

Cystic fibrosis (CF) lung disease is characterized by an intense mucopurulent process driven in large part by IL-1β (1). The neutrophil-dominant inflammatory response results in the accumulation of active neutrophil elastase (NE) in CF airway secretions. Both IL-1β and neutrophil elastase are highly predictive of bronchial destruction or bronchiectasis, which is the hallmark of CF lung disease (2, 3). Several key clinical studies have identified that this mucopurulent process appears early in infants, is much more severe than would be expected for the initial airway bacterial burden, and is sustained despite repeated or continuous antibiotic suppression (4, 5). These characteristics point to a probable link between the CF basic defect, deficient cystic fibrosis transmembrane conductance regulator (CFTR), and the inflammatory response. Despite the identification of several potential molecular pathways to explain why CFTR deficiency is associated with excessive airway inflammation, the lack of effective CFTR restoration in vivo had made it difficult to validate in vitro observations in patients. The arrival of highly effective modulators of CFTR, particularly the combination of elexacaftor, tezacaftor, and ivacaftor (ETI), in the clinical arena has changed this perspective and now allows clinical studies of potential inflammatory pathways to be validated in individuals with CF.
An emerging pathway relevant to CF monocytes and macrophages involves potassium (K+) efflux, inflammasome activation, and IL-1β release. IL-1β is activated by caspase-1, which cleaves pro–IL-1β (6). Pro–IL-1β mRNA is induced after the binding of LPS to the Toll-like receptor 4 receptor on monocytes and macrophages. The engagement of Toll-like receptor 4 by LPS also initiates the assembly of a key protein complex for caspase-1 activation known as NLRP3 (nucleotide-binding oligomerization domain, leucine-rich repeat–containing protein 3). NLRP3 activation requires NEK7 (NIMA-related kinase 7). The activated NLRP3 receptor recruits the adaptor protein ASC (apoptosis-associated speck-like protein containing CARD), which contains a pyrin domain and a caspase-1 recruitment domain (CARD) needed to recruit pro–caspase-1. Subsequently, in the presence of high (100 μM) extracellular ATP (eATP) concentration, the purine P2X7 receptor (P2X7R), which is abundantly expressed on monocytes and macrophages, initiates a series of steps involving cation influx, K+ efflux, macropore formation, and ultimately activation of the NLRP3 inflammasome complex and caspase-1, which is essential to cleave pro–IL-1β and release active IL-1β (7, 8) (Figure 1).
Figure 1.
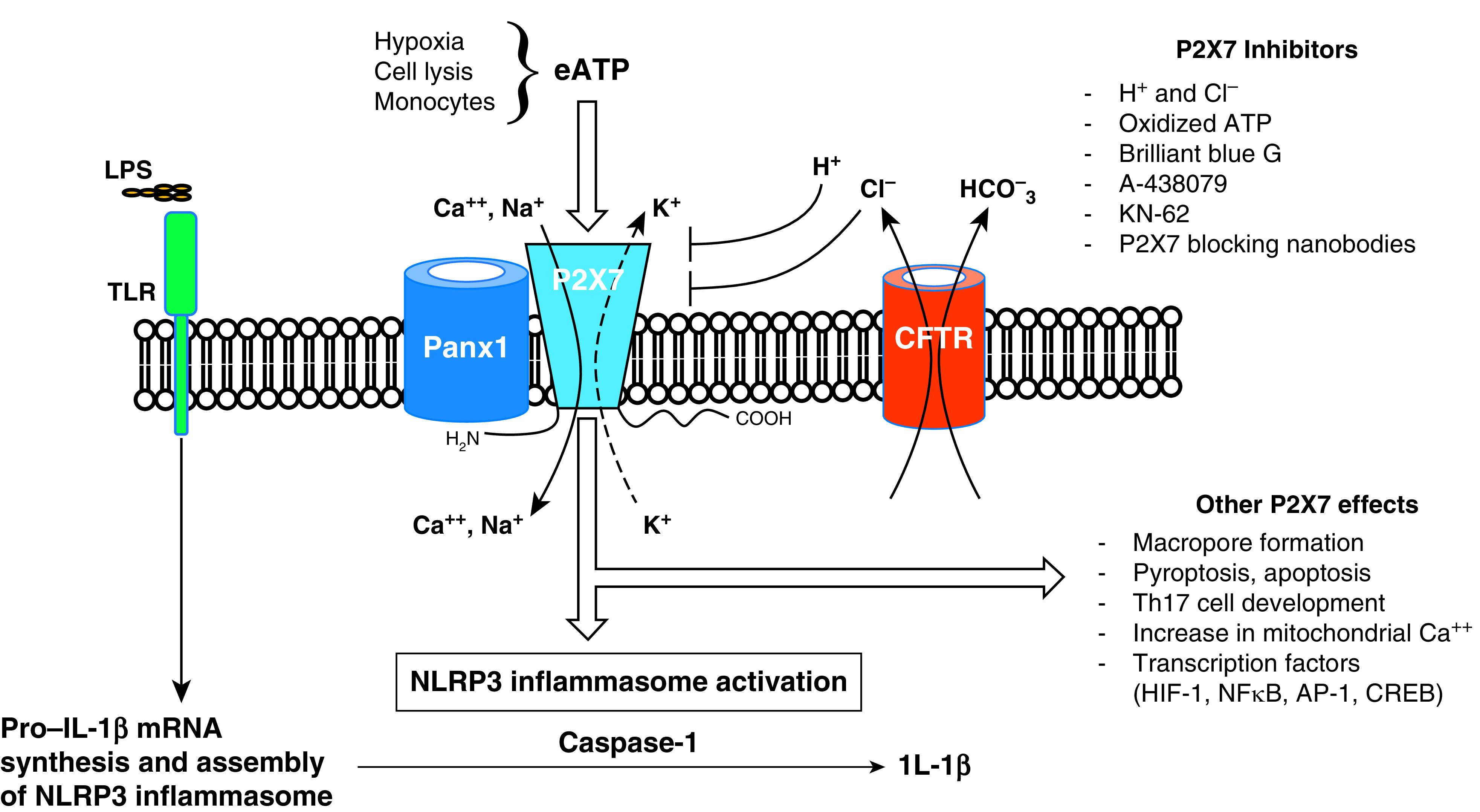
Monocyte P2X7 receptor signaling and nucleotide-binding oligomerization domain, leucine-rich repeat–containing protein 3 (NLRP3) inflammasome activation. LPS binding to Toll-like receptor 4 (TLR4) induces pro–IL-1β synthesis and NLRP3 inflammasome assembly. Hypoxia and/or cell lysis induces the release of a large amount of extracellular ATP (eATP) through Panx1, which engages the P2X7 receptor. In contrast to macrophages, monocytes alone can also release sufficient eATP to activate the NLRP3 inflammasome. Cation influx drives K+ efflux either through P2X7 or alternate channels, a key step in initiating NLRP3 inflammasome activation. Upon activation, the NLRP3 inflammasome converts pro–caspase-1 to caspase-1, which acts upon pro–IL-1β to produce IL-1β. Other effects of P2X7 engagement with eATP include a macropore formation that contributes to apoptosis and pyroptosis, T-helper cell type 17 (Th17) development, an increase in mitochondrial calcium, and modulation of several transcription factors that affect inflammation. Restoration of cystic fibrosis transmembrane conductance regulator (CFTR) function normalizes Cl− and P2X7-dependent K+ efflux, inhibits inflammasome activation, and decreases IL-1β release in CF monocytes. Several P2X7 inhibitors have been identified. AP-1 = adaptor protein complex 1; CREB = cAMP-responsible element-binding protein; HIF-1 = hypoxia-inducible factor-1; NF-κB = nuclear factor-κB.
The study published in this issue of the Journal by Gabillard-Lefort and colleagues (pp. 783–794) brings important new information to help us understand, at least in part, how excessive inflammation may be directly associated with CFTR dysfunction through the P2X7R–NLRP3–caspase-1–IL-1β or NLRP3 inflammasome axis (9). The authors initially validated their novel hypothesis that blood monocyte CFTR expression and function play a role in regulating the activation of the NLRP3 inflammasome through the engagement of P2X7R by eATP. The major sources of eATP in vivo are hypoxia and cell lysis, both of which are omnipresent in the CF airway. After these novel observations, the investigators sought to validate that the CFTR and P2X7R interactions occur in individuals with CF treated with the CFTR modulator ETI combination TRIKAFTA (Vertex Pharmaceuticals). The therapy not only restored monocyte CFTR expression and function but also decreased monocyte P2X7R expression, corrected K+ efflux, and normalized intracellular Ca2+. The antiinflammatory benefits of ETI therapy were reflected in decreased blood monocyte NLRP3 mRNA, protein, caspase-1, and IL-1β. Furthermore, TRIKAFTA therapy in individuals with CF normalized their plasma eATP and IL-1β levels.
The study by Gabillard-Lefort and colleagues presents compelling data connecting defective CFTR, P2X7R expression, and blood monocyte NLRP3 inflammasome activation by eATP and LPS, clearly representing an important advance in knowledge of links between CFTR and inflammation. However, the physiologic impact of these observations on CF lung inflammation could be affected by several factors. First, the reported experiments focused on monocytes derived from blood. As monocytes migrate to the lung and differentiate into macrophages, they acquire unique characteristics distinct from those of blood monocytes. Specifically, monocytes spontaneously release endogenous ATP in amounts sufficient to support LPS-induced IL-1β release. In contrast, macrophages do not spontaneously release ATP and require IFN-γ to release sufficient eATP to support LPS/P2X7R-mediated inflammasome activation and IL-1β release (10). However, the eATP released from hypoxia and cell lysis in the CF airway is likely sufficient to trigger macrophage P2X7R. Second, the CF airway milieu is very different from plasma. Inhibition of CFTR chloride conductance in monocytes led to a decrease in extracellular Cl− ion. Because extracellular Cl− suppresses P2X7R activation, CFTR deficiency in monocytes could in theory facilitate P2X7R activation. However, another potent inhibitor of P2X7R activation is the extracellular proton concentration (11). The airway surface liquid is generally more acidic in the absence of CFTR, thus making it difficult to predict how CFTR-dependent changes in Cl− efflux and proton concentration will affect P2X7R activation in CF airways. Thus, although there is strong evidence of a link between CFTR dysfunction and P2X7R in blood monocytes, the exact mechanisms by which ion fluxes are coordinated between CFTR, P2X7R, Panx1, and other ion channels remain unknown and could be an important area of investigation to better understand inflammation associated with CF lung disease.
In summary, the study by Gabillard-Lefort and colleagues identifies a novel therapeutic target of interest for controlling inflammation associated with CFTR deficiency. Attempts to develop new therapies focused on the end results of excessive inflammation in CF airways, such as free elastase and oxidative stress, have been disappointing. Such approaches do not affect the CFTR–P2X7R axis, and, upon eATP and LPS stimulation, the exaggerated inflammatory response will continue. However, several P2X7R antagonists and other agents affecting the P2X7R axis, such as secretory leukocyte protease inhibitor (12), have been identified and may represent novel antiinflammatory approaches in CF and other lung diseases associated with CFTR functional deficiency, such as chronic obstructive pulmonary disease. Importantly, the work by Gabillard-Lefort and colleagues reminds us that the most effective strategy to control the excessive inflammatory response responsible for airway destruction in CF is likely to be the correction of CFTR function in all individuals with CF.
Footnotes
Originally Published in Press as DOI: 10.1164/rccm.202201-0008ED on February 9, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Chen G, Sun L, Kato T, Okuda K, Martino MB, Abzhanova A, et al. IL-1β dominates the promucin secretory cytokine profile in cystic fibrosis. J Clin Invest . 2019;129:4433–4450. doi: 10.1172/JCI125669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, et al. AREST CF Investigators Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med . 2013;368:1963–1970. doi: 10.1056/NEJMoa1301725. [DOI] [PubMed] [Google Scholar]
- 3. Montgomery ST, Dittrich AS, Garratt LW, Turkovic L, Frey DL, Stick SM, et al. AREST CF Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J Cyst Fibros . 2018;17:715–722. doi: 10.1016/j.jcf.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 4. Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med . 1995;151:1075–1082. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 5. DeBoer EM, Kimbell JS, Pickett K, Hatch JE, Akers K, Brinton J, et al. AREST CF Lung inflammation and simulated airway resistance in infants with cystic fibrosis. Respir Physiol Neurobiol . 2021;293:103722. doi: 10.1016/j.resp.2021.103722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol . 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 7. Kopp R, Krautloher A, Ramírez-Fernández A, Nicke A. P2X7 interactions and signaling: making head or tail of it. Front Mol Neurosci . 2019;12:183. doi: 10.3389/fnmol.2019.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Linden J, Koch-Nolte F, Dahl G. Purine release, metabolism, and signaling in the inflammatory response. Annu Rev Immunol . 2019;37:325–347. doi: 10.1146/annurev-immunol-051116-052406. [DOI] [PubMed] [Google Scholar]
- 9.Gabillard-Lefort C, Casey M, Glasgow AMA, Boland F, Kerr O, Marron E, et al. Trikafta rescues CFTR and lowers monocyte P2X7R-induced inflammasome activation in cystic fibrosis Am J Respir Crit Care Med 2022205783–794 [DOI] [PubMed] [Google Scholar]
- 10. Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood . 2009;113:2324–2335. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Flittiger B, Klapperstück M, Schmalzing G, Markwardt F. Effects of protons on macroscopic and single-channel currents mediated by the human P2X7 receptor. Biochim Biophys Acta . 2010;1798:947–957. doi: 10.1016/j.bbamem.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 12. Zakrzewicz A, Richter K, Zakrzewicz D, Siebers K, Damm J, Agné A, et al. SLPI inhibits ATP-mediated maturation of IL-1β in human monocytic leukocytes: a novel function of an old player. Front Immunol . 2019;10:664. doi: 10.3389/fimmu.2019.00664. [DOI] [PMC free article] [PubMed] [Google Scholar]