

Abstract
Background
Previous studies have revealed the critical role of transglutaminase 2 (TGM2) as a potential therapeutic target in cancers, but the oncogenic roles and underlying mechanisms of TGM2 in gastric cancer (GC) are not fully understood. In this study, we examined the role and potential mechanism of TGM2 in GC.
Methods
Western blotting, immunohistochemistry, CCK8, colony formation and transwell assays were used to measure TGM2 expression in the GC cells and tissues and to examine the in vitro role of TGM2 in GC. Xenograft and in vivo metastasis experiments were performed to examine the in vivo role of TGM2 in GC. Gene set enrichment analysis, quantitative PCR and western blotting were conducted to screen for potential TGM2 targets involved in GC. Gain/loss‐of‐function and rescue experiments were conducted to detect the biological roles of STAT1 in GC cells in the context of TGM2. Co‐immunoprecipitation, mass spectrometry, quantitative PCR and western blotting were conducted to identify STAT1‐interacting proteins and elucidate their regulatory mechanisms. Mutations in TGM2 and two molecules (ZM39923 and A23187) were used to identify the enzymatic activity of TGM2 involved in the malignant progression of GC and elucidate the underlying mechanism.
Results
In this study, we demonstrated elevated TGM2 expression in the GC tissues, which closely related to pathological grade, and predicted poor survival in patients with GC. TGM2 overexpression or knockdown promoted (and inhibited) cell proliferation, migration, and invasion, which were reversed by STAT1 knockdown or overexpression. Further studies showed that TGM2 promoted GC progression by inhibiting STAT1 ubiquitination/degradation. Then, tripartite motif‐containing protein 21 (TRIM21) was identified as a ubiquitin E3 ligase of STAT1 in GC. TGM2 maintained STAT1 stability by facilitating the dissociation of TRIM21 and STAT1 with GTP‐binding enzymatic activity. A23187 abolished the role of TGM2 in STAT1 and reversed the pro‐tumor role of TGM2 in vitro and in vivo.
Conclusions
This study revealed a critical role and regulatory mechanism of TGM2 on STAT1 in GC and highlighted the potential of TGM2 as a therapeutic target, which elucidates the development of medicine or strategies by regulating the GTP‐binding activity of TGM2 in GC.
Keywords: TGM2, STAT1, TRIM21, ubiquitination, degradation, gastric cancer
List of Abbreviations
- GC
gastric cancer
- TGM2
transglutaminase 2
- TRIM21
tripartite motif‐containing protein 21
- IFN
interferon
- FBS
fetal bovine serum
- qPCR
quantitative polymerase chain reaction
- ELISA
enzyme‐linked immunosorbent assay
- EBV
epstein‐barr virus
- SDS‐PAGE
Sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- PVDF
polyvinylidene difluoride
- GSEA
gene set enrichment analysis
- Co‐IP
co‐immunoprecipitation
- MS
mass spectrometry
- NC
negative control
- ECM
extracellular matrix
- FBS
fetal bovine serum
- TMA
tissue microarray
- MOI
multiplicity of infection
- PFA
paraformaldehyde
- IRS
immunoreactive score
- IHC
immunohistochemistry
- H&E
hematoxylin and eosin
- CCK8
cell counting kit‐8
- PBS
phosphate‐buffered saline
- TBST
tris‐buffered saline containing 0.1% Tween
- TCGA
the Cancer Genome Atlas
- CIN
chromosomal instability
- GS
genomically stable
- MSI
microsatellite instable
- OS
overall survival
- EMT
Epithelial‐mesenchymal transition
- GTP
guanosine triphosphate
- ATP
adenosine triphosphate
1. BACKGROUND
According to the GLOBOCAN database, gastric cancer (GC) is the fifth most common malignant tumor and the fourth leading cause of cancer‐related mortality worldwide [1]. Over the past 35 years, gastric cancer incidence and mortality rates have been growing very fast in China [2]. Despite many advances have been made in diagnostic approaches and surgical procedures, the high incidence of recurrence and metastasis results in poor 5‐year survival rate of advanced stages of GC [3, 4, 5, 6]. Heterogeneity is a major challenge in the development of effective therapeutic strategies against GC, and current treatment methods are insufficient for its management. Recently, precision medicine and targeted therapy have become topics of increasing interest [7]. However, only a limited number of functional targets and their underlying mechanisms have been identified in GC. Hence, it is important to elucidate the molecular mechanisms of GC development, to facilitate the identification of therapeutic targets for developing effective strategies.
Transglutaminase 2 (TGM2) is a multifunctional enzyme involved in extracellular matrix degradation, apoptosis, and signal transduction [8, 9]. Aberrant TGM2 expression has been observed in multiple types of cancer cells [10, 11, 12, 13, 14, 15, 16], including ovarian cancer, breast cancer, pancreatic cancer, glioma, melanoma, lung cancer, colon cancer, and leukemia. Extracellular pools of TGM2 cross‐link and bind to numerous components of the extracellular matrix (ECM), such as fibronectin, vitronectin, collagen, fibrin, laminin, osteonectin, and osteopontin [17], leading to the speculation that TGM2 functions as a suppressor of cellular invasion [18].
In addition to this hypothesized tumor suppressor activity, cytosolic TGM2 was found to be associated with poor drug response, increased metastatic potential, and poor patient survival [10, 11, 13‐15], indicating that cytosolic TGM2 is a potential therapeutic target in cancer. However, the oncogenic roles and potential mechanisms of TGM2 in GC remain unclear. In this study, we examined the role and potential mechanism of cytosolic TGM2 in GC cells.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
Human GC cell lines HGC27 and KATOIII were obtained from Zhong Qiao Xin Zhou Biotech (Shanghai, China) and human GC cell lines AGS, MKN45, NCI‐N87 and MKN28 were obtained from Cellcook Biotech (Guangzhou, China). HGC27, MKN28, KATOIII, MKN45 and NCI‐N87 were cultured in RPMI‐1640 medium (Wisent, Shanghai, China) containing 10% fetal bovine serum (FBS; Wisent, Nanjing, China) and 1% penicillin/streptomycin (ThermoFisher Scientific, Massachusetts, USA). AGS cells were cultured in an F‐12K medium (Wisent, Nanjing, China) supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were incubated at 37°C in a humidified atmosphere containing 5% CO2. The information on the GC cell lines is listed in Supplementary Table S1.
2.2. Tissue specimens and tissue microarray (TMA)
TMA of 90 pairs of tumorous and adjacent non‐tumorous human gastric tissues (2009.12‐2010.6) were purchased from Outdo Biotech (HStmA180Su09; Shanghai, China). Regular follow‐ups were conducted on 90 patients until June 2016. A total of 24 pairs of GC and adjacent normal tissues were collected from patients with GC who underwent surgery at the First Affiliated Hospital of Nanjing Medical University (Nanjing, Jiangsu, China). All samples were frozen in liquid nitrogen and stored at ‐80°C until use. Plasma samples from 24 patients with GC (same as above) and 8 healthy controls were collected from the First Affiliated Hospital of Nanjing Medical University. None of the patients with GC received preoperative chemotherapy or radiotherapy, and the diagnoses were validated through pathological analysis. This study was approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University, and consent was obtained from all the patients.
2.3. Bioinformatics
The Cancer Genome Atlas (TCGA, http://www.cancergenome.nih.gov/) [19] was used for gene set enrichment analysis (GSEA) to analyze the expression of TGM2 in patients with GC and the effect of TGM2 on clinical survival. The Kaplan‐Meier plotter (http://www.kmplot.com/) was used to analyze the effect of tripartite motif‐containing protein 21 (TRIM21) on clinical survival.
2.4. Enzyme‐linked immunosorbent assay (ELISA)
TGM2 can be secreted extracellularly; therefore, we employed ELISA to test the TGM2 level in the plasma of patients with GC, as well as the supernatants and lysates of GC cells. Samples were collected for ELISA according to the manufacturer's instructions (ThermoFisher Scientific, Massachusetts, USA). Briefly, the standard sample was diluted two‐fold from a starting concentration of 200 ng/mL to 3.125 ng/mL, and plasma samples were diluted 1:2 for use. Standard and plasma samples were added to a 96‐well plate (100 μL/well) and incubated at 37°C for 90 min. After washing five times, biotin‐labeled secondary antibodies were added to the wells (100 μL/well) at a dilution of 1:1000 and incubated at 37°C for 60 min. After washing five times, horseradish peroxidase (HRP)‐conjugated streptavidin was added to the well (100 μL/well) at a dilution of 1:1000 and incubated at 37°C for 60 min. After five washes, 3,3′,5,5′‐tetramethylbenzidine coloring solution was added to the well (100 μL/well), developed for 5 min, and then stopped with stopping solution (50 μL/well). The plates were analyzed using a microplate reader at an OD450.
2.5. RNA isolation, reverse transcription, and quantitative PCR (qPCR)
Total RNA was extracted from tissues and cells using TRIzol reagent (ThermoFisher Scientific, Massachusetts, USA). Further, 500 ng of total RNA was reverse‐transcribed to cDNA in a final volume of 10 μL using the TRUEscriptRT kit (Proteinbio, Nanjing, China). Specific primers were synthesized commercially (Proteinbio, Nanjing, China), and qPCR was performed using Universal SYBR Green qPCR Supermix (US EVERBRIGHT, Suzhou, China) and a 7500 Real‐time PCR System (Applied Biosystems, Massachusetts, USA). The relative mRNA expression was normalized to that of β‐actin and calculated by the 2−ΔΔCT method. Each qPCR was performed in triplicates. The PCR primer sequences are listed in Supplementary Table S2.
2.6. Preparation of stable cells
TGM2‐overexpressing and TGM2 S171E‐overexpressing lentiviruses were constructed by GenePharma Biotechnology Co. Ltd (Shanghai, China). TGM2‐shRNA plasmids (shRNA‐1:ACAGCAACCTTCTCATCGAGT, shRNA‐2: TGAGAAATACCGTGACTGCCT, shRNA‐3: CCACCCACCATATTGTTTGAT) were constructed by Proteinbio Co. Ltd (Nanjing, China). Luciferase lentiviruses were constructed by Biosciences Inc. (Shanghai, China). Briefly, the cells (2 × 104) were seeded in a 24‐well plate until they reached ∼80% confluence after 24 h of culture. Cells were infected with lentiviral vectors at a multiplicity of infection of 20 or transfected with 5 μg of plasmids. After 48 h incubation, 0.5 μg/mL puromycin (MCE, New Jersey, USA) or 0.4 μg/mL G418 (MCE, New Jersey, USA) was added to the well based on the antibiotic resistance genes on the vectors. Then, the cells were incubated for 7 days with puromycin or for 14 days with G418 for stable cell line selection.
2.7. RNA interference and plasmid transfection
For RNA interference, small interfering RNAs (siRNAs) targeting STAT1 and TRIM21 were constructed by GenePharma Biotechnology Co. Ltd (Shanghai, China). Briefly, the cells (6 × 105) were seeded in a 6‐well plate and reached ∼80% confluence after 24 h of culture. A mixture of 50 nmol/L siRNA and 7.5 μL of Lipofectamine 3000 (ThermoFisher Scientific, Massachusetts, USA) was incubated at room temperature for 20 min and then added to the cells. Cells were collected after 48 h of culture.
For plasmid transfection, plasmids encoding FLAG‐coupled STAT1(STAT1‐FLAG), FLAG‐coupled TRIM21 (TRIM21‐FLAG), FLAG‐coupled TRIM21 with ZF_RING domain deleted (TRIM21_△RING‐FLAG), TGM2, STAT1, HA‐coupled ubiquitin (Ub‐HA) and relatively empty vectors were constructed by Miaolingbio (Wuhan, China). Briefly, the cells (6 × 105) were seeded in a 6‐well plate and reached ∼80% confluence after 24 h of culture. A mixture of 15 μg plasmid and 7.5 μL of Lipofectamine 3000 was incubated at room temperature for 20 min and then added to the cells. Cells were collected after 48 h of culture. The information on siRNA and plasmids used in this study is listed in Supplementary Table S3 and Supplementary Table S4, respectively.
2.8. Immunofluorescence assay
The cells were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature. Then, the cells were incubated with a blocking solution (Beyotime, Haimen, China) for 1 h at room temperature. Next, the cells were incubated with the primary antibody overnight at 4°C. The next day, the cells were washed three times with washing buffer (Beyotime, Haimen, China) at room temperature. They were then incubated with the secondary antibody (Abclonal, Wuhan, China) at a dilution of 1:150 in the dark for 1 h at room temperature. Finally, the cells were washed three times with washing buffer (Beyotime, Haimen, China) and counterstained using DAPI (Beyotime, Haimen, China). Cells were stored in the dark at 4°C until further investigation. If needed, protein expression and colocalization were analyzed quantitatively using the ImageJ software (National Institutes of Health, Bethesda, USA). The antibodies used in this study are listed in Supplementary Table S5.
2.9. Immunohistochemical and hematoxylin and eosin staining
Immunohistochemistry (IHC) was performed to determine TGM2 levels in the TMA. Briefly, the paraffin‐embedded section was de‐waxed in xylene and rehydrated in a graded alcohol series. Heat‐activated antigen retrieval was performed using a sodium citrate buffer (10 mmol/L, pH 6.0). The section was heated using the microwave method for 20 min. Endogenous peroxidase activity was suppressed by exposure to 3% hydrogen peroxide for 10 min. The section was then blocked with 5% BSA and was incubated with TGM2 antibody (Abcam, New York, USA) at a dilution of 1:50 at 37°C for 1 h, which was followed by the incubation with HRP‐conjugated secondary antibody (Abcam, New York, USA) at a dilution of 1:2000 at 37°C for 20 min. The section was visualized using DAB and counterstained with hematoxylin for microscopic examination.
Tumor tissues and lung tissues from mice were stained with hematoxylin and eosin (H&E) staining for pathological evaluation. Briefly, paraffin‐embedded sections were de‐waxed in xylene and rehydrated in a graded alcohol series. Sections were incubated with hematoxylin for 5 min at room temperature, and then moved into water to wash away floating color for 2 min. Sections were moved into a differentiation solution (1% hydrochloric acid alcohol) for a few seconds to 30 seconds. Sections were washed in water for 30‐60 min and then dipped into eosin for 2 min. The sections were then washed in water for further investigation.
The immunoreactive score (IRS) was used for expression assessment. Staining intensity was graded as 0, 1, 2, and 3 (no staining, weak, moderate and strong staining, respectively). The proportion of positive cells was scored as 1, 2, or 3 (<10%, 10%‐50%, or >50%, respectively). The following formula combined the two scores: IRS = positive rate score × intensity score.
2.10. Cell counting kit‐8 assay
Cell proliferation was detected using the Cell counting kit‐8 (CCK8) kit (MCE, New Jersey, USA) according to the manufacturer's instructions. Briefly, the cells were seeded in 96‐well plates (2000 cells/well), inoculated with complete medium and cultured for 96 h; 10 µL of CCK8 reagent and 90 µL of complete medium were mixed and added to each well every 24 h, and incubated at 37°C for 2 h. The plates were analyzed using a microplate reader at an OD450.
2.11. Colony formation assay
Cells were seeded in six‐well plates (500 cells/well) and cultured in a complete medium for 10‐14 days. The colonies were fixed with 4% PFA at room temperature for 15 min and stained with 0.1% crystal violet solution for 30 min.
2.12. Wound‐healing assay
The cells were seeded in a 6‐well plate (6 × 105 cells/well). After the cells reached confluence, the monolayer was wounded with a 200 µL pipette tip, and detached cells were removed with PBS. The cells were then cultured in a serum‐free medium to inhibit proliferation. Images were taken at 0 and 24 h, and the area of the wound was measured using ImageJ software. All experiments were performed in triplicates.
2.13. Transwell assay
Transwell assays were performed to assess the migration and invasion abilities of cells. For the migration experiment, 3 × 104 cells suspended in serum‐free medium were added to the upper chamber, and 10% FBS medium (750 µL) was added to the bottom chamber to induce cell migration to the other side of the membrane. After 36 h of culturing, the chamber was removed and fixed with 4% PFA for 30 min. The upper cells were wiped away with cotton swabs and stained with 0.1% crystal violet solution for 20 min. For the invasion experiment, Matrigel (BD Bioscience, San Diego, CA, USA) was added to the upper chamber at a dilution of 1:8. The treatment was the same as described above. The cells in five random fields of view were counted at 100× magnification. All experiments were performed in triplicates.
2.14. Flow cytometric analysis of cell apoptosis
Apoptosis was detected using an Annexin V‐Alexa Fluor647 kit according to the manufacturer's instructions (US Everbright, Suzhou, China). Briefly, the cells (6 × 105) were seeded in 6‐well plates. After 24 h culture, the cells were collected, resuspended in buffer, and stained with Annexin V‐Alexa Fluor647 (50 µg/mL) and propidium iodide (10 µg/mL) in the dark for 15 min before being subjected to a flow cytometer (LSR, BD Biosciences, San Diego, CA, USA).
2.15. Protein isolation and western blotting
GC cells and tissue samples were lysed in radioimmunoprecipitation assay lysis buffer (Beyotime, Haimen, China). Protein concentrations were measured using an enhanced BCA protein assay kit (Beyotime, Haimen, China). The proteins were subjected to 10% or 12.5% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Massachusetts, USA). The membranes were then blocked with blocking buffer (Beyotime, Haimen China) for 2 h at room temperature and incubated with primary antibodies overnight at 4°C. After washing with Tris‐buffered saline containing 0.1% Tween (TBST) three times (15 min/time), the membranes were incubated with HRP‐conjugated secondary antibodies for 1 h at room temperature. The membranes were washed again, and the protein expression levels were visualized with a Super ECL chemiluminescent substrate kit (US Everbright, Suzhou, China). If needed, the relative protein expression was measured using ImageJ software (National Institutes of Health, Bethesda, USA). The antibodies used in this study are listed in Supplementary Table S5.
2.16. Co‐immunoprecipitation (Co‐IP) and mass spectrometry (MS)
Co‐IP and MS were used to examine proteins coupled to STAT1. The cells were transfected with STAT1‐FLAG and vector plasmids. After 48 h, cells (1 × 107) were washed in PBS twice, and then 500 µL of NP40 lysis buffer (Beyotime, Haimen, China) containing 1× Protease Inhibitor Cocktail (MCE, New Jersey, USA) was added to the cell lysate. One‐tenth of the supernatant was saved as an input. To the remaining supernatant, 40 µL anti‐FLAG M2 affinity gels (Sigma‐Aldrich, Saint Louis, USA) were added, and the samples were incubated at 4°C overnight. The beads were washed five times with TBS buffer, eluted by adding 80 µL SDS loading buffer and boiled. Protein samples were analyzed by MS (OE Biotech, Shanghai, China). Briefly, the lyophilized peptide fractions were resuspended in ddH2O containing 0.1% formic acid, and 2 µL aliquots were loaded into a nanoViper C18 (Acclaim PepMap 100, 75 µm × 2 cm) trap column. Online Chromatography separation was performed using the Easy nLC 1200 system (ThermoFisher, Massachusetts, USA). The trapping and desalting procedures were performed with a volume of 20 µL 100% solvent A (0.1% formic acid). An elution gradient of 5%–38% solvent B (80% acetonitrile, 0.1% formic acid) for 60 min was used on an analytical column (Acclaim PepMap RSLC; 75 µm × 25 cm; C18‐2 µm 100 Å). Data‐dependent acquisition mass spectrum techniques were used to acquire tandem MS data on a Thermo Fisher Q Exactive mass spectrometer (Thermo Fisher, Massachusetts, USA) fitted with a Nano Flex ion source. Data were acquired using an ion spray voltage of 1.9 kV, and an interface heater temperature of 275°C. For a full MS survey scan, the target value was 3 × 106 and the scan ranged from 350 to 2,000 m/z at a resolution of 70,000 and a maximum injection time of 100 ms. For the MS2 scan, only spectra with a charge state of 2‐5 were selected for fragmentation by high‐energy collision dissociation with a normalized collision energy of 28. The MS2 spectra were acquired in the ion trap in rapid mode with an AGC target of 8000 and a maximum injection time of 50 ms. Dynamic exclusion was set for 25 s. Data were processed using ProteomeDiscover software (version 2.1). The antibodies used in this study are listed in Supplementary Table S5.
2.17. Co‐IP and western blotting
Co‐immunoprecipitation and western blotting were used to examine the ubiquitin levels of STAT1. Cells were transfected with STAT1‐FLAG, Ub‐HA or vector plasmids. After 48 h, cells (1 × 107) were washed in PBS twice (before collection, cells were treated with MG132 (10 µmol/L, for ∼2 h), then incubated with 500 µL of NP40 lysis buffer containing 1× protease inhibitor cocktail. One‐tenth of the supernatant was saved as an input. To the remaining supernatant, 40 µL anti‐FLAG M2 affinity gels were added to the cell supernatant, and the samples were incubated at 4°C overnight. Beads were then washed five times with TBS before being eluted by adding 80 µL SDS loading buffer and boiled for western blotting.
Two‐way Co‐IP and western blotting were used to examine the binding between STAT1 and TRIM21. The cells were divided into two parts. One part of cells was transfected with the STAT1‐FLAG or vector plasmids. After 48h, cells (1 × 107) were washed in PBS twice and then incubated with 500 µL NP40 lysis buffer containing 1× protease inhibitor cocktail. One‐tenth of the supernatant was saved as an input. To the remaining supernatant, 40 µL anti‐FLAG M2 affinity gels were added to the cell supernatant, and the samples were incubated at 4°C overnight. Beads were then washed five times with TBS before being eluted by adding 80 µL SDS loading buffer and boiled for western blotting. The remaining cells were incubated with anti‐TRIM21 antibody or IgG control at 4°C overnight, then Protein A+G agarose beads (Beyotime, Haimen, China) was added, and incubated at 4°C for another 2∼4h. Beads were then washed five times with TBS before being eluted by adding 80 µL SDS loading buffer and boiled for western blotting. The procedure of western blotting was the same as above. If needed, the relative protein expression was measured using ImageJ software. The antibodies used in this study are listed in Supplementary Table S5.
2.18. Mouse xenograft model and in vivo metastasis assay
Four‐week‐old female BALB/c nude mice were purchased from the Laboratory Animal Center of Nanjing Medical University and housed in an SPF facility with a 12/12 h day/night cycle and free access to chow and water. This animal study was approved by the Ethics Committee of the Laboratory Animal Management of Nanjing Medical University. The animals were terminated if the body weight loss was more than 20%‐25% or the tumor weight was over 10% of the body weight. All animals were euthanized.
For in vivo tumor growth study, nude mice were inoculated subcutaneously into the flanks with 3 × 106 GC cells suspended in 0.1 mL PBS. Tumor volumes were calculated as 1/2 (length ×width2) once per week. After 4 weeks, xenograft tumors were harvested after intraperitoneal anesthesia with ketamine and promethazine, fixed in 4% PFA, paraffin‐embedded, and analyzed by H&E staining.
To study the effect of A23187 on in vivo tumor growth, nude mice were inoculated subcutaneously into flanks with 3 × 106 stably cells suspended in 0.1 mL PBS. After 1 week, pre‐established tumor xenografts were treated with dimethyl sulfoxide or A23187 (1.0 mg/kg, 2×/week × 3) (MCE, New Jersey, China). After another 3 weeks, xenograft tumors were collected after intraperitoneal anesthesia with ketamine and promethazine, fixed in 4% PFA, paraffin‐embedded and analyzed by H&E staining.
In the nude mouse lung metastasis model, 2 × 106 luciferase‐labeled GC cells in 0.1 mL PBS were injected into nude mice through their tail veins. Four weeks later, nude mice were injected with 100 mg/kg D‐luciferin (Goldbio, USA) intraperitoneally and subjected to bioluminescent scans using an IVIS 100 Imaging System (Xenogen, Alameda, California, USA) after inhalation anesthesia with isoflurane. The lungs were removed after intraperitoneal anesthesia with ketamine and promethazine, fixed in 4% PFA, embedded in paraffin, and analyzed by H&E staining.
2.19. Statistical analysis
Statistical analysis in the current study was performed using GraphPad Prism (version 6.0) and SPSS software (version 22.0). Quantitative data are presented as mean ± standard deviation. Differences in the mean between the two groups were analyzed using the Student's t‐test. Pearson's χ2‐test was used to analyze the associations between TGM2 expression and clinicopathological features. The Kaplan–Meier test was applied to calculate survival rates, and log‐rank tests were used to examine differences in survival rates between the two groups. All statistical tests were two‐tailed exact tests with a significance level of P < 0.05.
3. RESULTS
3.1. TGM2 was upregulated in GC and associated with GC progression
We analyzed TGM2 expression and its clinical significance in GC based on the TCGA dataset (375 GC tissues and 32 adjacent normal tissues). Overall, TGM2 mRNA expression was significantly higher in GC tissues than in the adjacent normal tissues (P = 0.001) (Figure 1A). In the subcategory analysis, TGM2 mRNA was significantly higher in the diffuse subtype than in the intestinal subtype (P = 0.038; Figure 1B). Epstein‐Barr Virus (EBV)‐positive GC tissues showed the highest TGM2 mRNA expression, which was higher than any other molecular subtype, including chromosomal instability (CIN) (P = 0.002), genomically stable (GS; P = 0.001), and microsatellite instability (MSI) (P < 0.001; Figure 1C). TGM2 mRNA was higher in grade 3 GC tissues than in grade 2 GC tissues (P < 0.001; Figure 1D). Furthermore, TGM2 mRNA showed the highest expression level in stage III GC tissues, which was higher than that in stage I tissues (P = 0.004; Figure 1E). We also found that EBV‐positive GC tissues expressed higher levels of TGM2 mRNA than EBV‐negative GC tissues (P < 0.001; Figure 1F), and H. pylori‐positive GC tissues had higher TGM2 mRNA expression than H. pylori‐negative GC tissues (P = 0.048; Figure 1G). Furthermore, survival analysis indicated that higher TGM2 expression was associated with poor overall survival (OS) in patients with GC (P = 0.009; Figure 1H).
FIGURE 1.
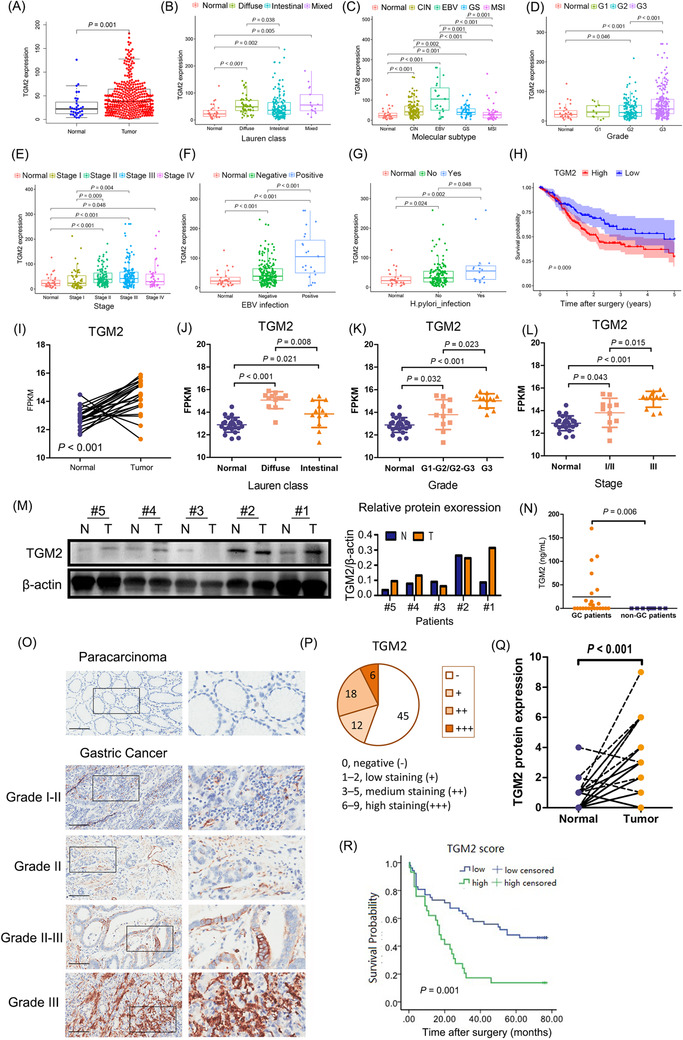
TGM2 was upregulated in GC and associated with GC progression. (A) TGM2 mRNA was significantly higher in GC tissues than the adjacent normal tissues (P = 0.001) based on the TCGA dataset (375 GC tissues and 32 adjacent normal tissues). (B) In the subcategory analysis, TGM2 mRNA was significantly higher in the diffuse subtype than in the intestinal subtype (P = 0.038). (C) In the subcategory analysis, EBV‐positive GC tissues showed the highest TGM2 mRNA expression, which was higher than any other molecular subtype, including CIN (P = 0.002), GS (P = 0.001) and MSI (P < 0.001). (D) In the subcategory analysis, TGM2 mRNA was higher in grade 3 GC tissues than in grade 2 GC tissues (P < 0.001). (E) In the subcategory analysis, TGM2 mRNA showed the highest expression level in stage III GC tissues, which was higher than that in stage I tissues (P = 0.004). (F) In the subcategory analysis, EBV‐positive GC tissues expressed higher levels of TGM2 mRNA than EBV‐negative GC tissues (P < 0.001). (G) In the subcategory analysis, H. pylori‐positive GC tissues had higher TGM2 mRNA expression than H. pylori‐negative GC tissues (P = 0.048). (H) Survival analysis based on the TCGA dataset indicated that higher TGM2 expression was associated with poor OS in patients with GC (P = 0.009). (I) RNA sequencing data of 24 pairs of GC tissues collected from our hospital were analyzed. And the results showed that TGM2 mRNA was generally higher in tumor tissues than in adjacent normal tissues (P < 0.001). (J) In the subcategory analysis, TGM2 mRNA was higher in the diffuse subtype than in the intestinal subtype (P = 0.008). (K) In the subcategory analysis, TGM2 mRNA was higher in grade 3 GC tissues than in grade1‐2/grade 2‐3 GC tissues (P = 0.023). (L) In the subcategory analysis, TGM2 mRNA was higher in stage III GC tissues than in stage I/II tissues (P = 0.015). (M) TGM2 protein expression was confirmed in five pairs of GC tissues by western blotting, which was selected based on TGM2 mRNA level (three pairs with a higher TGM2 mRNA in GC tissues than adjacent normal tissues, one pair with lower TGM2 mRNA in GC tissues than adjacent normal tissues, and one pair with a similar TGM2 mRNA level in GC tissues as adjacent normal tissues), the results showed that TGM2 protein expression pattern was in line with TGM2 mRNA pattern. Bar chart was the quantitative analysis of TGM2 protein. (N) TGM2 was detected in plasma samples from 24 patients with GC and 8 healthy participants by ELISA, and the results showed that TGM2 was detectable in 11 of 24 plasma samples from patients with GC but not in healthy plasma samples (P = 0.006) (O) TMA containing 90 pairs of GC tissues was used to analyze TGM2 protein expression by IHC. Representative micrographs of TGM2 protein expression in GC tissues and adjacent normal tissues in different pathological grades were displayed. Scale bar: 100 µm. (P) IHC assay showed that TGM2 expression was detectable in 36 out of 81 patients with GC, Circle chart summarized the numbers of patients with high/medium‐TGM2 expression and low/negative‐TGM2 expression. (Q) IHC assay showed that IRS was higher in GC tissues than in adjacent normal tissues (P < 0.001). (R) Totally, 81 out of 90 pairs of GC tissues in TMA were enrolled for analysis because of incomplete information, Kaplan‐Meier survival analysis indicated that higher TGM2 expression was associated with poor OS in patients with GC (P = 0.001). Abbreviations: GC, gastric cancer; TCGA, The database the Cancer Genome Atlas; TGM2 transglutaminase 2, ELISA enzyme‐linked immunosorbent assay; IHC, immunohistochemistry; IRS, immunoreactive score; TMA, tissue microarray; EBV, epstein‐barr virus; CIN, chromosomal instability; GS, genomically stable; MSI, microsatellite instable; OS, overall survival.
To verify the bioinformatics results, we analyzed the RNA sequencing data of 24 pairs of GC tissues collected at our hospital, and found that TGM2 mRNA was generally higher in tumor tissues than in the adjacent normal tissues (P <0.001) (Figure 1I), Then, the subcategory analysis showed that TGM2 mRNA was higher in the diffuse GC subtype than in the intestinal GC tissues (P = 0.008) (Figure 1J), TGM2 mRNA was higher in grade 3 GC tissues than in grade1‐2/grade 2‐3 GC tissues (P = 0.023) (Figure 1K), and TGM2 mRNA was higher in stage III GC tissues than in stage I/II GC tissues (P = 0.015) (Figure 1L). The results were consistent with those from the TCGA dataset. TGM2 protein expression was confirmed in five pairs of GC tissues, which were selected based on TGM2 mRNA level (three pairs with higher TGM2 mRNA in GC tissues than the adjacent normal tissues, one pair with lower TGM2 mRNA in GC tissues than the adjacent normal tissues, and one pair with similar TGM2 mRNA level in GC tissues as the adjacent normal tissues), and the results showed that TGM2 protein expression pattern was in line with TGM2 mRNA pattern (Figure 1M),. Moreover, TGM2 was detectable in 11 out of 24 plasma samples from patients with GC, but not in healthy plasma samples (P = 0.006) (Figure 1N), and further analysis indicated that there was no correlation between extracellular TGM2 and clinicopathological characteristics, including sex, pathological grade, lymph node metastasis, clinical stage and Lauren subtype (Table 1).
TABLE 1.
Association between extracellular TGM2 and clinicopathological characteristics in 24 patients with GC
| Extracellular TGM2 | ||||
|---|---|---|---|---|
| Characteristics | Positive | Negative | P value | |
| Total cases | 11 | 13 | N/A | |
| Sex (cases [%]) | 0.679 | |||
| Male | 8 (72.73) | 8 (61.54) | ||
| Female | 3 (27.27) | 5 (38.46) | ||
| Pathological grade (cases [%]) | 0.813 | |||
| G1‐G2/G2 | 3 (27.27) | 3 (23.08) | ||
| G2‐G3/G3 | 8 (72.73) | 10 (76.92) | ||
| Lymph node metastasis (cases [%]) | 0.239 | |||
| N0 | 6 (54.55) | 4 (30.77) | ||
| N1‐N3 | 5 (45.45) | 9 (69.23) | ||
| Clinical stage (cases [%]) | 0.239 | |||
| I‐II | 6 (54.55) | 4 (30.77) | ||
| III | 5 (45.45) | 9 (69.23) | ||
| Lauren subtype (cases [%]) | 0.682 | |||
| Intestinal | 5 (45.45) | 7 (53.85) | ||
| Diffuse | 6 (54.55) | 6 (46.15) | ||
Abbreviations: GC, gastric cancer; TGM2, transglutaminase 2; N/A, not applicable.
For TMA, 81 out of 90 pairs of tissues were enrolled for analysis because of incomplete information. As indicated in Figure 1O‐Q, IHC assays showed that TGM2 expression was detectable in 36 out of 81 GC tissues, and IRS was higher in GC tissues than in the adjacent normal tissues (P < 0.001). To investigate the clinical significance of TGM2 in GC, we classified the patients into TGM2 high/medium and TGM2 low/negative groups based on their expression values. TGM2 expression positively correlated with pathological grade (P = 0.043), tumor size (P = 0.047), and lymph node metastasis (P = 0.034) (Table 2). Kaplan–Meier survival curves revealed a negative association between TGM2 expression and OS (P = 0.001) (Figure 1R). These findings revealed that higher TGM2 expression indicated more advanced disease and poor OS in patients with GC.
TABLE 2.
Association between TGM2 protein expression and clinicopathological characteristics in 81 patients with GC
| TGM2 protein expression | ||||
|---|---|---|---|---|
| Characteristics | high/medium | low/negative | P value | |
| Total cases | 24 | 57 | N/A | |
| Sex (cases [%]) | 0.772 | |||
| Male | 18 (75.00) | 45 (78.95) | ||
| Female | 6 (25.00) | 12 (21.05) | ||
| Age (year, mean±SD) | 68.17 ± 11.17 | 65.84 ± 11.19 | 0.346 | |
| Pathological grade (cases [%]) | 0.043 | |||
| G1∼G2/G2 | 4 (16.67) | 19 (33.33) | ||
| G2∼G3/G3 | 20 (83.33) | 38 (66.67) | ||
| Tumor size (cm, mean±SD) | 6.55 ± 2.83 | 5.22 ± 2.33 | 0.047 | |
| Lymph node metastasis (cases [%]) | 0.034 | |||
| N0 | 3 (12.50) | 21 (36.84) | ||
| N1‐N3 | 21 (87.50) | 36 (63.16) | ||
| Clinical stage (cases [%]) | 0.355 | |||
| I | 1 (4.17) | 9 (15.79) | ||
| II | 6 (25.00) | 18 (31.58) | ||
| III | 16 (66.66) | 29 (50.88) | ||
| IV | 1 (4.17) | 1 (1.75) | ||
Abbreviations: GC, gsstric cancer; TGM2, transglutaminase 2; SD, standard deviation; N/A, not applicable.
3.2. TGM2 promoted GC malignant progression in vitro and in vivo
To further confirm the oncogenic properties of TGM2 in GC, we observed the biological function of TGM2 by infecting AGS and HGC27 cells with TGM2 overexpression lentivirus and transfected MKN28 cells with TGM2 shRNA plasmids after determining the expression levels of TGM2 in GC cells (Figure 2A). The efficiency of the gain/loss was assessed using western blotting and qPCR (Figure 2B‐C). And we further confirmed that the overexpression or knockdown caused changes in the expression of TGM2, mainly in cell lysates by ELISA (Figure 2D). We then observed that TGM2 overexpression promoted cell proliferation, and the downregulation of TGM2 led to significant inhibition of cell proliferation by colony formation and CCK8 assays (Figure 2E‐F). A flow cytometry assay indicated that TGM2 overexpression significantly decreased cell apoptosis rate. In contrast, the opposite result was observed after the silencing of TGM2 in GC cells (Figure 2G). In vivo, GC cells were inoculated subcutaneously into the flanks of nude mice. After 4 weeks, xenograft tumors were harvested for pathological evaluation and size and weight evaluation (Figure 2H). H&E staining confirmed the pathological features of the tumors (Figure 2I). And we observed that TGM2 overexpression in AGS and HGC27 cells increased subcutaneous tumor size and weight (Figure 2J‐L), indicating that TGM2 promoted GC cell proliferation.
FIGURE 2.
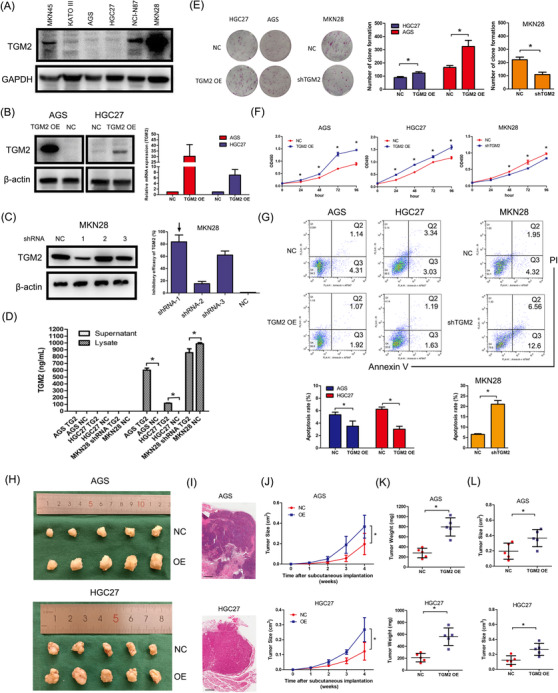
TGM2 promoted cell proliferation in GC. (A) TGM2 expression in GC cells was assessed using western‐blotting, and AGS, HGC27 and MKN28 cells were chosen for cell models based on TGM2 expression level, cell quality and cell biological features. (B) TGM2‐overexpressing GC cells were constructed using lentivirus infection in AGS and HGC27 cells, and the efficiency was assessed using western‐blotting and qPCR. (C) TGM2‐knocked down GC cell was constructed using plasmid transfection, and the efficiency was assessed using western blotting and qPCR. (D) TGM2 expression was detected in supernatants and lysates of TGM2‐overexpressing/TGM2‐knocked down GC cells by ELISA, and the results showed that TGM2 overexpression or knockdown caused changes in the expression of TGM2 mainly in cell lysates. *: P < 0.05. (E) The effect of TGM2 on cell proliferation was evaluated by colony formation assay. The results showed that TGM2 overexpression promoted cell proliferation in GC cells, and the downregulation of TGM2 led to significant inhibition of cell proliferation. Representative images of colony formation assays were displayed, and the bar charts showed the differences of clone number between TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells. *: P<0.05. (F) The effect of TGM2 on cell proliferation was evaluated by CCK8 assay. The results showed that TGM2 overexpression promoted cell proliferation in GC cells, and the downregulation of TGM2 led to significant inhibition of cell proliferation. *: P<0.05. (G) The effect of TGM2 on cell apoptosis was evaluated by flow cytometry. The results showed that TGM2 overexpression significantly decreased the rate of cell apoptosis. In contrast, the opposite result was observed after the silencing of TGM2 in GC cells. The bar charts showed the differences in apoptosis rates between TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells. *: P<0.05. (H) Mouse xenograft model was used to evaluate the effect of TGM2 on cell proliferation in vivo; images of transplanted subcutaneous tumors from mouse models were displayed. (I) Representative micrographs of transplanted subcutaneous tumors by H&E staining were displayed. Scale bar: 500 µm. (J) Tumor volume curve in mice was evaluated for 4 weeks since subcutaneous implantation. The results showed that TGM2 overexpression increased subcutaneous tumor growth in AGS and HGC27 cells. *: P<0.05. (K) Tumor weight in mice was evaluated. The results showed that TGM2 overexpression increased subcutaneous tumor growth in AGS and HGC27 cells. *: P<0.05. (L) Tumor volume in mice was evaluated. The results showed that TGM2 overexpression increased subcutaneous tumor growth in AGS and HGC27 cells. *: P<0.05. Abbreviations: GC gastric cancer, TGM2 transglutaminase 2, ELISA enzyme‐linked immunosorbent assay, H&E hematoxylin and eosin, NC negative control, OE overexpression, CCK8 cell counting kit‐8, qPCR quantitative polymerase chain reaction
To verify whether TGM2 affects the metastatic capability of GC cells, we performed wound‐healing and transwell assays. As illustrated in Figure 3A‐B, the overexpression of TGM2 markedly promoted cell migration and invasion, and more GC cells migrated or invaded the lower side of the membrane, whereas the knockdown of TGM2 had the opposite effect. As epithelial‐mesenchymal transition (EMT) is closely related to cell migration and invasion, we performed western blotting to examine the EMT markers. As shown in Figure 3C. TGM2 overexpression upregulated N‐cadherin (a mesenchymal marker) in AGS and HGC27 cells and vimentin (a mesenchymal marker) in HGC27 cells. The epithelial marker E‐cadherin was expressed at lower levels in these two cell types. In contrast, when TGM2 was knocked down in the MKN28 cells, N‐cadherin and vimentin were downregulated, whereas E‐cadherin expression did not change markedly. The above results demonstrate that TGM2 promotes the metastasis of GC cells, and EMT is likely to be involved in the positive effects of TGM2 on GC metastasis. Next, we examined the role of TGM2 in metastasis in vivo. In the tail vein metastasis model, we observed that the knockdown of TGM2 in MKN28 cells significantly reduced the luminescence density of lung‐metastatic lesions. However, overexpression of TGM2 in AGS cells significantly increased the luminescence density of lung‐metastatic lesions. (Figure 3D). Further, H&E staining validated the metastatic lesions in the lungs (Figure 3E). The in vivo results indicated that TGM2 promotes the metastatic capacity of GC cells. Considering all the evidence, we conclude that TGM2 promotes oncogenic behavior in GC.
FIGURE 3.
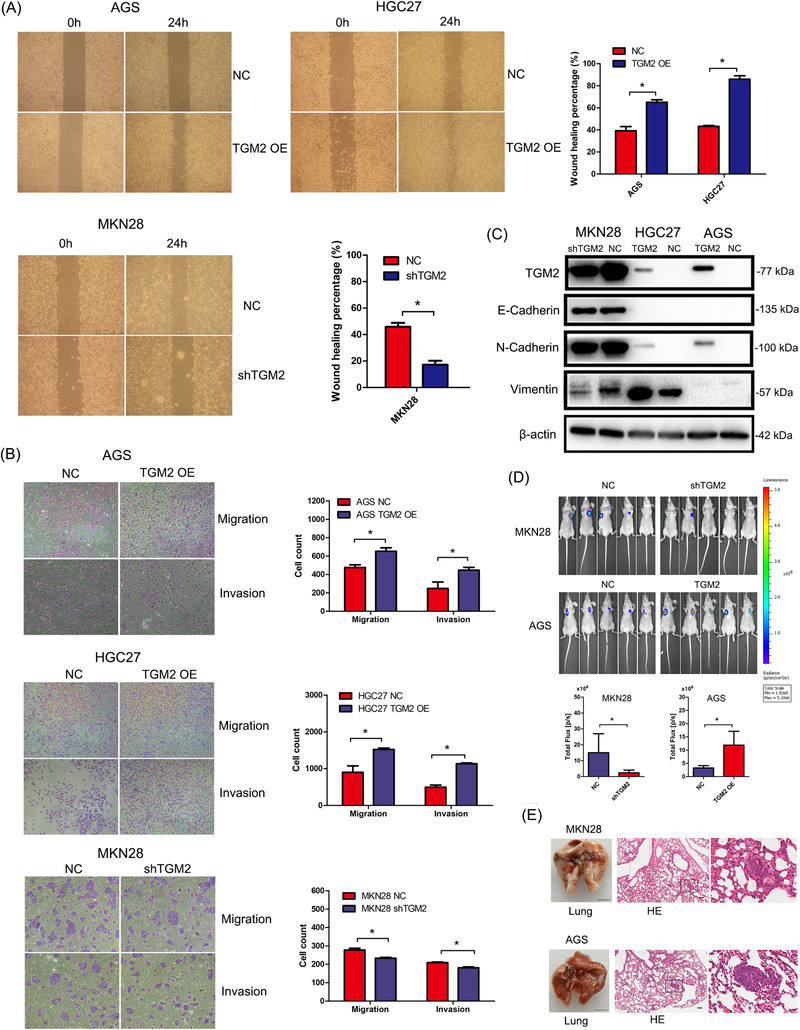
TGM2 promoted cell metastasis in GC. (A) The effect of TGM2 on cell migration was evaluated using wound‐healing assay, and the results showed that TGM2 overexpression markedly promoted cell migration, and more GC cells migrated to the lower side of the membrane, whereas the knockdown of TGM2 had the opposite effect. Representative micrographs of wound healing assay were displayed, the bar charts showed the differences of migratory area between TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells. *: P<0.05. (B) The effect of TGM2 on cell migration and invasion was evaluated using transwell assay, and the results showed that TGM2 overexpression markedly promoted cell migration and invasion, and more GC cells migrated/invaded to the lower side of the membrane, whereas the knockdown of TGM2 had the opposite effect. Representative micrographs of transwell assay were displayed. The bar charts showed the differences of migrated or invaded cell numbers between TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells. *: P<0.05. (C) The EMT markers were examined in TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells. Using western blotting. The results showed that TGM2 overexpression upregulated N‐cadherin (a mesenchymal marker) in AGS and HGC27 cells and vimentin (a mesenchymal marker) in HGC27 cells. The epithelial marker E‐cadherin was expressed at lower levels in these two cell types. In contrast, N‐cadherin and vimentin were downregulated when TGM2 was knocked down in MKN28 cells, whereas E‐cadherin expression did not change markedly. (D) The effect of TGM2 in metastasis in vivo was evaluated in BALB/c nude mice by injecting 2 × 106 luciferase‐labeled stably transfected GC cells through their tail veins. Four weeks later, nude mice were injected with 100 mg/kg D‐luciferin and subjected to bioluminescent scans using an IVIS 100 Imaging System. The results showed that the knockdown of TGM2 in MKN28 cells significantly reduced the luminescence density of lung‐metastatic lesions. However, TGM2 overexpression in AGS cells significantly increased the luminescence density of metastatic lung lesions. Representative images of lung‐metastatic lesions were displayed, the bar charts showed the luminescence density of lung‐metastatic lesions between TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells. *: P<0.05. (E) The lungs were removed, then fixed in 4% PFA and paraffin‐embedded for H&E staining. The results of H&E staining confirmed the histomorphology of the metastatic lesions in the lungs. Representative images of the lung were displayed. Scale bar: 0.5 cm. And the representative micrographs of cancerous lesions in the lungs detected by H&E staining were displayed. Scale bar: 100 µm. Abbreviations: GC, gastric cancer; TGM2, transglutaminase 2; H&E, hematoxylin and eosin; NC, negative control; OE, overexpression; PFA, paraformaldehyde.
3.3. TGM2 promoted GC development by regulating STAT1
To explore the mechanism underlying TGM2 in GC development, we conducted GSEA using a dataset (375 patients with GC) from TCGA and found that several cytokine and inflammatory pathways are involved in the role of TGM2 in GC (Figure 4A), including cytokine‐cytokine receptor interaction, toll‐like receptor signaling pathway, and nod‐like receptor signaling pathway. The enrichment plot of cytokine‐cytokine receptor interactions is shown in Figure 4B as an example. We then examined several inflammatory signaling molecules by western blotting and found that STAT1 and its activated form (pSTAT1) were both upregulated in TGM2‐overexpressing GC cells, and the opposite results were observed in TGM2‐knocked down GC cells (Figure 4C). We isolated the cytoplasmic and nuclear components from cell lysates and found that TGM2 overexpression increased the level of STAT1 and pSTAT1 in both the cytoplasm and nuclei of AGS and HGC27 cells. The opposite results were observed when TGM2 was knocked down in MKN28 cells. (Figure 4D). Furthermore, immunofluorescence showed that TGM2 was localized mainly in the cytoplasm of HGC27 and MKN28 cells and mainly in the nuclei of AGS cells. TGM2 overexpression upregulated STAT1 levels in the AGS and HGC27 cells, and the knockdown of TGM2 reduced STAT1 levels in the MKN28 cells. (Figure 4E). To determine whether TGM2 promoted GC development by regulating STAT1, we knocked down STAT1 in TGM2‐overexpressing cells using STAT1 siRNA or transfected TGM2‐knocked down cells with STAT1 plasmids (Figure 4F), and observed that the downregulation of STAT1 reversed the promoting effect of TGM2 on cell proliferation, migration, and invasion, and the upregulation of STAT1 rescued the inhibitory effect of TGM2 knockdown on cell proliferation, migration, and invasion (Figure 4G‐H). Altogether, these results indicate that TGM2 promotes cell proliferation and metastasis by regulating STAT1 in GC cells.
FIGURE 4.
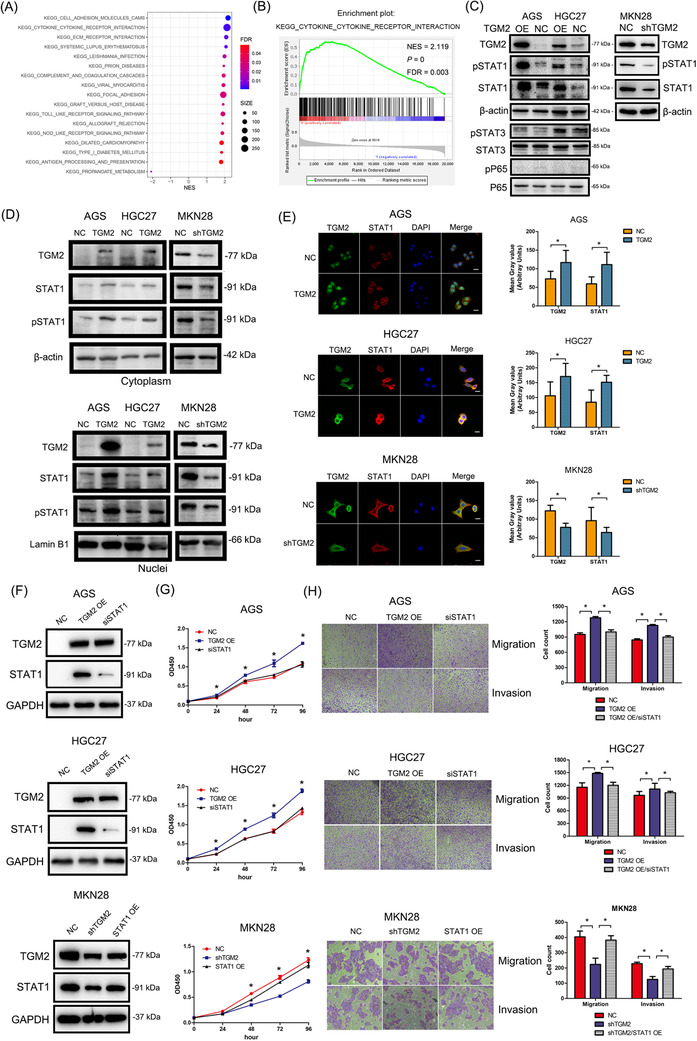
TGM2 promoted GC development by regulating STAT1. (A) GSEA was conducted to explore the mechanism underlying TGM2 in GC development using a dataset (375 patients with GC) from TCGA, and several cytokine and inflammatory pathways were found that are involved in the role of TGM2 in GC, including cytokine‐cytokine receptor interaction, toll‐like receptor signaling pathway, and nod‐like receptor signaling pathway. Bubble chart of GSEA was displayed. (B) The enrichment plot of cytokine‐cytokine receptor interaction was displayed as an example (NES = 2.119, FDR = 0.003, P = 0) (C) Several typical inflammatory signaling molecules were examined by western blotting. The results showed that STAT1 and STAT3 signaling pathways were involved. And STAT1 and its activated form (pSTAT1) were both upregulated in TGM2‐overexpressing GC cells, and the opposite results were observed in TGM2‐knocked down GC cells, which triggered our interest for further study. (D) The cytoplasmic and nuclear components were isolated from cell lysates for western blotting, the results showed that TGM2 overexpression increased the level of STAT1 and pSTAT1 in both cytoplasm and nuclei of AGS and HGC27 cells and the opposite results were observed when TGM2 was knocked down in MKN28 cells. (E) Immunofluorescence was conducted to examine the expression and localization of STAT1 and TGM2. The results showed that TGM2 was localized mainly in the cytoplasm of HGC27 and MKN28 cells and mainly in the nuclei of AGS cells. And TGM2 overexpression upregulated STAT1 levels in the AGS and HGC27 cells, and the knockdown of TGM2 reduced STAT1 level in the MKN28 cells. Representative micrographs of immunofluorescence were displayed. The bar charts showed the differences of gray values between TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells. *: P<0.05; scale bar: 20 µm. (F) For rescue assay, STAT1 was knocked down in TGM2‐overexpressing AGS and HGC27 cells using STAT1 siRNA, and upregulated in TGM2‐knocked down MKN28 cells using STAT1 plasmids. And western blotting confirmed the efficiency of the gain/loss in these GC cells. (G) Rescue assay on cell proliferation was evaluated by CCK8 assay. The results showed that the downregulation of STAT1 reversed the promoting effect of TGM2 on cell proliferation. *: P<0.05. (H) Rescue assay on cell migration and invasion were evaluated by transwell assay. The results showed that the upregulation of STAT1 rescued the inhibitory effect of TGM2 knockdown on cell migration and invasion. Bar charts showed the differences of migrated or invaded cell numbers between STAT1‐overexpressing/STAT1‐ knocked down GC cells and NC cells. *: P<0.05. Abbreviations: GC, gastric cancer; TGM2, transglutaminase 2; CCK8, cell counting kit‐8; NC, negative control; OE, overexpression; NES, normalized enrichment score; FDR, false discovery rate.
3.4. TGM2 promoted STAT1 stability by reducing its ubiquitination/degradation in GC cells
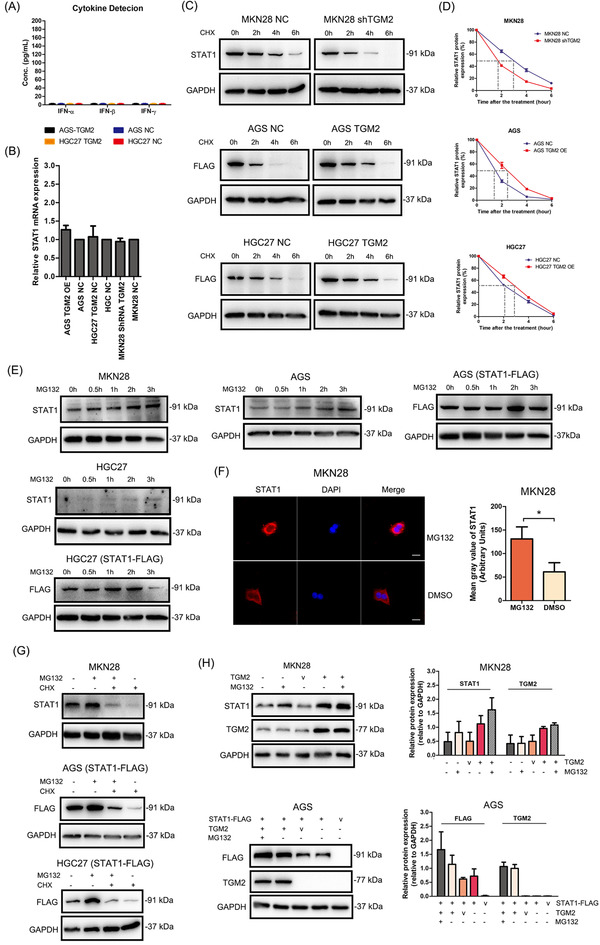
Next, we examined how TGM2 regulates STAT1 expression in GC cells. The interferon (IFN) family, especially IFN‐γ, is a well‐known cytokine that can activate the JAK/STAT1 signaling pathway; therefore, we first detected IFN‐α, IFN‐β, and IFN‐γ levels in the supernatants of GC cells by ELISA, and failed to observe detectable secretion of IFNs (Figure 5A), which indicated that TGM2‐induced STAT1 upregulation may not be associated with IFNs stimulation and that some other mechanisms may be involved in the activation of the STAT1 pathway. We then examined the effect of TGM2 on STAT1 mRNA (Figure 5B), TGM2 expression had no effect on the mRNA level of STAT1 in these cells. We speculated that TGM2 might affect STAT1 expression at the post‐transcriptional level. We examined STAT1 levels after CHX (20 µmol/L) treatment in GC cells and found that CHX reduced STAT1 levels in a time‐dependent manner, and TGM2 prolonged the half‐life of endogenous STAT1 in MKN28 cells and exogenous STAT1 in AGS and HGC27 cells (Figure 5C‐D). Next, we observed that MG132 treatment (10 µmol/L) increased the endogenous and exogenous STAT1 levels (Figure 5E). Then, MKN28 was used as a cell model to observe STAT1 levels after MG132 treatment for 2 h by immunofluorescence, as shown in Figure 5F, MG132 treatment increased STAT1 levels mainly in the cytoplasm. We also found that CHX treatment reversed the effect of MG132 on STAT1 expression in GC cells (Figure 5G). In addition, we found that transiently‐transfected TGM2 plasmid increased the endogenous and exogenous STAT1 levels, and MG132 treatment can increase the endogenous and exogenous STAT1 levels more in the context of TGM2 expression in GC cells (Figure 5H). Finally, Co‐IP results showed that MG132 treatment increased the ubiquitin levels of the endogenous and exogenous STAT1 in GC cells (Figure 5I), and TGM2 overexpression reduced the ubiquitin level of STAT1 in AGS cells, while the opposite results were observed when TGM2 was knocked down in MKN28 cells (Figure 5J).
FIGURE 5.
TGM2 promoted STAT1 stability by reducing its ubiquitination/degradation in GC cells (A) IFN‐α, IFN‐β and IFN‐γ levels in the supernatants of GC cells were examined by ELISA, and no detectable IFNs were observed. (B) The effect of TGM2 on STAT1 mRNA was examined by qPCR. And the results showed that TGM2 expression had no effect on the mRNA level of STAT1 in GC cells. The bar chart showed the differences in STAT1 mRNA between TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells. (C) MKN28 cells were treated with CHX (20 µmol/L) for 2 h, 4 h and 6 h to examine the changes of endogenous STAT1 levels by western blotting. AGS and HGC27 cells transfected with STAT1‐FLAG plasmids were treated with CHX (20 µmol/L) for 2 h, 4 h and 6 h to examine the changes in exogenous STAT1 levels by western blotting. The results showed that CHX reduced STAT1 levels in a time‐dependent manner, and TGM2 prolonged the half‐life of endogenous and exogenous STAT1 in GC cells. (D) The curve charts showed the half‐life of STAT1 in the context of TGM2 expression. (E) MKN28, AGS and HGC27 cells were treated with MG132 (10 µmol/L) for 0.5 h, 1 h, 2 h and 3 h to examine the changes of endogenous STAT1 levels by western‐blotting. And AGS and HGC27 cells transfected with STAT1‐FLAG plasmids were treated with MG132 (10 µmol/L) for 0.5 h, 1 h, 2 h and 3 h to examine the changes of exogenous STAT1 levels by western‐blotting. The results showed that MG132 treatment increased the endogenous and exogenous STAT1 levels in GC cells. (F) MKN28 was used as a cell model to observe STAT1 levels after MG132 (10 µmol/L) treatment for ∼2 h by immunofluorescence, and the result showed that MG132 treatment increased STAT1 levels mainly in the cytoplasm. Representative micrographs of immunofluorescence were displayed. The bar charts showed the quantitative analysis of STAT1 protein levels between MG132‐treated cells and DMSO‐treated cells. *: P<0.05; scale bar: 20 µm. (G) GC cells were treated with CHX (20 µmol/L) combined with MG132 (10 µmol/L) or either one for ∼2 h to examine the changes in endogenous and exogenous STAT1 levels by western blotting. The results showed that CHX treatment reversed the effect of MG132 on the endogenous and exogenous STAT1 levels in GC cells. (H) GC cells were transiently transfected with TGM2 plasmid to examine the changes in endogenous and exogenous STAT1 levels in GC cells by western blotting. The results showed that transient TGM2 overexpression increased the endogenous and exogenous STAT1 levels, and MG132 treatment (10 µmol/L, ∼2 h) increased the endogenous and exogenous STAT1 levels more in the context of TGM2 expression in GC cells. The bar charts showed the quantitative analysis of STAT1 and TGM2 protein levels. (I) GC cells were transiently transfected with Ub‐HA plasmid, and Co‐IP and western blotting examined the ubiquitin level of endogenous and exogenous STAT1 in GC cells. The results showed that MG132 treatment (10 µmol/L, ∼2 h) increased the ubiquitin level of the endogenous and exogenous STAT1 in GC cells. (J) GC cells were transiently transfected with Ub‐HA plasmid, and the changes in ubiquitin level of STAT1 in GC cells in the context of TGM2 overexpression were examined by Co‐IP and western blotting. The results showed that TGM2 overexpression reduced the ubiquitin level of STAT1 in AGS cells, while the opposite results were observed when TGM2 was knocked down in MKN28 cells. Abbreviations: GC, gastric cancer; TGM2, transglutaminase 2; IFN, interferon; NC, negative control; v, vector; Co‐IP, co‐immunoprecipitation; IP, immunoprecipitation; Ub, ubiquitin.
3.5. TRIM21 was the ubiquitin E3 ligase of STAT1 in GC cells
To identify the ubiquitin E3 ligase of STAT1, we transfected the STAT1‐FLAG plasmid into GC cells. Co‐IP and MS analyses were conducted, and TRIM21 was identified as a component of the mixture, which was considered to be a ubiquitin E3 ligase (Figure 6A‐B). We further examined the binding of TRIM21 and STAT1 in GC cells by two‐way Co‐IP and confirmed that there was mutual binding between TRIM21 and STAT1 (Figure 6C). In addition, the MKN28 cell line was used as a cell model to observe the colocalization of STAT1 and TRIM21 by immunofluorescence because of the high expression of both STAT1 and TRIM21. The results showed that TRIM21 and STAT1 colocalized mainly in the cytoplasm of GC cells (Figure 6D). Pearson's coefficient and overlap coefficient are all >0.7, indicating a high colocalization ratio of TRIM21 and STAT1. We then constructed a TRIM21 plasmid with the ZF_RING domain deleted (TRIM21_△RING‐FLAG, the active site of TRIM21, which contains the site to combine the substrate and E2 enzyme), and found that TRIM21_△RING failed to bind to STAT1 (Figure 6E‐G). The above results indicated a direct binding of TRIM21 and STAT1.
FIGURE 6.
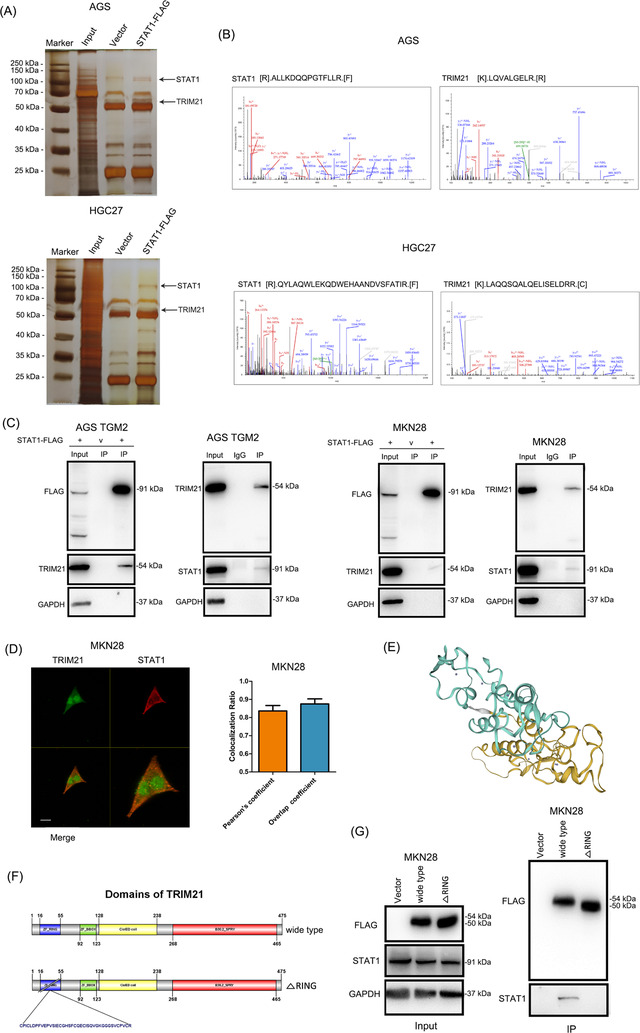
Mutual binding between TRIM21 and STAT1 in GC cells (A) STAT1‐FLAG plasmid was transfected into GC cells for Co‐IP. The proteins pulled down were analyzed by silver staining. The images of silver staining were displayed, and the locations of target proteins were marked with black arrows. (B) The secondary structure of STAT1 and TRIM21 in the mass spectrum were displayed. (C) The binding between TRIM21 and STAT1 was examined by two‐way Co‐IP and western blotting, and the results confirmed a mutual binding between TRIM21 and STAT1. (D) The MKN28 cell line was used as a cell model to observe the colocalization of STAT1 and TRIM21 by immunofluorescence because of the high expression of both STAT1 and TRIM21. that TRIM21 and STAT1 colocalized mainly in the cytoplasm of GC cells. The bar chart showed the quantitative analysis of the colocalization ratio of TRIM21 and STAT1. Scale bar: 20 µm. (E) The image of 3D structure of TRIM21 was displayed. (F) Schematic diagram of TRIM21 domains and the construction of TRIM21 plasmid with the ZF_RING domain deleted (TRIM21_△RING). (G) Cells were transfected with TRIM21‐FLAG and TRIM21_△RING‐FLAG plasmids, Co‐IP and western blotting confirmed that TRIM21 was bound to STAT1 at the ZF_RING domain, indicating the direct binding of TRIM21 and STAT1. Abbreviations: GC, gastric cancer; TGM2, transglutaminase 2; Co‐IP, co‐immunoprecipitation; IP, immunoprecipitation.
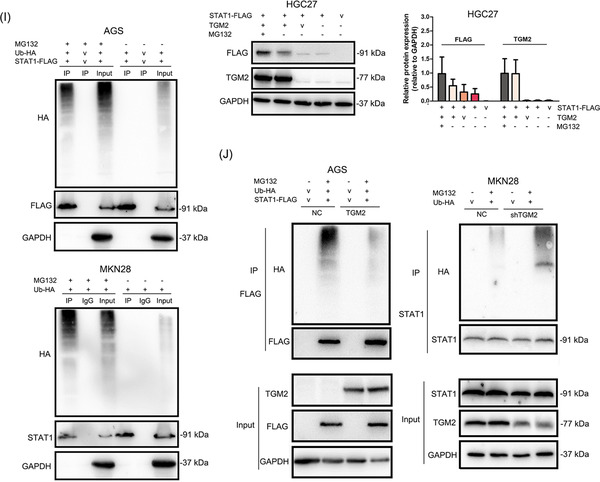
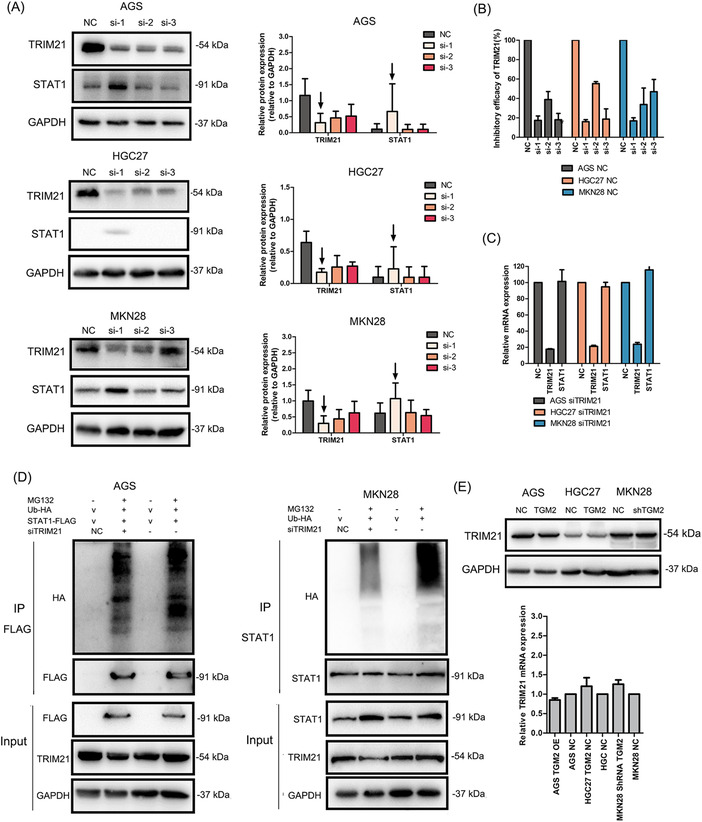
We then knocked down TRIM21 in GC cells by transfecting TRIM21 siRNAs and observed that TRIM21 si‐1 showed the highest efficacy by western blotting and qPCR (Figure 7A‐B). Furthermore, the knockdown of TRIM21 with TRIM21 si‐1 increased STAT1 protein levels (Figure 7A). Further study showed that TRIM21 affected STAT1 expression at the post‐translational level (Figure 7C). Then, Co‐IP and western blotting revealed that the knockdown of TRIM21 reduced the ubiquitin level of the exogenous and endogenous STAT1 in GC cells (Figure 7D). The above results indicated that TRIM21 is a ubiquitin E3 ligase of STAT1 in GC cells.
FIGURE 7.
TRIM21 was the E3 ubiquitin ligase of STAT1 and TGM2 promoted the dissociation of TRIM21 and STAT1 (A) TRIM21 was knocked down in GC cells by transfecting siRNAs. And western blotting confirmed that TRIM21 si‐1 showed the highest inhibitory efficacy on TRIM21 protein. What's more, TRIM21 si‐1 increased STAT1 protein levels in GC cells. The bar chart showed the quantitative analysis of TRIM21 and STAT1 protein levels. (B) TRIM21 was knocked down in GC cells by transfecting siRNAs. The knockdown efficacy was evaluated by qPCR, and the results showed that TRIM21 si‐1 showed the highest inhibitory efficacy on TRIM21 mRNA. (C) The effect of TRIM21 knockdown on STAT1 mRNA levels was examined by qPCR, and the results showed that TRIM21 knockdown had no effect on STAT1 mRNA levels. (D) GC cells were transiently transfected with Ub‐HA plasmids to examine the effect of TRIM21 knockdown on the ubiquitin level of STAT1 by Co‐IP and western blotting. The results showed that TRIM21 knockdown reduced the ubiquitin level of the exogenous and endogenous STAT1 (E) TRIM21 expression in TGM2‐overexpressing/TGM2‐knocked down GC cells and NC cells were evaluated by western blotting and qPCR. The results showed that TGM2 had no effect on TRIM21 expression at both the mRNA and protein levels. (F) The colocalization of TRIM21 and STAT1 was examined using immunofluorescence. The results showed that TGM2 overexpression/knockdown upregulated/downregulated STAT1 levels, while had no effect on TRIM21 level in GC cells. TGM2 expression didn't affect the colocalization capacity of TRIM21 and STAT1. The representative images of immunofluorescence were displayed. The upper bar chart showed the quantitative analysis of STAT1 and TRIM21 protein levels, *: P<0.05. The lower bar chart showed the quantitative analysis of colocalization ratio of TRIM21 and STAT1. (G) Co‐IP and western blotting showed that TGM2 facilitated the dissociation of TRIM21 and STAT1. And TGM2 was not a component in the Co‐IP mixture. Bar chart showed the quantitative analysis of TRIM21 and STAT1 protein levels. (H) The role of TRIM21 in the prognosis of patients with GC was analyzed with an online bioinformatics tool (The Kaplan‐Meier plotter), and the result indicated that TRIM21 showed anti‐tumor properties, which prolonged the postoperative OS (P = 0.0056) and first progression (P = 0.0025) in patients with GC. Abbreviations: GC, gastric cancer; TGM2, transglutaminase 2; v, vector; NC, negative control; OS, overall survival; FP, first progression; Co‐IP, co‐immunoprecipitation; IP, immunoprecipitation.
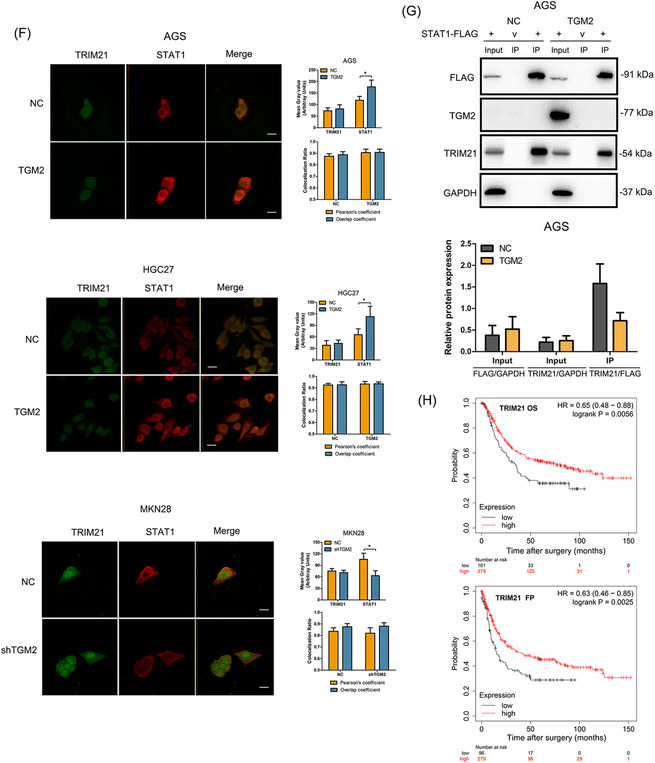
Next, qPCR and western blotting further confirmed that TGM2 had no effects on TRIM21 expression at both the mRNA and protein levels (Figure 7E). Therefore, we speculated that TGM2 might regulate the binding capacity of TRIM21 and STAT1, thus affecting the stability of STAT1 in GC cells. To verify this, we first examined the colocalization of TRIM21 and STAT1 using immunofluorescence. As shown in Figure 7F, TGM2 overexpression/knockdown upregulated/downregulated STAT1 level, while had no effect on TRIM21 level in GC cells. Furthermore, TGM2 overexpression/knockdown did not affect the colocalization ratio of TRIM21 and STAT1, indicating that immunofluorescence may not reflect the binding capacity of TRIM21 and STAT1. We then performed Co‐IP and western blotting and found that TGM2 overexpression facilitated the dissociation of TRIM21 and STAT1 (Figure 7G), which was consistent with our speculation. We also found that TGM2 was not a component in the Co‐IP mixture (Figure 7G), indicating TGM2 was not bound to STAT1. In addition, we analyzed the role of TRIM21 in the prognosis of patients with GC using an online bioinformatics tool (The Kaplan‐Meier plotter), and found that TRIM21 showed anti‐tumor properties, which prolonged the postoperative OS and first progression in patients with GC (Figure 7H). We speculated that TRIM21 might maintain the malignant progression of GC by facilitating STAT1 ubiquitination/degradation.
3.6. Cytosolic TGM2 promoted STAT1 stability with guanosine triphosphate ‐dependent enzymatic activity
TGM2 is a type of enzyme that exhibits two kinds of enzymatic activity: Ca2+‐dependent activity by binding Ca2+ and guanosine triphosphate (GTP)‐dependent activity by binding GTP (Figure 8A). Theoretically, the concentration of exocytic Ca2+ is 100‐fold more than that in cells; therefore, we speculated that cytosolic TGM2 might function in a GTP‐dependent manner. To verify this, we used two small molecular compounds, ZM39923 (which inhibits the Ca2+‐binding ability of TGM2) and A23187 (also named calcimycin, acting as a Ca2+ carrier to increase the concentration of cytosolic Ca2+) to treat GC cells. We observed that ZM39923 had no effect on the mRNA levels of TGM2 and STAT1, increased the expression of STAT1 protein in a dose‐dependent manner, and had no effect on TGM2 protein levels (Figure 8B). Further study showed that ZM39923 relieved the binding of TRIM21 and STAT1 and reduced the ubiquitin levels of STAT1 (Figure 8C), indicating that TGM2 maintains STAT1 stability in a Ca2+‐independent manner. It was reported that 171S in TGM2 was the key site for GTP‐binding, and the S171E mutation in TGM2 abolished GTP‐binding ability (Figure 8D). We then constructed TGM2 S171E overexpression cell lines and found that TGM2 S171E overexpression had no effect on STAT1 expression at both the mRNA and protein levels (Figure 8E‐G). Co‐IP and western blotting revealed that TGM2 S171E overexpression did not affect the binding of TRIM21 and STAT1 and had no effect on the ubiquitin level of STAT1 (Figure 8H). We also examined the effect of A23187 on STAT1 and found that A23187 caused an upstream fluctuation of TGM2 and STAT1 at the mRNA level and reduced STAT1 protein levels in a dose‐dependent manner but had no effect on TGM2 protein levels (Figure 8I). Co‐IP and western blotting revealed that A23187 reversed the dissociation of TRIM21 and STAT1 in the context of TGM2 and increased the ubiquitination of STAT1 (Figure 8J). The results above indicated that cytosolic TGM2 promoted the stability of STAT1 in a GTP‐binding‐dependent manner. Next, we examined the roles of ZM39923 and A23187 in tumor progression in vitro and found that ZM39923 promoted the proliferation, migration, and invasion of GC cells, while A23187 had an inhibitory role on GC cell proliferation, migration, and invasion (Figure 8K‐N). Finally, we treated xenograft mice with A23187 and found it inhibited tumor growth (Figure 8O‐R). Based on the collective data, we concluded that cytosolic TGM2 was prone to maintaining STAT1 stability by suppressing TRIM21‐mediated ubiquitination/degradation of STAT1 in a GTP‐binding‐dependent manner, thus leading to GC malignant progression (Figure 9).
FIGURE 8.
Cytosolic TGM2 promoted STAT1 stability with GTP‐dependent enzymatic activity (A) Schematic diagram of TGM2 domains was displayed (blue numbers refer to Ca2+‐binding sites, red numbers refer to GTP‐binding sites). (B) Cells were treated with ZM39923 at different concentrations (10 nmol/L, 20 nmol/L, 40 nmol/L) for 24 h to evaluate the effect of ZM39923 on STAT1 protein levels using western blotting. And the results showed that ZM39923 increased the expression of STAT1 protein in a dose‐dependent manner and had no effect on TGM2 protein levels. The bar charts showed the quantitative analysis of TGM2 and STAT1 protein levels. (C) Cells were treated with ZM39923 at different concentrations (10 nmol/L, 20 nmol/L, 40 nmol/L) for 24 h to evaluate the effect of ZM39923 on the mRNA levels of STAT1 mRNA by qPCR. And the results showed that ZM39923 had no effect on the mRNA levels of TGM2 and STAT1. (D) Cells were treated with ZM39923 (20 nmol/L) for 24h, Co‐IP and western blotting examined the effect of ZM39923 on the binding ability of TRIM21 and STAT1. The result showed that ZM39923 relieved the binding of TRIM21 and STAT1, leading to reduced ubiquitination of STAT1. (E) Schematic diagram of the construction of TGM2 S171E was displayed. (F) Cells were infected with TGM2 S171E lentivirus, and its effect of on STAT1 expression was examined by western blotting, and the result showed that TGM2 S171E overexpression had no effect on STAT1 protein levels. The bar chart showed the quantitative analysis of STAT1 protein levels. (G) Cells were infected with TGM2 S171E lentivirus, and its effect of on STAT1 expression was examined by qPCR. The result showed that TGM2 S171E overexpression had no effect on STAT1 mRNA levels. (H) The effect of S171E mutation in TGM2 on the binding ability of TRIM21 and STAT1 was examined using Co‐IP and western blotting, and the results showed that TGM2 S171E had no effect on the binding ability of TRIM21 and STAT1, as well as the ubiquitin level of STAT1. (I) Cells were treated with A23187 at different concentrations (2 µmol/L, 4 µmol/L, 8 µmol/L, 10 µmol/L) for 24h to evaluate the effect of A23187 on STAT1 expression by qPCR and western blotting, and the results showed that A23187 caused an upstream fluctuation of TGM2 and STAT1 at the mRNA level and reduced STAT1 protein levels in a dose‐dependent manner, but had no effect on TGM2 protein levels. (J) Cells were treated with A23187 (4 µmol/L) for 24h to examine the effect of A21187 on the binding ability of TRIM21 and STAT1 by Co‐IP and western blotting, and the result showed that A23187 reversed the dissociation of TRIM21 and STAT1 in the context of TGM2 and increased ubiquitination of STAT1 (K) The effect of A23187 (4 µmol/L) and ZM39923 (20 nmol/L) on GC cell proliferation were evaluated by colony formation assay. The result showed that ZM39923 promoted cell proliferation while A23187 had an inhibitory role in GC cell proliferation. Representative micrographs of colony formation assay were displayed. The bar chart showed the difference in clone number between ZM29923‐treated/A23187‐treated cells and NC cells. *: P<0.05. (L) The effect of A23187 (4 µmol/L) and ZM39923 (20 nmol/L) on GC cell proliferation was evaluated by CCK8 assay. The result showed that ZM39923 promoted cell proliferation while A23187 had an inhibitory role in GC cell proliferation. *: P<0.05. (M) The effect of A23187 (4 µmol/L) and ZM39923 (20 nmol/L) on cell migration were evaluated by wounding healing assay. The result showed that ZM39923 promoted cell migration, while A23187 had an inhibitory role on cell migration. Representative micrographs of wound healing assay were displayed. The bar charts showed the differences in the migratory area between ZM29923‐treated/A23187‐treated cells and NC cells. *: P<0.05. (N) Effect of A23187(4 µmol/L) and ZM39923 (20 nmol/L) on cell invasion was evaluated by transwell assay. The result showed that ZM39923 promoted cell invasion while A23187 had an inhibitory role on cell invasion. Representative micrographs of transwell assay were displayed, the bar charts showed the differences of invaded cell numbers between ZM29923‐treated/A23187‐treated cells and NC cells. *: P<0.05. (O) Mouse xenograft model was used to evaluate the effect of A12387 on cell proliferation in vivo; images of transplanted subcutaneous tumors from mouse models treated with A23187 and DMSO were displayed. (P) Representative micrographs of transplanted subcutaneous tumors by H&E staining were displayed. Scale bar: 500 µm. (Q) Tumor volume in mice was evaluated. The results showed that A23187 inhibited tumor growth. *: P <0.05 (R) Tumor weight in mice was evaluated. The results showed that A23187 inhibited tumor growth. *: P <0.05. Abbreviations: GC, gastric cancer; TGM2, transglutaminase 2; GTP, guanosine triphosphate; v, vector; NC, negative control; CCK8, cell counting kit‐8.
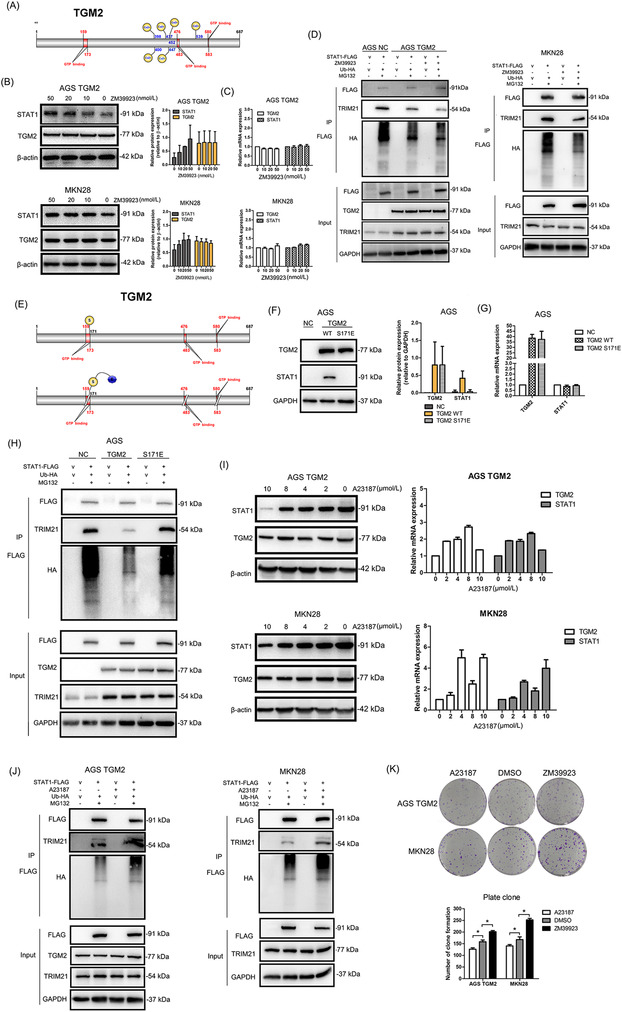
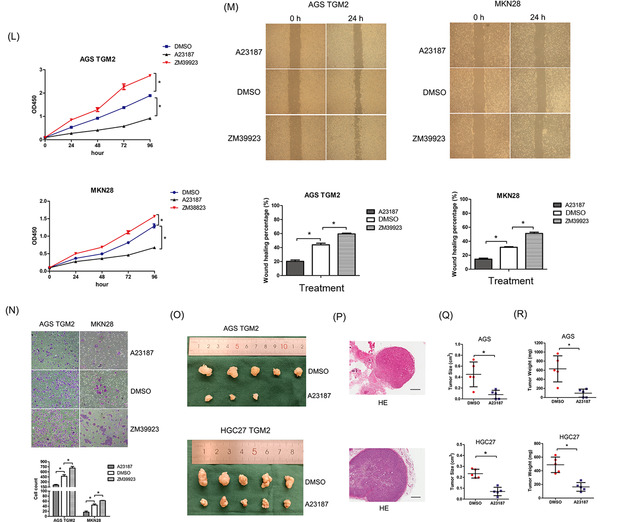
FIGURE 9.

Schematic model on the proposed role of TGM2 in regulating TRIM21‐mediated ubiquitination/degradation of STAT1 in GC malignant progression. Cytosolic TGM2 upregulates STAT1 stability by facilitating the dissociation of TRIM21 and STAT1 in the GTP‐binding manner, thus promoting GC malignant progression. And A23187 treatment shows anti‐tumor efficacy by increasing the intracellular Ca2+, which favors relieving the binding of TGM2 and GTP and reverses the role of TGM2 in STAT1 ubiquitination/degradation. Abbreviations: GC, gastric cancer; TGM2, transglutaminase 2; GTP, guanosine triphosphate; Ub, ubiquitin.
4. DISCUSSION
The oncogenic role and mechanism of TGM2 have been reported in several types of cancer but have seldom been reported in GC. In this study, we demonstrated that elevated TGM2 expression in GC promotes the malignant progression of gastric cancer and is associated with poor clinical prognosis. Furthermore, we revealed that TGM2 plays an oncogenic role in GC by inhibiting TRIM21‐medicated ubiquitination/degradation of STAT1 in a GTP‐binding‐dependent manner. A23187 can abolish the role of TGM2 in STAT1 and reverse the pro‐tumor role of TGM2 in GC by indirectly targeting GTP‐binding activity. To the best of our knowledge, this is the first report to reveal a novel regulatory mechanism of TGM2 on STAT1 in GC, highlighting its potential as a therapeutic target.
In this study, TGM2 was found to be involved in anti‐apoptotic and EMT processes, which is consistent with previous reports on other tumor types [12, 20, 21]. In addition, secretory TGM2 was only detectable in the plasma of some patients with GC, and no correlation was found between secretory TGM2 levels and clinicopathological characteristics. TGM2 is a predominantly cytosolic protein that is also present in the nucleus. However, TGM2, when secreted, can catalyze protein cross‐linking of the extracellular matrix, such as FN1 and SPP1, resulting in the irreversible formation of scaffolds to stabilize the integrity of dying cells before their clearance by phagocytosis, thereby preventing the leakage of harmful intracellular components [22]. We speculated that TGM2 might be secreted from GC cells under special conditions to reconstruct the tumor microenvironment, thus promoting GC development.
GSEA revealed that TGM2 in GC is closely associated with cytokine‐related and inflammatory pathways. STAT1 attracted our interest because TGM2 regulates STAT1 activation but also its expression. Thus far, most studies have focused on STAT1 activation and its downstream mechanism [23, 24, 25, 26, 27]. In this study, we focused on STAT1 expression and upstream regulatory mechanisms to improve our understanding of the role and mechanism of STAT1 in tumors. STAT1 is widely recognized as an essential component of IFN‐signaling that mediates several cellular functions in response to stimulation by cytokines, growth factors, and hormones. Previous studies have shown that STAT1 plays both oncogenic and tumor‐suppressive roles in various types of cancer [28, 29, 30]. In GC, the opposite results have also been reported. The IFNγ/STAT1 pathway acts as a suppressor to inhibit tumor invasion and metastasis by regulating EMT in GC [31], while the IFNγ/STAT1 signaling pathway was also reported to be associated with HER‐2 and could predict the poor prognosis of patients with GC [32]. In this study, STAT1 was found to play an oncogenic role in facilitating the malignant progression in GC. It should be noted that the expression and activation of STAT1 in this study were not associated with IFN‐signaling but were regulated by TGM2 at the post‐translational level, which may be a key influence on the role of STAT1 in tumors. The opposite tumoricidal and protumor effects of STAT1 may be attributed to differences in a tumor‐specific context, stimulator signal, and microenvironmental cues [33].
Ubiquitination is a common post‐translational modification of proteins that participate in the cell cycle, differentiation, survival, and death. In this study, we report for the first time that TGM2 maintains STAT1 stability by inhibiting its ubiquitination/degradation, revealing a novel regulatory mechanism of STAT1 in the context of TGM2. Then, using Co‐IP/MS, mutation, and western blotting, (TRIM21 was first identified and confirmed as the ubiquitin E3 ligase of STAT1 in GC cells. TRIM21 is a RING finger domain‐containing ubiquitin E3 ligase involved in innate immunity, systemic lupus erythematosus, Sjögren's syndrome, and tumors [34]. TRIM21 has been reported to have cancer‐promoting and anticancer effects in various tumors. In breast cancer, TRIM21 modulates EMT by mediating the stability of Snail, and high TRIM21 expression of TRIM21 is associated with longer OS in breast cancer [35]. TRIM21 promotes hepatocarcinogenesis by suppressing the p62‐Keap1‐Nrf2 antioxidant pathway [36]. To date, few studies have revealed the role of TRIM21 in GC, and only one recently published study showed that TRIM21 improved apatinib treatment sensitivity in GC by suppressing EZH1 stability, and the downregulation of TRIM21 expression was closely linked to higher recurrence and lower OS rate among patients with GC [37]. In this study, we found that TRIM21 functions as a ubiquitin E3 ligase of STAT1 and promotes STAT1 degradation in GC, explaining the analysis results from the KM plotter that high TRIM21 expression was associated with an improved survival prognosis. Our findings are in accordance with a recent report [37], indicating the anti‐tumor role of TRIM21 in GC.
Previous studies[38, 39] have reported that the expression of TRIM21 is induced by IFNs during viral infection and in autoimmune diseases. In our study, IFNs were not involved in the role of TGM2 on GC progression, which is consistent with that TGM2 had no effect on the expression of TRIM21. It may be because that TGM2 is not the inducer of IFNs in GC. We then found that the knockdown of TRIM21 effectively increased STAT1 levels at the post‐translational level, which triggered our speculation that TGM2 may facilitate the dissociation of TRIM21 and STAT1. We then performed Co‐IP and proved that TGM2 promoted the dissociation of TRIM21 and STAT1. The enzymatic activity of TGM2 is regulated by binding to Ca2+ or GTP. TGM2 can act as a protein‐glutamine gamma‐glutamyltransferase, thus mediating the cross‐linking of proteins by binding Ca2+ [8, 40‐42]. TGM2 can also act as protein disulfide isomerase, GTP/adenosine triphosphate (ATP) hydrolase, and serine/threonine kinase by binding to GTP [43, 44]. Therefore, the binding form of TGM2 that is involved in the regulation of STAT1 in GC cells deserves further elucidation, which will favor the development of strategies targeting TGM2 in GC.
Under physiological conditions, protein cross‐linking activity is inhibited by binding to GTP, and this inhibition is relieved by binding to Ca2+ in response to various stresses [45, 46, 47]. Because the intracellular Ca2+ concentration is 100‐fold lower than the extracellular Ca2+ concentration, we speculate that TGM2 may function in a GTP‐binding‐dependent manner in GC cells. To verify this, we treated GC cells with ZM39923 (an inhibitor that destroys the binding of Ca2+ to TGM2) or A23187 (a Ca2+ carrier that increases the intracellular Ca2+ concentration). We observed that ZM39923 treatment promoted the dissociation of TRIM21 and STAT1, leading to reduced ubiquitination of STAT1, and improved STAT1 protein levels. Inhibition of the Ca2+‐binding ability of TGM2 has been reported to increase the sensitivity of TGM2 to GTP [27]. These results indicate that TGM2 regulates STAT1 ubiquitination/degradation in a GTP‐binding‐dependent manner. To further confirm this, we constructed an S171E mutation in TGM2, which was reported to lose its GTP‐binding ability [48, 49]. We found that TGM2 S171E failed to upregulate STAT1 protein levels because it cannot facilitate the dissociation of TRIM21 and STAT1 to reduce the ubiquitination process. Considering that TGM2 was not bound to STAT1, therefore, it is not possible that TGM2 mediates the dissociation of STAT1 and TRIM21 by directlybinding to STAT1. Here, we speculated that TGM2 might act as a GTP hydrolase to provide energy to facilitate the dissociation of TRIM21 and STAT1.
The opposite results were observed for A23187. A23187 can increase the intracellular Ca2+ concentration, which further relieves the GTP‐binding activity of TGM2, thus facilitating STAT1 degradation. Although the STAT1 protein was decreased upon A23187 treatment, the mRNA of STAT1 was increased, which may be a rescue response in GC cells. Furthermore, in vitro and animal model results proved that A23187 can reduce GC malignant behaviors and inhibit tumor growth. The findings of this study indicate that the regulation of the GTP‐binding enzymatic activity of TGM2 may be a promising strategy for the management of TGM2‐positive patients with GC.
The present study has some limitations. First, owing to the heterogeneity in GC cell lines, such as AGS and HGC27 are prone to subcutaneous tumors, and MKN28 and AGS are prone to metastatic tumors in animal models, the role and mechanism of TGM2 in GC have not been investigated in all three GC cell strains selected at first. Secondly, although A23187 can inhibit GC growth in vitro and in vivo, it may not be the optimal inhibitor because it inhibits the GTP‐binding ability of TGM2 indirectly. What's more, the inhibitory role of A23187 on cell growth may be general. Therefore, other potential compounds against GTP‐binding directly deserved investigation and development. Finally, some underlying mechanisms have not been well verified, including whether TGM2 acts as a GTP hydrolase to promote the dissociation of TRIM21 and STAT1 in GC.
5. CONCLUSIONS
Our study revealed a novel role and mechanism of TGM2 in the malignant progression of GC, indicating that TGM2 is a potential therapeutic target in GC, and regulators of the GTP‐binding ability of TGM2 may be a promising strategy in the treatment of GC.
AUTHORS CONTRIBUTION
Lu Zhang and Zekuan Xu designed the study. Lu Zhang, Qingya Li, and Penghui Xu performed the in vitro experiments. Jing Yang and Zhe Xuan conducted the in vivo experiments. Qingya Li and Jianghao Xu analyzed the data. Lu Zhang, Qingya Li, and Zekuan Xu wrote the manuscript. All authors read and approved the final version of the manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
FUNDING
This study was funded by the National Natural Science Foundation of China (81802996, 81871946, and 82072708), Special Foundation for National Science and Technology Basic Research Program of China (2019FY101104), the Program for Development of Innovative Research Team in the First Affiliated Hospital of NJMU; the Priority Academic Program Development of Jiangsu Higher Education Institutions (JX10231801), Jiangsu Key Medical Discipline (General Surgery)(ZDXKA2016005), Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University.
CONSENT FOR PUBLICATION
Not applicable.
DECLARATIONS: ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The specimens of patients with gastric cancer were conducted with permission from the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University (permit number: 2022‐SR‐386). The tissue samples were obtained with written informed consent from each patient. All animal experiments were conducted according to animal protocols approved by Nanjing Medical University and were approved by the Ethics Committee of Nanjing Medical University (permit number:IACUC‐2203020).
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We are indebted to all the authors and our colleagues for the fruitful suggestions and discussions.
Zhang L, Li Q, Yang J, Xu P, Xuan Z, Xu J, et al. Cytosolic TGM2 promotes malignant progression in gastric cancer by suppressing the TRIM21‐mediated ubiquitination/degradation of STAT1 in a GTP binding‐dependent modality. Cancer Commun. 2023;43:123–149. 10.1002/cac2.12386
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49. [DOI] [PubMed] [Google Scholar]
- 2. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang XS, et al. Global surveillance of cancer survival 1995‐2009: analysis of individual data for 25,676,887 patients from 279 population‐based registries in 67 countries (CONCORD‐2). Lancet. 2015;385(9972):977–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oh SC, Sohn BH, Cheong JH, Kim SB, Lee JE, Park KC, et al. Clinical and genomic landscape of gastric cancer with a mesenchymal phenotype. Nat Commun. 2018;9(1):1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pasechnikov V, Chukov S, Fedorov E, Kikuste I, Leja M. Gastric cancer: prevention, screening and early diagnosis. World J Gastroenterol. 2014;20(38):13842–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang FH, Zhang XT, Li YF, Tang L, Qu XJ, Ying JE, et al. The Chinese Society of Clinical Oncology (CSCO): Clinical guidelines for the diagnosis and treatment of gastric cancer, 2021. Cancer Commun (Lond). 2021;41(8):747–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsieh YF, Liu GY, Lee YJ, Yang JJ, Sandor K, Sarang Z, et al. Transglutaminase 2 contributes to apoptosis induction in Jurkat T cells by modulating Ca2+ homeostasis via cross‐linking RAP1GDS1. PLoS One. 2013;8(12):e81516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lauzier A, Charbonneau M, Paquette M, Harper K, Dubois CM. Transglutaminase 2 cross‐linking activity is linked to invadopodia formation and cartilage breakdown in arthritis. Arthritis Res Ther. 2012;14(4):R159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Antonyak MA, Li B, Regan AD, Feng Q, Dusaban SS, Cerione RA. Tissue transglutaminase is an essential participant in the epidermal growth factor‐stimulated signaling pathway leading to cancer cell migration and invasion. J Biol Chem. 2009;284(27):17914–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mann AP, Verma A, Sethi G, Manavathi B, Wang H, Fok JY, et al. Overexpression of tissue transglutaminase leads to constitutive activation of nuclear factor‐kappaB in cancer cells: delineation of a novel pathway. Cancer Res. 2006;66(17):8788–95. [DOI] [PubMed] [Google Scholar]
- 12. Verma A, Guha S, Wang H, Fok JY, Koul D, Abbruzzese J, et al. Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin Cancer Res. 2008;14(7):1997–2005. [DOI] [PubMed] [Google Scholar]
- 13. Singer CF, Hudelist G, Walter I, Rueckliniger E, Czerwenka K, Kubista E, et al. Tissue array‐based expression of transglutaminase‐2 in human breast and ovarian cancer. Clin Exp Metastasis. 2006;23(1):33–9. [DOI] [PubMed] [Google Scholar]
- 14. Park KS, Kim HK, Lee JH, Choi YB, Park SY, Yang SH, et al. Transglutaminase 2 as a cisplatin resistance marker in non‐small cell lung cancer. J Cancer Res Clin Oncol. 2010;136(4):493–502. [DOI] [PubMed] [Google Scholar]
- 15. Fok JY, Ekmekcioglu S, Mehta K. Implications of tissue transglutaminase expression in malignant melanoma. Mol Cancer Ther. 2006;5(6):1493–503. [DOI] [PubMed] [Google Scholar]
- 16. Dyer LM, Schooler KP, Ai L, Klop C, Qiu J, Robertson KD, et al. The transglutaminase 2 gene is aberrantly hypermethylated in glioma. J Neurooncol. 2011;101(3):429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aeschlimann D, Thomazy V. Protein crosslinking in assembly and remodelling of extracellular matrices: the role of transglutaminases. Connect Tissue Res. 2000;41(1):1–27. [DOI] [PubMed] [Google Scholar]
- 18. Jones RA, Kotsakis P, Johnson TS, Chau DY, Ali S, Melino G, et al. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006;13(9):1442–53. [DOI] [PubMed] [Google Scholar]
- 19. Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kubo H, Shimizu M, Taya Y, Kawamoto T, Michida M, Kaneko E, et al. Identification of mesenchymal stem cell (MSC)‐transcription factors by microarray and knockdown analyses, and signature molecule‐marked MSC in bone marrow by immunohistochemistry. Genes Cells. 2009;14(3):407–24. [DOI] [PubMed] [Google Scholar]
- 21. Obinata A, Osakabe K, Yamaguchi M, Morimoto R, Akimoto Y. Tgm2/Gh, Gbx1 and TGF‐beta are involved in retinoic acid‐induced transdifferentiation from epidermis to mucosal epithelium. Int J Dev Biol. 2011;55(10‐12):933–43. [DOI] [PubMed] [Google Scholar]
- 22. Priglinger SG, May CA, Neubauer AS, Alge CS, Schonfeld CL, Kampik A, et al. Tissue transglutaminase as a modifying enzyme of the extracellular matrix in PVR membranes. Invest Ophthalmol Vis Sci. 2003;44(1):355–64. [DOI] [PubMed] [Google Scholar]
- 23. Liu C, Li X, Hao Y, Wang F, Cheng Z, Geng H, et al. STAT1‐induced upregulation of lncRNA KTN1‐AS1 predicts poor prognosis and facilitates non‐small cell lung cancer progression via miR‐23b/DEPDC1 axis. Aging (Albany NY). 2020;12(9):8680–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malilas W, Koh SS, Kim S, Srisuttee R, Cho IR, Moon J, et al. Cancer upregulated gene 2, a novel oncogene, enhances migration and drug resistance of colon cancer cells via STAT1 activation. Int J Oncol. 2013;43(4):1111–6. [DOI] [PubMed] [Google Scholar]
- 25. Huang R, Faratian D, Sims AH, Wilson D, Thomas JS, Harrison DJ, et al. Increased STAT1 signaling in endocrine‐resistant breast cancer. PLoS One. 2014;9(4):e94226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang XW, Li QH, Xu ZD, Dou JJ. STAT1‐induced regulation of lncRNA ZFPM2‐AS1 predicts poor prognosis and contributes to hepatocellular carcinoma progression via the miR‐653/GOLM1 axis. Cell Death Dis. 2021;12(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tabolacci C, De Martino A, Mischiati C, Feriotto G, Beninati S. The Role of Tissue Transglutaminase in Cancer Cell Initiation, Survival and Progression. Med Sci (Basel). 2019;7(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kachroo P, Lee MH, Zhang L, Baratelli F, Lee G, Srivastava MK, et al. IL‐27 inhibits epithelial‐mesenchymal transition and angiogenic factor production in a STAT1‐dominant pathway in human non‐small cell lung cancer. J Exp Clin Cancer Res. 2013;32:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lo UG, Pong RC, Yang D, Gandee L, Hernandez E, Dang A, et al. IFNgamma‐Induced IFIT5 Promotes Epithelial‐to‐Mesenchymal Transition in Prostate Cancer via miRNA Processing. Cancer Res. 2019;79(6):1098–112. [DOI] [PubMed] [Google Scholar]
- 30. Varikuti S, Oghumu S, Elbaz M, Volpedo G, Ahirwar DK, Alarcon PC, et al. STAT1 gene deficient mice develop accelerated breast cancer growth and metastasis which is reduced by IL‐17 blockade. Oncoimmunology. 2017;6(11):e1361088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jiang L, Liu JY, Shi Y, Tang B, He T, Liu JJ, et al. MTMR2 promotes invasion and metastasis of gastric cancer via inactivating IFNgamma/STAT1 signaling. J Exp Clin Cancer Res. 2019;38(1):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lv H, Zhang J, Sun K, Nie C, Chen B, Wang J, et al. Corrigendum: Expression of Human Epidermal Growth Factor Receptor‐2 Status and Programmed Cell Death Protein‐1 Ligand Is Associated With Prognosis in Gastric Cancer. Front Oncol. 2021;11:672599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon‐Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol. 2018;9:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alomari M. TRIM21 ‐ A potential novel therapeutic target in cancer. Pharmacol Res. 2021;165:105443. [DOI] [PubMed] [Google Scholar]
- 35. Jin Y, Zhang Y, Li B, Zhang J, Dong Z, Hu X, et al. TRIM21 mediates ubiquitination of Snail and modulates epithelial to mesenchymal transition in breast cancer cells. Int J Biol Macromol. 2019;124:846–53. [DOI] [PubMed] [Google Scholar]
- 36. Wang F, Zhang Y, Shen J, Yang B, Dai W, Yan J, et al. The Ubiquitin E3 Ligase TRIM21 Promotes Hepatocarcinogenesis by Suppressing the p62‐Keap1‐Nrf2 Antioxidant Pathway. Cell Mol Gastroenterol Hepatol. 2021;11(5):1369–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ping M, Wang S, Guo Y, Jia J. TRIM21 improves apatinib treatment in gastric cancer through suppressing EZH1 stability. Biochem Biophys Res Commun. 2022;586:177–84. [DOI] [PubMed] [Google Scholar]
- 38. Sjostrand M, Ambrosi A, Brauner S, Sullivan J, Malin S, Kuchroo VK, et al. Expression of the immune regulator tripartite‐motif 21 is controlled by IFN regulatory factors. J Immunol. 2013;191(7):3753–63. [DOI] [PubMed] [Google Scholar]
- 39. Jones EL, Laidlaw SM, Dustin LB. TRIM21/Ro52 ‐ Roles in Innate Immunity and Autoimmune Disease. Front Immunol. 2021;12:738473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanchan K, Ergulen E, Kiraly R, Simon‐Vecsei Z, Fuxreiter M, Fesus L. Identification of a specific one amino acid change in recombinant human transglutaminase 2 that regulates its activity and calcium sensitivity. Biochem J. 2013;455(3):261–72. [DOI] [PubMed] [Google Scholar]
- 41. Willis WL, Wang L, Wada TT, Gardner M, Abdouni O, Hampton J, et al. The proinflammatory protein HMGB1 is a substrate of transglutaminase‐2 and forms high‐molecular weight complexes with autoantigens. J Biol Chem. 2018;293(22):8394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maeda A, Nishino T, Matsunaga R, Yokoyama A, Suga H, Yagi T, et al. Transglutaminase‐mediated cross‐linking of WDR54 regulates EGF receptor‐signaling. Biochim Biophys Acta Mol Cell Res. 2019;1866(2):285–95. [DOI] [PubMed] [Google Scholar]
- 43. Lesort M, Tucholski J, Miller ML, Johnson GV. Tissue transglutaminase: a possible role in neurodegenerative diseases. Prog Neurobiol. 2000;61(5):439–63. [DOI] [PubMed] [Google Scholar]
- 44. Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin‐binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148(4):825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mian S, el Alaoui S, Lawry J, Gentile V, Davies PJ, Griffin M. The importance of the GTP‐binding protein tissue transglutaminase in the regulation of cell cycle progression. FEBS Lett. 1995;370(1‐2):27–31. [DOI] [PubMed] [Google Scholar]
- 46. Hwang KC, Gray CD, Sivasubramanian N, Im MJ. Interaction site of GTP binding Gh (transglutaminase II) with phospholipase C. J Biol Chem. 1995;270(45):27058–62. [DOI] [PubMed] [Google Scholar]
- 47. Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5(12):e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Antonyak MA, Jansen JM, Miller AM, Ly TK, Endo M, Cerione RA. Two isoforms of tissue transglutaminase mediate opposing cellular fates. Proc Natl Acad Sci U S A. 2006;103(49):18609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iismaa SE, Wu MJ, Nanda N, Church WB, Graham RM. GTP binding and signaling by Gh/transglutaminase II involves distinct residues in a unique GTP‐binding pocket. J Biol Chem. 2000;275(24):18259–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.