
FIGURE 3.
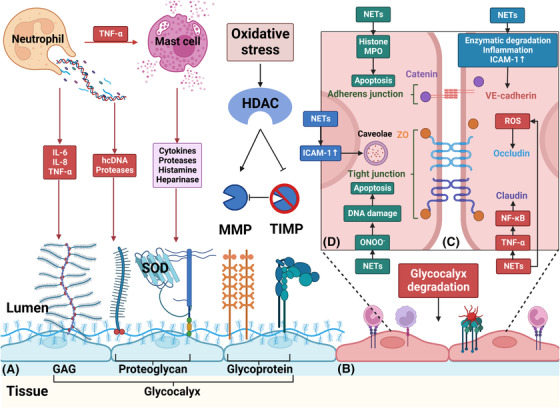
Neutrophils and NETs damage endothelial cells’ glycocalyx and increase endothelial permeability. (A) During sepsis, NETs can directly cause glycocalyx degradation through hcDNA and proteases. Additionally, a considerable number of inflammatory cytokines, such as IL‐6, IL‐8 and TNF‐α, induced by neutrophils and NETs can also damage glycocalyx. Moreover, mast cells are activated by TNF‐α and release cytokines, proteases, histamine and heparinase, further degrading glycocalyx. Furthermore, oxidative stress‐induced HDAC can upregulate MMP expression and inhibit tissue inhibitors of MMPs (TIMP1 and TIMP3), thereby activating MMPs to promote the shedding of the glycocalyx. (B) After degradation of the glycocalyx, endothelial cell adhesion molecules are exposed, triggering further inflammation, rolling, and adhesion of leukocytes and platelets. (C) Para‐endothelial permeability increases mainly due to junction cleavage. TNF‐α is shown to cause disruption of claudin through NF‐κB activation. ROS causes the redistribution of occludin, limiting its association with ZO‐1. VE‐cadherin is susceptible to enzymatic degradation. Additionally, inflammatory mediators promote VE‐cadherin phosphorylation and dissociation from p120 catenin and induce VE‐cadherin endocytosis via several signalling pathways. Upregulated ICAM‐1 can also promote VE‐cadherin phosphorylation. (D) ICAM‐1 can also increase trans‐endothelial permeability through caveolin‐1 phosphorylation, a major component of caveolae. However, endothelial cell apoptosis is the main cause of increased trans‐endothelial permeability. NETs contribute to apoptosis through MPO and histone. Moreover, the formation of ONOO− during oxidative stress also induces cell death via DNA damage. GAG, glycosaminoglycan; hcDNA, histone‐complexed DNA; HDAC, histone deacetylase; MMP, matrix metalloproteinase; ONOO‐, peroxynitrite; SOD, superoxide dismutase; VE‐cadherin, vascular endothelial‐cadherin; ZO, zonula occluding