
Fig. 1: Three families segregating recessive loss-of-function DPP9 germline variants.
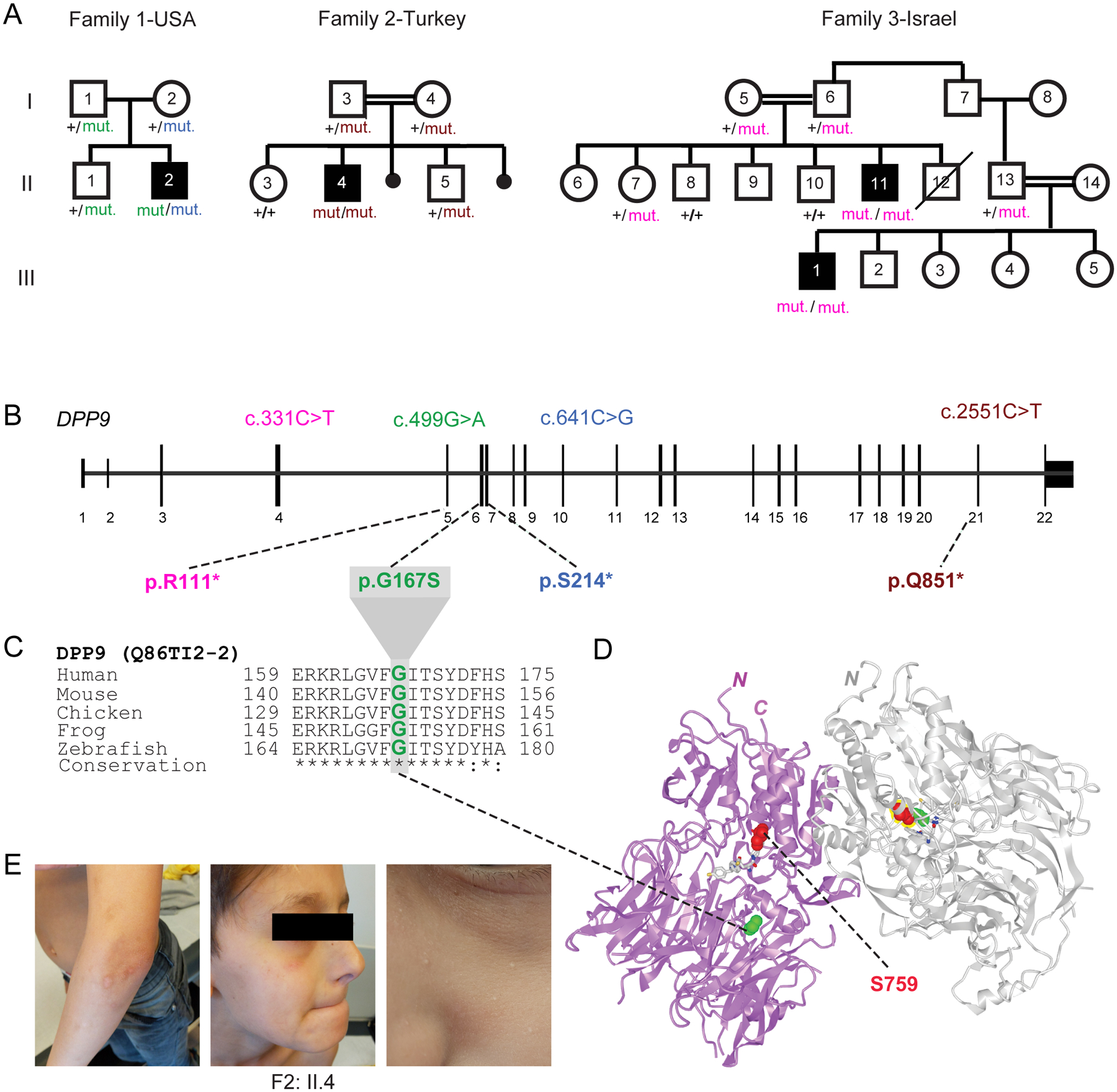
(A) Pedigrees of three families harboring germline DPP9 mutations. Proband from Family 1 (II.2, USA) is compound heterozygous, with paternal p.G167S and maternal p.S214* germline mutations. Proband from Family 2 (II.4, Turkey) carries a homozygous recessive p.Q851* mutation. Affected cousins from Family 3 (II.11 and III:1, Israel) carry a homozygous recessive p.R111* variant.
(B) Exon-intron genomic organization of DPP9 with positions of identified germline mutations.
(C) Phylogenetic conservation of the Gly167 residue, which is mutated in Family 1, across vertebrate species.
(D) Protein structure of a DPP9 homodimer (PDB ID: 6EOR) depicting location of p.G167 on the periphery of the substrate binding site and p.S759 in the catalytic residue.
(E) Photographs of proband from Family 2 (II.4) depicting recurrent HSV facial lesions, milia, and skin pigmentation anomalies on his arms.