
Fig. 1.
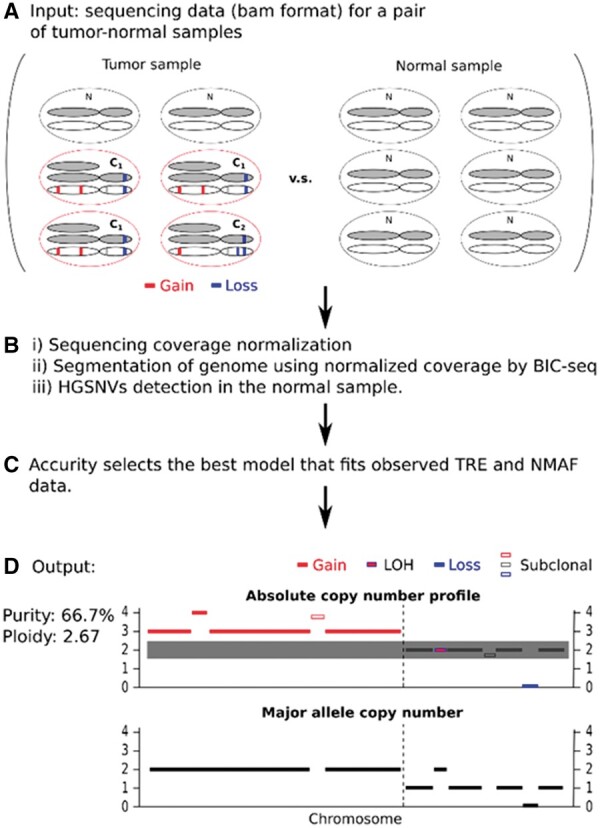
Workflow of Accurity. (A) DNA from a pair of matching tumor-normal samples is extracted and profiled using a WGS technology. In the tumor sample, about 2/3 of all cells are cancerous. About ¾ of all cancer cells belong to clone C1 and the rest belong to clone C2. Clone C1 and C2 share substantial SCNAs as they evolve from a single ancestral clone. (B) We i) normalize the sequencing coverage data to correct the GC bias; ii) use BIC-seq to partition the 22 autosomes of the cancer genome into segments according to sequencing coverage; iii) call heterozygous germline single nucleotide variants (HGSNVs) for the normal sample using our approximate variant caller. We calculate tumor read enrichment (TRE) for all cancer genome segments in bins of 500 bp and the normalized major allele fraction (NMAF) of all HGSNVs. (C) Accurity conducts an autocorrelation analysis on the TRE distribution to derive an initial periodicity estimate and fits a joint Hierarchical Gaussian Mixture (HGM) model on TRE and NMAF data according to Bayesian Information Criterion (BIC). (D) Accurity outputs tumor purity and tumor cell ploidy estimates, an absolute copy number profile and a major allele copy number profile for the cancer genome. Clonal segments are assigned with integers (solid bars) and subclonal segments are assigned with non-integers (hollow bars)