

Abstract

The synthesis of aryl–alkynyl compounds is usually achieved via Sonogashira catalysis, but this is inefficient for fluorinated aryls. An alternative method reported by Shirakawa and Hiyama, using alkynylstannanes and hemilabile PN ligands, works apparently fine for conventional aryls, but it is also poor for fluorinated aryls. The revision of the unusual literature cycle reveals the existence and nature of unreported byproducts and uncovers coexisting cycles and other aspects that explain the reasons for the conflict. This knowledge provides a full understanding of the real complexity of these aryl/alkynylstannane systems and the deviations of their evolution from that of a classic Stille process, providing the clues to design several very efficient alternatives for the catalytic synthesis of the desired ArF–alkynyl compounds in almost quantitative yield. The same protocols are also very efficient for the catalytic synthesis of alkynyl–alkynyl’ hetero- and homocoupling.
Introduction
Since its discovery, Stille catalysis has proven to be an excellent method for C–C coupling.1 Although less frequently used nowadays, it has an advantage that it can be applied to reagent-bearing groups that would not stand the reaction conditions of other name reactions. The mechanisms of the different steps of the classic cycle (Scheme 1, cycle I), namely, Ar–X oxidative addition, Bu3SnR transmetalation, and Ar–R reductive elimination, have been extensively studied.2
Scheme 1. Classic Stille Cycle (I) and the Alternative Pathway (II) Proposed by Shirakawa and Hiyama for Aryl–Alkynyl Coupling, Represented for a PN Ligand.

About 20 years ago, Shirakawa and Hiyama proposed that a different mechanism operated for the case of alkynyl stannanes (Scheme 1, cycle II, R = alkynyl, shortened as Alk from now on) and was particularly efficient with iminophosphine or aminophosphine chelating ligands (PN), but inefficient with PPh3 or diphosphines. In this cycle, the roles of the electrophile and the nucleophile on Pd, are altered relative to cycle I: instead of the Ar–X electrophile, the alkynyl stannane acts as the oxidant on Pd0(PN), formed in situ from the precatalyst (μ-Cl)2[Pd(π-allyl)]2 and an iminophosphine or aminophosphine, to give [Pd(Alk)(SnBu3)(PN)] (2).3 Then, Ar–X produces an Ar/SnBu3 exchange by an unexplained mechanism.4,5 The formation of 2 with PN ligands was experimentally ascertained by its NMR observation,6 but the operativity of the subsequent Ar/SnBu3 exchange proposed has never been demonstrated nor denied. Crociani et al. reported that using [Pd0(PN)(η2-dimethylfumarate)] as catalyst complex 2 was not observed and the classic cycle I, initiated by oxidative addition with Ar–X, was operating.7 Our interest in C–C couplings involving fluorinated aryls led us in the past to test the Sonogashira catalysis, the classic method for Ar–Alk coupling, but we found in preliminary experiments that, for ArF = C6F3Cl2-3,5, this process was quite inefficient: from (C6F3Cl2)–I and Bu3Sn–C≡C–Ph ([PdCl2(PPh3)2]/CuI, NEt3, 80 °C, dioxane, 24 h), it produced 90% conversion, but only 2% was the desired product and 88% was (C6F3Cl2)–H, confirming that, as it often happens, fluorinated aryls are a different challenge.8
We tried then the Stille reaction with alkynyl stannanes.9 The 1:1 coupling of ArF–I with PhC≡CSnBu3, in tetrahydrofuran (THF) at 50 °C, catalyzed by 5% of [Pd(ArF)I(PPh3)2] (either cis or trans) showed that the catalysis follows cycle I but is very inefficient, producing less than 5% of ArF–Alk in 3 h. The catalytic problem was identified with the fact that the trans-to-cis isomerization, required to give coupling, was extremely slow, and eventually, trans-[Pd(Alk)(ArF)(PPh3)2] became a catalyst trap. Curiously, the nonanalyzed Shirakawa’s results reported for conventional aryls and 2 PPh3 instead of PN were even worse (about 1% cross-coupling), as if their reaction, like ours, failed to be efficient in cycle I with PPh3.
The positive results reported by Shirakawa and Hiyama for Ar–Alk coupling of conventional aryls, using PN ligands and supposedly following cycle II, provided yields in the range of 86–93% (18–29 h in THF at 50 °C, 5 mol % of the (μ-Cl)2[Pd(π-allyl)]2 precatalyst, i.e., 10% Pd).5 They looked for an attractive alternative to an inefficient Sonogashira. However, when we checked the reaction of IC6F3Cl2-3,5 with Bu3Sn–C≡C–Ph using [Pd(Alk)(SnBu3)(PN)] (2) (10% 2, in THF, 50 °C, 24 h) as catalyst, the result was not only somewhat disappointing but also puzzling: 50% C6F3Cl2–Alk, 28% C6F3Cl2–SnBu3 and 22% C6F3Cl2–H.
Considering all the apparently contradictory data of the previous information, this study has two targets: (i) to find out what is exactly going on in the reactions involving aryl electrophiles and alkynylstannane nucleophiles and why they are inefficient for fluorinated aryls and (ii) to develop efficient catalytic protocols for catalytic synthesis of ArF–Alk compounds, potential precursors of many other fluorinated species.
Results and Discussion
Section A: Mechanistic Stoichiometric Studies
In this section, ArF stands for C6F3Cl2-3,5. With the previous information, it looked still possible that the use of chelating PN ligands might prevent the formation of Pd traps such as trans-[Pd(Alk)(ArF)L2]9 and derive cycle I to a different coupling pathway, that is, Shirakawa and Hiyama cycle II. For this reason, we recently studied the behavior of some chelating ligands in the context of Stille ArF–Alk couplings intentionally frustrated by the absence of the ArF–I oxidant (Scheme 2).10
Scheme 2. Reaction Sequence Monitored for the Reactions of [Pd(ArF)X(P–L)] [X = OTf (a); X = I (i)] with an Excess of PhC≡CSnBu3 (c), Pd:Sn = 1:20, in THF, in the Absence of ArF–I.

In the presence of the alynylstannane, the palladium(II) complexes [Pd(ArF)X(P–L)] (X = I or OTf; P–L = dppe, and ortho-C6H4(PPh2)(CH2–NMe2)) follow the classic transmetalation + coupling evolution that reproduces part of cycle I and eventually leads to 2 (labeled as type h in Scheme 2) which is the product of the first step of the proposed cycle II. Fortunately, we have been able to prepare cleanly complex [Pd(Alk)(SnBu3)(PN)] (2) according to eq 1. The X-ray diffraction structure (Figure 1) confirmed the isomer suggested by the 31P NMR data.11
 |
1 |
Figure 1.

X-ray structure of 2. H atoms and Bu groups are omitted for clarity. Relevant distances (Å) and angles (°): Pd1–Sn1 = 2.5569(3), Pd1–P1 = 2.2659(8), Pd1–N1 = 2.291(3), and Pd1–C1 = 1.993(3); C1–C2 = 1.210(5); C1–Pd1–Sn1 = 74.97(10), P1–Pd1–Sn1 = 101.75(2).
With complex 2 in hand, we could test the dark point in cycle II: how does compound 2 behave in the presence of ArFI? The stoichiometric reaction 2 + ArF–I in neat THF (the usual solvent in the catalysis of Shirakawa and Hiyama and in the work of Crociani)4,7 afforded, at completion (2 days at 25 °C), 80% ArFSnBu3 and 15% hydrolysis product ArFH but only 5% of ArF–Alk (Scheme 3 and Figure S1). In addition, the nonfluorinated complex [Pd(Alk)I(PN)] (3) was observed by 31P NMR. This result does not support the second step of cycle II, at least as a simple process.
Scheme 3. 19F Containing Products in the Stoichiometric Reactions of 2 + ArF–I at 25 °C.

Isolated yield.
Using THF/hexane = 2/5 (v/v) as the solvent, the result was strikingly different: [Pd(ArF)I(PN)] (1), which is only sparingly soluble in this mixture, was isolated in high yield (≈85%), proving that 2 can be transformed into 1 via reductive elimination of 2 to Alk–SnBu3 + Pd0(PN), followed by oxidative addition of ArF–I to Pd0(PN). This is an obvious pathway for compound 2 to re-enter cycle I in the form of 1. Small amounts of ArF–SnBu3 and ArF–H were also found in the filtrate. Furthermore, we confirmed that the stoichiometric reaction of 1 + 2 in THF at room temperature leads quickly to ArF–SnBu3 and traces of ArF–H. This explains the abundant formation of ArF–SnBu3 in THF. The conclusion is that two reactivity patterns from 2 + ArF–I coexist, one that produces 1 and can bring the reaction to cycle I, yielding ArF–Alk, and another one, so far unknown, which yields ArFSnBu3, a product absent from the proposed cycle II. In the studied conditions, this second pathway is much faster than cycle I. Taking into account the reactivity mentioned above and the species observed in our previous study in ref (10) (Scheme 2), the reactivity model shown in Scheme 4 can be proposed.
Scheme 4. Proposed Reaction Pathways to Explain the Competitive Formation of ArF–SnBu3, ArF–H, and ArF–Alk.

The reactions in Scheme 4 line A account for the formation of 1 from 2 and ArF–I. In THF/hexane, complex 1 is only scarcely soluble and precipitates. However, in THF, the coexistence in a solution of 1 and 2 leads to the formation of ArF–SnBu3 and ArF–H by the sequence of lines B/C (Scheme 4).12 Kinetic competition of hydrolysis and Sn–C reductive elimination on I2 yields ArF–H and abundant ArF–SnBu3. The fact that solutions of 1 or ArF–I in wet THF are perfectly stable for days at 50 °C (temperature of catalytic conditions) confirms that the formation of ArF–H requires also the presence of 2 and the formation of I2 (reaction in Scheme 4, line B). The scarce solubility of 1 in THF/hexane reduces its presence to a very small concentration in this mixture and, consistently, limits the formation of ArF–SnBu3 and ArF–H to a very small percentage. Finally, the direct sequence of line A followed by line D completes cycle I and produces a small percentage of ArF–Alk in arduous competition with the faster destructive competence of processes B and C. The competitive formation of the fluorinated compounds ArF–SnBu3, ArF–H, and ArF–Alk in THF was 19F NMR monitored at 10 °C and fitted using the COPASI software (Figure 2).13
Figure 2.

Concentration vs time 19F NMR monitoring data (triangles) and COPASI fitting (continuous lines) of the F-containing species in the reaction of 2 with ArF–I in THF at 10 °C.
Being I1 unobservable because of its minute concentration, experimental parameters for the 2 ⇋ I1 equilibrium cannot be obtained and reasonable ΔG0 and ΔG‡ values from density functional theory (DFT) calculations in THF (Figure 3) were used for the COPASI fitting (full details of the DFT work on the model of Scheme 4, including optimized structures of I2Me and TS2Me in Figure S5, are given in the Supporting Information).14
Figure 3.

Optimized structures of I1Me (left) and TS1Me (right), using SnMe3 instead of SnBu3. Selected distances are given in Å. ΔGTHF values relative to 2Me (with SnMe3) are given in kcal mol–1.
The main curves of Figure 2 show clearly an autocatalytic effect. Note that the initial concentration of 1 in this experiment is zero until a small amount is formed following process A in Scheme 4. The coexistence of 1 and 2 opens autocatalytic reactivity of pathway B + C, which accelerates and becomes eventually dominant.
In conclusion, our stoichiometric study reveals the existence of an ignored pathway starting with 2 and producing efficiently ArF–SnBu3 instead of ArF–Alk. Under the specific conditions of this stoichiometric study (Alk–SnBu3:ArF–I:Pd = 0:1:1 at 10 °C), pathway A + D of Scheme 4 which reproduces the reaction sequence of the classic Stille cycle I, is comparatively slow, whereas the sequence B + C, requiring the previous coexistence of 1 and 2 (achieved via A), is highly competitive but produces ArF–SnBu3 and ArF–H, not ArF–Alk. Interestingly, in the stoichiometric reaction at a higher temperature (50 °C, the temperature used by Shirakawa and Hiyama in the catalytic experiments), the percentage of the ArF–Alk (13%) and ArF–H (22%) increases moderately in the detriment of ArF–SnBu3 (65%) (Figure S2). The ArF–Alk versus undesired products’ ratio increases almost 3 times from 0.052 at 25 °C to 0.149 at 50 °C, supporting higher rate acceleration with the temperature of the desired pathway A + D (Stille reaction). It seems that this tendency with temperature might explain the high coupling percentages of Ar–Alk products (yields in the order 80–90%) obtained for conventional aryls by Shirakawa and Hiyama in catalytic conditions (50 °C), but we will soon discuss that it is not that simple. Shirakawa and Hiyama did not report the existence and nature of the other products formed that account for the 100% conversions, but it is reasonable to think that Ar–SnBu3 products were formed, as supported by our results in the next section.
Section B: Catalytic Studies for ArF–C≡C–R Stille Coupling
The catalytic conditions differ from the stoichiometric experiment in that the reagents AlkSnBu3 and ArFI are in large excess relative to the Pd catalyst (e.g., 100:100:10). The Pd0(PN) molecules formed in the initial coupling (whether from 1 or from 2 as a catalyst) have to be recycled. The peculiarity of this reaction is that both reagents are able to produce the required oxidative addition. This gives rise to two competitive recycling processes in the second and subsequent turnovers: reoxidation with ArF–I follows the cycle on the left in Scheme 5 to create 1, whereas recycling of Pd0(PN) via oxidative addition with Alk–SnBu3 (Scheme 5, right) yields 2. This produces the coexistence of 1 and 2 and consequently opens pathway B + C in Scheme 4 to the undesired formation of ArFSnBu3.
Scheme 5. Two Cycles Forming ArF–SnBu3 and 3 by Autocatalytic Recycling of Pd0(PN).

At first sight, the two cycles in Scheme 5 look self-destructive since each turnover recovers one Pd in the form of 1 or 2, producing one ArF–SnBu3 molecule, whereas one Pd is apparently lost in the form of the out-of-cycle complex 3. However, complex 3 can be recovered as Pd0(PN) in a sequence that would be part of an alternative cycle I starting with Alk–I and ArF–SnBu3 instead of ArF–I and Alk–SnBu3 (Scheme 6). We call this alternative the transposed catalysis and will come back to it later on. The cycles in Scheme 5 are crucial to understand the behavior of this peculiar catalytic system involving alkynylstannanes because (i) the existence of both cycles, re-entering the catalyst via Pd0(PN) oxidative addition with Alk–SnBu3 or with ArF–I explains the mechanism of 2 ↔ 1 conversion along the catalysis; (ii) the coexistence of 1 and 2 activates pathway B + C that causes the formation of 3 and undesired ArF–SnBu3; (iii) at the same time, the formation of 3 opens an alternative transposed Stille cycle that makes the undesired ArF–SnBu3 a useful reagent to produce ArF–Alk (Scheme 6). Consequently, it is a matter of finding how to improve the step rates leading to the desired evolution to ArF–Alk via 1 and 3(15) or reduce the rates leading to ArF–SnBu3 from 2.
Scheme 6. Alternative transposed Stille Cycle Based on the Formation of 3 in Scheme 5.

The results of our first set of catalytic experiments to produce Ph-C≡C-(C6F3Cl2-3,5) are summarized in Table 1. The results in the absence of additives, using 10% of 2 or 1 as catalysts (entries 1 and 2), were promising: working at 50 °C, yields of ArF–Alk in the range of 50–58% were achieved. The rest was undesired ArF–H and ArF–SnBu3. The yields required improvement, but the data were mechanistically meaningful. It is striking that, with complex 1 being an in-cycle species of the classic Stille mechanism (according to Scheme 1) and complex 2 being a disturbing species foreign to Stille cycle I, the results of Stille (ArF–Alk) versus undesired (ArF–SnBu3 + ArF–H) yields are not very different (50/50 with 2, 58/42 with 1). This is due to the 2 ↔ 1 ↔ 3 conversion that, after a way, creates similar catalyst conditions.
Table 1. Catalytic Results of the ArF–Alk Coupling with Different Catalysts and Additivesa.

| entry | catalyst | additives (mol %) | ArFAlk | ArFSnBu3 | ArFH |
|---|---|---|---|---|---|
| 1 | 2 (10%) | 50 | 28 | 22 | |
| 2 | 1 (10%) | 58 | 33 | 9 | |
| 3 | 2 (10%) | 1% AsPh3 | 85 | 6 | 9 |
| 4 | 1 (10%) | 1% AsPh3 | 88 | 2 | 10 |
| 5 | 1 (10%) | 10% AsPh3, | 79 | 1 | |
| 6 | 4 (10%) | <1 | 5 | ||
| 7 | 1 (10%) | 100% LiCl | 51 | 18 | 31 |
| 8 | 2 (10%) | 1% AsPh3, 100% LiCl | 90 | 10 | |
| 9 | 1 (10%) | 1% AsPh3, 100% LiCl | >99b | ||
| 10 | 1 (2%) | 0.2% AsPh3, | 39 | 3 | 13 |
| 11 | 1 (2%) | 0.2% AsPh3, 100% LiCl | 96 | 4 | |
| 12 | 1 (10%) | 1% AsPh3, 110% LiCl | 98c | 2 | 0 |
19F NMR yields of each product.
Analogous results are obtained for the reaction at 40 °C after 48 h (98%) or replacing LiCl with CsF.
Reaction with 4-FC6H4I. 110 mol % of Alk–SnBu3 and LiCl are used because the first turnover from 1 can form up to 10% of PhC≡C–C6F3Cl2-3,5 instead of PhC≡C–C6H4F-4.
The improved competitivity of the Stille cycle observed in entries 1 and 2 (estimated by the yield in ArF–Alk vs ArF–SnBu3 + ArF–H) is clearly way larger than expected from the temperature effect observed in the stoichiometric studies: 5% ArF–Alk at 25 °C and only 13% at 50 °C in the stoichiometric reaction, versus 50–58% ArF–Alk at 50 °C in catalysis. In the stoichiometric reaction 2 + ArF–I, the evolution to produce ArFSnBu3 through reactions B and C (Scheme 4) is, according to the results, largely dominant over the pathways A + D or 3 + ArFSnBu3 (Scheme 6).15 In contrast, in catalytic conditions, the high concentrations of ArFI and AlkSnBu3, the cycles in Scheme 5 and the Pd0(PN) recycling, get into play, substantially improving the competitivity of cycle I. Although 2 dominates initially in entry 1 and 1 does the same in entry 2, when the 2 ↔ 1 ↔ 3 conversions via Scheme 5 adjust their concentrations, the two catalyses come to similar success, casually close to 50% in the catalytic conditions used.
First Catalytic Improvement
In spite of the remarkably better ArF–Alk formation found in catalytic conditions, the percentage of reaction (via the Stille cycle with 1 or 3) with a fluorinated aryl such as C6F3Cl2-3,5 is unsatisfactory. Fortunately, in any coupling catalysis, the initial catalyst is converted to Pd0 in the first turnover. If we could modify the Pd0(PN) ephemeral intermediate to the one that could be oxidized by ArF–I but not by Alk–SnBu3, the undesired cycle in Scheme 5 (right), producing undesired ArF–SnBu3, should disappear and, using 1 as a catalyst, the evolution after the first turnover should be derived to the desired cycle I. Similarly, starting with catalyst 2, all of it would be converted to 1 after the first turnover and be ready to follow Stille cycle I.
For the sake of simplicity, we have so far represented the Pd0 species produced upon coupling as Pd0(PN), but it is unrealistic that this unsaturated molecule can survive without immediately capturing potential coordinating L ligands in solution (e.g., L = THF, OH2, or triple bonds of the different alkynyl-containing molecules) to give Pd0(PN)Ln. In order to improve the catalytic results, we should simply find some appropriate coordinating L molecule as an additive that facilitates the oxidative addition by ArF–I of the corresponding Pd0(PN)Ln as much as possible. After some unsuccessful trials, we were glad to see that the addition of AsPh3 in a largely substoichiometric proportion relative to the Pd catalyst (AsPh3:Pd = 1:10) is enough to quite efficiently quench the formation of complex 2, producing a clear increase in the percentage of ArF–Alk (>85%, Table 1, entries 3 and 4). The catalytic results confirm that the presence of substoichiometric AsPh3 makes the choice of catalyst 1 or 2 (entries 3 and 4) almost indifferent because if catalyst 2 is used, it only exists during the initial turnover.
This terrific effect supports that AsPh3 coordinates with Pd0(PN) in preference to the other potential ligands in solution to give Pd0(PN)(AsPh3), perhaps in equilibrium with Pd0(PN)(AsPh3)2. The former, more electron-rich than Pd0(PN) (hence more easily oxidizable) and less hindered for the approximation of the ArF–I bond to the Pd atom than Pd0(PN)(AsPh3)2, is likely to be the most reactive one. Additionally, the presence of AsPh3 is able to prevent the formation of I1 (Scheme 4), and hence of 2, hampering the kinetic competition of Alk–SnBu3 for oxidative addition. The computed equilibrium constant for L displacement of η2-alkynylstannane by AsPh3 in Pd0(PN)L is ca. Keq ≈ 30 at 25 °C, which supports the higher stability of Pd0(PN)(AsPh3) compared to I1. On the other hand, the formation of 1 by the oxidative addition of ArF–I to Pd0(PN) is ca. 35 kcal mol–1 more favorable than the formation of 2 by the oxidative addition of Alk–SnBu3 to Pd0(PN). Although AsPh3 modifies the Pd0(PN) species to Pd0(PN)(AsPh3), it is not consumed in the catalytic synthesis, and its role is catalytic.
Note that AsPh3:Pd = 1:10 is largely substoichiometric relative to the total concentration of Pd but, at the same time, largely overstoichiometric relative to the nonobservable concentration of Pd0(PN) just released during reductive elimination. In contrast, the addition of stoichiometric AsPh3 (Table 1, entry 5) is less efficient, reflecting the usual coupling retardation that almost any added ligand produces at the transmetalation step of Stille reactions.16 For AsPh3, this effect is moderate because it is not a strong ligand for PdII, but entry 5 warns that the addition of AsPh3 should be substoichiometric (the proportion has not been optimized).
Second Catalytic Improvement
In the catalytic reactions discussed so far, the experimental observations were mainly based on the very informative 19F NMR spectra. Analysis of the 31P NMR spectra of solutions after catalysis identified a signal with tin satellites corresponding to [PdI(SnBu3)(PN)] (4). This compound was independently synthesized for unambiguous identification by X-ray diffraction (eq 2 and Figure 4). Complex 4 is the product of the oxidative addition of I–SnBu3 (the byproduct of the Stille transmetalation) to Pd0(PN), and it turns out to be an irreversible Pd trap that precludes its re-entrance into the catalytic cycle. In fact, it was tested as a possible Pd precatalyst (Table 1, entry 6) and produced only negligible conversion.
 |
2 |
Figure 4.
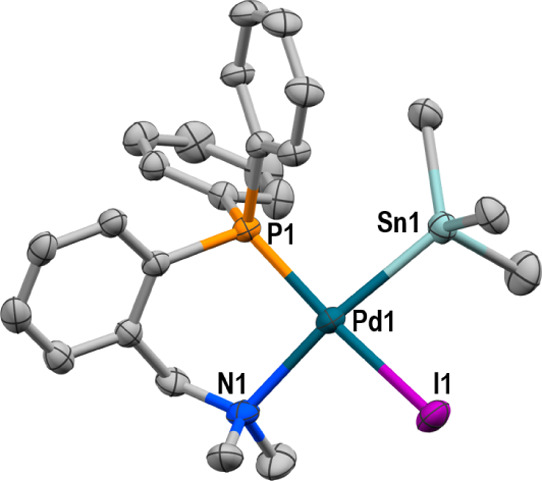
X-ray structure of 4. Hydrogen atoms and Bu groups are omitted for clarity. Relevant distances (Å) and angles (°): Pd1–Sn1 = 2.5884(6), Pd1–N1 = 2.313(5), and I1–Pd1–Sn1 = 83.94(2).
The accumulative formation of 4 along the catalysis can eventually reduce the active catalyst concentration to inefficient figures. LiCl (stoichiometric relative to the reactants) was added in order to transform I–SnBu3 into Cl–SnBu3, less able to oxidize Pd0(PN).17 This additive showed only moderate efficiency when alone (entry 7 vs entries 1 and 2), but in combination with 1% AsPh3, it brought the ArF–Alk yield to 90% in the reaction with precatalyst 2 (entry 8) and to quantitative yield in the reaction with catalyst 1 (entry 9). The addition of LiCl (CsF has a similar effect) becomes more relevant for a lower percentage of the catalyst: with 2% 1 + 0.2% AsPh3, the catalysis only reaches 39% yield of ArF–Alk before the catalytic activity expires, while the combination 2% 1 + 0.2% AsPh3 + stoichiometric LiCl increases this yield to 96%. Finally, entry 12 in Table 1 shows that the use of the two additives is also efficient for more conventional (less fluorinated) aryls, such as C6H4F-4, which yields 98% of 4-FC6H4–Alk and (as suspected for unreported data in the Shirakawa and Hiyama results) 2% of 4-FC6H4–SnBu3. This result suggests that the coupling yields reported by Shirakawa and Hiyama can also be substantially improved by the use of these additives.
Failed or Partially Frustrated Attempts of Catalysis Improvement
In addition to the results in Table 1, we tested the reaction in entry 9 using ArF–Cl instead of ArF–I. It was unsuccessful, even at reflux in THF, due to its higher barrier for oxidative addition. Also, [Pd0(PN)(η2-dmfu)] (X-ray structure in Figure S7), which worked well with conventional aryls as reported by Crociani,7 proved inactive for ArF–I + Alk–SnBu3 catalysis because, in contrast with Ar–I, ArF–I does not undergo oxidative addition.
A very disappointing result was that when the best catalytic conditions for C6F3Cl2–I were applied to other fluorinated aryl iodides, significantly higher difficulty was found, depending on their fluorination (Table 2, entry 1). The almost quantitative results with 4-F-C6H4–I (entry 2) show that this aryl practically displays the behavior not far from what we call “conventional” aryls, but for other fluorinated aryls, a very significant drop of yield is observed. The effect is particularly high when one or (more markedly) the two positions ortho to the ipso-C atom are fluorinated (entries 3 and 4).
Table 2. Catalytic ArF–Alk Results of ArF–I + PhC≡C–SnBu3 Coupling Catalyzed by Complex 1, with Different Fluorinated Aryl Groupsa.
The specific problem of highly fluorinated aryls is that, due to the high group electronegativity, their nucleophilic reagents (e.g., C6F5–SnBu3) are weaker than conventional aryls. On the other hand, the corresponding electrophilic reagents producing the oxidative addition (e.g., C6F5–I) are also less reactive than their nonfluorinated congeners.18,19 For electron-rich Pd0 complexes, even C6F5–I reacts sufficiently well, but the poor electron density of Pd0(PN) worsens the problem. The fact that increasing the reaction temperature to 90–100 °C (entries 5 and 7) very significantly improves the conversion indicates that more fluorinated reagents are finding higher energetic barriers in the oxidative addition or in other catalytic steps. Yet, at the limit of fluorination (C6F5, entry 7), an acceptable 70% yield of the coupling product C6F5–C≡C–Ph can be achieved with just 2% of catalyst 1, although at 90 °C and suffering 27% of hydrolysis.
More Practical Alternative?: The “Transposed” Catalysis
The solutions so far applied to improve the catalysis are based on trying to make the oxidative addition reaction ArFI + Pd0(PN) more efficient than Alk–SnBu3 + Pd0(PN). Obviously, in the mechanistic study in Section A, we could not alter the combination of reagents used by Shirakawa and Hiyama, Ar–I + Alk–SnBu3, but for catalysis, this restriction does not hold. Moreover, in Scheme 5, we have found that the initial conversion 2 ↔ 1 and the coexistence of both complexes in solution lead to the formation of [Pd(Alk)I(PN)] (3) and ArF–SnBu3, also able to follow a Stille cycle. Both Stille cycles, C6F3Cl2–I + AlkSnBu3 (cycle I in Scheme 1) and its transposed version (Scheme 6) are able to produce the desired ArF–Alk product. It is probably hopeless to approach a mechanistic study on so many products, steps, and barriers. From a practical view, it is more reasonable simply to check the catalytic efficiency when the groups (ArF and Alk) transpose their roles.
A recently published DFT study at the ZORA-BLYP/TZ2P level has shown that the activation energy for the oxidative addition of C(spn)–X bonds (n = 1–3; X = H, Me, Cl) to Pd0Ln is lower for a lower number of C substituents. Hence, it is the lowest for alkynyl groups (n = 1).20 Assuming that this will hold for X = SnBu3, the oxidative addition of the Alk–SnBu3 bond (n = 1) should be favored by this effect compared to ArF–I (n = 2). Moreover, the entropically disfavored associative step of oxidative addition should be better compensated by the stronger coordination to Pd0 of the C≡C triple bond compared to ArF. We suggest that these two circumstances concur to facilitate the oxidative addition of Alk–SnBu3 and create the alkynylstannane problem when combining ArF–I (only weak π-donors) and alkynylstannanes (stronger π-donors), which is not observed for other stannane reagents. According to this analysis, the lower oxidative addition barriers in a transposed Stille cycle, with Csp atoms versus Csp2 and the easier coordination to Pd0(PN), should facilitate the Alk–I oxidative addition, easing the direct formation of [Pd(Alk)I(PN)] (3). A subsequent transmetalation reaction with ArF–SnBu3 should continue the transposed Stille cycle.
Complex 3 was prepared from 2 taking advantage of the easy reductive elimination of AlkSnBu3 to I1 (eq 3) to be used as a catalyst. Complex [Pd(Alk)Cl(PN)] (5), used also in Table 3, can be prepared similarly using Cl–Alk instead of I–Alk (see full characterization and synthetic details in the Supporting Information, including the X-ray structure of 5 in Figure S6).
 |
3 |
Table 3. Catalytic Results of the Transposed ArF–Alk Coupling Using Alk–X (X = Cl, I), ArF–SnBu3, and Catalysts 2, 3, or 5a.

| entry | Pd catalyst | additives | ArF–Alk | ArF–Sn | ArF–H |
|---|---|---|---|---|---|
| 1 | 3 (10%) * | 81 | 18 | 1 | |
| 2 | 3 (10%) *,b,c | AsPh3, LiCl | >99 | ||
| 3 | 3 (10%) *,c | LiCl | >99 | ||
| 4 | 3 (2%) *,b | AsPh3 | 68 | 29 | 3 |
| 5 | 3 (2%) *,c | LiCl | 98 | 2 | |
| 6 | 5 (2%) ** | 31 | 65 | 4 | |
| 7 | 5 (2%) **,b | AsPh3 | 97 | 3 | |
| 8 | 2 (10%) **,d | 21 | 51 | 28 | |
| 9 | 2 (10%)**,b | AsPh3 | 89 | 6 | 5 |
| 10 | 2 (2%) **,b | AsPh3 | 82 | 12 | 6 |
ArF = C6F3Cl2-3,5.
Substoichiometric AsPh3 (10 mol % with respect to the Pd catalyst).
Stoichiometric LiCl (100 mol %).
Only 49% conversion. *Alk–I is used as a reactant and the reaction is left for 24 h at 50 °C. **Alk–Cl is used as a reactant and the reaction is left for 36 h at 70 °C.
The results of several transposed catalyses with 2, 3, or 5 as catalysts are presented in Table 3, and the catalytic cycles applied in each case are illustrated in Scheme 7. The analysis of the results reveals a bonus in the transposed protocol and provides a finer appreciation of the role of each additive.
Scheme 7. In Situ Formation of Catalysts 3 and 5 from 2 and Routes for the Formation of the Alk–Alk Byproduct and the Heterocoupling Product Alk–ArF.

Li salts used only for catalyst 3. ArF = C6F3Cl2.
Scheme 7 is represented assuming the formation of Pd0(PN)(AsPh3) after the first turnover in the presence of substoichiometric AsPh3. This is not the case for several entries (for instance, with 10 mol % of catalyst 3, entries 1 and 2 show that the use of stoichiometric LiCl, either with or without substoichiometric AsPh3, increases the yield of C6F3Cl2–Alk from 81% to quantitative). With 2% catalyst and 0.2% AsPh3 (entries 4 and 5), only 68% conversion to ArF–Alk is achieved, and stoichiometric LiCl is still needed to keep the catalyst active, hampering the formation of [PdI(SnBu3)(PN)] (4). Practically, a quantitative yield (98%) is obtained with 2% of 3 in 24 h at 50 °C. The addition of AsPh3 seems almost unnecessary or little effective in reactions with Alk–I (entries 1–5) but is required (as well as a higher temperature) for the otherwise slow oxidative addition with Alk–Cl (entries 6–10). This is clearly observed when comparing entries 6 and 7, with 5 as a catalyst.
Catalyst 5, with X = Cl, was prepared because should the oxidative addition be active with Alk–Cl, the transmetalation byproduct would be the nondisturbing Cl–SnBu3 instead of I–SnBu3. Then, we could spare the stoichiometric LiCl, making the reaction significantly more atom-economic. As the oxidative addition of chlorides has a higher barrier than iodides, Alk–Cl, with [Pd(Alk)Cl(PN)] (5) as the catalyst (entry 6), notably slowed the catalytic rate, but the addition of substoichiometric AsPh3 and increase of the reaction temperature had again a clear accelerating effect (entry 7), providing 97% yield of ArF–Alk in 36 h at 70 °C with only 2% 5 and 0.2% AsPh3.21 Although the increase in temperature causes a slight increase of hydrolysis, this method is probably the cleanest and more practical catalytic protocol.
With 2 as the catalyst (Table 2, entries 8–10), the initial turnover on 2 stoichiometrically produced AlkSnBu3 (up to 10%), which reacted with the oxidative addition product [Pd(Alk)Cl(PN)] (5) to give [Pd(Alk)2(PN)] and then Alk–Alk. The formation of the latter was confirmed by GC–MS (Scheme 7, cycle pathway above). To compensate for this loss of Alk–Cl reagent, its proportion was increased in the percentage of catalyst being used (e.g., 110 mol % of Alk–Cl if 10% of the catalyst is used). Again, the catalysis with 2 was comparatively slow in the absence of AsPh3 (entry 8, only 49% conversion and 21% yield), but it worked reasonably well with 10 mol % of 2 and 1 mol % of AsPh3 (up to 89% yield) or with 2 mol % of 2 and 0.2 mol % of AsPh3 (82% yield). This proves efficient in situ formation, in catalytic conditions, of Pd0(PN)(AsPh3)n and therefrom 5 (Scheme 7, upper cycle).
Table 4 shows the catalytic results of the transposed catalysis for the challenging C6F5 aryl. It produces C6F5–Alk in high yield (87%) and with little hydrolysis (Table 4, entry 2) using Alk–Cl, 10 mol % of 5, and 1 mol % of AsPh3. The amount of the catalyst can be reduced to 2% operating at 90 °C, with a significant reduction of conversion.
Table 4. Catalytic Results of C6F5–Alk Couplings Using Alk–Cl and C6F5–SnBu3 Catalyzed by 5a.

| entry | t (h) | T °C | catalyst | SM | C6F5–Alk | C6F5–H |
|---|---|---|---|---|---|---|
| 1b | 24 | 70 | 5 (10%) | 32 | 61 | 7 |
| 2b | 36 | 70 | 5 (10%) | 0 | 87 | 13 |
| 3b | 36 | 90 | 5 (2%) | 21 | 72 | 7 |
SM = starting material.
Substoichiometric AsPh3 (10 mol % with respect to the Pd catalyst).
Finally, since all the previous catalytic reactions use the hemilabile PN ligand, which presumably facilitates the transmetalation and reductive elimination steps by easy N dissociation,22 we decided to check the activity of [PdCl2(Ph-PEWO-F)] (6), bearing a phosphine-olefin ligand, also hemilabile in PdII and designed to facilitate difficult couplings.23 Because it makes Pd0 oxidation to PdII more difficult, C6F5–I was used. An in situ formed Buchwald complex with tBuXPhos, which displays a similar ability to promote challenging couplings,24 was also tested (Table 5).
Table 5. Catalytic Results of C6F5–Alk Couplings Using C6F5–I and Bu3Sn–C≡CPh or the Transposed Combination Using Alk–Cl, Catalyzed by [PdCl2(Ph-PEWO-F)] (6) or {[PdCl2(CH3CN)2] + tBuXPhos} (7)a,b.
| entry | R′–Sn | R–X | Cat | SM | R–R′ | C6F5H |
|---|---|---|---|---|---|---|
| 1c,d | PhCC–Sn | C6F5–I | 6 (5%) | 5 | 85 | 6e |
| 2d | C6F5–Sn | PhCC–Cl | 6 (5%) | 30 | 30 | 40 |
| 3c,f | PhCC–Sn | C6F5–I | 7 (10%) | <1 | 86 | 14 |
| 4f | C6F5–Sn | PhCC–Cl | 7 (10%) | 58 | 1 | 41 |
SM = starting material.
Reaction conditions: 24 h, 80 °C, 1,4-dioxane. R–X (1 equiv).
Stoichiometric LiCl (100 mol %).
R′–SnBu3 (1.1 equiv).
4% of unknown Pd(C6F5) species.
R′–SnBu3 (1.2 equiv.)
The results using Alk–Cl and C6F5–SnBu3 (entries 2 and 4) were unexpectedly disappointing, with high hydrolysis percentages, but those using C6F5–I and Alk–SnBu3 (entries 1 and 3) were very satisfactory as an alternative that works well in dioxane at 80 °C only with stoichiometric LiCl.
Stille Catalysis for the Heterocoupling of Alkynyls
The positive results in entries 9 and 10 of Table 3 and GC–MS confirmed that the formation of Alk–Alk products, indicating that the homocoupling of alkynyls (upper cycle of Scheme 7) can be promoted with our PN ligand platform, is an invitation to test the Stille catalysis for the heterocoupling of alkynyls. There are many reports for efficient and selective catalytic homo- and heterocoupling of alkynyls,25 but a Stille process, in case it is needed for reasons of compatibility with sensitive groups, is lacking. Table 6 collects some tests for the catalytic synthesis, as an example, of the unsymmetrical 1,3-diyne tBuC≡C–C≡CPh using tBuC≡C–I, Bu3Sn–C≡CPh, and 2 as precatalyst.
Table 6. Catalytic Results of Alk–Alk′ Couplings Using tBu–C≡C–I and Bu3Sn–C≡CPh Catalyzed by 2.

| entry | catalyst (%) | additivesa | tBuC2–C2Ph | tBuC2–C2tBu | tBuC2–I |
|---|---|---|---|---|---|
| 1 | 10 | AsPh3, LiCl | 92 | 8 | |
| 2 | 10 | LiCl | 92 | 8 | |
| 3 | 10 | 91 | 9 | ||
| 4 | 2 | LiCl | 91 | 9 | |
| 5 | 2 | 58 | 6 | 36 |
1 mol % AsPh3 and 100 mol % LiCl when specified.
The conversions are quantitative for entries 1–4, with high selectivity toward heterocoupling, over the homocoupling of the electrophile (92:8 or 91:9 by GC–MS). Since in this case, 2 only acts as a precursor of the Pd0 species and the oxidative addition is fast, the effect of AsPh3 is negligible (entry 1 vs entry 2). The effect of LiCl is unnoticed with 10 mol % of the catalyst (entry 2 vs 3) and becomes evident only with 2 mol % of the catalyst in the absence of LiCl protection (entry 5 vs 4). Needless to say, this method can be applied to the synthesis of symmetric dialkynes if using reagents with Alk = Alk′.
Conclusions
Our study demonstrates that catalytic cycle II in Scheme 1, so far accepted to be a mechanistic exception operating in Stille reactions with alkynylstannanes, must be discarded as such because reacting [Pd(Alk)(SnBu3)(PN)] (2) with ArF–I, it mainly produces ArF–SnBu3 instead of Alk–ArF. However, an alternative Stille cycle starting with the transposed products ArF–SnBu3 + Alk–X as reagents (Scheme 6) may also form Alk–ArF in moderate to high yield depending on the exact nature of Alk–X (X = I, Cl). Remarkably, the direct Stille cycle I (Scheme 1) starting with Ar–I, Alk–SnBu3, and Pd0(PN) can be made preferred by preventing the formation of undesired Pd–Sn species using special protocols. These are absolutely necessary for fluorinated aryls: (i) the addition of substoichiometric percentages of AsPh3 (AsPh3: Pd = 1:10) to give Pd0(PN)(AsPh3) blocks the formation of 2, making the direct Stille catalysis more competitive; (ii) the addition of stoichiometric LiCl relative to the Sn reagent (Cl/Sn = 1:1) hinders the formation of [PdI(SnBu3)(PN)] (4), a Pd trap. The use of the two additives brings the catalytic results of aryl fluorinated alkynes and, of course, those of conventional aryls, close to quantitative; (iii) a more radical solution to the problems in catalysis associated with the use of alkynylstannanes is to avoid its use, employing instead the transposed combination of reagents. Moreover, with Alk–Cl as the electrophile (e.g., Alk–Cl + ArF–SnBu3), this method skips the need for LiCl. This procedure works great with just a minimal substoichiometric percentage of AsPh3 as an additive (2 mol % of the Pd catalyst and 0.2 mol % of AsPh3) and even without AsPh3 in some cases. In summary, the classic Stille catalysis rules the Ar–Alk couplings, provided that the particular idiosyncrasy of alkynylstannanes as potential oxidative addition reagents is understood and accordingly dealt with.
The mechanisms discussed in this study for ArF reagents are shared by the conventional aryls but only produce very problematic results when fluorinated aryls are involved due to the more challenging oxidative addition of ArF–X electrophiles (in the direct Stille reaction) and the very low nucleophilicity of ArF–SnBu3 nucleophiles (in the transposed Stille reaction), compared to their congeners with conventional aryls.
In this general study, the reaction temperatures and times or the minimum percentages of catalysts or additives have not been exhaustively optimized and there is space for further improvement in specific cases. The problems and solutions in this work could be applied to make other R–Alk Stille couplings feasible, as shown for the efficient synthesis of unsymmetrical 1,3-diynes. We wish to remark on the splendid catalytic effect of AsPh3 which, in substoichiometric amounts relative to the metal catalyst, precludes the formation of undesired intermediates in kinetically efficient concentrations. In this respect, we refer the reader to a related case (different in its intimate details) occurring in gold catalysis.26
Acknowledgments
We thank the Spanish MINECO (project PID2020-118547GB-I00) and the Junta de Castilla y León (project VA224P20) for financial support. We acknowledge the DJEI/DES/SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support. M.N.P.-D is thankful for the funding provided by the Irish Research Council (GOIPD/2020/701) and the University of Valladolid (Margarita Salas program, ref. CONVREC-2021-221).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c10842.
Synthesis and full characterization of the metal complexes, NMR spectra (1H, 13C, 19F, 31P, and 119Sn), kinetic and microkinetic details, DFT section, and catalytic experiments (PDF)
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- For reviews, see:; a Espinet P.; Echavarren A. M. The mechanisms of the Stille reaction. Angew. Chem., Int. Ed. 2004, 43, 4704–4734. 10.1002/anie.200300638. [DOI] [PubMed] [Google Scholar]; b Cordovilla C.; Bartolomé C.; Martínez-Ilarduya J. M.; Espinet P. The Stille reaction, 38 years later. ACS Catal. 2015, 5, 3040–3053. 10.1021/acscatal.5b00448. [DOI] [Google Scholar]
- a Casado A. L.; Espinet P. Mechanism of the Stille Reaction. 1. The Transmetalation Step. Coupling of R1I and R2SnBu3 Catalyzed by trans-[PdR1IL2] (R1 = C6Cl2F3; R2 = Vinyl, 4-Methoxyphenyl; L = AsPh3). J. Am. Chem. Soc. 1998, 120, 8978–8985. 10.1021/ja9742388. [DOI] [Google Scholar]; b Casado A. L.; Espinet P.; Gallego A. M. Mechanism of the Stille reaction. 2. Couplings of aryl triflates with vinyltributyltin. Observation of intermediates. A more comprehensive scheme. J. Am. Chem. Soc. 2000, 122, 11771–11782. 10.1021/ja001511o. [DOI] [Google Scholar]; c Casado A. L.; Espinet P.; Gallego A. M.; Martínez-Ilarduya J. M. Snapshots of a Stille reaction. Chem. Commun. 2001, 339–340. 10.1039/b008811k. [DOI] [Google Scholar]
- Although not of general knowledge at that time, the in situ formation of their catalyst from (μ-Cl)2[Pd(allyl)]2 produces unsaturated (allyl–alkynyl) byproducts that can facilitate the Ar–Alk reductive elimination step:; a Albéniz A. C.; Espinet P.; Martín-Ruiz B. The Pd-Catalyzed Coupling of Allyl Halides and Tin Aryls: Why the Catalytic Reaction Works and the Stoichiometric Reaction Does Not. Chem.—Eur. J. 2001, 7, 2481–2489. . [DOI] [PubMed] [Google Scholar]; b Pérez-Rodríguez M.; Braga A. A. C.; Garcia-Melchor M.; Pérez-Temprano M. H.; Casares J. A.; Ujaque G.; de Lera A. R.; Álvarez R.; Maseras F.; Espinet P. C–C Reductive Elimination in Palladium Complexes, and the Role of Coupling Additives. A DFT Study Supported by Experiment. J. Am. Chem. Soc. 2009, 131, 3650–3657. 10.1021/ja808036j. [DOI] [PubMed] [Google Scholar]
- Shirakawa E.; Yoshida H.; Hiyama T. On the catalytic cycle of the palladium-catalyzed cross-coupling reaction of alkynylstannane with aryl iodide. Tetrahedron Lett. 1997, 38, 5177–5180. 10.1016/s0040-4039(97)01121-0. [DOI] [Google Scholar]
- Shirakawa E.; Hiyama T. The palladium-iminophosphine catalyst for the reactions of organostannanes. J. Organomet. Chem. 1999, 576, 169–178. 10.1016/s0022-328x(98)01056-0. [DOI] [Google Scholar]
- The carbostannylation of alkynes does occur via alkyne insertion in these [Pd(Alk)(SnBu3)(PN)] species. See:Shirakawa E.; Yoshida H.; Kurahashi T.; Nakao Y.; Hiyama T. Carbostannylation of Alkynes Catalyzed by an Iminophosphine–Palladium Complex. J. Am. Chem. Soc. 1998, 120, 2975–2976. 10.1021/ja974206k. [DOI] [Google Scholar]
- They use η2-(dimethyl fumarate)(iminophosphine)palladium (0) complexes as catalysts, which do not undergo oxidative addition of the Sn–Alk bond. Consequently, the hypothetical feasibility of cycle II could not be investigated and there is no explicit opinion about its operativity or nonoperativity:; a Crociani B.; Antonaroli S.; Beghetto V.; Matteoli U.; Scrivanti A. Mechanistic study on the cross-coupling of alkynyl stannanes with aryl iodides catalyzed by η2-(dimethyl fumarate)palladium(0) complexes with iminophosphine ligands. Dalton Trans. 2003, 2194–2202. 10.1039/B300020F. [DOI] [Google Scholar]; b Crociani B.; Antonaroli S.; Canovese L.; Uguagliati P.; Visentin F. Kinetic Studies of the Oxidative Addition and Transmetallation Steps Involved in the Cross-Coupling of Alkynyl Stannanes with Aryl Iodides Catalysed by η2-(Dimethyl fumarate)(iminophosphane)palladium(0) Complexes. Eur. J. Inorg. Chem. 2004, 2004, 732–742. 10.1002/ejic.200300376. [DOI] [Google Scholar]
- Ponce-de-León J.; Espinet P. Selective synthesis of fluorinated biaryls by [MCl2(PhPEWO-F)] (M = Ni, Pd) catalysed Negishi cross-coupling. Chem. Commun. 2021, 57, 10875–10878. 10.1039/D1CC04915A. [DOI] [PubMed] [Google Scholar]
- Pérez-Temprano M. H.; Gallego A. M.; Casares J. A.; Espinet P. Stille Coupling of Alkynyl Stannane and Aryl Iodide, a Many-Pathways Reaction: The Importance of Isomerization. Organometallics 2011, 30, 611–617. 10.1021/om100978w. [DOI] [Google Scholar]
- Gallego A. M.; Peñas-Defrutos M. N.; Marcos-Ayuso G.; Martin-Álvarez J. M.; Martínez-Ilarduya J. M.; Espinet P. Experimental study of speciation and mechanistic implications when using chelating ligands in aryl-alkynyl Stille coupling. Dalton Trans. 2020, 49, 11336–11345. 10.1039/d0dt02335c. [DOI] [PubMed] [Google Scholar]
- Other examples of Pd–Sn bonded structures in:; a Cabon Y.; Reboule I.; Gebbink R. J. M. K.; Deelman B.-J. Oxidative addition of Sn-C bonds on Palladium(0): identification of palladium-stannyl species and a facile synthetic route to diphosphinostannylene-palladium complexes. Organometallics 2010, 29, 5904–5911. 10.1021/om1007067. [DOI] [Google Scholar]; b Das D.; Pratihar S.; Roy S. Heterobimetallic Pd–Sn catalysis: a Suzuki, tandem ring-closing sequence toward indeno[2,1-b]thiophenes and indeno[2,1-b]indoles. Org. Lett. 2012, 14, 4870–4873. 10.1021/ol3021995. [DOI] [PubMed] [Google Scholar]
- Scheme 4 shows also that in reaction B, [Pd(Alk)I(PN)] (3) is generated, which eventually produces variable amounts of Alk–Alk by reaction with AlkSnBu3 and the catalytically inactive [PdI2(PN)]. See the Supporting Information for the complete kinetic model and characterization details.
- Hoops S.; Sahle S.; Gauges R.; Lee C.; Pahle J.; Simus N.; Singhal M.; Xu L.; Mendes P.; Kummer U. COPASI--a Complex Pathway Simulator. Bioinformatics 2006, 22, 3067–3074. 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- All the computed structures can be found here: http://dx.doi.org/10.19061/iochem-bd-6-102. It is worth commenting that TS1 displays a considerable elongation of the Pd–N bond. This distortion may not be accessible to a strongly chelating ligand, dppe, for instance. Thus, the ″special″ behavior found for these PN ligands is associated with their hemilabile character and might be found also in other hemilabile ligands. See for instance:; a Slone C. S.; Weinberger D. A.; Mirkin C. A. The Transition Metal Coordination Chemistry of Hemilabile Ligands. Prog. Inorg. Chem. 1999, 48, 233–350. 10.1002/9780470166499.ch3. [DOI] [Google Scholar]; b Braunstein P.; Naud F. Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems. Angew. Chem., Int. Ed. 2001, 40, 680–699. . [DOI] [PubMed] [Google Scholar]; c Stradiotto M.; Lundgren R. J.; Buchwald S. L.; Milstein D.. Ligand Design in Metal Chemistry: Reactivity and Catalysis; Wiley, 2016. [Google Scholar]
- The nucleophilicity of ArF in ArFSnBu3 is low, and effective participation of the transposed combination of reagents in Scheme 6 can require higher temperatures and concentrations than for conventional arylstannanes.
- For transmetalations hindered by addition of ligand, see:; a Gazvoda M.; Virant M.; Pinter B.; Košmrlj J. Mechanism of copper-free Sonogashira reaction operates through palladium-palladium transmetallation. Nat. Commun. 2018, 9, 4814–4822. 10.1038/s41467-018-07081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pérez-Temprano M. H.; Casares J. A.; de Lera A. R.; Álvarez R.; Espinet P. Strong metallophillic interactions in the Palladium arylation by gold aryls. Angew. Chem., Int. Ed. 2012, 51, 4917–4920. 10.1002/anie.201108043. [DOI] [PubMed] [Google Scholar]
- For the positive effect of LiCl, see:; a del Pozo J.; Carrasco D.; Pérez-Temprano M. H.; García-Melchor M.; Álvarez R.; Casares J. A.; Espinet P. Stille coupling involving bulky groups feasible with gold cocatalyst. Angew. Chem., Int. Ed. 2013, 52, 2189–2193. For CsF see: 10.1002/anie.201209262. [DOI] [PubMed] [Google Scholar]; b Mee S. P. H.; Lee V.; Baldwin J. E. Stille Coupling Made Easier-The Synergic Effect of Copper(I) Salts and the Fluoride Ion. Angew. Chem., Int. Ed. 2004, 43, 1132–1136. For a review see: 10.1002/anie.200352979. [DOI] [PubMed] [Google Scholar]; c Eckert P.; Sharif S.; Organ M. G. Salt to taste: the critical roles played by inorganic salts in organozinc formation and in the Negishi reaction. Angew. Chem., Int. Ed. 2020, 60, 12224–12241. 10.1002/anie.202010917. [DOI] [PubMed] [Google Scholar]
- The oxidative addition starts by coordination of the aryl ring to Pd, and presumably, the lower the π-donor ability of the fluorinated aryl, the more thermodynamically unfavorable this initial step.
- It is remarkable and unexpected that C6F3Cl2–I works so much better than C6F5–I. While both aryls are electronically very similar in the C6-C1-C2 positions, the carbon atoms bonded to Cl must be substantially richer in electron density and the Cl atoms may also act as weak σ-donors. We hypothesize that these aspects may facilitate the aryl coordination for C6F3Cl2–I and its subsequent oxidative addition.
- Hansen T.; Sun X.; Dalla Tiezza M.; van Zeist W.-J.; Poater J.; Hamlin T. A.; Bickelhaupt F. M. C(spn)–X (n = 1–3) Bond Activation by Palladium. Chem.—Eur. J. 2022, 28, e202103953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAUTION: The reaction can be carried out in a Schlenk tube with Young's tap, which well supports the overpressure of THF (b. p. 66 °C) at 70 °C. Alternatively, dioxane can be used with a 95% yield.
- In fact, the catalysis does not work with strong P-P chelating ligands such as dppe.
- Gioria E.; del Pozo J.; Lledós A.; Espinet P. Understanding the Use of Phosphine-(EWO) Ligands in Negishi Cross-Coupling: Experimental and Density Functional Theory Mechanistic Study. Organometallics 2021, 40, 2272–2282. 10.1021/acs.organomet.1c00001. [DOI] [Google Scholar]
- Gioria E.; del Pozo J.; Martínez-Ilarduya J. M.; Espinet P. Promoting Difficult Carbon-Carbon Couplings: Which Ligand Does Best?. Angew. Chem., Int. Ed. 2016, 55, 13276–13280. 10.1002/anie.201607089. [DOI] [PubMed] [Google Scholar]
- For Pd-catalyzed examples of preparation of unsymmetrical 1,3-diynes, see:; a Shi W.; Luo Y.; Luo X.; Chao L.; Zhang H.; Wang J.; Lei A. Investigation of an Efficient Palladium-Catalyzed C(sp)–C(sp) Cross-Coupling Reaction Using Phosphine–Olefin Ligand: Application and Mechanistic Aspects. J. Am. Chem. Soc. 2008, 130, 14713–14720. 10.1021/ja8049436. [DOI] [PubMed] [Google Scholar]; b Chinchilla F.; Nájera C. Chemicals from Alkynes with Palladium Catalysts. Chem. Rev. 2014, 114, 1783–1826. 10.1021/cr400133p. [DOI] [PubMed] [Google Scholar]; c Toledo A.; Funes-Ardoiz I.; Maseras F.; Albéniz A. C. Palladium-Catalyzed Aerobic Homocoupling of Alkynes: Full Mechanistic Characterization of a More Complex Oxidase-Type Behavior. ACS Catal. 2018, 8, 7495–7506. 10.1021/acscatal.8b01540. [DOI] [Google Scholar]
- Bartolomé C.; Ramiro Z.; Peñas-Defrutos M. N.; Espinet P. Some Singular Features of Gold Catalysis: Protection of Gold(I) Catalysts by Substoichiometric Agents and Associated Phenomena. ACS Catal. 2016, 6, 6537–6545. 10.1021/acscatal.6b01825. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.