Funae Y, Shimada N, Kamataki T, and Kuroiwa Y (1996b) Role of human cytochrome P4502A6 in C-oxidation of nicotine. Drug Metab Dispos 24:1212–1217. [PubMed] [Google Scholar]
- Pellegrini M, Marchei E, Rossi S, Vagnarelli F, Durgbanshi A, García-Algar O, Vall O, and Pichini S (2007) Liquid chromatography/electrospray ionization tandem mass spectrometry assay for determination of nicotine and metabolites, caffeine and arecoline in breast milk. Rapid Commun Mass Spectrom 21:2693–2703 10.1002/RCM.3137. [DOI] [PubMed] [Google Scholar]
- Perez-Paramo YX, Chen G, Ashmore JH, Watson CJW, Nasrin S, Adams-Haduch J, Wang R, Gao YT, Koh WP, Yuan JM, et al. (2019) Nicotine-N’-oxidation by flavin monooxygenase enzymes. Cancer Epidemiol Biomarkers Prev 28:311–320 10.1158/1055-9965.EPI-18-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A, Xia Z, Chen G, Lazarus P (2017a) Exemestane potency is unchanged by common nonsynonymous polymorphisms in CYP19A1: results of a novel anti-aromatase activity assay examining exemestane and its derivatives. Pharmacol Res Perspect 5:e00313 10.1002/prp2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A, Xia Z, Chen G, Lazarus P (2017b) In vitro metabolism of exemestane by hepatic cytochrome P450s: impact of nonsynonymous polymorphisms on formation of the active metabolite 17β-dihydroexemestane. Pharmacol Res Perspect 5:e00314 10.1002/prp2.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pianezza ML, Sellers EM, Tyndale RF (1998) Nicotine metabolism defect reduces smoking. Nature 393:750. [DOI] [PubMed] [Google Scholar]
- Pitarque M, von Richter O, Oke B, Berkkan H, Oscarson M, and Ingelman-Sundberg M (2001) Identification of a single nucleotide polymorphism in the TATA box of the CYP2A6 gene: impairment of its promoter activity. Biochem Biophys Res Commun 284:455–460 10.1006/bbrc.2001.4990. [DOI] [PubMed] [Google Scholar]
- Rangiah K, Hwang W-T, Mesaros C, Vachani A, Blair IA (2011) Nicotine exposure and metabolizer phenotypes from analysis of urinary nicotine and its 15 metabolites by LC-MS. Bioanalysis 3:745–761 10.4155/BIO.11.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter T, Mürdter TE, Heinkele G, Pleiss J, Tatzel S, Schwab M, Eichelbaum M, Zanger UM (2004) Potent mechanism-based inhibition of human CYP2B6 by clopidogrel and ticlopidine. J Pharmacol Exp Ther 308:189–197 10.1124/jpet.103.056127. [DOI] [PubMed] [Google Scholar]
- Scott SA, Sangkuhl K, Stein CM, Hulot JS, Mega JL, Roden DM, Klein TE, Sabatine MS, Johnson JA, Shuldiner AR; Clinical Pharmacogenetics Implementation Consortium (2013) Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther 94:317–323 10.1038/clpt.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesardic D, Boobis AR, Murray BP, Murray S, Segura J, de la Torre R, Davies DS (1990) Furafylline is a potent and selective inhibitor of cytochrome P450IA2 in man. Br J Clin Pharmacol 29:651–663 10.1111/j.1365-2125.1990.tb03686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova IF, Poulos TL (2013) Understanding the mechanism of cytochrome P450 3A4: recent advances and remaining problems. Dalton Trans 42:3116–3126 10.1039/c2dt31833d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP (1994) Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414–423. [PubMed] [Google Scholar]
- Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M (2006) A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 79:103–113 10.1016/j.clpt.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Stresser DM, Perloff ES, Mason AK, Blanchard AP, Dehal SS, Creegan TP, Singh R, Gangl ET (2016) Selective Time- and NADPH-Dependent Inhibition of Human CYP2E1 by Clomethiazole. Drug Metab Dispos 44:1424–1430 10.1124/dmd.116.070193. [DOI] [PubMed] [Google Scholar]
- Taavitsainen P, Juvonen R, Pelkonen O (2001) In vitro inhibition of cytochrome P450 enzymes in human liver microsomes by a potent CYP2A6 inhibitor, trans-2-phenylcyclopropylamine (tranylcypromine), and its nonamine analog, cyclopropylbenzene. Drug Metab Dispos 29:217–222. [PubMed] [Google Scholar]
- Tsai MC, Gorrod JW (1999) Determination of nicotine and its metabolites in biological fluids: in vitro studies, in Analytical Determination of Nicotine and Related Compounds and their Metabolites, Elsevier, Amsterdam, Netherlands DOI: 10.1016/b978-044450095-3/50016-4. [DOI] [Google Scholar]
- van Waateringe RP, Mook-Kanamori MJ, Slagter SN, van der Klauw MM, van Vliet-Ostaptchouk JV, Graaff R, Lutgers HL, Suhre K, El-Din Selim MM, Mook-Kanamori DO et al. (2017) The association between various smoking behaviors, cotinine biomarkers and skin autofluorescence, a marker for advanced glycation end product accumulation. PLoS One 12:e0179330 10.1371/journal.pone.0179330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bahr C, Spina E, Birgersson C, Ericsson O, Göransson M, Henthorn T, Sjöqvist F (1985) Inhibition of desmethylimipramine 2-hydroxylation by drugs in human liver microsomes. Biochem Pharmacol 34:2501–2505 10.1016/0006-2952(85)90533-7. [DOI] [PubMed] [Google Scholar]
- Walsky RL, Obach RS, Gaman EA, Gleeson JP, Proctor WR (2005) Selective inhibition of human cytochrome P4502C8 by montelukast. Drug Metab Dispos 33:413–418. [DOI] [PubMed] [Google Scholar]
- Wang P-F, Neiner A, Kharasch ED (2019) Efavirenz Metabolism: Influence of Polymorphic CYP2B6 Variants and Stereochemistry. Drug Metab Dispos 47:1195–1205 10.1124/DMD.119.086348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka H, Nakajima M, Fukami T, Sakai H, Nakamura A, Katoh M, Takamiya M, Aoki Y, Yokoi T (2005) CYP2A6 AND CYP2B6 are involved in nornicotine formation from nicotine in humans: interindividual differences in these contributions. Drug Metab Dispos 33:1811–1818 10.1124/dmd.105.006254. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, Nakajima M, Nishimura K, Yoshida R, Fukami T, Katoh M, Yokoi T (2004) Metabolic profile of nicotine in subjects whose CYP2A6 gene is deleted. Eur J Pharm Sci 22:419–425 10.1016/j.ejps.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Yildiz D (2004) Nicotine, its metabolism and an overview of its biological effects. Toxicon 43:619–632 10.1016/j.toxicon.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Yokota H, Ohgiya N, Ishihara G, Ohta K, Yuasa A (1989) Purification and properties of UDP-glucuronyltransferase from kidney microsomes of β-naphthoflavone-treated rat. J Biochem 106:248–252 10.1093/oxfordjournals.jbchem.a122839. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Ingelman-Sundberg M, Lauschke VM (2017) Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin Pharmacol Ther 102:688–700 10.1002/cpt.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu AZX, Zhou Q, Cox LS, Ahluwalia JS, Benowitz NL, Tyndale RF (2013) Variation in trans-3′-hydroxycotinine glucuronidation does not alter the nicotine metabolite ratio or nicotine intake. PLoS One 8:e70938 10.1371/journal.pone.0070938. [DOI] [PMC free article] [PubMed] [Google Scholar]