

Abstract

PTEN (phosphatase and tensin homolog deleted on chromosome 10) is a tightly regulated dual-specificity phosphatase and key regulator of the PI3K/AKT/mTOR signaling pathway. PTEN phosphorylation at its carboxy-terminal tail (CTT) serine/threonine cluster negatively regulates its tumor suppressor function by inducing a stable, closed, and inactive conformation. Germline PTEN mutations predispose individuals to PTEN hamartoma tumor syndrome (PHTS), a rare inherited cancer syndrome and, intriguingly, one of the most common causes of autism spectrum disorder (ASD). However, the mechanistic details that govern phosphorylated CTT catalytic conformational dynamics in the context of PHTS-associated mutations are unknown. Here, we utilized a comparative protein structure network (PSN)-based approach to investigate PTEN CTT phosphorylation-induced conformational dynamics specific to PTEN-ASD compared to PTEN-cancer phenotypes. Results from our study show differences in structural flexibility, inter-residue contacts, and allosteric communication patterns mediated by CTT phosphorylation, differentiating PTEN-ASD and PTEN-cancer phenotypes. Further, we identified perturbations among global metapaths and community network connections within the active site and inter-domain regions, indicating the significance of these regions in transmitting information across the PSN. Together, our studies provide a mechanistic underpinning of allosteric regulation through the coupled interplay of CTT phosphorylation conformational dynamics in PTEN-ASD and PTEN-cancer mutations. Importantly, the detailed atomistic interactions and structural consequences of PTEN variants reveal potential allosteric druggable target sites as a viable and currently unexplored treatment approach for individuals with different PHTS-associated mutations.
Introduction
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a tumor suppressor gene frequently mutated somatically in tumors as well as in the germline of patients. Importantly, germline PTEN mutations predispose individuals to a rare inherited cancer syndrome, PTEN hamartoma tumor syndrome (PHTS), and are one of the most common monogenic causes of autism spectrum disorder (ASD).1,2 PHTS is an autosomal dominant disorder variably characterized by macrocephaly, hamartomatous overgrowths, and malignant neoplasia, especially of the breast (85% lifetime risk in females), thyroid (35% lifetime risk), and endometrium (28% lifetime risk).1,3 At present, clinical management guidelines assign enhanced surveillance for component cancers in all PHTS-affected individuals and a low threshold to evaluate children for ASD. Currently, it is impossible to predict at the individual level (vs population estimates) who will develop cancer or ASD. Therefore, the identification and characterization of different PTEN mutations further elucidate the underlying mechanisms of disease and are of clinical importance for PTEN-informed cancer-risk assessment and medical management. Moreover, early interventions in PHTS improve the outcomes and prevent severe/advanced disease.4
Recently, we identified distinct PTEN protein structural differences in PHTS-affected individuals with cancer compared to those with ASD.5,6 We showed that germline PTEN mutations within the active site and inter-domain interface affect both the thermodynamic stability and structural integrity and hence the conformation of the protein required for catalysis and/or protein–protein interactions (PPIs).5 Furthermore, we identified distinct structural communication pathway signatures for ASD versus cancer-associated PTEN mutations and a potential basis for allosteric communication formed by the inter-domain region of PTEN.6 These data suggest dynamic allostery may be involved in regulating the PTEN function and reveal distinct disease states that contribute to autism or cancer and thus offer insight into PTEN’s potential druggability, which may be different for ASD versus cancer outcomes.
PTEN comprises 403 amino acids that govern an N-terminal PIP2 binding domain (PBD, residues 1–15), a dual-specificity phosphatase domain (residues 16–185), and a C2 domain (residues 190–350) which contains an intrinsically disordered D-loop (IDR, residues 282–313), followed by a flexible carboxy-terminal tail (CTT, residues 351–403) [Figure 1]. Though the initial PTEN X-ray crystal structure lacks the PBD, IDR, and CTT regions (PDB: 1D5R),7 structural characterizations reveal tight interactions between the phosphatase domain and the C2 domain (Figure 1A). We recently constructed a full-length PTEN structure that includes the CTT and its phospho-regulatory site (Figure 1B).8,9 Studies over the past 2 decades provide a valuable framework to further understand PTEN regulation by CTT phosphorylation and its role in stability, as well as PPIs and their implication in disease.10−13 Moreover, biochemical and biophysical studies have revealed that phosphorylation of the serine/threonine cluster (Ser380, Thr382, Thr383, and Ser385) on the PTEN CTT alters the conformational state from an “open” active state to a “closed” autoinhibited state, resulting in reduced membrane localization and phosphatase activity14 (Figure 1C,D). This closed conformation precludes productive CTT binding partner interactions, thereby inducing pathological conformations [“conformational phenotypes”]11,15 that further impair activity and disrupt downstream signaling pathways.12,13 Our full-length PTEN structure shows the phospho-tail in the proposed conformationally closed state8 (Figure 1B). However, the mechanistic details that govern phosphorylated CTT catalytic conformational dynamics and allosteric communication at atomic resolution in the context of PHTS-associated mutations are unknown.
Figure 1.

PTEN domains and structure. (A) Crystal structure of PTEN (PDBID: 1D5R) with an N-terminal catalytic phosphatase domain, PD (cyan, residues 16–185); a membrane-binding C2 domain, C2D (orange, residues 190–350); and marked residues denoting missing regions. (B) In silico model of the full-length PTEN structure. The missing regions in PTEN, the PBD (blue, residues 1–15), the IDR (green, residues 282–312), and the CTT (magenta, residues 351–403) were constructed using the I-TASSER program. (C) In silico full-length model of PTEN with the phosphorylated Ser/Thr cluster in the CTT (magenta) indicated. Inset illustrates the four phosphorylated residues, pSer380, pThr382, pThr383, and pSer385. (D) Schematic of PTEN in “open” and phosphorylation-induced “closed” autoinhibited states. (E) In silico full-length model of PTEN depicting germline mutations PTEN-Y68H (ASD, blue), PTEN-G132D (ASD, magenta), PTEN-M134R (cancer, green), PTEN-R173C (ASD and/or cancer, orange), and active site residues D92, C124, and R130 (red licorice).
Here, we utilized a protein structure network (PSN)-based approach to investigate PTEN CTT phosphorylation-induced conformational dynamics specific to PTEN-ASD and PTEN-cancer phenotypes. We posit that CTT phosphorylation influences structural communication and allosteric coupling in PHTS-associated mutations. We performed explicit solvent molecular dynamics (MD) simulations on full-length (unphosphorylated CTT versus phosphorylated CTT), wild-type (WT) PTEN, and germline PTEN mutations [PTEN-ASD (Y68H and G132D), PTEN-cancer (M134R), and PTEN-ASD-and/or-cancer (R173C)] [Figure 1E] that exhibited the most exaggerated disease-specific effects in our preliminary studies.5,6 Results from our study show differences in structural flexibility, inter-residue contacts, and allosteric communication patterns mediated by CTT phosphorylation in PTEN-ASD and PTEN-cancer phenotypes. Further, we identified conformational changes within the active site which may have an effect on the catalytic function. Moreover, the study highlights perturbations among global metapaths and community network connections within the active site and inter-domain regions, indicating the significance of these regions in transmitting information across the PSN. Our studies identify the mechanistic underpinning of allosteric regulation through the coupled interplay of CTT phosphorylation conformational dynamics in PTEN-ASD and PTEN-cancer mutations. Importantly, the detailed atomistic interactions and structural consequences of PTEN variants reveal potential allosteric druggable target sites as a viable and currently unexplored treatment approach for individuals with different PHTS-associated mutations.
Materials and Methods
Selection of ASD-Associated and Cancer-Associated Mutations for Structural Analysis
The germline PTEN missense mutations [PTEN-ASD (Y68H and G132D), PTEN-cancer (M134R), and PTEN-ASD-and/or-cancer (R173C)] [Figure 1E] were selected based on exhibiting the most exaggerated disease-specific effects from our previous studies.5,6 This prospective study was performed within the framework of the “Molecular Mechanisms Involved in Cancer Predisposition” study (Cleveland Clinic Institutional Review Board approved IRB no. 8458) and conducted with informed consent and in accordance with the World Medical Association Declaration of Helsinki.
Preparation of the Full-Length In Silico PTEN Model
The full-length WT PTEN model was generated previously8 using the I-TASSER webserver16 (Figure 1B). The CHARMM-GUI webserver17 was used to construct PTEN mutant structures (Y68H, G132D, M134R, and R173C) [Figure 1E] from the WT PTEN model and to introduce phosphorylated CTT Ser/Thr cluster sites (S380/T382/T383/S385) to phosphorylated (P-WT) PTEN (Figure 1C), P-Y68H, P-G132D, P-M134R, and P-R173C systems. Each of the systems was subsequently refined using Schrödinger Protein Preparation Wizard18 and Prime Wizard.19
Atomistic MD Simulations
All-atom MD simulations were conducted using GROMACS 2020.2 simulation software20 with the CHARMM36m forcefield21 and a 2 fs timestep in explicit solvent. All systems were neutralized with the addition of Na ions inside a cubic box under periodic boundary conditions using the TIP3P solvent model. Each system (WT PTEN, P-WT, Y68H, P-Y68H, G132D, P-G132D, M134R, P-M134R, R173C, and P-R173C) was subjected to energy minimization via the steepest descent method. Total minimization was carried out until convergence, where the maximum atomic force was <1000 kJ/mol nm. The minimized structures were subjected to a preequilibrium simulation for 350 ps with restrained backbones until the solvent reached 303 K. In the production runs, we employed the Nose–Hoover Langevin22 thermostat with a time constant of 1.0 ps to maintain the constant temperature at 303 K. The pressure was iso-tropically maintained at 1 atm using the Parrinello–Rahman barostat23 with a time constant of 5 ps and a compressibility of 4.5 × 10–5 bar–1. Lennard Jones interactions were smoothly shifted to zero from 1.0 to 1.2 nm, and long-range electrostatic interactions were evaluated using the Particle Mesh Ewald24 method. For each structure production run, 1000 ns each (1 μs) of unrestrained NPT was performed at 303 K, where bond lengths were constrained using the linear constraint solver (LINCS)25 algorithm (Table S1, see Supporting Information). Our simulations closely followed the same protocol as in our previous works.8,9,26 Coordinates were saved every 20 ps, leading to 50,000 configurations for each system. Trajectory analyses were conducted using GROMACS utilities, VMD27 or PyMOL.28
Clustering Analysis
We utilized the GROMACS clustering analysis to perform hierarchical cluster analyses and selected representative (centroid) structures from the most populated cluster of each trajectory ensemble. We used the GROMOS clustering algorithm29 with Cα of 0.30 nm root-mean-square deviation (rmsd) cutoff to determine similar clusters.
Residue–Interaction Network Analysis
The centroid structure from clustering analyses for the WT PTEN, P-WT PTEN, and each mutant PTEN structure was submitted to the residue interaction network generator30 to construct the interactive residue interaction network (RIN) as previously described.6 Each RIN was constructed considering amino acid residues as nodes that are connected by non-covalent interactions and edges represented by contacts between atoms. We utilized the RINs for WT PTEN, P-WT PTEN, and each mutant PTEN structure to analyze the residue–residue interaction of PTEN and visualized it with Cytoscape 3.7.2.31 For each system, two important network centrality measures were computed, degree (connectivity) and betweenness. In a network, a node corresponds to a given protein, and an unweighted and undirected edge corresponds to an interaction between those two proteins. Degree centrality was defined as the number of interactions between residues and is reflected by the size of the node (the expansion in size of a node and progression toward red color is directly proportional to its degree connectivity). Betweenness centrality (Bk) of a node k is the number of times that a node is included in the shortest path between each pair of nodes, normalized by the total number of pairs, and is defined as
| 1 |
where N denotes the number of shortest paths between two nodes. Betweenness centrality measures the importance of a node to the connection of different parts of a network—nodes with these characteristics are considered as potential mediating sites of structural communication and allosteric regulation.32
Protein-Structure Network and Global Metapath Analysis
The long-range communication and allosteric networks were characterized by a mixed PSN and an elastic network model–normal mode analysis (ENM–NMA) approach utilized in previous studies to investigate structural and allosteric communication pathways.33−38 The centroid structure from clustering analyses for the WT PTEN, P-WT PTEN, and each mutant PTEN structure was utilized as input to the WebPSN interface (http://webpsn.hpc.unimo.it) as previously described.6 The mixed PSN-ENM method (WebPSN) involves multiple steps consisting of network features (i.e., nodes, hubs, links, and so forth) that are computed by building a protein structure graph, and the shortest communication pathways in which the conformational dynamics contribute in terms of both correlated motions and path occurrence are acquired from a single, high-resolution structure.34,37 The strength of interaction between residues (nodes) i and j (Iij) is evaluated as a percentage given by eq 1
| 2 |
where Iij is the interaction percentage of nodes i and where j is the number of side-chain atom pairs within a given cutoff (4.5 Å), and Ni and Nj are, respectively, the normalization factors (NF) for residues i and j; the NF account for the difference in size of different nodes and their propensity to make the maximum number of contacts with other nodes in protein structures. The algorithm defines all possible communication paths between selected node pairs and filters the results to the cross-correlation of atomic motions, as derived from ENM-NM. Filtering consists of retaining only the shortest path(s) that contain at least one residue correlated (i.e., with a cross-correlation value ≥ 0.6) with either one of the two extremes (i.e., the first and last residues in the path). Metapaths made of the most recurrent nodes and links in the path pool (i.e., global metapaths) infer a coarse picture of the structural communication in the considered system. In detail, metapaths are made of nodes ≥5% of the considered path pool (i.e., “frequent nodes”) and of links, satisfying both the conditions of being present in one of the paths and of connecting “frequent nodes.” A comparison of metapaths (i.e., difference network) between WT PTEN and P-WT PTEN as well as unphosphorylated and phosphorylated CTT mutant structures was useful to infer commonalities and differences in the structural communication of two functionally different states33,34,38,39 induced by mutation and phosphorylation states.
Computed network parameters include the following: (1) Hubs, which are nodes with the highest degree of connectivity, (2) shortest path length, which is the minimum number of links required to span through one node to another in a PSN, and (3) community, which is the region within a network where nodes are more connected to each other. All network parameters were visualized with VMD.27
Allosteric Coupling Intensities
Sites with strong dynamic allosteric residue coupling to functional sites regardless of distance separation, likely contribute to function.40 Allosteric coupling propagates between distal sites of residues through regions of higher coupling density.41 The centroid structure from clustering analyses for the WT PTEN, P-WT PTEN, and each mutant PTEN structure was utilized as input to the Ohm interface (https://dokhlab.med.psu.edu).41 Each centroid structure was generated, whereby only protein atoms (including hydrogen) were saved to individual structure files. The Ohm algorithm first extracted all the atom-wise contacts from the three-dimensional protein structure to generate an average atom-contacts matrix.41 Two atoms within 3.4 Å are counted as a contact, and distances between every two atoms in the protein structure are calculated
| 3 |
where Cij is the number of atom contacts between residue i and residue j, and a and b are atoms in residues i and j, respectively. a and b cannot
be backbone atoms simultaneously if |i – j| = 1. r0 is the distance cutoff, 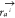 is the position of atom a in residue i, and
is the position of atom a in residue i, and 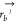 is the position of atom b in residue j. H(x) is the heaviside step function. Subsequently, the number of contacts
each atom in residue i forms with atoms in residue j is determined by dividing the number of contacts between
residues i and j by the number of
atoms in residue i. Likewise, we divide the number
of contacts between residues i and j by the number of atoms in residue j to evaluate
how many contacts each atom in residue j forms with
atoms in residue i
is the position of atom b in residue j. H(x) is the heaviside step function. Subsequently, the number of contacts
each atom in residue i forms with atoms in residue j is determined by dividing the number of contacts between
residues i and j by the number of
atoms in residue i. Likewise, we divide the number
of contacts between residues i and j by the number of atoms in residue j to evaluate
how many contacts each atom in residue j forms with
atoms in residue i
| 4 |
where Nij is the number of average atom-contacts of residue i with respect to residue j; Cij is the number of contacts between residue i and residue j; Ci is the number of atoms in residue i; and Cj is the number of atoms in residue j. Cij is always equal to Cji, whereas Nij is not necessarily equal to Nji.
All protein residue pair distances were calculated, which initialized a distance matrix M(i,j), where i and j are residue indices. The distance between two residues is defined as the minimum distance between their atoms. For each residue, its neighbors G(i) are identified by a distance threshold of 4.5 Å. Next, vector D (direction) is initialized with a size of N, the number of residues. D(i) is then assigned by the index of the neighboring residue with a higher allosteric coupling intensity (ACI) than residue i. If no neighbor with a higher ACI exists, then D(I) is assigned as −1. Each residue with a direction value of −1 represents an allosteric hotspot.
Structural Visualization and Analysis
VMD27 and PyMOL28 were used for simulation visualization. Graphs were generated in the GRACE graphical plotting program (https://plasma-gate.weizmann.ac.il/Grace/).42
Results and Discussion
CTT Phosphorylation Impacts Structural Stability and Conformational Flexibility
MD simulations on full-length in silico models were performed for WT PTEN, PTEN-Y68H (ASD), PTEN-G132D (ASD), PTEN-M134R (cancer), and PTEN-R173C (ASD and/or cancer) unphosphorylated systems, and CTT-phosphorylated systems P-WT-PTEN, P-Y68H, P-G132D, P-M134R, and P-R173C. First, rmsd’s of backbone atoms from the initial structure were computed to find the overall stability and convergence of simulations (Figure S1A,B). The unphosphorylated systems of WT PTEN (black) and Y68H (blue) fluctuate initially and then become more stable from 400 ns onward, indicating the trajectory convergence. In contrast, the G132D (magenta), M134R (green), and R173C (orange) systems drift from the initial structure for relatively long periods, displaying sudden jumps in rmsd before finally equilibrating at ∼750 ns (Figure S1A). The computed rmsd’s for the phosphorylated CTT systems of P-WT PTEN (black), P-Y68H (blue), P-G132D (magenta), P-M134R (green), and P-R173C (orange) fluctuate initially and become stable after 100 ns, much sooner than the unphosphorylated systems (Figure S1B). However, for P-M134R, the system drifts until ∼400 ns, and a sudden jump is seen at ∼430 ns. A higher degree of fluctuation in CTT regions in the G132D and M134R compared to the P-G132D and P-M134R indicates that phosphorylation further stabilizes the structures, as is evident in the lower rmsd values seen for all CTT phosphorylated systems in Figure S1B.
Next, the radius of gyration (Rg) was computed to determine the overall structural compactness of the simulations (Figure 2A,B). The average Rg calculated for the unphosphorylated CTT systems was found to be the same (∼23 Å) [Figure 2A]. However, for M134R, a sudden jump to ∼28 Å is seen around 500 ns, which is maintained until ∼700 ns, suggesting a major conformational change before settling to ∼23 Å. Similarly, the average Rg calculated for the phosphorylated CTT systems was found to be the same (∼23 Å), whereas in the case of P-G132D the Rg increases to ∼26 Å, but then undergoes a sudden compaction back to the initial conformation (Figure 2B).
Figure 2.

Conformational effects of PTEN CTT phosphorylation for each system. Time-evolution backbone Rg profiles for (A) unphosphorylated CTT systems and (B) phosphorylated CTT systems. (C) Backbone superimposition of representative centroid structures of unphosphorylated CTT systems (left panel) and phosphorylated systems (right panel) onto WT PTEN (black) reveals CTT conformational changes [Y68H (black)/P-Y68H (blue), G132D (black)/P-G132D (magenta), M134R (black)/P-M134R (green), and R173C (black)/P-R173C (orange)]. Approximately 50,000 frames each representing a 20 ps timestep were utilized for analysis.
Furthermore, we superimposed the representative centroid conformations of each system to observe the structural changes as shown in Figure 2C and found major changes occurred in the CTT comparing the unphosphorylated systems (left panel) to the phosphorylated systems (right panel). The unphosphorylated CTT (left panel) structures reveal the tails all cluster near the C2 domain and inter-domain regions of PTEN. However, the unphosphorylated CTT in both the G132D and M134R models appears collapsed toward the bottom of the C2 domain. This may explain the sudden jump in Rg which led to the drastic conformational change.
The impact of CTT phosphorylation on the flexibility of PTEN was further explored via the calculation of RMS fluctuations (RMSFs) of protein backbone atoms obtained from MD simulations (Figure 3). The atomic positional RMSFs describe the flexibility of an individual residue and determine the conformational dynamics profile signature for each system. No noticeable changes in the RMSF distributions were seen in WT PTEN. The RMSF distributions for each of the unphosphorylated CTT mutant systems reveal only moderate fluctuations within the C2 domain, with increased fluctuations in the CBR3 loop, IDR, and CTT regions for the Y68H, G132D, and R173C systems (Figure 3A). Modest fluctuations in the CBR3 loop, Cα2, and IDR, with a rather large increase in the CTT region, were seen for the M134R system. Similarly, the G132D system had a large increase in the CTT region. The RMSF distributions for each of the phosphorylated CTT mutant systems show an increase in fluctuations across the PD and C2 domains for the P-WT PTEN and P-Y68H systems (Figure 3B). Specifically, both systems reveal increased fluctuations in the PBD, CBR3 loop, IDR, and CTT. A moderate increase in fluctuation in the IDR is seen in the P-G132D system, with a high increase in the CTT region. Likewise, the P-M134R system also reveals a large increase in the CTT region. In contrast, the CTT of the P-R173C system has a modest increase with similar fluctuation in the CBR3 loop and IDR regions. Structural mapping of the conformational dynamic profiles further highlighted similarities across WT PTEN/P-WTPTEN, Y68H/P-Y68H, and R173C/P-R173C structures, while demonstrating a visible increase in the IDR and CTT flexibility and thus stability for (Figure 3C,D). In contrast, the G132D/P-G132D and M134R/P-M134R systems only show an increase in flexibility in the CTT region with a more pronounced effect in the phosphorylated CTT systems, a characteristic also seen in the RMSF distributions (Figure 3A,B). An interesting observation of the comparative dynamic analysis is an increase in the conformational mobility of the PBD in the P-Y68H systems, suggesting that CTT phosphorylation has long-range effects which impact the dynamic flexibility of this region in this mutation (Figure 3D).
Figure 3.

Conformational flexibility profile from MD simulations for unphosphorylated and phosphorylated CTT systems. Time-evolution profile of RMSF backbone atoms with respect to the initial structure for (A) unphosphorylated CTT and (B) phosphorylated CTT systems for WT PTEN/P-WT PTEN (black), Y68H/P-Y68H (blue), G132D/P-G132D (magenta), M134R/P-M134R (green), and R173C/P-R173C (orange) mutants. RMSF conformational mobility metric mapped onto the structure for (C) unphosphorylated CTT and (D) phosphorylated CTT systems.
CTT Phosphorylation Mediates Long-Range Conformational Changes in the Active Site
The PTEN PD contains the catalytic active site delimited in part by the WPD loop (residues 88–98), the highly conserved signature motif, HCxxGxxR (P loop, residues 123–130), and the extended TI loop (residues 160–171).7 Within the catalytic active site, Asp92 (D92), Cys124 (C124), and Arg130 (R130) are important for the coordination of its main lipid substrate, PIP3, whereby D92 serves as a general acid, and R130 facilitates the migration of a phosphate group from the 3′ region of the inositol head group of PIP3 to the side chain of C124.7 The position of these three critical active site residues is essential for their interaction with PIP3 lipid substrate and the overall function of PTEN.43 Previous studies showed that missense PTEN mutations in these active site residues result in a severe reduction of the phospholipid phosphatase activity of PTEN.44 Therefore, to determine whether CTT phosphorylation induces long-range conformational changes of the active site region, we analyzed the conformational dynamics of the WPD- and P-loop for each system (Figures 4A,B, S1C,D) and quantified the interaction distances between D92, C124, and R130 Cα atoms throughout each of the MD simulation trajectories (Figure 4C,D). We found that the per-residue rmsd’s for the P- and WPD-loops were higher (>30 Å) in the M134R system compared to the WT PTEN and other mutant systems (Figure S1C). Similarly, the P-M134R and P-G132D displayed a higher per-residue-rmsd in the P-loop throughout the entire simulation, which is also seen in the full rmsd (Figure S1B,D), suggesting that fluctuations in this region dominate the overall conformation of the structure. Additionally, we found that the interaction distance between D92 and R130 Cα atoms for both the WT and P-WT PTEN systems remains the same, with a slightly higher frequency of interaction for the P-WT PTEN system (Figures 4C and S2A). However, the D92-R130 interaction distance slightly increased for the P-Y68H when compared to the Y68H unphosphorylated CTT system, with a higher frequency of interaction for the Y68H system (Figures 4C and S2A). In contrast, the D92-R130 interaction distance markedly decreased for the P-G132D, P-M134R, and P-R173C phosphorylated CTT systems yet with a slightly higher frequency of interaction compared to their respective unphosphorylated CTT systems (Figures 4C and S2A). We also found a higher (>30 Å) per-residue rmsd’s in the WPD loop and P loop, for the P-R173C and P-M13R and P-G132D, respectively, compared to the WT PTEN and other mutant systems (Figure S1D), suggesting that fluctuations in these regions dominate the overall conformation of the structure.
Figure 4.

Long-range conformational dynamics of the catalytic active site. The conformational dynamics of the active site WPD- and P-loops for (A) unphosphorylated CTT and (B) phosphorylated CTT systems are colored with WT PTEN/P-WTPTEN (black), Y68H/P-Y68H (blue), G132D/P-G132D (magenta), M134R/P-M134R (green), and R173C/P-R173C (orange). Distance interaction calculated between Cα atoms of catalytic residues (C) D92-R130 and (D) C124-R130 with respect to their initial structure. Inset illustrates D92, C124, and R130 residue positions in sticks within the active site WPD- and P-loops.
Furthermore, we show that the interaction distance between C124 and R130 Cα atoms slightly decreased for the P-WT system, whereas the P-Y68H and P-G132D remained the same, with a higher frequency of interaction for the P-WT system and a lower frequency of interaction for the P-Y68H and P-G132D compared to their respective unphosphorylated CTT systems (Figures 4D and S2B). In contrast, the C124-R130 interaction distance increased in the P-M134R and P-R173C systems, with a slightly higher frequency of interaction for P-M134R and a lower frequency of interaction in P-R173C compared to their respective unphosphorylated CTT systems (Figure S2B). Overall, it suggests that CTT phosphorylation influences the interaction between C124 and R130, which contributes toward the orientation and dynamics of the P- and WPD-loops in the (P-G132D, P-M134R, and P-R173C).
CTT Phosphorylation Influences Communication in the Active Site
To determine how CTT phosphorylation-induced conformational dynamics elicit long-range structural communication and allosteric propagation that influence the active site, we examined the RIN connectivity of the catalytic triad (D92, C124, and R130) in each of the unphosphorylated and phosphorylated CTT systems. RINs have been utilized to identify critical nodes (residues) in the inter-residue communication network with a high degree of connectivity (>4 edges, interactions)45 and are crucial for structural stability, signal propagation, and protein function.46 We found that the catalytic triad RIN was distinctly different for each of the unphosphorylated systems compared to their respective CTT phosphorylated systems (Figure S3A,B). Specifically, the catalytic triad for the P-WT PTEN shows both an increase in connectivity and expansion of critical active site hub residues compared to its unphosphorylated CTT system, suggesting CTT phosphorylation plays a key role in structural communication while increasing signal propagation to residues within the active site. In contrast, the catalytic triad for the P-Y68H shows a decrease in connectivity, but a slight expansion in communication with connection to N372 in the CTT, suggesting phosphorylation diminishes signal propagation and possible catalytic function. Similarly, the catalytic triad for the P-G132D shows a decrease in connectivity but an expansion to V133 and N69. Interestingly, the catalytic triads for the P-M134R and P-R173C RINs both show a marked increase in connectivity and diminished expansion compared to their respective unphosphorylated CTT systems, suggesting CTT phosphorylation mediates rapid signal propagation to a small network of core residues. The distinct differences in connectivity distribution demonstrate that CTT phosphorylation plays a vital role in conformational dynamics and elicits strong network connectivity with active site residues, suggesting a role in phosphorylation-induced allosteric communication as seen in our previous studies.5,6,9 The presence of clinically relevant mutations (Y68H, G132D, M134R, R173C) modulates how the effects of phospho-regulation on the catalytic site, potentially resulting in differences in catalytic activity and regulation.
Community Structure Analysis Reveals Regions Correlated with CTT Phosphorylation
A community network analysis approach33,34,36−38 was applied to identify regions (“communities”) within the structure network of each system that closely correlate with CTT phosphorylation. Incidentally, communities are sets of highly interconnected vertices such that nodes (amino acid residues) belonging to the same communities are densely linked to each other and poorly connected to nodes outside the community.33 The communities are correlated with each other through critical nodes that establish contacts for long-range allosteric mechanisms.47 The PSN is divided into communities based on the flow of allosteric information that passes through each pair of nodes (edge). This is defined as the number of shortest pathways that pass through the edge and is measured by the edge betweenness parameter.34 Representative community networks obtained from the MD simulations are shown in Figure 5. The residues in the active site region of WT PTEN consist of the largest community shown as C1 red module, whereas this region consists of the third largest community in C3 shown as blue module in the P-WT PTEN system. Interestingly, the largest community in the P-WT PTEN module is located in the β-sandwich of the C2 domain. For the Y68H and G132D systems, the active site region resides in the C2 module. However, in the P-Y68H, the active site region shifts to the C3 module, whereas in the P-G132D, it remains in the C2 community. The active site region in the M134R is also in the C2 module, whereas in the P-M134R system, this region not only shifts to the largest community (C1), but this module also expands and extends through the inter-domain region into the C2 domain. The residues in the active site region of R173C locate within the C1 module, whereas in the P-R173C system, this module (C1) extends and dominates throughout the phosphatase domain and inter-domain region. A rearrangement of modules was also observed throughout each structure when comparing the unphosphorylated CTT (Figure 5A) with phosphorylated CTT systems (Figure 5B), which indicates the perturbation affects allosteric communications at a global level. Moreover, the shift of the active site region from the larger community (C1 or C2) to a smaller community (C3) in WT PTEN/P-WT PTEN and Y68H/P-Y68H is indicative of a loss in the importance of this region in the network connection. In contrast, the shift of the active site region to larger expanded communities in the M134R/P-M134R and R173C/P-R173C systems indicates the significance of these dense network of residues in transmitting information and is thus critical for long-range allosteric communication.
Figure 5.

Long-range community network analysis. Communities are mapped onto the protein structures of (A) unphosphorylated CTT and (B) phosphorylated CTT systems. The nodes and link communities are shown, colored according to their size, with red indicating the largest community (C1).
Previously, we identified phosphorylation-induced allosteric coupling between the inter-domain region and “adjacent hydrophobic site” (AHS, framed by residues K13, R47, A126, G127, K128, T160, and G165)48 neighboring the active site in the phosphatase of WT PTEN.9 To further interrogate the effect CTT phosphorylation has on PTEN-ASD and PTEN-cancer mutations, we used Ohm41 to identify regions that influence allosteric coupling (Figure S4). The analysis revealed that allosteric coupling was enhanced across all phosphorylated CTT systems compared to the unphosphorylated systems. Specifically, the pα3, pα5, pα6 (Motif 1), and IDR regions, which are adjacent to the active site and constitute a large portion of the inter-domain region, show an increased ACI in the phosphorylated CTT systems. Interestingly, all of the phosphorylated CTT systems, with the exception of P-Y68H, reveal increased ACIs in the N-terminal region and AHS of PTEN. Overall, these results suggest that positions exhibiting increased coupling around the active site and N-terminal region are critical to the long-range allosteric communication pathway. These results also support our previous in silico computational data5,6,8,9 and experimental findings highlighting the N-terminal region as a critical region in PTEN regulation.
CTT Phosphorylation Reveals Distinct Global Communication Pathways
To further investigate the long-range allosteric structural communication pattern within each of the unphosphorylated and phosphorylated CTT systems, we analyzed the metapath and mapped residues participating in each path using a mixed PSN and ENM approach (Figure 6). Previously, we identified distinct structural communication pathway signatures for ASD versus cancer-associated PTEN mutations and a potential basis for allosteric communication formed by the inter-domain region of PTEN.6 Here, the effects of CTT phosphorylation reveal distinct changes in the long-range, global metapaths. The global metapath in the WT PTEN reveals the most recurrent links, which begin in the ATP-B motif (residues 60–73), passing through the active site and inter-domain region into the C2 domain. The P-WT PTEN system shares a similar global metapath; however, the most recurrent links dominate the inter-domain region. The ASD-associated global metapaths of P-Y68H and P-G132D traverse through nodes made up of the most recurrent links in the AHS and active site within the phosphatase domain and a several nodes in the inter-domain region. In contrast to their Y68H and G132D unphosphorylated CTT systems, which show the global metapath predominantly in the phosphatase domain.
Figure 6.

Long-range allosteric communication mapped onto the protein structures of (A) unphosphorylated CTT and (B) phosphorylated CTT systems. The global metapaths are shown with color indicating occurrence.
Interestingly, though M134R and R173C have a global metapath that extends from the phosphatase domain across the inter-domain and into the β-sandwich of the C2 domain, the P-M134R and P-R173C systems have less recurrent links and a more clustered global metapath, concentrated in their core (inter-domain interface). This supports our previous results where cancer-associated mutations demonstrated a long-range communication pathway within the inter-domain region of the protein and underscores the critical functional roles of the Met134 (M134) and the highly conserved Arg173 (R173) positions in allosteric communication.6,26 M134 mutations result in compromised lipid phosphatase activity49,50 and are associated with breast cancer49 and Bannayan–Riley Ruvalcaba syndrome,49,51 supporting its functional importance. Moreover, R173, which forms a rich hydrogen bond network with the inter-domain region, is among the eight most frequently mutated PTEN residues in PHTS-cancer.7
To infer differences in the structural communication, we computed the difference network between metapaths, which identified commonalities and differences in the structural communication of the functionally different stated induced by mutation and phosphorylation state (Figure S5). Collectively, the metapaths are quite different, as they only share few core nodes in common. The switch in communication from the unphosphorylated to the phosphorylated CTT systems can be clearly seen in the comparison between metapaths. Remarkably, the metapaths in the phosphorylated CTT states share the strong involvement of the inter-domain region. Interestingly, a connection with the PIP2-binding domain plays a crucial role in the metapaths of P-Y68H and P-G132D. Phosphorylation-induced network perturbations in the inter- and intra-domain communications were also analyzed (Table S2). The length of the shortest communication paths in each of the phosphorylated CTT states was decreased compared to the unphosphorylated systems, which reveals that core residues are key players in small-world communication, with the exception of P-M134R and P-R173C. Overall, our results indicate the distinct long-range communication path mediated by CTT phosphorylation in PTEN-ASD and PTEN-cancer mutations. Notably, it further emphasizes the inter-domain region as a critical region that participates in the long-range communication.
Conclusions
Our comparative protein structural network analysis investigated the distinct differences in structural dynamics, flexibility, and allosteric network communication in WT PTEN, P-WT PTEN, and unphosphorylated and phosphorylated CTT germline PTEN mutations PTEN-Y68H (ASD), PTEN-G132D (ASD), PTEN-M134R (cancer), and PTEN-R173C (ASD and/or cancer). To date, there have been no reports aimed at unraveling the mechanistic details that govern phosphorylated CTT catalytic conformational dynamics and allosteric communication at atomic resolution in the context of PHTS-associated mutations. Our results reveal the mechanistic underpinnings of allosteric regulation through the coupled interplay of CTT phosphorylation conformational dynamics in PHTS-ASD and PHTS-cancer mutations. We identified conformational changes within the active site which may have an effect on catalytic function (Figures 4 and S1–S3). Moreover, the study highlights perturbations among global metapaths and community network connections within the active site and inter-domain regions, indicating the significance of these regions in transmitting information across PSN (Figures 5 and 6, S5). These results are also consistent with regions identified in our previously published work.5,6,9 Notably, our results lend insight into diminutive structural changes which may provide an understanding toward PHTS-associated pathological conformations (“conformational phenotypes”)11,15 and preponderance of disease.13 While the use of conventional MD simulations is critical to assess the effects of CTT phosphorylation conformational dynamics in PHTS-associated mutations, a possible limitation is that the initial conformations from each of the MD simulations might bias the conformational space. While our simulations appear to converge nicely, enhanced sampling simulations combining weighted ensemble with metadynamics may increase efficiency in simulating rare events, and provide an ability for full sampling of the energetic landscape.52 Overall, we believe the current work provides a basis for empirical studies to successfully modulate conformational states of PTEN function in individuals with different PTEN mutations. Altogether, our study reveals the detailed structural consequences of PTEN CTT phosphorylation and serves as a future basis for designing novel personalized therapeutics to attenuate PHTS-associated ASD and/or cancer phenotypes.
Acknowledgments
We thank Ann Tushar for her critical review of the manuscript. This study was funded, in part, by the Ambrose Monell Foundation PTEN-Switch Grant and a grant allocation of computing time from the Ohio Supercomputing Center (PCCF0020) [both to C.E.]. I.N.S. is funded, in part, by the Ambrose Monell Cancer Genomic Medicine Fellowship, the NIH National Cancer Institute T32 5T32CA59366-22, and the NIH National Institute of General Medical Sciences (NIGMS) Maximizing Opportunities for Scientific and Academic Independent Careers (MOSAIC) K99/R00 grant—1K99GM143552-01. C.E. is an American Cancer Society Clinical Research Professor and the Sondra J. and Stephen R. Hardis Endowed Chair of Cancer Genomic Medicine at the Cleveland Clinic.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.2c06776.
Time evolution of backbone RMSD profiles, frequency of interaction distances calculated for catalytic residues D92-R130 and C124-R130 for each simulation system, catalytic triad (D92, C124, and R130) residue interaction network maps for each simulation system, comparison of allosteric coupling intensities and global metapaths for CTT systems, comparison of the full-length in silico PTEN structure simulation systems, and a comparison of metapaths for each PSN-ENM PTEN structure (PDF)
Author Contributions
C.E. and I.N.S. conceived and designed the study. I.N.S. set up, performed, analyzed, and produced visualizations for simulations. I.N.S., J.D., and C.E. interpreted all results. I.N.S., J.D., and C.E. wrote the manuscript. All authors critically revised the manuscript and gave the final approval.
The authors declare no competing financial interest.
Notes
The GROMACS software necessary for running the MD simulations is publicly available at https://manual.gromacs.org/2020.2/. The WebPSN and Ohm software utilized for carrying out PSN (community network and metapath) and allosteric coupling intensity analyses are publicly available at http://webpsn.hpc.unimo.it/wpsn3.php and https://dokhlab.med.psu.edu. Usage instructions and documentation for both GROMACS and WebPSN are provided at the respective weblinks. The complete MD trajectories used (a) to plot the rmsd time series and (b) Rg time series, (c) to plot the RMSF time series, (d) to plot distance distributions, (e) to map RMSF structure profiles, (i) to generate PSN community maps, (j) to generate PSN metapaths, (k) and to generate allosteric coupling intensities are available from the author upon request.
Supplementary Material
References
- Tan M. H.; Mester J. L.; Ngeow J.; Rybicki L. A.; Orloff M. S.; Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler M. G.; Dasouki M. J.; Zhou X. P.; Talebizadeh Z.; Brown M.; Takahashi T. N.; Miles J. H.; Wang C. H.; Stratton R.; Pilarski R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C. PTEN: one gene, many syndromes. Hum. Mutat. 2003, 22, 183–198. 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]; a Tan M. H.; Mester J.; Peterson C.; Yang Y.; Chen J. L.; Rybicki L. A.; Milas K.; Pederson H.; Remzi B.; Orloff M. S.; et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 2011, 88, 42–56. 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehia L.; Keel E.; Eng C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. 10.1146/annurev-med-052218-125823. [DOI] [PubMed] [Google Scholar]
- Smith I. N.; Thacker S.; Jaini R.; Eng C. Dynamics and structural stability effects of germline PTEN mutations associated with cancer versus autism phenotypes. J. Biomol. Struct. Dyn. 2018, 37, 1766–1782. 10.1080/07391102.2018.1465854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I. N.; Thacker S.; Seyfi M.; Cheng F.; Eng C. Conformational Dynamics and Allosteric Regulation Landscapes of Germline PTEN Mutations Associated with Autism Compared to Those Associated with Cancer. Am. J. Hum. Genet. 2019, 104, 861–878. 10.1016/j.ajhg.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. O.; Yang H.; Georgescu M. M.; Di Cristofano A.; Maehama T.; Shi Y.; Dixon J. E.; Pandolfi P.; Pavletich N. P. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. 10.1016/S0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- Jang H.; Smith I. N.; Eng C.; Nussinov R. The mechanism of full activation of tumor suppressor PTEN at the phosphoinositide-enriched membrane. iScience 2021, 24, 102438. 10.1016/j.isci.2021.102438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I. N.; Dawson J. E.; Krieger J.; Thacker S.; Bahar I.; Eng C. Structural and Dynamic Effects of PTEN C-Terminal Tail Phosphorylation. J. Chem. Inf. Model. 2022, 62, 4175–4190. 10.1021/acs.jcim.2c00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F.; Ramaswamy S.; Nakamura N.; Sellers W. R. Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol. 2000, 20, 5010–5018. 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Chen Z.; Dempsey D. R.; Thomas S. N.; Hayward D.; Bolduc D. M.; Cole P. A. Molecular Features of Phosphatase and Tensin Homolog (PTEN) Regulation by C-terminal Phosphorylation. J. Biol. Chem. 2016, 291, 14160–14169. 10.1074/jbc.M116.728980. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nanda H.; Heinrich F.; Lösche M. Membrane association of the PTEN tumor suppressor: Neutron scattering and MD simulations reveal the structure of protein-membrane complexes. Methods 2015, 77-78, 136–146. 10.1016/j.ymeth.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Davé V.; Iakoucheva L. M.; Malaney P.; Metallo S. J.; Pathak R. R.; Joerger A. C. Pathological unfoldomics of uncontrolled chaos: intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. 10.1021/cr400713r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby C. A.; Dixon J. E. Pten. Annu. Rev. Biochem. 2014, 83, 641–669. 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- Malaney P.; Uversky V. N.; Davé V. Identification of intrinsically disordered regions in PTEN and delineation of its function via a network approach. Methods 2015, 77–78, 69–74. 10.1016/j.ymeth.2014.10.005. [DOI] [PubMed] [Google Scholar]; a Xu W.; Zhen Z.; Zhou S. F.; Lu N. Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors. Drug Des., Dev. Ther. 2014, 8, 1745–1751. 10.2147/DDDT.S71061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc D.; Rahdar M.; Tu-Sekine B.; Sivakumaren S. C.; Raben D.; Amzel L. M.; Devreotes P.; Gabelli S. B.; Cole P. Phosphorylation-mediated PTEN conformational closure and deactivation revealed with protein semisynthesis. Elife 2013, 2, e00691 10.7554/eLife.00691. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Masson G. R.; Perisic O.; Burke J. E.; Williams R. L. The intrinsically disordered tails of PTEN and PTEN-L have distinct roles in regulating substrate specificity and membrane activity. Biochem. J. 2016, 473, 135–144. 10.1042/BJ20150931. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Masson G. R.; Williams R. L. Structural Mechanisms of PTEN Regulation. Cold Spring Harbor Perspect. Med. 2020, 10, a036152. 10.1101/cshperspect.a036152. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Silva A.; Yunes J. A.; Cardoso B. A.; Martins L. R.; Jotta P. Y.; Abecasis M.; Nowill A. E.; Leslie N. R.; Cardoso A. A.; Barata J. T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Invest. 2008, 118, 3762–3774. 10.1172/JCI34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R.; Tsai C. J.; Jang H. Protein ensembles link genotype to phenotype. PLoS Comput. Biol. 2019, 15, e1006648 10.1371/journal.pcbi.1006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]; a Lee J.; Cheng X.; Swails J. M.; Yeom M. S.; Eastman P. K.; Lemkul J. A.; Wei S.; Buckner J.; Jeong J. C.; Qi Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Friesner R. A.; Xiang Z.; Honig B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]; a Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J.; Honig B.; Shaw D. E.; Friesner R. A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Abraham M. J. M.; Murtola R.; Schulz S.; Páll J. C.; Smith B.; Hess E.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmüller H.; MacKerell A. D. Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Brooks B. R. Self-guided Langevin dynamics simulation method. Chem. Phys. Lett. 2003, 381, 512–518. 10.1016/j.cplett.2003.10.013. [DOI] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic transitions in single-crystals - a new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. 10.1002/(Sici)1096-987x. [DOI] [Google Scholar]
- Dawson J. E.; Smith I. N.; Martin W.; Khan K.; Cheng F.; Eng C. Shape Shifting: The Multiple Conformational Substates of the PTEN N-terminal PIP2 Binding Domain. Protein Sci. 2022, 31, e4308 10.1002/pro.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Schrödinger . The PyMOL Molecular Graphics System; Schrödinger, LLC, 2010.
- Daura X.; Gademann K.; Jaun B.; Seebach D.; van Gunsteren W. F.; Mark A. E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem., Int. Ed. 1999, 38, 236–240. . [DOI] [Google Scholar]
- Piovesan D.; Minervini G.; Tosatto S. C. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016, 44, W367–W374. 10.1093/nar/gkw315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgatti A. P. E.; Everett M. G. A Graph-theoretic perspective on centrality. Soc. Network. 2006, 28, 466–484. 10.1016/j.socnet.2005.11.005. [DOI] [Google Scholar]
- Fanelli F.; Felline A.; Raimondi F.; Seeber M. Structure network analysis to gain insights into GPCR function. Biochem. Soc. Trans. 2016, 44, 613–618. 10.1042/BST20150283. [DOI] [PubMed] [Google Scholar]
- Felline A.; Seeber M.; Fanelli F. webPSN v2.0: a webserver to infer fingerprints of structural communication in biomacromolecules. Nucleic Acids Res. 2020, 48, W94–W103. 10.1093/nar/gkaa397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S.; Akif M. Comparative protein structure network analysis on 3CL(pro) from SARS-CoV-1 and SARS-CoV-2. Proteins 2021, 89, 1216–1225. 10.1002/prot.26143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi F.; Felline A.; Fanelli F. Catching Functional Modes and Structural Communication in Dbl Family Rho Guanine Nucleotide Exchange Factors. J. Chem. Inf. Model. 2015, 55, 1878–1893. 10.1021/acs.jcim.5b00122. [DOI] [PubMed] [Google Scholar]
- Raimondi F.; Felline A.; Seeber M.; Mariani S.; Fanelli F. A Mixed Protein Structure Network and Elastic Network Model Approach to Predict the Structural Communication in Biomolecular Systems: The PDZ2 Domain from Tyrosine Phosphatase 1E As a Case Study. J. Chem. Theory Comput. 2013, 9, 2504–2518. 10.1021/ct400096f. [DOI] [PubMed] [Google Scholar]
- Seeber M.; Felline A.; Raimondi F.; Mariani S.; Fanelli F. WebPSN: a web server for high-throughput investigation of structural communication in biomacromolecules. Bioinformatics 2015, 31, 779–781. 10.1093/bioinformatics/btu718. [DOI] [PubMed] [Google Scholar]
- Felline A.; Ghitti M.; Musco G.; Fanelli F. Dissecting intrinsic and ligand-induced structural communication in the beta3 headpiece of integrins. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 2367–2381. 10.1016/j.bbagen.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Ose N. J.; Butler B. M.; Kumar A.; Kazan I. C.; Sanderford M.; Kumar S.; Ozkan S. B. Dynamic coupling of residues within proteins as a mechanistic foundation of many enigmatic pathogenic missense variants. PLoS Comput. Biol. 2022, 18, e1010006 10.1371/journal.pcbi.1010006. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Weinkam P.; Pons J.; Sali A. Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 4875–4880. 10.1073/pnas.1116274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Jain A.; McDonald L. R.; Gambogi C.; Lee A. L.; Dokholyan N. V. Mapping allosteric communications within individual proteins. Nat. Commun. 2020, 11, 3862. 10.1038/s41467-020-17618-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner P.; Stambulchik E.. GRaphing, Advanced Computation and Exploration of data, 1981. https://plasma-gate.weizmann.ac.il/Grace/ Online (accessed Oct 01, 2012).
- Chia J. Y.; Gajewski J. E.; Xiao Y.; Zhu H. J.; Cheng H. C. Unique biochemical properties of the protein tyrosine phosphatase activity of PTEN-demonstration of different active site structural requirements for phosphopeptide and phospholipid phosphatase activities of PTEN. Biochim. Biophys. Acta 2010, 1804, 1785–1795. 10.1016/j.bbapap.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Myers M. P.; Stolarov J. P.; Eng C.; Li J.; Wang S. I.; Wigler M. H.; Parsons R.; Tonks N. K. the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 9052–9057. 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Xiao Y.; Yeong Chit Chia J.; Gajewski J. E.; Sio Seng Lio D.; Mulhern T. D.; Zhu H. J.; Nandurkar H.; Cheng H. C. PTEN catalysis of phospholipid dephosphorylation reaction follows a two-step mechanism in which the conserved aspartate-92 does not function as the general acid--mechanistic analysis of a familial Cowden disease-associated PTEN mutation. Cell Signal 2007, 19, 1434–1445. 10.1016/j.cellsig.2007.01.021. [DOI] [PubMed] [Google Scholar]; b Yehia L.; Ngeow J.; Eng C. PTEN-opathies: from biological insights to evidence-based precision medicine. J. Clin. Invest. 2019, 129, 452–464. 10.1172/JCI121277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishveshwara S.; Ghosh A.; Hansia P. Intra and inter-molecular communications through protein structure network. Curr. Protein Pept. Sci. 2009, 10, 146–160. 10.2174/138920309787847590. [DOI] [PubMed] [Google Scholar]
- Böde C.; Kovács I. A.; Szalay M. S.; Palotai R.; Korcsmáros T.; Csermely P. Network analysis of protein dynamics. FEBS Lett. 2007, 581, 2776–2782. 10.1016/j.febslet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Sethi A.; Eargle J.; Black A. A.; Luthey-Schulten Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6620–6625. 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y.; Stec B.; Redfield A. G.; Weerapana E.; Roberts M. F. Phospholipid-binding sites of phosphatase and tensin homolog (PTEN): exploring the mechanism of phosphatidylinositol 4,5-bisphosphate activation. J. Biol. Chem. 2015, 290, 1592–1606. 10.1074/jbc.M114.588590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figer A.; Kaplan A.; Frydman M.; Lev D.; Paswell J.; Papa B.; Goldman E.; Friedman E. Germline mutations in the PTEN gene in Israeli patients with Bannayan-Riley-Ruvalcaba syndrome and women with familial breast cancer. Clin Genet. 2002, 62, 298–302. 10.1034/j.1399-0004.2002.620407.x. [DOI] [PubMed] [Google Scholar]
- Mighell T. L.; Evans-Dutson S.; O’Roak B. J. A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am. J. Hum. Genet. 2018, 102, 943–955. 10.1016/j.ajhg.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Post K. L.; Belmadani M.; Ganguly P.; Meili F.; Dingwall R.; McDiarmid T. A.; Meyers W. M.; Herrington C.; Young B. P.; Callaghan D. B.; et al. Multi-model functionalization of disease-associated PTEN missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat. Commun. 2020, 11, 2073. 10.1038/s41467-020-15943-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-García M. J.; Eggenschwiler J. T.; Caspary T.; Alcorn H. L.; Wyler M. R.; Huangfu D.; Rakeman A. S.; Lee J. D.; Feinberg E. H.; Timmer J. R.; et al. Analysis of mouse embryonic patterning and morphogenesis by forward genetics. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 5913–5919. 10.1073/pnas.0501071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman D. M.; Chong L. T. Weighted Ensemble Simulation: Review of Methodology, Applications, and Software. Annu. Rev. Biophys. 2017, 46, 43–57. 10.1146/annurev-biophys-070816-033834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.