

Abstract
The microbial cell wall is essential for maintenance of cell shape and resistance to external stressors1. The primary structural component of the cell wall is peptidoglycan, a glycopolymer with peptide crosslinks located outside of the cell membrane1. Peptidoglycan biosynthesis and structure are responsive to shifting environmental conditions such as pH and salinity2–6, but the mechanisms underlying such adaptations are incompletely understood. Precursors of peptidoglycan and other cell surface glycopolymers are synthesized in the cytoplasm and then delivered across the cell membrane bound to the recyclable lipid carrier undecaprenyl phosphate7 (C55-P, also known as UndP). Here we identify the DUF368-containing and DedA transmembrane protein families as candidate C55-P translocases, filling a critical gap in knowledge of the proteins required for the biogenesis of microbial cell surface polymers. Gram-negative and Gram-positive bacteria lacking their cognate DUF368-containing protein exhibited alkaline-dependent cell wall and viability defects, along with increased cell surface C55-P levels. pH-dependent synthetic genetic interactions between DUF368-containing proteins and DedA family members suggest that C55-P transporter usage is dynamic and modulated by environmental inputs. C55-P transporter activity was required by the cholera pathogen for growth and cell shape maintenance in the intestine. We propose that conditional transporter reliance provides resilience in lipid carrier recycling, bolstering microbial fitness both inside and outside the host.
Subject terms: Microbial genetics, Bacterial physiology, Pathogens, Bacteriology
Members of the DUF368-containing and DedA transmembrane protein families have conditional roles in undecaprenyl phosphate translocation in Gram-negative and Gram-positive bacteria and may have a widely conserved function in the biogenesis of microbial cell surface glycopolymers.
Main
Phosphorylated undecaprenyl (C55) lipids have an essential role as recyclable carrier molecules in microbial cell surface glycopolymer biogenesis7. During peptidoglycan biosynthesis, the sugar-linked pentapeptide subunits of peptidoglycan that are assembled in the bacterial cytoplasm are covalently linked to C55-P for transmembrane transport7 (Fig. 1a). After the carrier-linked peptidoglycan precursor (lipid II) is moved from the inner to outer leaflet of the membrane by MurJ8, subsequent incorporation of the muropeptide into polymerized peptidoglycan leaves behind C55 pyrophosphate (C55-PP). Carrier recycling is initiated by hydrolysis of C55-PP to C55-P by membrane-associated phosphatases including UppP (also known as BacA) and the PAP2-domain proteins PgpB, YbjG and LpxT7. Then, C55-P is presumably flipped back into the cytosolic face of the membrane to complete recycling. Preliminary structural studies have proposed that UppP may also function as a C55-P translocase9,10, but the protein(s) responsible for C55-P internalization have not yet been identified. C55-P recycling is a key step in the biosynthesis of peptidoglycan as well as other cell surface glycopolymers, including wall teichoic acids, certain lipopolysaccharide modifications and capsules7. Given its wide-ranging and critical role in cell surface maintenance, C55-P recycling is considered to be an important target for antimicrobial therapies, and naturally occurring antibiotics that inhibit this process have been identified11.
Fig. 1. VCA0040 is required for cell shape maintenance of V. cholerae at alkaline pH.

a, Usage and recycling of C55-P in bacteria. Sites of antibiotic action relevant to Fig. 3 are indicated with red bars. Dashed arrows represent multiple enzymatic and/or transport steps. The grey hexagon represents variable sugars linked to C55-P for downstream glycopolymer assembly. LPS, lipopolysaccharide; PG, peptidoglycan. b, Conservation of DUF368 in selected bacterial phyla (upright) and genera (italic). The coloured segments and associated labels denote selected phyla with substantial DUF368 conservation. The fraction of sequenced unique clade genomes encoding a DUF368-containing protein is indicated. c,d, Predicted ribbon (c) and electrostatic surface coloured (d) structures of VCA0040. Colour scale, −10 to 10 kcal (mol e−). e,f, Growth (e) and morphology (f) of wild-type (WT), Δvca0040 and Δvca0040 + vca0040 (chromosomally complemented) V. cholerae in LB medium. g, Medium pH of V. cholerae overnight LB cultures. h, V. cholerae growth in M9 medium buffered to the indicated pH. i, The effects of buffered spent supernatant (cell-free medium after 24 h of growth at 30 °C from a 1:1,000 culture) on V. cholerae morphology in log phase. j, The effects of pH on V. cholerae growth (top) and morphology (bottom) during log phase in LB medium buffered with 50 mM Na2HPO4. NB, unbuffered medium. e,g,h, Data are mean ± s.d. from n = 3 cultures per strain or condition. f,i,j, Representative images from n = 3 cultures per strain or condition. Scale bars, 3 μm (f) and 5 μm (i,j).
Vibrio cholerae, the Gram-negative causal agent of the diarrheal disease cholera, is exposed to dramatic changes in pH and ion concentrations as it enters, transits and exits the host gastrointestinal tract. A variety of sensing and signalling networks enable V. cholerae to adapt to changing environments. For example, the pathogen responds to altered salinity and pH by using a sodium motive force (SMF) instead of a proton motive force (PMF) to power protein secretion and flagellar-dependent motility, and regulate virulence gene expression12,13. The peptidoglycan composition of V. cholerae is thought to be influenced by environmental inputs, but the effects of pH on V. cholerae cell wall biology are unknown14.
DUF368 impacts V. cholerae alkaline fitness
A recent in vivo transposon-insertion sequencing screen for determinants of intestinal colonization in a contemporary V. cholerae clinical isolate identified numerous genes not previously linked to V. cholerae pathogenesis, including several loci of unknown function15 (Extended Data Fig. 1a). One of these genes, vca0040 (N900_RS14215), was selected for further study because similar datasets16–18 suggested a universal requirement for this locus in V. cholerae intestinal colonization. VCA0040 is predicted to be a multi-pass inner membrane protein and contains the widely conserved domain of unknown function DUF368 (also known as PF04018) (Fig. 1b and Extended Data Fig. 1b–e). The predicted structural model of VCA0040 has a large putative cleft, characteristic of domains with ligand binding and/or transport activity (Fig. 1c,d and Extended Data Fig. 2). A V. cholerae strain lacking VCA0040 (Δvca0040) exhibited a stationary phase growth defect (Fig. 1e and Extended Data Fig. 3a) that was accompanied by a distinct cell shape phenotype, where Δvca0040 cells became large spheres (Fig. 1f and Extended Data Fig. 3b,c). These spherical V. cholerae were viable and gave rise to normal rod-shaped daughter cells (Extended Data Fig. 3d).
Extended Data Fig. 1. Identification and conservation of VCA0040.

(a): Infant rabbit transposon-insertion sequencing data reproduced from Hubbard et al. (ref. 15) identifying vca0040 (red) as a V. cholerae intestinal colonization determinant. The tcp operon, known to be critical for colonization, is highlighted in blue. One representative rabbit (#403) is shown. Inverse p-values were obtained by the Mann-Whitney U test without adjustments for multiple comparisons. Plotted thresholds are the same as Fig. 4a. (b and c): Domain structure, locus tags, accession numbers (b), and alignment (c) of V. cholerae and S. aureus DUF368-containing proteins. Alignment was performed with T-COFFEE. IN: predicted cytosol-facing sequence. HEL: predicted transmembrane helix sequence. OUT: predicted external-facing sequence. (d and e): Phylogenetic conservation of DUF368 domains (PF04018). (d): Family-level conservation (blue lines) grouped by phylum (colored arcs). Selected well-represented phyla are labeled. Blue lines are not scaled and do not provide information on the number or diversity of within-family DUF368 sequences. (e): Family-level breakdown of DUF368 conservation.
Extended Data Fig. 2. Structural modeling of DUF368-containing proteins.

The V. cholerae (left) and S. aureus (right) DUF368-containing proteins were modeled using AlphaFold2 and visualized with ChimeraX. (a): Multiple sequence alignment (MSA) coverage (left) and pLDDT (confidence score, right) per position for each protein from AlphaFold2. (b): Overall ribbon structures, colored from N (violet) to C (red) termini. (c and d): Electrostatic surface potential (c) and lipophilicity (d) maps generated using default ChimeraX settings. V. cholerae panels are reproduced from Fig. 1c,d for clarity. Electrostatic colour scale: -10 to 10 kcal/(mol·e). Lipophilicity colour bar: relative molecular lipophilicity as calculated by ChimeraX. (e): MSA coverage, pLDDT plot, and ribbon and electrostatic surface views of the DUF368 protein from Haloferax volcanii (HVO_RS12060).
Extended Data Fig. 3. Growth and morphology of Δvca0040 V. cholerae.

(a): CFU plating time course of WT and Δvca0040 V. cholerae. Onset of stationary phase occurs after 6 h. (b): Phase-contrast imaging of WT, Δvca0040 and Δvca0040 + vca0040 V. cholerae in overnight cultures in the indicated conditions. (c): Phase-contrast imaging time course of the indicated strains grown in LB at 30 °C for the indicated amount of time. (d): Timelapse imaging of Δvca0040 cells immobilized on an agarose pad from a 30 °C overnight culture. Images were acquired over 3 h of growth. (e): Imaging of V. cholerae treated with normal, boiled (10 min) or filtered (<3K MW cutoff) cell-free spent supernatant for 4 h at 30 °C. (f): Imaging of V. cholerae treated with vehicle (DMSO) or 3 mM of D- or L-Ala in fresh LB for 4 h at 30 °C. a: mean ± SD of n = 3 independent cultures per strain. b, c, e, f: representative images of n = 3 independent cultures per strain/condition. d: representative timelapse of a single cell representative of >20 sphere-shaped bacteria across n = 3 separate fields of view from a single overnight culture. Scale bars: 3 (b,c,f) or 5 (d,e) µm.
We reasoned that a stationary phase-specific factor could be triggering the shape defect in Δvca0040 cells. Accordingly, exposure of exponentially growing Δvca0040 cells to cell-free spent supernatants from stationary phase cultures rapidly induced sphere formation (Extended Data Fig. 3e). Heat-labile and high-molecular-weight factors, as well as d-amino acids—which modulate stationary phase peptidoglycan composition19—were excluded as candidates (Extended Data Fig. 3e,f). In LB cultures, V. cholerae entry into stationary phase is accompanied by media alkalinization (Fig. 1g). Since minimal shape defects were observed in neutral buffered M9 medium (Extended Data Fig. 3b), we hypothesized that Δvca0040 cells were alkaline-sensitive. Buffering M9 media to different pH values revealed a growth defect at pH 8, but not at pH 6 or pH 7, and acidification of the cell-free spent supernatant ablated the sphere-induction phenotype (Fig. 1h,i). In buffered LB, Δvca0040 cells exhibited growth and cell shape defects at pH above 8, but not at pH values of up to 7 (Fig. 1j). Thus, the DUF368-containing protein VCA0040 is required for V. cholerae cell shape integrity and growth in alkaline conditions.
Altered peptidoglycans in DUF368 mutants
DUF368 domains are almost universally conserved across the Vibrionaceae and are present in thousands of additional Gram-negative, Gram-positive and archaeal species that inhabit a wide range of niches (Supplementary Tables 1 and 2). Heterologous expression of Gram-positive (Staphylococcus aureus) or archaeal (Haloferax volcanii) DUF368 homologues at least partially complemented the alkaline growth defect of Δvca0040 V. cholerae cells (Extended Data Fig. 4a,b). Both homologues showed strong predicted structural similarity to VCA0040, suggesting that DUF368-containing protein function is conserved not only between Gram-negative and Gram-positive species, but across microbial kingdoms (Extended Data Fig. 2).
Extended Data Fig. 4. DUF368 functional conservation and effects on PG composition and crosslinking.

(a and b): Alkaline growth of Δvca0040 V. cholerae bearing pBAD18 vectors expressing (a) SAOUHSC_00846 from S. aureus or (b) HVO_RS12060 from H. volcanii. (c-e): Composition and crosslinking analysis in the indicated V. cholerae strains at the indicated pH in M63 media. Suppressor: Δvca0040/ΔsecDF1. (f and g): Same as c-e for S. aureus grown in the indicated conditions. a, b: representative data from n = 3 cultures per strain/condition. c-g: mean ± SD from n = 3 cultures per strain/condition. Statistical analysis was performed with one-way ANOVAs with Tukey’s multiple comparison tests (c-e) or unpaired student’s two-tailed t-tests (f, g).
Since peptidoglycan is required for maintenance of bacterial cell shape, we hypothesized that DUF368 functions affect the cell wall. The Δvca0040 mutant had 1.5–2× less peptidoglycan than the wild type, and modest cross-linking defects with concurrent accumulation of the peptidoglycan precursor UDP-N-acetylmuramyl pentapeptide (UDP-M5), suggestive of a peptidoglycan biosynthesis defect upstream of cross-linking (Fig. 2a,b and Extended Data Fig. 4c–e). These phenotypes were present at neutral pH and were exacerbated by exposure to alkaline conditions. Consistent with the idea that DUF368 function is conserved, S. aureus lacking SAOUHSC_00846 also had an alkaline growth defect, reduced peptidoglycan quantity, accumulation of UDP-M5 and decreased cross-linking (Fig. 2c–e and Extended Data Fig. 4f,g). We concluded from these data that DUF368-containing proteins are conditionally required to maintain peptidoglycan production and composition.
Fig. 2. DUF368 function is conserved and necessary for peptidoglycan maintenance.

a,b, Total peptidoglycan (a) and intracellular UDP-M5 (b) in V. cholerae grown in M63 minimal medium at the indicated pH. Suppressor, Δvca0040/ΔsecDF1. c, Alkaline growth of ΔSAOUHSC_00846 (Δ0846) S. aureus in the indicated conditions and with the indicated expression vectors. Representative results from n = 3 independent experiments per condition. d,e, Total peptidoglycan (d) and intracellular UDP-M5 (e) in S. aureus grown in tryptic soy broth (TSB) with 100 mM bicine at the indicated pH. a,b,d,e, n = 3 cultures per strain or condition; data are mean ± s.d. a,b, One-way ANOVA with Tukey’s multiple comparison test. d,e, Unpaired Student’s two-tailed t-test.
Impaired C55-P recycling in DUF368 mutants
Two additional observations focused our investigation of DUF368 domains on cell wall assembly. First, we observed that although most DUF368-containing proteins consist primarily of one or two DUF368 domains, several microorganisms encode dual-domain proteins with DUF368–PAP2, PAP2–DUF368 or DUF368–BacA architectures (Supplementary Table 3). Second, during sphere formation in Δvca0040 V. cholerae, there was an approximately tenfold induction of pgpB, which encodes a C55-PP phosphatase20 (Extended Data Fig. 5a–d and Supplementary Table 4). Despite these associations of DUF368 with C55-related processes, the Δvca0040 mutant exhibited only minor (up to 4×) increases in minimum inhibitory concentrations (MICs) of an array of peptidoglycan-targeting antibiotics, probably because most cell wall-acting compounds cannot penetrate the Gram-negative outer membrane (Supplementary Table 5). Accordingly, MIC screening in S. aureus revealed that the ΔSAOUHSC_00846 mutant was far more sensitive (more than 64×) to amphomycin than the wild type in alkaline conditions (Fig. 3a and Supplementary Table 5). Amphomycin is a Ca2+-dependent lipopeptide antibiotic that specifically binds C55-P in the outer leaflet of the cytoplasmic membrane and inhibits its recycling21–23 (Fig. 1a). Consistent with our previous data (Fig. 2d,e), amphomycin is known to induce UDP-M5 accumulation and decrease peptidoglycan cross-linking in S. aureus24. The ΔSAOUHSC_00846 mutant was additionally moderately sensitized to tunicamycin, which inhibits the first committed and C55-P-dependent steps of wall teichoic acid and peptidoglycan synthesis as well as bacitracin, which binds C55-PP and prevents C55-P generation on the extracytosolic face of the membrane25,26 (Figs. 1a and 3a). MICs for other peptidoglycan-targeting molecules such as fosfomycin were minimally changed (Fig. 3a and Supplementary Table 5). To test DUF368 function in a Gram-negative organism, we heterologously expressed SAOUHSC_00846 or vca0040 in the outer membrane-permeable—and thus amphomycin-sensitized—Escherichia coli strain lptD421327 (Supplementary Table 5). Expression of either DUF368-containing protein in lptd4213 E. coli conferred amphomycin resistance (Supplementary Table 5). Since E. coli lacks an endogenous DUF368-containing protein, this result indicates that DUF368 function is sufficient for resistance to a surface C55-P-targeting antibiotic.
Extended Data Fig. 5. Additional characterization of bacteria lacking DUF368-containing proteins.

(a-d): RNAseq of Δvca0040 V. cholerae, with schematic (a), confirmation of sphere formation via imaging at sample collection (b), principal component analysis of RNAseq data (c), and MA plot of analyzed sequences (d) (n = 3 independent cultures per strain). An arbitrary fold change (FC) cutoff of 4 was implemented during RNAseq analysis, and genes with FC values beneath the p-value threshold of 0.05 are highlighted in red. pgpB and vca0040 are labeled. (e and f): UPLC traces (e) and quantification of C55-P relative abundance (f) in V. cholerae grown at the indicated pH. (g): Ampho-FL-stained S. aureus grown at pH 6, 7, or 8.5. (h): Quantification of ampho-FL staining signal at different pHs as shown in g. Symbols represent the mean ampho-FL intensity of n = 1–8 individual bacterial clusters in independent cultures. Single pH 8.5 cultures were used as controls as a new batch of ampho-FL was used for this experiment. e, g: representative data of n = 3 cultures per strain/condition (except for n = 1 culture in pH 8.5 in g). f, h: n = 3 independent cultures. Statistical analysis was performed with (d) Wald tests in the DESeq2 pipeline with multiple comparison adjustments or (f, h) unpaired student’s two-tailed t-tests. Scale bars, 5 µm.
Fig. 3. C55-P recycling is impaired in bacteria lacking DUF368-containing proteins.

a, Fold change (FC) in MIC for ΔSAOUHSC_00846 S. aureus in the indicated conditions. Amphomycin*, amphomycin with 1 mM CaCl2. b, Ultra high performance liquid chromatography (UPLC) traces of purified C55-OH (a) and C55-P (b) standards (top), wild-type S. aureus lipid extracts spiked with each standard (middle) or wild-type S. aureus treated with 50 μg ml−1 amphomycin (bottom). Rt, retention time. c, UPLC traces of lipid extracts of wild-type or mutant S. aureus grown in TSB pH 7 (top) or pH 8.5 (bottom). d,e, Relative abundance of C55-OH (d) and C55-P (e) under same conditions as in c. f, UPLC trace of lipid extracts of complemented ΔSAOUHSC_00846 S. aureus grown in TSB pH 8.5. g,h, Raw peak proportions of C55-OH (g) and C55-P (h) in each strain grown at pH 8.5. i, S. aureus stained with ampho-FL at pH 8.5. Scale bars, 5 μm. DIC, differential interference contrast image. j, Quantification of ampho-FL signal. Symbols represent the mean ampho-FL intensity of 1 to 8 individual bacterial clusters in independent cultures. k, Proposed model of disrupted C55-P recycling and the associated consequences in bacteria lacking C55-P translocase activity. b,c,f,i, Representative data from n = 3 cultures per strain or condition. d,e,g,h,j, Data are mean ± s.d. from n = 3–5 cultures per strain or condition. Each point is an independent culture. d,e, Unpaired student’s two-tailed t-test. g,h,j, One-way ANOVA with Tukey’s multiple comparison test.
The peptidoglycan composition and antibiotic susceptibility data indirectly suggested that C55-P recycling was impaired in the ΔSAOUHSC_00846 mutant, specifically in C55-P re-internalization. To further investigate this idea, we quantified C55 species in membrane lipid extracts from wild-type and ΔSAOUHSC_00846 cultures. Amphomycin treatment of wild-type S. aureus led to marked accumulation of both C55-P and the Gram-positive-restricted C55-P precursor C55-OH28 (undecaprenol, also known as UndOH) (Fig. 3b). This phenotype was replicated in ΔSAOUSHC_00846 S. aureus cells grown in alkaline media, but not in neutral conditions (Fig. 3c–e) or in a complemented strain (Fig. 3f–h), providing direct evidence that C55-P homeostasis is linked to SAOUSHC_00846. We also observed similar alkaline-dependent increases in C55-P (but not C55-OH) in Δvca0040 V. cholerae (Extended Data Fig. 5e,f). To specifically quantify C55-P in the outer leaflet of the cell membrane, we synthesized fluorescein-conjugated amphomycin (ampho-FL) and used it to label live bacterial cells29. Ampho-FL-labelled ΔSAOUHSC_00846 S. aureus exhibited increased signal relative to wild-type bacteria in alkaline media, demonstrating that surface C55-P levels are conditionally elevated in the mutant (Fig. 3i,j and Extended Data Fig. 5g,h). Together, these observations suggest that the alkaline accumulation of surface C55-P in DUF368-containing protein mutants provokes a compensatory—but ultimately insufficient—increase in C55-P synthesis, resulting in surface glycopolymer production defects and impaired growth (Fig. 3k). These findings are consistent with a model in which DUF368-containing proteins are C55-P recycling transporters that are active in alkaline conditions.
Genetic interactions between DUF368 and DedA
As C55-P recycling is thought to be an essential function and DUF368 mutants were conditionally viable, we next carried out a synthetic transposon screen to define the genetic network of vca0040. Many of the identified synthetic sick or lethal interactions were related to cell envelope homeostasis (Fig. 4a and Supplementary Table 6). The strongest interaction of vca0040 was with N900_RS16280 (also known as vca0534), which encodes a homologue of the E. coli DedA family member YghB (Fig. 4b). V. cholerae express two other DedA proteins, but these were not hits in this screen. DedA family transmembrane proteins (also known as PF09335 (SNARE-associated Golgi protein)) are conserved in all three domains of life, are generally required for cell envelope homeostasis, and are suspected to mediate PMF-dependent transport, but their specific substrates are unknown30,31. Similar to DUF368, although most DedA-containing sequences encode only that domain, there are many instances of domain architectures in which DedA is fused to the C55-PP phosphatase domain PAP2 (Supplementary Table 3). The synthetic lethality of vca0040 and yghB was validated by genetic depletion and reciprocal synthetic transposon screening (Fig. 4c, Extended Data Fig. 6a and Supplementary Table 7), and overexpression of YghB rescued the alkaline defect of Δvca0040 V. cholerae (Extended Data Fig. 6b).
Fig. 4. DedA family members are additional C55-P translocase candidates.

a, A synthetic transposon screen in Δvca0040 V. cholerae, showing log2 mean read fold changes (MFC; threshold ±2) and inverse Mann–Whitney U test P-values (threshold 100). Pooled data from two independent transposon libraries. b, Domain content and predicted structure of V. cholerae VCA0534 (also known as YghB). c, Growth of ΔyghB V. cholerae vca0040 depletion strains. Ara, arabinose; Glc, glucose. d, Spontaneous suppressors of ΔSAOUHSC_00846 map to two S. aureus DedA family proteins, with predicted structures (right) and specific promoter mutations selected for validation (bold). e, Rescue of ΔSAOUHSC_00846 by expression of SAOUHSC_00901 (0901) or SAOUHSC_2816 (2816) from hybrid promoters containing the IPTG-inducible promoter Pspac linked to 25-bp (construct 1) or 200-bp (construct 2) native promoter stretches with the indicated suppressor mutations (*). f, Fold change in amphomycin MIC for wild-type and mutant S. aureus relative to the MIC for the wild type at pH 7 (150 μg ml−1). c,e, Representative results from n = 3 independent experiments per condition. f, Data are mean ± s.d. from n = 3 cultures per strain or condition.
Extended Data Fig. 6. Genetic interactions between DedA and DUF368 proteins.

(a): Synthetic transposon-insertion screen in ΔyghB V. cholerae. P-values were obtained by the Mann-Whitney U (MWU) test without adjustment for multiple comparisons. MFC: mean fold change in read coverage between ΔyghB and WT. (b): Growth of Δvca0040 V. cholerae expressing vectors with arabinose-inducible genes on regular LB (left), LB pH 9 (middle) and LB pH 9 + arabinose (right) plates. (c): Growth of V. cholerae lacking one or both of vca0040 and/or yghB on LB pH 6 plates. LB was buffered with 100 mM MES and pH-adjusted with HCl. a: pooled data from n = 2 independent transposon libraries. b, c: representative images from n = 3 cultures per strain/condition.
Interactions between DUF368 and DedA proteins were also observed in S. aureus. In alkaline-enriched spontaneous suppressors of ΔSAOUHSC_00846 S. aureus, we found frequent promoter (and no coding region) mutations in the genes encoding the two S. aureus DedA proteins (SAOUHSC_00901 and SAOUHSC_02816) (Fig. 4d). Incorporating these mutations into an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible system expressing either DedA protein rescued ΔSAOUHSC_00846 even without induction, suggesting that the isolated mutations increase gene expression and compensate for the absence of SAOUHSC_00846, as in V. cholerae (Fig. 4e). These genetic studies suggest that DedA family members are also required for C55-P translocation, in agreement with recent reports that eukaryotic DedA proteins are associated with lipid transport and bacterial DedA mutants are defective in C55 carrier-dependent lipopolysaccharide modifications31–33.
We hypothesized that similar to DUF368-containing proteins, DedA family members function conditionally. Although deletion of SAOUHSC_00901 did not affect amphomycin MICs, the ΔSAOUHSC_02816 mutant exhibited moderate acid-dependent amphomycin sensitivity, strengthening the association of DedA family members with C55-P translocation and suggesting that they function at lower pH ranges than their DUF368-containing counterparts (Fig. 4f). The sensitivity of ΔSAOUHSC_02816 to other antibiotics was unaltered at any pH, highlighting the specificity of conditional amphomycin sensitization (Supplementary Table 5). The concept of pH-dependent translocase contributions is also supported by the alkaline amphomycin sensitivity of lptd4213 E. coli, which has only DedA paralogues and no cognate DUF368-containing protein (Supplementary Table 5). A strain lacking yghB and vca0040 was viable at acidic pH, suggesting at least one other V. cholerae protein can carry out C55-P translocation, or that C55-P may spontaneously traverse the membrane in acidic conditions (Extended Data Fig. 6c).
Na+ shapes C55-P translocase biology
Analyses of spontaneous suppressors of Δvca0040 V. cholerae, which have distinctive colony morphology (Extended Data Fig. 7a) revealed that the requirement of VCA0040 for V. cholerae fitness is controlled by Na+ concentration in addition to pH. Whole-genome sequencing of suppressor colonies lacking cell shape defects revealed four suppressor genes, three of which (secD1, secF1 and ppiD) function in Sec-mediated protein secretion (Extended Data Fig. 7b). The fourth—yfgO—encodes a protein of unknown function from the AI-2E transporter family, members of which have recently been implicated in peptidoglycan biosynthesis and Na+/H+ antiport34,35. Deletions of suppressor genes rescued the shape and peptidoglycan defects of Δvca0040 V. cholerae, even though stationary culture pH was alkaline in all suppressors (Fig. 2a,b and Extended Data Fig. 7c,d). In V. cholerae, SecD–SecF is duplicated as SecD1–SecF1 and SecD2–SecF2, ancillary complexes that differentially couple SecYEG activity to the SMF or PMF, respectively36 (Extended Data Fig. 7e). The activity of at least one SecD–SecF pair was required for V. cholerae viability, and deletion of secD2 and secF2 did not ameliorate the stationary phase cell shape defect of Δvca0040 (Extended Data Fig. 7c,f,g). We initially hypothesized that altered Sec substrate specificity could explain the identification of SecD1 and SecF1 as suppressors. However, the ΔsecDF1 V. cholerae proteome was unchanged from that of wild-type cells, aside from the expected increases in SecD2 and SecF2, suggesting that the suppressive effects of secD1 and secF1 loss were attributable to reduced Na+ flux (Extended Data Fig. 7h and Supplementary Table 8). Indeed, increasing Na+ but not K+ concentration induced alkaline growth and peptidoglycan defects in Δvca0040 V. cholerae (Fig. 2a,b and Extended Data Fig. 7i–k). The requirement of increased Na+ concentration for the alkaline defect of DUF368-deficient V. cholerae appeared to be species-specific, as ΔSAOUHSC_00846 S. aureus, although alkaline-sensitive, was not similarly rescued by Na+ depletion (Fig. 2c). Of note, unlike Δvca0040, a ΔyghB V. cholerae mutant was not able to grow without Na+ at pH values above 6 (Extended Data Fig. 7l,m), suggesting that the presence of Na+ divergently affects distinct C55-P translocases. Together, these data illustrate that multiple environmental inputs can control C55-P translocase requirements and potentially function in different microorganisms.
Extended Data Fig. 7. Contribution of Na+ to Δvca0040 alkaline susceptibility.

(a): Representative colonies of WT, Δvca0040, and suppressor V. cholerae on LB+X-gal plates. White arrows indicate suppressors. (b): Spontaneous suppressor mutations of Δvca0040 V. cholerae. SecD1, SecF1 and PpiD are part of the V. cholerae protein secretion machinery. Colored text indicates suppressors from a secondary screen in ΔsecD2/Δvca0040 (blue) or ΔsecF2Δvca0040 (red) V. cholerae. Asterisk: stop codon. (c): Phase-contrast imaging of overnight 30 °C LB cultures of the indicated strains in the Δvca0040 background. Scale bars, 3 μm. (d): Overnight 30 °C LB culture pH of the indicated strains. (e): SecDF1 and SecDF2 functions in V. cholerae. Possible SecDF functions: coupling ion import to SecA ATPase stimulation (bottom arrow) or Sec substrate guidance into the periplasm (top arrow). (f): Synthetic lethality of secD2 in ΔsecD1 V. cholerae using the pAM299 suicide vector for Ara-dependent secD2 expression. (g): Growth of the indicated strains in M63 media with added NaCl or KCl. (h): Waterfall plot comparing ΔsecDF1 to WT V. cholerae proteome ranked by protein fold change (FC). (i): Growth of Δvca0040 V. cholerae in [Na+]-varying (top) or [K+]-varying (bottom) M63 media at pH 6, 7 or 8. (j and k): Growth of WT (j) or Δvca0040 (k) V. cholerae in M63 pH 8 with a range of [NaCl]. 100mM NaCl curves from i are replicated in j and k for comparison. (l and m): Growth of the indicated V. cholerae strains on non-buffered (l) or pH 6 (m) LB with regular (~170 mM) or no added NaCl. a: representative image of n = 11 independent suppressor strains. c, f, l, m: representative data of n = 3 cultures per strain/condition. d: means ± SD of n = 3 cultures per strain. g, i, j, k: mean ± SD of at least 3 technical replicates of single cultures (representative of n = 3 cultures per strain/condition). h: pooled data comparing n = 4 independent cultures of each strain.
DUF368 enables V. cholerae pathogenesis
We used an infant (postnatal day (P)1–P2) rabbit model that mimics severe human cholera37 to investigate the role of VCA0040 in pathogenesis. Typically, this model uses alkaline inocula to buffer the acidic stomach environment. To exclude inoculum effects, we carried out mixed infections with inocula comprising a 1:1 mix of wild-type and Δvca0040 V. cholerae at pH 7 or pH 9 (Fig. 5a). Regardless of inoculum pH, the Δvca0040 mutant had a profound competitive defect (100–1,000x) compared with the wild type (Fig. 5b). The pH of the inoculum did not affect total colonization or accumulation of diarrhea-like caecal fluid, the primary marker of disease in this model (Extended Data Fig. 8a,b). In single infections with fluorescent wild-type or Δvca0040 V. cholerae, the mutant exhibited severe colonization defects (around 1,000-fold) in all intestinal segments (Fig. 5c,d). These values overestimate the true colonization of the mutant, because rabbits from each group were co-housed to exclude litter effects, enabling substantial transmission from wild-type-infected to Δvca0040-infected rabbits (Extended Data Fig. 8c). Transmission probably explains why caecal fluid accumulation in Δvca0040-infected rabbits was lower than in wild-type rabbits but did not reach statistical significance (Fig. 5e). Caecal fluid collected from both mixed and singly infected animals was strongly alkaline (pH 8.5–9) (Fig. 5f). Imaging of caecal fluid samples from Δvca0040-infected rabbits revealed large, fluorescent, spherical cells similar to those observed in alkaline cultures of the Δvca0040 mutant (Extended Data Fig. 8d). Similarly, when freshly grown rod-shaped wild-type or Δvca0040 V. cholerae were incubated in cell-free caecal fluid samples. Δvca0040—but not wild-type—cells became spherical when exposed to caecal fluid, suggesting that VCA0040 function is needed to maintain V. cholerae shape in the alkaline in vivo milieu (Fig. 5g). An infection defect was not detected for ΔSAOUHSC_00846 S. aureus in a mouse intravenous infection–organ abscess model, in which the pathogen is unlikely to encounter alkaline environments (Extended Data Fig. 9).
Fig. 5. DUF368 is required for V. cholerae pathogenesis.

a,b, Schematic (a) and intestinal competitive indices (b) in mixed-infection models (n = 6 rabbits in each group). SI, small intestine. c–e, Schematic (c), number of intestinal colony-forming units (CFU) (d) and fluid accumulation ratio (FAR) (e) in single infections (n = 3 rabbits (wild type) and n = 8 rabbits (Δvca0040)). CF, caecal fluid; pSI, proximal small intestine; mSI, medial small intestine; dSI, distal small intestine. Open circles indicate rabbits with limit of detection (LOD) measurements where the true CFU burden is at least (for upper LOD circles) or at most (for lower LOD circles) the plotted value. Note two Δvca0040 animals had insufficient caecal fluid accumulation for FAR calculation and were assigned LOD values (arbitrary 25 μl volume). b,d, Data are geometric mean. d, Two-tailed Mann–Whitney U tests. e, Data are mean ± s.d. analysed with an unpaired Student’s two-tailed t-test. f, pH of caecal fluid from infected animals. Data are mean ± s.d. g, Incubation of laboratory-grown V. cholerae with filtered caecal fluid samples from three different rabbits. Representative images from n = 3 ex vivo incubations with independent caecal fluid samples. Scale bars, 3 μm. h, Proposed model for microbial C55-P translocation and its integrated control by environmental inputs, the presence of other translocases, and non-translocase-related environmental adaptation mechanisms.
Extended Data Fig. 8. Supplemental data for infant rabbit infections.
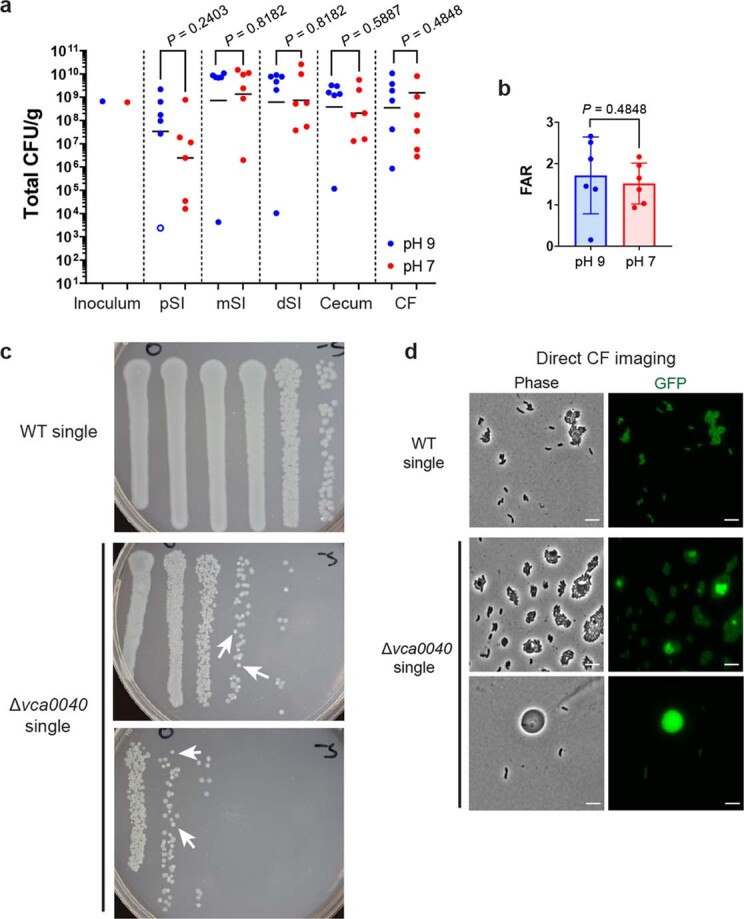
(a): Total CFU in mixed infections with differing inoculum pH (n = 6 each for pH 7 and 9). (b): Fluid accumulation ratio (FAR) in mixed infections with differing inoculum pH. (c): Representative small intestinal CFU plates from single infections showing transmission of WT V. cholerae (greyish translucent colonies) from WT-infected rabbits to Δvca0040-infected rabbits in the same litter. Note that Δvca0040 colonies (white arrows) are lighter and smaller than WT colonies, making them visually distinguishable and countable. (d): Direct imaging of GFP+ V. cholerae in single infection CF samples. Large GFP-bright spheres are likely Δvca0040 cells in the presence of transmitted, rod-shaped WT cells. Scale bars, 3 μm. a: geometric means analyzed with two-tailed Mann-Whitney U tests. b: mean ± SD analyzed with an unpaired student’s two-tailed t-test. c, d: representative images from n = 3 WT and n = 7 Δvca0040 singly-infected rabbits.
Extended Data Fig. 9. Intravenous (IV) infections of mice with S. aureus.

(a): Schematic of IV infections and sample harvesting workflow. TSA counts indicate total S. aureus CFU burden, whereas TSAK counts indicate ΔSAOUHSC_00846 burden. N = 5 mice were used per timepoint (n = 10 total). (b): Raw CFU counts from TSA and TSA + kanamycin (TSAK) plates. In the absence of a competitive defect, the TSAK counts should be 50% of those on the TSA plates (and thus are very close together on the log10-plotted graph). (c): Ratio of ΔSAOUHSC_00846 (KanR) colony-forming units (CFU) to total S. aureus CFU at Day 2 (left) and Day 5 (right) post-infection. The dotted line indicates the mean starting proportion from n = 2 technical replicate plates of the inoculum. Open circles represent the limit of detection (LOD). Note that samples with CFU counts below the LOD were not plotted. b: geometric means. c: means ± SD.
Discussion
We propose that DUF368-containing and DedA family proteins are C55-P translocases that provide functional resiliency in this crucial step of envelope maintenance (Fig. 5h). The identification of C55-P transport proteins provisionally fills a major gap in the C55 lipid carrier recycling pathway. As our genetic and phenotypic data do not definitively exclude transport-independent activities, biochemical and structural studies are required to confirm whether these two families indeed carry out this function. The apparent pH and ion dependency of the contributions of these candidate translocases to fitness suggests that these proteins could be energetically controlled, since a major effect of pH on the microbial cell is the governance of transmembrane ionic gradients. For example, DUF368-containing proteins appear to contribute most in alkaline conditions where the PMF is low38, suggesting that if C55-P translocation is energy-dependent, DUF368-mediated transport may depend on the SMF or another non-PMF energy gradient. It is also possible that protonation states of C55-P, which are probably pH-dependent, determine the maximally active translocase or potential contribution of spontaneous C55-P scrambling. Characterizing the potential energetic reliance and directionality of multiple C55-P translocases will be an important topic of future study.
The conditionality of DUF368-containing and DedA family proteins, and perhaps additional as-yet-unidentified translocases, could support C55-P flux in diverse microbial niches (Fig. 5h). The sensitivity of Δvca0040 V. cholerae to increased sodium ion concentration and the apparent enrichment of DUF368 in other known halophiles such as S. aureus and the archaeal class Halobacteria (including H. volcanii) suggests that regardless of their energetic input, DUF368-containing proteins may facilitate microbial adaptation to high-salt environments (Supplementary Table 1). Specifically, the sodium and pH-dependence of the contribution of VCA0040 to V. cholerae physiology may reflect the robust capacity of the cholera pathogen to colonize the human small intestine, where alkaline conditions and millimolar-scale sodium concentrations are likely to occur39. Alongside the variable conservation of redundant translocases (Extended Data Fig. 10a and Supplementary Table 9), additional inputs and adaptation mechanisms likely also shape how C55-P recycling contributes to microbial fitness. For example, although vca0040 is required only in alkaline conditions in V. cholerae, this gene is essential in the related pathogen Vibrio parahaemolyticus even at neutral pH, despite the conservation of yghB and suppressor loci, suggesting that the pH setpoint at which DUF368 family activity is dominant is not universal40 (Extended Data Fig. 10b). Although DUF368-containing proteins—which have been renamed polyprenyl phosphate transporter (PopT) proteins in concurrently reported work41—are restricted to bacteria and archaea, DedA family members are widely present in eukaryotes, including humans. Thus, our findings may affect the understanding of polyprenyl phosphate (for example, dolichol phosphate) translocation in all kingdoms of life.
Extended Data Fig. 10. Distribution of putative C55-P translocases in bacteria and genetic analysis of a DUF368 protein in V. parahaemolyticus.

(a): Venn diagram of Annotree species-level outputs for PFAM queries PF04018 (DUF368), PF09335 (DedA), and PF02673 (BacA/UppP). Out of 30,255 bacterial species in the Annotree database, 29,416 (97.2%) are predicted to contain at least one of the three proteins. A list of species in each Venn diagram segment is listed in Supplementary Table 9. This is likely also an overestimate of the number of species lacking any of these three domains due to sequence divergence and potentially inadequate PFAM annotation. For example, the obligate intracellular pathogen Rickettsia rickettsii (GCA_000018225.1) is in the triply-absent group, but upon manual curation is known to have peptidoglycan and has an annotated DedA coding sequence, perhaps reflecting a PFAM annotation lag. Interestingly, however, Mycoplasma species, which do not have lipopolysaccharide or peptidoglycan (and thus may not be dependent on C55-P translocation) appear to be genuinely triply-absent except for a select few members with only a DUF368-containing protein. (b): Growth of V. parahaemolyticus lacking the DUF368-containing protein VPA1624 at neutral and acidic pH. VPA1624 genetically interacts with the V. parahaemolyticus secDF1 loci, as deletion of these suppressors originally identified in V. cholerae rescues the vpa1624-depleted strain. Ara: arabinose, Glc: glucose. Representative data of n = 3 independent cultures per strain/condition.
Methods
Bacterial strains, media and growth conditions
All V. cholerae strains used in this study are derivatives of HaitiWT, a spontaneous streptomycin-resistant variant of a clinical isolate from the 2010 Haiti cholera epidemic42. All S. aureus strains used in this study are derivatives of HG003 (itself a derivative of NCTC8325). Strain and plasmid information is listed in Supplementary Table 10. All constructed strains were verified by Sanger sequencing of the targeted locus (Genewiz).
The following media were used in this study: lysogeny broth (LB) Miller (10 g l−1 NaCl) (BD Biosciences, USA), M9 minimal media (12.8 g l−1 Na2HPO4·7H2O, 3 g l−1 KH2PO4, 0.5 g l−1 NaCl, 1 g l−1 NH4Cl, 1 mM MgSO4 and 10 µM CaCl2) (lab-made), M63 minimal media (2 g l−1 (NH4)2SO4, 13.6 g l−1 KH2PO4, 0.5 mg l−1 FeSO4·7H2O and 1 mM MgSO4) (US Biologicals), TSB and tryptic soy agar (TSA) (BD Biosciences). M9 was supplemented with 0.4% glucose and pH-adjusted with NaOH. M63 was supplemented with 2% glucose and pH-adjusted with KOH. In general, LB was buffered with 50 mM Na2HPO4 or 100 mM Tris and pH-adjusted with NaOH or HCl, and TSB was buffered with 100 mM bicine. Variations in buffering are noted in figure legends. Plates were used at 1.5% final agar concentration. Where necessary, the following antibiotics or supplements were used: streptomycin (200 μg ml−1), carbenicillin (50 µg ml−1), kanamycin (50 µg ml−1), neomycin (50 ug ml−1), targocil (10 μg ml−1), erythromycin (10 μg ml−1), diaminopimelic acid (DAP, 0.3 mM), 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-gal, 60 µg ml−1), arabinose (0.2%) or IPTG (1 mM).
For routine culture, V. cholerae were grown in non-buffered LB at 37 °C and S. aureus were grown in non-buffered TSB at 37 °C. Cultures for spent supernatant analysis in V. cholerae were obtained by subculturing 37 °C-grown overnight cultures 1:1,000 in fresh media and growing for 24 h at 30 °C. To collect spent supernatants, cultures were centrifuged for 10 min at 5,000g at 4 °C. Supernatants were transferred to a new tube, re-centrifuged, and sterile filtered with 0.22 µm syringe filters into new tubes. Supernatants were stored at 4 °C for future use. Cultures used for pH quantification were centrifuged for 10 min at 5,000g before measurement. All pH measurements, including for media and buffer formulation, were performed with a pH meter (Thermo ORION) freshly calibrated to two appropriate pH standards from 4, 7 and 10.
Bioinformatic methods
Information on the loci, genomes, and accession numbers for genes and proteins from this study is available in Supplementary Table 10.
Genomic locus correspondence in V. cholerae
During our study, which mainly used a contemporary pandemic strain of V. cholerae (HaitiWT, also known as KW3, assembly accession GCA_001318185.1), we noted several annotation inconsistencies with genes ostensibly conserved in the reference shotgun assembly (GCA_000006745.1) of N16961 V. cholerae. Compared to newer long-read assemblies (GCA_003063785.1 and GCA_900205735.1), the original assembly of N16961 has ~50 more ORFs annotated as pseudogenes due to frameshifts that are not included in coding sequence tables. Three of these were directly related to our study: (1) lacZ, a widely used reporter gene (VC2338), (2) secF2 (VCA0692), and (3) yghB (VCA0534). lacZ is known to be functional in N16961, and re-sequencing of our laboratory N16961 stock and examination of newer N16961 assemblies confirmed that secF2 and yghB are in fact intact in this strain and lack the predicted frameshift. The ‘pseudogene’ annotation of secF2 led to its recent usage as a safe-harbour locus for genome editing in V. cholerae43, and caution is probably required for this method. To resolve the issues of difference between strain genomes, a batch BLAST dictionary assigning putative VC locus tags (the conventional V. cholerae gene naming system) to annotated HaitiWT loci is given in Supplementary Table 11.
Phylogenetic and domain architecture analysis
To identify sequences with annotated DUF368 domains, Annotree44 was used with the Pfam identifier PF04018 at a cut-off e-value of 1x10−30. The results from this search are included in Supplementary Table 1. Taxa with ‘_X’ names were manually collated into a single group for ease of analysis. In a related effort, to identify annotation-independent homologues of vca0040, we used a HMMER search with the VCA0040 sequence from wild-type V. cholerae (Supplementary Table 2). To analyse the relative distributions of PF04018, PF09335 (DedA), and PF02673 (BacA), we used Annotree at a cut-off e-value of 1 × 10−15. Differential species lists with all possible conservation combinations were visualized with a Venn diagram generator (https://bioinformatics.psb.ugent.be/webtools/Venn/) and are provided in Supplementary Table 9.
To assess domain architecture, we used InterProScan (https://www.ebi.ac.uk/interpro) with DUF368 (PF04018) or DedA (PF09335) as queries and manually examined identified domain structures. Sequences with domain architectures relevant to this study were exported and collated in Supplementary Table 3.
Structural modelling
Structural prediction of DUF368 and DedA family proteins in this study was performed with AlphaFold2 on the CoLabFold publicly accessible interface45. Sequences were modelled as monomers using mmseqs2 for multiple sequence alignment. Structures were ranked by pLDDT and the top-ranked structures were visualized with ChimeraX (UCSF).
Cloning, vectors and strain construction
V. cholerae
Cloning of expression vectors and deletion plasmids was performed by standard isothermal assembly techniques with 25-bp overhangs on each fragment (HiFi DNA Assembly Kit, NEB). V. cholerae mutants were generated by allelic exchange as previously described with the suicide vector pCVD442 bearing 500–700 bp upstream and downstream homology arms of the targeted locus46,47. Either SM10λpir or MFDλpir E. coli (a DAP auxotroph) were used as the donor strain with identical conjugation conditions apart from DAP supplementation. Single crossover transconjugants were isolated by selective plating on LB + streptomycin/carbenicillin and passaged in 10% sucrose overnight at room temperature to select for double crossover events. Cells were plated on LB + streptomycin, re-patched onto LB + streptomycin and LB + streptomycin/carbenicillin, and streptomycin- and carbenicillin-resistant colonies were screened by colony PCR for successful deletion. For deletions involving vca0040, vca0040 was always deleted last and at least two clones per strain were stocked and verified for the correct phenotype to guard against spontaneous suppressor formation. For native vca0040 expression from the ectopic chromosomal site, the full coding sequence along with 350 bp upstream was cloned to include the native promoter and inserted at a known neutral genomic location in V. cholerae48. For expression of SAOUHSC_00846 from the vca0040 locus in Δvca0040 V. cholerae, we used allelic exchange to replace the vca0040 coding sequence with a SAOUHSC_00846 sequence codon-optimized for V. cholerae. For depletion of yghB from Δvca0040 V. cholerae, we used pAM299, a suicide vector which replaces the native allele of targeted gene with an arabinose-inducible copy49. For overexpression studies, we used pBAD18 for plasmid-based arabinose induction50.
S. aureus
S. aureus ΔSAOUHSC_00846 was generated with a one-step allelic exchange using the pTarKO plasmid bearing 1,000 bp upstream and downstream homology arms of the targeted locus flanking a kanamycin cassette as previously described51. In brief, the assembled pTarKO plasmid was electroporated into electrocompetent S. aureus RN4220 tarOoff and selected for double crossovers on TSB with kanamycin, neomycin and targocil. The kanamycin insertion in SAOUHSC_00846 was then transduced to S. aureus HG003 with phage phi85. An identical protocol was used to generate ΔSAOUHSC_02816. For ΔSAOUHSC_0901, the strain NE1150, which carries an erythromycin-resistance-marked transposon insertion in SAOUHSC_0090152, was used as the donor for transduction into HG003. For overexpression studies, we used pLOW for plasmid-based IPTG induction53. Assembled plasmids were electroporated into RN4220 wild type and transduced to S. aureus HG003 ∆SAOUHSC_00846 with phage phi85.
E. coli lptD4213 arabinose suppressor selection
E. coli MC4100 and its derivative lptD4213 are sensitive to arabinose due to the araD139 mutation. To enable the use of the pBAD arabinose expression system, spontaneous arabinose-resistant mutants of MC4100 lptd4213 were isolated as previously described54. In brief, overnight cultures of lptD4213 E. coli were plated (~10 µl) on LB + 0.4% arabinose plates and incubated at 37 °C. Putative suppressor colonies were purified on a new LB + 0.4% arabinose plate. Suppressor colonies were then screened for growth on LB + 0.4% arabinose and no growth on M9 minimal agar + 0.2% arabinose, in order to select for strains that are resistant to arabinose, but unable to use arabinose as carbon source (that is, ara−/r, but not ara+ colonies). To transform E. coli with various vectors, competent cells were heat shocked according to a standard protocol and selected on LB supplemented with the appropriate antibiotic(s).
V. parahaemolyticus
V. parahaemolyticus RIMD2210633 mutants were constructed with a similar allelic exchange system to V. cholerae as previously described, with the suicide vector pDM440.
Growth assays
Growth curves in liquid medium
For automated A600 measurements, 1 ml of a saturated 37 °C LB overnight V. cholerae culture was washed once with 1 ml fresh media (specific to the experiment) and resuspended in 1 ml fresh media. Resuspended bacteria were diluted to a starting dilution of 1:4,000 by performing a 1:100 dilution into 1 ml fresh media and a subsequent 1:40 dilution into 195 µl of specific media aliquoted into sterile 96-well plates (Corning). At least three technical replicates per strain and media condition were run per plate along with at least three blank media wells. Growth curves were performed in a BioTek Epoch2 spectrophotometer with shaking and A600 readings were taken every 10 min for 20–24 h. Gen5 version 3.08 (BioTek) was used for data collection. Data for each condition were averaged across technical and biological replicates corrected against the baseline blank A600 values.
Overexpression vector plate dilution assays on solid medium
V. cholerae strains were grown overnight at 37 °C in LB with addition of 50 µg ml−1 carbenicillin for strains with pBAD18. The cultures were diluted 1:100 into 5 ml of LB (supplemented with 50 µg ml−1 carbenicillin and 0.2% arabinose for strains with pBAD18) and grown at 37 °C to an OD600 of ~0.7. Cells were normalized to an A600 of 0.1 and then serially tenfold diluted 6 times. Five microlitres of the dilution series were plated on LB agar 100 mM Tris pH 9 (with 0.2% arabinose, 0.2% glucose, or no sugar added) and incubated at 30 °C for 18–24 h.
S. aureus strains were grown overnight at 37 °C in TSB with addition of 10 µg ml−1 erythromycin for strains with pLOW. The cultures were diluted 1:100 into 5 ml of TSB (supplemented with 10 µg ml−1 erythromycin and 1 mM IPTG for strains with pLOW) and grown at 37 °C to an A600 of ~0.7. Cells were normalized to an A600 of 0.1 and then serially tenfold diluted 6 times. Five microlitres of the dilution series were plated on TSA 100 mM Tris pH 9 (with or without 1 mM IPTG) and incubated at 37 °C for 18–24 h.
Live and single timepoint phase-contrast microscopy
For single timepoint imaging of live cells, samples from the indicated cultures were concentrated as necessary and immobilized on 0.8% agarose pads in sterile PBS on glass slides (Gene Frames, Thermo) and dried before coverslip placement. For time-lapse imaging of live cells, samples were spotted on 0.8% agarose pads in sterile LB before imaging in a temperature-controlled chamber at a rate of 1 frame per minute for 3 h of growth. Cells were imaged with a Nikon Eclipse Ti microscope equipped with an Andor NeoZyla camera and a 100× oil phase 3 1.4-numerical-aperture (NA) objective using NIS Elements AR software version 4.6 (Nikon). Image analysis was performed with ImageJ version 1.53. Images in manuscript figures are representative of at least 10 fields of view (FOVs) from the same sample (>200 cells) and multiple independent replicate cultures as indicated.
Sphere formation and incubation assays
To induce sphere formation, V. cholerae 37 °C overnight cultures (where Δvca0040 cells are still largely rod-shaped) were back-diluted 1:100 into the indicated supernatant or fresh media and grown for 4 h shaking at 200 rpm at 30 °C. Then, cells were imaged as described above. For the d/l-Ala treatment assay, overnight cultures were expanded in fresh LB for 90 min prior to spike-in of the amino acid for 1 h.
MIC assays
V. cholerae
To quantify MICs for various antimicrobial agents, 37 °C overnight cultures of V. cholerae were diluted 1:100,000 in fresh media. Diluted cultures were used to inoculate 96-well plates containing 12 twofold dilutions of the indicated agent in LB medium at a ratio of 50 µl culture:50 µl medium. Four technical replicates were performed per strain per dilution. Plates were incubated for 24 h at 37 °C and MIC values were read as the first dilution where no turbidity was observed. For repeat assays, MICs were performed with independent overnight cultures.
S. aureus and E. coli
Overnight cultures (at 37 °C) of S. aureus or E. coli were diluted to A600 = 0.01 and then further diluted 1:100 in fresh TSB media. Diluted cultures were added to 96-well plates containing twofold dilutions of the indicated agent in TSB medium or TSB medium 5 mM Tris pH 8.5 at a ratio of 75 µl culture:75 µl media. Plates were incubated for 24 h at 30 °C (37 °C for E. coli) with shaking. MIC values were read off as the first dilution where no turbidity was observed.
For E. coli lptD4213 vector overexpression MICs, overnight cultures were diluted 1:100 into non-buffered LB + carbenicillin + 0.2% arabinose. The culture was outgrown for 1.5–2 h and diluted to A600 = 0.01. This was further diluted to A600 = 0.0001 and 75 µl of the culture was added to 75 µl of serial dilution of the drug. Diluted cultures were added to 96-well plates containing 2-fold dilutions of the indicated agent in is LB 5 mM Tris pH 8.5, 1 mM CaCl2, with 0.2% glucose or 0.2% arabinose at a ratio of 75 µl culture: 75 µl media. Plates were incubated and read as described above.
Peptidoglycan characterization
Preparation of V. cholerae and S. aureus sacculi
For V. cholerae, strains were grown overnight at 37 °C in LB + 200 µg ml−1 streptomycin. For each sample, 500 µl of overnight culture were collected and centrifuged at 5,000g for 5 min, washed once with 500 µl of the corresponding media, and resuspended in 500 µl of the corresponding media. This culture was then added to 50 ml of M63 media (pH 7 with 100 mM NaCl, pH 8 with 100 mM NaCl or pH 8 with 0 mM NaCl) supplemented with 2% glucose and 200 µg ml−1 streptomycin and incubated at 30 °C for approximately 6 h until the A600 reached ~0.5. Bacteria were collected by centrifugation at 6,000g for 10 min at 4 °C and resuspended in 1.5 ml of 1× PBS. The resuspension was added dropwise into 1.5 ml of boiling 5% SDS solution. The mixture was boiled for 1 h and stirred for a further 2 h after the heat was turned off.
For S. aureus, strains were grown overnight at 37 °C in 3 ml TSB. 500 µl of the overnight culture was added to 100 ml of TSB without pH adjustment or TSB + 100 mM bicine pH 8.5 and incubated at 37 °C for ~2 h until the A600 reached 0.5. The bacterial cells were collected by centrifugation at 5,000g for 10 min at 4 °C and resuspended in 1.5 ml of 1× PBS. Resuspended samples were boiled as for V. cholerae.
Total peptidoglycan and cross-linking quantification from sacculi samples
Peptidoglycan was extracted from boiled samples as described previously for Gram-negative and Gram-positive bacteria55. Once boiled, cell wall material was pelleted by ultracentrifugation and washed with water. Clean sacculi were digested with muramidase (100 μg ml−1) and soluble muropeptides reduced using 0.5 M sodium borate pH 9.5 and 10 mg ml−1 sodium borohydride. The pH of the samples was then adjusted to 3.5 with phosphoric acid. UPLC analyses were performed on a Waters UPLC system equipped with an ACQUITY UPLC BEH C18 Column, 130 Å, 1.7 μm, 2.1 mm × 150 mm (Waters Corporation) and identified by A204 nm. Muropeptides were separated using a linear gradient from buffer A (0.1% formic acid in water) to buffer B (0.1% formic acid in acetonitrile). Identification of individual peaks was assigned by comparison of the retention times and profiles to validated chromatograms. The relative amount of each muropeptide was calculated by dividing the peak area of a muropeptide by the total area of the chromatogram. The abundance of peptidoglycan (total peptidoglycan) was assessed by normalizing the total area of the chromatogram to the A600. The degree of cross-linking was calculated as described previously56.
Intracellular UDP-M5 quantification
For V. cholerae, strains were grown overnight at 37 °C in LB + 200 µg ml−1 streptomycin. For each sample, 500 µl of overnight culture were collected and centrifuged at 5,000g for 5 min, washed once with 500 µl of the corresponding media, and resuspended in 500 µl of the corresponding media. Eighty microlitres of this resuspension was then added to 8 ml of M63 media (pH 7 with 100 mM NaCl, pH 8 with 100 mM NaCl or pH 8 with 0 mM NaCl) supplemented with 2% glucose and 200 µg ml−1 streptomycin and incubated at 30 °C for approximately 6 h until the A600 reached ~0.5. Bacteria cells were collected by centrifugation at 5,000g for 10 min at 4 °C, washed twice with 1 ml of ice cold 0.9% NaCl, and resuspended in 200 µl of milliQ water. The resuspension was boiled for 30 min, centrifuged at 20,000g for 15 min, and the supernatant was filtered and analysed.
For S. aureus, strains were grown overnight at 37 °C in 3 ml TSB. Eighty microlitres of the overnight culture was added to 8 ml of TSB without pH adjustment or TSB + 100 mM bicine pH 8.5 and incubated at 37 °C for ~2 h until the A600 reached ~0.5. Cultures were then processed as for V. cholerae.
Quantification of soluble UDP-M5 muropeptide levels by liquid chromatography–mass spectrometry (LC–MS) of filtered supernatants was performed as previously described57. Detection and characterization of soluble muropeptides by LC–MS was performed on an UPLC system interfaced with a Xevo G2/XS Q-TOF mass spectrometer (Waters Corporation) using previously reported conditions57. UDP-M5 levels were quantified by integrating peak areas from extracted ion chromatograms (EICs) of the corresponding m/z value. UDP-M5 abundance was normalized to culture A600 as for total peptidoglycan.
Quantification of C55-OH and C55-P in bacterial membrane lipid extracts
Preparation of S. aureus or V. cholerae membranes
S. aureus or V. cholerae cells were grown overnight at 37 °C in 10 ml TSB or LB pH 7, respectively. Cells were washed, collected by centrifugation, resuspended in the experimental medium (for example, TSB or LB at a given pH) and inoculated at a 1:200 dilution into 200 ml of the experimental medium. Once an A600 of 0.6 was reached, cells were collected by centrifugation, resuspended in 10 ml of phosphate-buffered saline (PBS), and disrupted in a French press. Cell debris was discarded, and the supernatant centrifuged in a Beckman Optima Max TL ultracentrifuge for 15 min at 270,000g. Lipids from the membrane pellets were then extracted as described below.
Extraction of lipids from membrane fractions
S. aureus and V. cholerae membrane lipids were extracted largely as previously described58. Membrane pellets were resuspended in 500 µl of PBS. Then, 1.25 ml of methanol and 625 µl of chloroform were added. The suspension was vortexed for 2 min at room temperature, and the homogenates were centrifuged at 7,100g for 10 min at 4 °C. 625 µl of chloroform and 625 µl of PBS were added to the supernatants and they were vortexed and centrifuged at 7,100g for 10 min at 4 °C to separate the chloroform phase from the PBS–methanol phase. The chloroform phase was then isolated and vacuum dried. Dried pellets containing the purified membrane lipids were resuspended in 300 µl of the UPLC mobile phase solvents (50% H2O with 0.1 % formic acid + 50% isopropanol with 0.1 % formic acid). To ensure that this method is capable of extracting C55-OH and C55-P, 200 nmol of each lipid standard (Larodan) was added to fresh purified membrane samples before subjecting them to the extraction protocol.
Detection of C55-OH and C55-P by UPLC
Lipids extracts resuspended in UPLC mobile phase solvents were analysed by UPLC on a reverse-phase C18 column Kinetex C18 UPLC Column 1.7 µm particle size, 100 Å pore size, 50 × 2.1 mm. Separation of the lipids was accomplished using a gradient of H2O with 0.1% formic acid for solvent A and isopropanol with 0.1 % formic acid for solvent B, a flow rate of 0.4 ml/min with a linear gradient over 8 min, column temperature 60 °C and a wavelength of 210 nm. Identification of C55-OH and C55-P was based on the retention time of standards and their abundances were calculated relative to the total peak area of each chromatogram. For calculating mutant-to-wild type ratios, samples from independent cultures grown on the same day were randomly paired. Empower3 chromatography data software (Waters) was used for UPLC data collection and analysis.
Ampho-FL synthesis and staining
To synthesize ampho-FL, 5 mg of amphomycin (Cayman Chemical) was dissolved in 200 µl of dimethyl formamide (DMF) and combined with 7 µl of triethylamine. Separately, 3 mg of fluorescein-C5,6-NHS (Thermo Fisher, USA) was dissolved in 200 µl of DMF and then added to the amphomycin solution. The reaction mixture was stirred at room temperature in the dark for 24 h. The solution was diluted in one equal volume of DMSO and purified by reverse-phase HPLC (Agilent 1260 Infinity) using a C18 stationary phase column (Luna 5 µM C18(2) 100 Å, 250 × 10 mm). HPLC conditions were as follows—phase A: water (0.1% formic acid); phase B: acetonitrile (0.1% formic acid). Phase B: 0–2 min, 50%; 2–15 min, linear gradient 50%–100%, 15–17 min, 100%. The wavelength of the detector was set at 254 nm. The flow rate was 4.7 ml min−1. The mixture of conjugated fluorescein-C5,6 products eluted at 9 min. HPLC eluates were collected in a 50 ml round-bottom flask, concentrated by rotatory evaporation, transferred with DMSO to a tared microcentrifuge tube and lyophilized, resulting in 1.7 mg of ampho-FL (M+1 = 1,649.23).
For ampho-FL labelling, overnight cultures of S. aureus HG003 wild type or ∆SAOUHSC_00846 were diluted 1:100 in fresh TSB media buffered at pH 6 (100 mM MES), 7 (100 mM Tris), or 8.5 (100 mM bicine). The new cultures were grown to A600 = 0.5. 1 ml of the culture was centrifuged at 1,000g for 5 min. The pellet was resuspended in 500 µl of 1× Tris-buffered saline (TBS) pH 9. Ninety-eight microlitres of the mixture was transferred to a new tube and combined with 2 µl of 10 mg ml−1 ampho-FL conjugate in DMSO. The mixture was incubated in the dark at room temperature for 10 min, washed three times with 500 µl of 1× TBS pH 9, and resuspended in 100 µl of 1× TBS pH 9. Ten microlitres bacteria was then added to a 0.8% agarose pad in TBS pH 9 containing 1 μg ml−1 propidium iodide and imaged as described above. To avoid bias in FOV selection, the pad was scanned with the FITC channel off to select FOVs based solely on cell density. Bacterial cells were imaged using DIC at a 30 ms exposure and ampho-FL signal was captured with the FITC filter at a 1 s exposure. Fluorescence intensity of ampho-FL was quantified using ImageJ version 1.53. FITC signal profiles were generated by drawing bisecting lines through dividing, propidium iodide-negative cells. The FITC signal was recorded at the background to the left of the cells (A), at the left wall peak (B), at the middle walls (C), at the right wall peak (D), and at the background to the right of the cells (E). The average signal was calculated by (B + C + D)/4 − (A + E)/2.
Transposon-insertion sequencing
Generation of transposon libraries in the Δvca0040 and ΔyghB backgrounds was performed as previously described for HaitiWT V. cholerae15. Strains were conjugated to SM10λpir E. coli bearing the donor transposon vector pSC189. Due to the moderate conjugation defect of Δvca0040, we used a 5× concentration of the conjugation reactions compared to the other libraries. Reactions were plated on 245 mm2 LB + streptomycin/kanamycin agar plates to isolate V. cholerae transconjugants and incubated overnight at 30 °C. Two independent Δvca0040 and ΔyghB libraries were generated, consisting of approximately 200,000 colonies each and stocked at an A600 of ~10 in LB + 25% glycerol. For synthetic transposon-insertion sequencing analyses, a frozen aliquot of each library was thawed and used for genomic DNA extraction with the Wizard kit (Promega). DNA libraries were prepared in an identical manner to previous transposon-insertion sequencing experiments from our group and sequenced on an in-house MiSeq platform (lllumina)18. Reads were trimmed, mapped, and processed as previously described with the Con-ARTIST transposon-insertion sequencing analysis pipeline using Python version 3.0 and Matlab version 918. To identify synthetic interaction loci, we used the wild type as the ‘input’ library and the mutant as the ‘output’ library during analysis.
RNA sequencing
For transcriptomic analyses, triplicate wild-type and Δvca0040 V. cholerae 37 °C overnight cultures were back-diluted 1:100 into fresh LB medium and grown at 30 °C for 8 h with shaking. At 8 h, samples were checked by microscopy to ensure onset of sphere formation in the mutant samples, at which point 2 ml of each culture was spun down (5 min at 5000g at room temperature). RNA was extracted with Trizol reagent (Sigma-Aldrich) per the manufacturer’s instructions. Isolated RNA was then DNase-treated and re-isolated with ethanol precipitation according to a standard protocol. RNA samples were quality checked with a Bioanalyzer to confirm RIN values > 6. Library preparation and sequencing was performed by the Microbial Genome Sequencing Center (Pittsburgh). RNA-sequencing analysis was performed largely as described59. Reads were mapped to the V. cho\lerae KW3 genome (NCBI assembly GCA_001318185.1) with Bowtie2 (version 2.4.1)60. A read matrix for each sample was then generated with featureCounts from Rsubread (version 2.4.3)61. Read matrices were then analysed with DESeq2 (version 1.30.1) using default settings in R (version 4.0.3) to identify differentially expressed genes. Data shrinkage was performed with ashr62.
Multiplexed tandem mass tag proteomics
For proteomic analyses, triplicate 37 °C wild-type and ΔsecDF1 V. cholerae overnight cultures were grown as described in ‘RNA sequencing’. At 8 h of growth, whole-cell pellet (WCP) samples were prepared by centrifuging 1 ml of cells for 5 min at 5,000g at room temperature, washing once in fresh LB, flash frozen and kept at −80 °C. Mass spectrometry analysis was performed by the Thermo Fisher Center for Multiplexed Proteomics (TCMP) at Harvard Medical School according to standard protocols. WCP samples were subjected to a total proteomics workflow with fractionation. Pelleted cells were first lysed in 8 M urea, 200 mM 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid (EPPS) and 1% SDS with phosphatase and protease inhibitors. Then, samples were reduced with DTT and alkylated with iodoacetamide. Alkylated proteins were precipitated with methanol/chloroform and resuspended in 200 mM EPPS pH 8 and digested sequentially with 1:50 LysC and 1:100 trypsin. Peptides were labelled with TMT16 reagents, pooled, and fractionated by a basic reverse-phase protocol into 12 fractions. Fractions were then dried, cleaned on a C18-packed stage tip, and eluted into an mass spectrometry sample vial for analysis. Samples were resuspended in 5% acetonitrile/5% formic acid and analysed by LC–MS3 on an Orbitrap Lumos mass spectrometer using a 180 min MS3 method. Peptides were detected (MS1) and quantified (MS3) in the Orbitrap, and sequenced (MS2) in the ion trap. MS2 spectra were searched with the COMET algorithm against the V. cholerae KW3 proteome, its reversed complement, and known contaminants. Spectral matches were filtered to a 1% false-discovery rate using the target-decoy strategy combined with linear discriminant analysis. Proteins were quantified from peptides with a summed signal to noise threshold of >150 and isolation specificity of >0.5.
Spontaneous suppressor isolation and sequencing
V. cholerae
To ensure independent isolation of spontaneous suppressors, the Δvca0040 strain was re-derived 11 times from different conjugation reactions. From each colony PCR- and cell shape defect-verified Δvca0040 clone, colonies with mutant morphology (small and completely blue) on LB + streptomycin/X-gal plates were re-streaked onto new LB + streptomycin/X-gal plates and grown for 36–48 h at 37 °C. This process was repeated once. From the tertiary plates, a single colony with wild-type morphology (large with a white halo) was re-streaked to confirm a stable suppressor phenotype. Suppressors were checked by microscopy of 30 °C overnight cultures to confirm cell shape defect reversion and verified by colony PCR to confirm the absence of vca0040 from their genome. Validated suppressors were grown overnight, and genomic DNA was extracted as described above. Library preparation and sequencing were either performed in-house or outsourced to the Microbial Genome Sequencing Center (Pittsburgh). For in-house whole-genome sequencing, gDNA was tagmented and barcoded with the Nextera XT library preparation kit (Illumina), quality checked by BioAnalyzer and sequenced on a MiSeq platform to at least 20–50× depth of the V. cholerae genome (~4 Mbp). Genome assembly and variant identification was performed with CLC Genomics Workbench 12 (Qiagen, Germany). Variants were filtered against an assembled HaitiWT isolate re-sequenced with the same workflow. Mutations were assigned as suppressors if they were present in >90% of reads and did not occur in a known poorly mapping or highly repetitive region such as a tRNA locus. For the secondary screen in secDF2- Δvca0040 bacteria, the Δvca0040 deletion was re-derived three independent times in the ΔsecD2 or ΔsecF2 background. Suppressors were isolated and sequenced identically to those in the parental Δvca0040 background.
S. aureus
Multiple 2 ml cultures of S. aureus HG003 ∆SAOUHSC_00846 were grown in TSB overnight at 37 °C. One-hundred and ninety microlitres from each independent overnight culture was then separately plated on TSA 100 mM Tris pH 9 plates (where the mutant is entirely inhibited for growth) and grown at 37 °C for 24 h. Suppressors were confirmed by re-streaking on TSA 100 mM Tris pH 9 plates. Validated suppressors were grown overnight, and genomic DNA was extracted as described above. Library preparation, sequencing, and variant identification were performed as described above for V. cholerae.
Infant rabbit V. cholerae infections and caecal fluid imaging
Infant rabbit oral infections with V. cholerae were performed as described previously15. One- to two-day-old New Zealand White rabbits (Charles River Laboratories) were housed with their dams for the duration of the experiment in a temperature- and humidity-controlled facility with 12-h light/dark cycles (16–22 °C, 50% humidity). Inocula were prepared by centrifuging a late exponential phase culture (A600 0.6–0.8) V. cholerae and resuspending in 2.5% sodium bicarbonate. For varying the pH of the inocula, sodium bicarbonate was pH-adjusted to pH 9 or 7 with NaOH or HCl immediately before resuspension. Infected kits were were orally gavaged with 109 CFU V. cholerae in a 500 μl gavage volume, returned to their dam, and monitored for 16–20 h post-infection, when they were sacrificed by isoflurane inhalation and intracardiac injection of 20 mEq potassium chloride. Small intestinal segments and the caecum were isolated by dissection, homogenized by bead beading in PBS, and dilutions were plated on appropriate agar plates for colony enumeration. Plates were counted after an overnight incubation at 30 °C. LOD measurements were calculated by imputing a single colony at the lowest (lower bound) or highest (higher bound) dilution where a colony could reasonably have been identified, meaning that LOD values are samples where the true value is either at most (for lower bound LODs) or at least (for higher bound LODs) the limit value. During dissection of the caecum, a 28G needle was used to extract crude caecal fluid. A sample of crude caecal fluid was taken for CFU plating and imaging, and the remainder was centrifuged at 21,000g for 2 min. The pellet was discarded and supernatants were frozen at −20 °C for downstream analyses. For caecal fluid incubation assays, overnight cultures of V. cholerae were back-diluted 1:100 in caecal fluid and grown for 4 h at 30 °C shaking before imaging.
Mouse S. aureus intravenous infection
S. aureus HG003 (wild type) and KanR-marked ΔSAOUHSC_00846 were grown overnight at 37 °C. Cultures were mixed at a 1:1 ratio, combined 1:1 with 50% glycerol, and stored at frozen at −80 °C in several aliquots. For infections, an aliquot was thawed and diluted in PBS to a density of ~3 × 107 CFU ml−1. One-hundred microlitres was injected into the tail vein of 8- to 9-week-old female Swiss Webster mice. Mice were monitored daily and housed in a temperature- and humidity-controlled facility with 12-h light/dark cycles (20–24 °C, 50% humidity). At days 2 and 5 post-infection, mice were sacrificed by isoflurane inhalation and cervical dislocation and the heart, lungs, right kidney, liver and spleen were collected. Organs were homogenized with stainless steel beads in PBS and plated on TSA and TSA + kanamycin (TSAK) and incubated overnight to enumerate total ΔSAOUHSC_00846 CFU, respectively. LOD calculations were performed as for infant rabbit experiments.
Animal use statement
All animal work in this study was performed in accordance with the NIH Guide on Use of and Care for Laboratory Animals and with the approval of the Brigham and Women’s Hospital IACUC (protocol no. 2016N000334 for infant rabbits and protocol no. 2016N000416 for mice).
Statistics and reproducibility
Statistical tests used and replicate information are indicated in the figure legends, relevant methods sections and the Reporting summary. Unless otherwise indicated, experiments were performed with at least three independent biological replicates and either representative results or aggregate data (mean ± s.d.) are shown. Sample sizes were not statistically predetermined. Investigators were not blinded to sample identity throughout the study, and randomization of animals in infection experiments was performed as described in the Reporting Summary. All replicates resulted in similar results. Statistical analyses were performed in Prism 9 (Graphpad) and Microsoft Excel 2020.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-022-05569-1.
Supplementary information
Bacterial and archaeal clades with PF04018-containing proteins. Annotree was used to plot and identify species with an annotated DUF368 domain. Each tab of the spreadsheet is a different level of bacterial classification (Phylum>Order>Family>Genus>Species). Archaeal clades are combined on the tab “Archaea”.
Homologues of VCA0040 in bacteria. HMMER was used to identify homologous sequences to VCA0040 in the Uniprot database. Each species is listed once.
Notable domain architectures of DUF368- and DedA-containing proteins. Sequences were obtained by searching InterProScan with the PF family designations for DUF368 (PF04018) and DedA (PF09335).
RNAseq of Δvca0040 V. cholerae during sphere formation. For the top 40 up-regulated genes in Δvca0040, manual curation of subcellular localization was performed with SignalP and pSortb. P-values were obtained through the Wald test performed by DESeq2 with adjustments for multiple comparisons.
MIC data for V. cholerae, S. aureus and E. coli. Data for each organism is separated into individual worksheets. Note that S. aureus data is split into two worksheets, one for WT versus Δ0846 characterization (“SA”) and one for WT versus Δ0846, Δ2816 and Δ0901 (“SA_dedA”). All V. cholerae data is under “VC” and all E. coli lptd4213 data is under “EC_lptd4213”.
Synthetic transposon-insertion screening in Δvca0040 V. cholerae. Note that intergenic regions (IG_) and regions with <5 informative sites (possible transposon insertion sites) were not analyzed. Hits on the second tab were thresholded at an inverse MWU p-value >100 before sorting by mean log2 fold change.
Synthetic transposon-insertion screening in ΔyghB V. cholerae. Note that this dataset has not been filtered as for Supplementary Table 6.
Multiplexed comparative proteomics of whole-cell WT and ΔsecDF1 V. cholerae. Fold change was calculated by dividing the average normalized relative abundance of proteins in ΔsecDF1 by that of WT.
Conservation of PF04018, PF09335, and PF02763 in bacterial species. Spreadsheet was generated by Annotree queries for all possible combinations of the three protein families. The “Master” sheet lists all species-level organisms with the indicated combination of domains. The “Model Species” sheet lists selected microorganisms of particular interest and can be used to look up any given species in the reference table. Species of interest should be manually confirmed for presence or absence by BLAST to guard against misannotation or non-annotation issues.
Strains and vectors and genomic information used in this study. Any listed strains or vectors may be requested from the lead contact (mwaldor@research.bwh.harvard.edu).
BLAST dictionary for assignment of putative VC names to HaitiWT V. cholerae loci. All coding sequences from HaitiWT were used as queries in a batch BLAST of the N16961 V. cholerae proteome. The top hit by e-value was selected and VC name automatically assigned. However, as batch BLAST outputs a hit regardless of confidence, manual curation is still required for specific loci when using this dictionary.
Acknowledgements
This work was supported by The National Institutes of Health (R01AI-042347 to M.K.W. and F31AI156949 to K.H.), Howard Hughes Medical Institute (M.K.W.), National Sciences and Engineering Research Council of Canada (PGSD3-487259-2016 to B.S.), Swedish Research Council (F.C.), Knut och Alice Wallenbergs Stiftelse (F.C.), Laboratory of Molecular Infection Medicine Sweden (F.C.) and Kempe Foundation (F.C.) We thank C. Wegler in the Cava laboratory for technical assistance with C55-P analysis; members of the Waldor laboratory for comments and discussions during the study; D. Rudner for coordinating submission; the Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School, especially R. Rodrigues, for assistance with proteomics; and the Microbial Genome Sequencing Center (MiGS) for assistance with RNA sequencing and whole-genome sequencing. Schematics and model figures in Figs. 1, 3 and 5 and Extended Data Figs. 5, 7 and 9 were generated with BioRender.com. This article is subject to HHMI’s Open Access to Publications policy. HHMI lab heads have previously granted a nonexclusive CC BY 4.0 license to the public and a sublicensable license to HHMI in their research articles. Pursuant to those licenses, the author-accepted manuscript of this article can be made freely available under a CC BY 4.0 license immediately upon publication.
Extended data figures and tables
Source data
Author contributions
B.S., V.S. and M.K.W. conceived the study. B.S., V.S. and E.B. designed experiments with supervision from F.C. and M.K.W. B.S. performed in vitro and in vivo V. cholerae experiments, as well as bioinformatic analyses and data visualization. V.S. performed in vitro S. aureus experiments, E. coli lptd4213 experiments, and synthesized ampho-FL. E.B. performed biochemical peptidoglycan and C55-P extraction and quantification. F.G.Z. performed in vitro experiments and aided in data replication. K.H. identified V. cholerae spontaneous suppressors, performed the in vivo S. aureus experiment, and aided in data replication. All authors participated in data analysis and interpretation. B.S. and M.K.W. wrote the manuscript with input and editing from all other authors.
Peer review
Peer review information
Nature thanks Carol Gross, Anant Menon and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Data availability
Sequence data were deposited to the NCBI Sequence Read Archive under BioProject accession codes PRJNA868324 (V. cholerae suppressor genome sequencing), PRJNA868332 (V. cholerae RNA sequencing), PRJNA877769 (V. cholerae transposon-insertion sequencing) and PRJNA877773 (S. aureus suppressor genome sequencing). Proteomics data were deposited in MassIVE under the accession code MSV000090217. All other data are freely available without restriction from the corresponding authors upon request. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Brandon Sit, Veerasak Srisuknimit, Emilio Bueno
Contributor Information
Felipe Cava, Email: felipe.cava@umu.se.
Matthew K. Waldor, Email: mwaldor@research.bwh.harvard.edu
Extended data
is available for this paper at 10.1038/s41586-022-05569-1.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-022-05569-1.
References
- 1.Egan AJF, Errington J, Vollmer W. Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol. 2020;18:446–460. doi: 10.1038/s41579-020-0366-3. [DOI] [PubMed] [Google Scholar]
- 2.Mueller, E. A. & Levin, P. A. Bacterial cell wall quality control during environmental stress. mBio10.1128/mBio.02456-20 (2020). [DOI] [PMC free article] [PubMed]
- 3.Vijaranakul U, Nadakavukaren MJ, de Jonge BL, Wilkinson BJ, Jayaswal RK. Increased cell size and shortened peptidoglycan interpeptide bridge of NaCl-stressed Staphylococcus aureus and their reversal by glycine betaine. J. Bacteriol. 1995;177:5116–5121. doi: 10.1128/jb.177.17.5116-5121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madiraju MVVS, Brunner DP, Wilkinson BJ. Effects of temperature, NaCl, and methicillin on penicillin-binding proteins, growth, peptidoglycan synthesis, and autolysis in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 1987;31:1727–1733. doi: 10.1128/AAC.31.11.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mainardi J, et al. Resistance to cefotaxime and peptidoglycan composition in Enterococcus faecalis are influenced by exogenous sodium chloride. Microbiology. 1998;144:2679–2685. doi: 10.1099/00221287-144-10-2679. [DOI] [PubMed] [Google Scholar]
- 6.Pazos M, Peters K, Vollmer W. Robust peptidoglycan growth by dynamic and variable multi-protein complexes. Curr. Opin. Microbiol. 2017;36:55–61. doi: 10.1016/j.mib.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Workman SD, Strynadka NCJ. A slippery scaffold: synthesis and recycling of the bacterial cell wall carrier lipid. J. Mol. Biol. 2020;432:4964–4982. doi: 10.1016/j.jmb.2020.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Sham L-T, et al. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014;345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Ghachi M, et al. Crystal structure of undecaprenyl-pyrophosphate phosphatase and its role in peptidoglycan biosynthesis. Nat. Commun. 2018;9:1078. doi: 10.1038/s41467-018-03477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Workman SD, Worrall LJ, Strynadka NCJ. Crystal structure of an intramembranal phosphatase central to bacterial cell-wall peptidoglycan biosynthesis and lipid recycling. Nat. Commun. 2018;9:1159. doi: 10.1038/s41467-018-03547-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Koirala B, Hernandez Y, Zimmerman M, Brady SF. Bioinformatic prospecting and synthesis of a bifunctional lipopeptide antibiotic that evades resistance. Science. 2022;376:991–996. doi: 10.1126/science.abn4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Häse CC, Barquera B. Role of sodium bioenergetics in Vibrio cholerae. Biochim. Biophys. Acta. 2001;1505:169–178. doi: 10.1016/S0005-2728(00)00286-3. [DOI] [PubMed] [Google Scholar]
- 13.Juárez O, Barquera B. Insights into the mechanism of electron transfer and sodium translocation of the Na+-pumping NADH:quinone oxidoreductase. Biochim. Biophys. Acta. 2012;1817:1823–1832. doi: 10.1016/j.bbabio.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvarez L, Hernandez SB, Cava F. Cell wall biology of Vibrio cholerae. Annu. Rev. Microbiol. 2021;75:151–174. doi: 10.1146/annurev-micro-040621-122027. [DOI] [PubMed] [Google Scholar]
- 15.Hubbard TP, et al. A live vaccine rapidly protects against cholera in an infant rabbit model. Sci. Transl. Med. 2018;10:eaap8423. doi: 10.1126/scitranslmed.aap8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamp HD, Patimalla-Dipali B, Lazinski DW, Wallace-Gadsden F, Camilli A. Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog. 2013;9:e1003800. doi: 10.1371/journal.ppat.1003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu Y, Waldor MK, Mekalanos JJ. Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe. 2013;14:652–663. doi: 10.1016/j.chom.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchard JR, et al. ARTIST: high-resolution genome-wide assessment of fitness using transposon-insertion sequencing. PLoS Genet. 2014;10:e1004782. doi: 10.1371/journal.pgen.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam H, et al. d-Amino acids govern stationary phase cell wall remodeling in bacteria. Science. 2009;325:1552–1555. doi: 10.1126/science.1178123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Touzé T, Blanot D, Mengin-Lecreulx D. Substrate specificity and membrane topology of Escherichia coli PgpB, an undecaprenyl pyrophosphate phosphatase. J. Biol. Chem. 2008;283:16573–16583. doi: 10.1074/jbc.M800394200. [DOI] [PubMed] [Google Scholar]
- 21.Schneider T, et al. The lipopeptide antibiotic Friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob. Agents Chemother. 2009;53:1610–1618. doi: 10.1128/AAC.01040-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubinchik E, et al. Mechanism of action and limited cross-resistance of new lipopeptide MX-2401. Antimicrob. Agents Chemother. 2011;55:2743–2754. doi: 10.1128/AAC.00170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka H, et al. Studies on bacterial cell wall inhibitors. II. Inhibition of peptidoglycan synthesis in vivo and in vitro by amphomycin. Biochim. Biophys. Acta. 1977;497:633–640. doi: 10.1016/0304-4165(77)90283-5. [DOI] [PubMed] [Google Scholar]
- 24.Singh M, Chang J, Coffman L, Kim SJ. Solid-state NMR characterization of amphomycin effects on peptidoglycan and wall teichoic acid biosyntheses in Staphylococcus aureus. Sci. Rep. 2016;6:31757. doi: 10.1038/srep31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell J, et al. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 2011;6:106–116. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Storm DR, Strominger JL. Complex formation between bacitracin peptides and isoprenyl pyrophosphates. The specificity of lipid–peptide interactions. J. Biol. Chem. 1973;248:3940–3945. doi: 10.1016/S0021-9258(19)43823-4. [DOI] [PubMed] [Google Scholar]
- 27.Sampson BA, Misra R, Benson SA. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics. 1989;122:491–501. doi: 10.1093/genetics/122.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker BR, Ives CM, Bray A, Caffrey M, Cochrane SA. Undecaprenol kinase: function, mechanism and substrate specificity of a potential antibiotic target. Eur. J. Med. Chem. 2021;210:113062. doi: 10.1016/j.ejmech.2020.113062. [DOI] [PubMed] [Google Scholar]
- 29.Tiyanont K, et al. Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc. Natl Acad. Sci. USA. 2006;103:11033–11038. doi: 10.1073/pnas.0600829103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doerrler WT, Sikdar R, Kumar S, Boughner LA. New functions for the ancient DedA membrane protein family. J. Bacteriol. 2013;195:3–11. doi: 10.1128/JB.01006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okawa F, et al. Evolution and insights into the structure and function of the DedA superfamily containing TMEM41B and VMP1. J. Cell Sci. 2021;134:jcs255877. doi: 10.1242/jcs.255877. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S, Doerrler WT. Members of the conserved DedA family are likely membrane transporters and are required for drug resistance in Escherichia coli. Antimicrob. Agents Chemother. 2014;58:923–930. doi: 10.1128/AAC.02238-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panta PR, Doerrler WT. A Burkholderia thailandensis DedA family membrane protein is required for proton motive force dependent lipid a modification. Front. Microbiol. 2020;11:618389. doi: 10.3389/fmicb.2020.618389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong P, et al. A UPF0118 family protein with uncharacterized function from the moderate halophile Halobacillus andaensis represents a novel class of Na+ (Li+)/H+ antiporter. Sci. Rep. 2017;7:45936. doi: 10.1038/srep45936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamsås GA, et al. CozEa and CozEb play overlapping and essential roles in controlling cell division in Staphylococcus aureus. Mol. Microbiol. 2018;109:615–632. doi: 10.1111/mmi.13999. [DOI] [PubMed] [Google Scholar]
- 36.Ishii E, et al. Nascent chain-monitored remodeling of the Sec machinery for salinity adaptation of marine bacteria. Proc. Natl Acad. Sci. USA. 2015;112:E5513–E5522. doi: 10.1073/pnas.1513001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ritchie, J. M., Rui, H., Bronson, R. T. & Waldor, M. K. Back to the future: studying cholera pathogenesis using infant rabbits. mBio10.1128/mBio.00047-10 (2010). [DOI] [PMC free article] [PubMed]
- 38.Krulwich TA, Sachs G, Padan E. Molecular aspects of bacterial pH sensing and homeostasis. Nat. Rev. Microbiol. 2011;9:330–343. doi: 10.1038/nrmicro2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Molla AM, Rhman M, Sarker SA, Sack DA, Molla A. Stool electrolyte content and purging rates in diarrhea caused by rotavirus, enterotoxigenic E. coli and V. cholerae in children. J. Pediatr. 1981;98:835–838. doi: 10.1016/S0022-3476(81)80863-3. [DOI] [PubMed] [Google Scholar]
- 40.Hubbard TP, et al. Genetic analysis of Vibrio parahaemolyticus intestinal colonization. Proc. Natl Acad. Sci. USA. 2016;113:6283–6288. doi: 10.1073/pnas.1601718113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roney, I. J. & Rudner, D. Z. Two broadly conserved families of polyprenyl-phosphate transporters. Nature10.1038/s41586-022-05587-z (2022). [DOI] [PMC free article] [PubMed]
- 42.Chin C-S, et al. The origin of the Haitian cholera outbreak strain. N. Engl. J. Med. 2011;364:33–42. doi: 10.1056/NEJMoa1012928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dalia TN, Chlebek JL, Dalia AB. A modular chromosomally integrated toolkit for ectopic gene expression in Vibrio cholerae. Sci Rep. 2020;10:15398. doi: 10.1038/s41598-020-72387-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mendler K, et al. AnnoTree: visualization and exploration of a functionally annotated microbial tree of life. Nucleic Acids Res. 2019;47:4442–4448. doi: 10.1093/nar/gkz246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jumper J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sit, B. Insights into Vibrio cholerae Vaccine Development and Physiology From Small Animal Models of Intestinal Colonization and Disease. PhD thesis, Harvard University (2021).
- 47.Sit B, et al. Oral immunization with a probiotic cholera vaccine induces broad protective immunity against Vibrio cholerae colonization and disease in mice. PLoS Negl. Trop. Dis. 2019;13:e0007417. doi: 10.1371/journal.pntd.0007417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abel S, et al. Sequence tag-based analysis of microbial population dynamics. Nat. Methods. 2015;12:223–226. doi: 10.1038/nmeth.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Möll A, et al. A d,d-carboxypeptidase is required for Vibrio cholerae halotolerance. Environ. Microbiol. 2015;17:527–540. doi: 10.1111/1462-2920.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee W, et al. Antibiotic combinations that enable one-step, targeted mutagenesis of chromosomal genes. ACS Infect. Dis. 2018;4:1007–1018. doi: 10.1021/acsinfecdis.8b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fey PD, et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio. 2013;4:e00537–12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liew ATF, et al. A simple plasmid-based system that allows rapid generation of tightly controlled gene expression in Staphylococcus aureus. Microbiology. 2011;157:666–676. doi: 10.1099/mic.0.045146-0. [DOI] [PubMed] [Google Scholar]
- 54.Englesberg E, et al. l-Arabinose-sensitive, l-ribulose 5-phosphate 4-epimerase-deficient mutants of Escherichia coli. J. Bacteriol. 1962;84:137–146. doi: 10.1128/jb.84.1.137-146.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alvarez L, Hernandez SB, de Pedro MA, Cava F. Ultra-sensitive, high-resolution liquid chromatography methods for the high-throughput quantitative analysis of bacterial cell wall chemistry and structure. Methods Mol. Biol. 2016;1440:11–27. doi: 10.1007/978-1-4939-3676-2_2. [DOI] [PubMed] [Google Scholar]
- 56.Alvarez, L., Cordier, B., Van Teeffelen, S. & Cava, F. Analysis of Gram-negative bacteria peptidoglycan by ultra-performance liquid chromatography. Bio. Protoc.10, e3780. (2020). [DOI] [PMC free article] [PubMed]
- 57.Hernández SB, Dörr T, Waldor MK, Cava F. Modulation of peptidoglycan synthesis by recycled cell wall tetrapeptides. Cell Rep. 2020;31:107578. doi: 10.1016/j.celrep.2020.107578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barreteau, H. et al. Quantitative high-performance liquid chromatography analysis of the pool levels of undecaprenyl phosphate and its derivatives in bacterial membranes. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.877, 213–220 (2009). [DOI] [PubMed]
- 59.Warr AR, Kuehl CJ, Waldor MK. Shiga toxin remodels the intestinal epithelial transcriptional response to enterohemorrhagic Escherichia coli. PLoS Pathog. 2021;17:e1009290. doi: 10.1371/journal.ppat.1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 62.Stephens M. False discovery rates: a new deal. Biostatistics. 2017;18:275–294. doi: 10.1093/biostatistics/kxw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial and archaeal clades with PF04018-containing proteins. Annotree was used to plot and identify species with an annotated DUF368 domain. Each tab of the spreadsheet is a different level of bacterial classification (Phylum>Order>Family>Genus>Species). Archaeal clades are combined on the tab “Archaea”.
Homologues of VCA0040 in bacteria. HMMER was used to identify homologous sequences to VCA0040 in the Uniprot database. Each species is listed once.
Notable domain architectures of DUF368- and DedA-containing proteins. Sequences were obtained by searching InterProScan with the PF family designations for DUF368 (PF04018) and DedA (PF09335).
RNAseq of Δvca0040 V. cholerae during sphere formation. For the top 40 up-regulated genes in Δvca0040, manual curation of subcellular localization was performed with SignalP and pSortb. P-values were obtained through the Wald test performed by DESeq2 with adjustments for multiple comparisons.
MIC data for V. cholerae, S. aureus and E. coli. Data for each organism is separated into individual worksheets. Note that S. aureus data is split into two worksheets, one for WT versus Δ0846 characterization (“SA”) and one for WT versus Δ0846, Δ2816 and Δ0901 (“SA_dedA”). All V. cholerae data is under “VC” and all E. coli lptd4213 data is under “EC_lptd4213”.
Synthetic transposon-insertion screening in Δvca0040 V. cholerae. Note that intergenic regions (IG_) and regions with <5 informative sites (possible transposon insertion sites) were not analyzed. Hits on the second tab were thresholded at an inverse MWU p-value >100 before sorting by mean log2 fold change.
Synthetic transposon-insertion screening in ΔyghB V. cholerae. Note that this dataset has not been filtered as for Supplementary Table 6.
Multiplexed comparative proteomics of whole-cell WT and ΔsecDF1 V. cholerae. Fold change was calculated by dividing the average normalized relative abundance of proteins in ΔsecDF1 by that of WT.
Conservation of PF04018, PF09335, and PF02763 in bacterial species. Spreadsheet was generated by Annotree queries for all possible combinations of the three protein families. The “Master” sheet lists all species-level organisms with the indicated combination of domains. The “Model Species” sheet lists selected microorganisms of particular interest and can be used to look up any given species in the reference table. Species of interest should be manually confirmed for presence or absence by BLAST to guard against misannotation or non-annotation issues.
Strains and vectors and genomic information used in this study. Any listed strains or vectors may be requested from the lead contact (mwaldor@research.bwh.harvard.edu).
BLAST dictionary for assignment of putative VC names to HaitiWT V. cholerae loci. All coding sequences from HaitiWT were used as queries in a batch BLAST of the N16961 V. cholerae proteome. The top hit by e-value was selected and VC name automatically assigned. However, as batch BLAST outputs a hit regardless of confidence, manual curation is still required for specific loci when using this dictionary.
Data Availability Statement
Sequence data were deposited to the NCBI Sequence Read Archive under BioProject accession codes PRJNA868324 (V. cholerae suppressor genome sequencing), PRJNA868332 (V. cholerae RNA sequencing), PRJNA877769 (V. cholerae transposon-insertion sequencing) and PRJNA877773 (S. aureus suppressor genome sequencing). Proteomics data were deposited in MassIVE under the accession code MSV000090217. All other data are freely available without restriction from the corresponding authors upon request. Source data are provided with this paper.