

Abstract
The reported primary dementia-protective benefits of angiotensin II type 1 receptor (AT1R) blockers (ARB) are believed, at least in part, to arise from systemic effects on blood pressure. However, there is a specific and independently regulated brain renin-angiotensin system (RAS). Brain RAS acts mainly through three receptor subtypes; AT1R, AT2R, and AT4R. The AT1R promotes inflammation and mitochondrial reactive oxygen species generation. AT2R increases nitric oxide. AT4R is essential for dopamine and acetylcholine release. It is unknown whether ARB use is associated with changes in the brain RAS. Here, we compared the impact of treatment with ARB on not cognitively impaired individuals and individuals with Alzheimer’s dementia using postmortem frontal-cortex samples of age- and sex-matched participants (70–90 years old, n = 30 in each group). We show that ARB use is associated with higher brain AT4R, lower oxidative stress, and amyloid-β burden in NCI participants. In AD, ARB use was associated with lower brain AT1R but had no impact on inflammation, oxidative stress, or amyloid-β burden. Our results may suggest a potential role for AT4R in the salutary effects for ARB on the brains of not cognitively impaired older adults.
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s11357-022-00639-8.
Keywords: Angiotensin receptor blocker, AT4R, Alzheimer’s disease, Oxidative stress, Inflammation, Brain
Introduction
Alzheimer’s dementia (AD) is a complex neurodegenerative disease characterized by the accumulation of neuritic plaques and neurofibrillary tangles. It is the most common cause of dementia in older persons [1]. In the USA, AD affects more than 6.1 million people age 65 and older, and this number is projected to increase to 13.8 million by 2060 [2]. Aging is a key risk factor in AD development, partly through chronic inflammation, mitochondrial dysfunction, and oxidative stress [3, 4]. Although multiple potential biosignatures have been proposed, no clear aging-related etiological mechanisms have been identified.
The renin-angiotensin system (RAS), a central regulator of blood pressure and sodium balance, is involved (via activation of the angiotensin II type 1 receptor (AT1R)) in several molecular mechanisms that are linked to AD, including chronic inflammation, oxidative stress damage, mitochondrial decline, and amyloid-β production [5–8]. Angiotensin (Ang) II type 1 receptor blockers (ARB) have established renal, cardiovascular, and blood pressure benefits and are in widespread clinical use. The primary dementia-protective benefits of ARB are believed to arise, at least partly, from their anti-hypertensive systemic effects. However, there is also a brain-specific RAS [5]. Brain RAS acts mainly through three receptor subtypes; AT1R, AT2R, and AT4R [9]. The AT1R promotes inflammation and mitochondrial reactive oxygen species (ROS) generation. AT2R increases nitric oxide (NO). AT4R, the most recently discovered angiotensin receptor, is essential for dopamine and acetylcholine release and mediates memory consolidation [10–13]. AT4R is distributed throughout the brain, but more predominantly in the hippocampus, neocortex, basal ganglia, and amygdala [5, 9, 14]. Recently, higher levels and activity of AT1R have been shown in the postmortem brains of older individuals with Alzheimer’s dementia [15]. The presence of brain-specific RAS and higher brain AT1R levels in AD prompt the question of whether brain AT1R blockade contributes to the salutary effects of ARB. In this study, we compared the impact of treatment with ARB, respectively, on not cognitively impaired individuals and individuals with Alzheimer’s dementia using postmortem frontal-cortex samples of age- and sex-matched participants (70–90 years old, n = 30 in each group). We examined the effects of ARB on brain RAS and brain health biomarkers, including oxidative stress and inflammation, as well as outcomes such as tangle density, amyloid-β burden, and cognitive function.
Methods
Postmortem frontal cortex brain samples of age- and sex-matched NCI individuals who used ARB (n = 30) and did not use ARB (n = 30) and AD participants who used ARB (n = 30) and did not use ARB (n = 30) were studied. Protein levels of the brain functional RAS receptors AT1R, AT2R, and AT4R were quantified using western blot technique. Then, to determine the association between ARB and molecular mechanisms linked to AD, brain protein carbonyl (PC) levels as a marker of oxidative stress and serum and brain cytokines (Interleukin 6 (IL-6), IL-1β, Interferon-gamma (IFN-γ), and Tumor necrosis factor α (TNF-α)) as markers of inflammation were measured and compared between the groups. Finally, we assessed the impact of ARB on AD neuropathologies, neurofibrillary tangles, amyloid-β burden, as well as cognitive function.
Human brain samples and protein extraction
Postmortem frontal cortex brain samples came from participants in the Rush Memory and Aging Project [16]. The study was approved by an Institutional Review Board of Rush University Medical Center. All participants signed informed consent, Anatomic Gift Act, and a repository consent to allow their data to be repurposed. Expert neurologists made a clinical AD diagnosis by reviewing all clinical data blinded to all neuroimaging and blood tests results and postmortem data. Participants in the ARB use group were selected if there was a record of taking any ARB in the last follow-up visit. Participants who have never used ARB or angiotensin-converting enzyme inhibitors in any visit were included in the non-ARB group. Medication was recorded for the previous 2 weeks before each follow-up visits of Rush MAP participants. To obtain brain tissue postmortem, under sterile conditions, gray matter from midfrontal cortices was excised and kept at − 80 °C without any fixation until analyzed. Methodologies that have been used to obtain postmortem biological specimens were explained in detail [17]. Midfrontal cortex has been selected based on accessibility, availability, and its interconnections with limbic structures, as well as cortical association areas, that are consistently damaged in AD [18]. For protein extraction, 25 mg of the fresh frozen sample was homogenized in a lysis buffer (T-PER, Pierce, MA) using Bullet Blender, a high-throughput homogenizer (Thomas Scientific, NJ). Total protein content was quantified using BCA assay (Pierce, MA).
Measurement of angiotensin receptor levels
Protein levels of AT1R, AT2R, and AT4R were measured by western blot. Fifteen µg of extracted proteins were resolved by 4% to a 12% acrylamide gradient SDS-PAGE midi gel allowing for protein analysis of 24 participants at a time. Internal control was included with every gel for correction of exposure time differences. Samples were then transferred to a nitrocellulose membrane using standard conditions. Membranes were blocked with 5% skim milk in TBS-T at room temperature for 1 h. Each membrane was then incubated overnight with one of the following rigorously validated, well-characterized primary antibodies at 4 °C. The following antibodies were used: AT1R (sc-515884; Santa Cruz, CA) [19], AT2R (sc-9040; Santa Cruz, CA) [20, 21], and AT4R (HPA043642; Sigma-Aldrich, MO) [22]. Specificity validation of AT1R and AT2R antibodies were made by previous studies studying Chinese hamster ovary cells and naïve HEK-293 T cells lacking AT1R and AT2R [20, 23, 24]. Membranes were then washed, incubated with horseradish peroxidase (HRP)-conjugated secondary antibody. Normalization was done with Actin. After adding a chemiluminescent substrate, the signal was visualized, digitized, and quantified using a Kodak Gel Logic 2200 image workstation with its associated software. Western blot imaging analysis revealed two distinct proteins with different molecular masses identified by the AT4R antibody. This result is consistent with other reports of AT4R quantification [25]. Multiple reasons could have led to this including the presence of 3 isoforms due to alternative splicing as well as posttranslational modifications (glycosylation) of AT4R protein [25–27]. In our analysis, we have used the most prominent band that was seen around 160 kDa in the majority of our participants.
Measurement of brain oxidative stress
Brain protein carbonyl levels were measured by OxiSelect ELISA according to the manufacturer's protocol (Cell Biolabs, CA) [28].
Measurement of brain and serum cytokines
Pro-inflammatory cytokines were measured from participants’ serum and brain tissue using Mesoscale 4-Plex cytokine assays (K15052D; Meso Scale Diagnostics, MD). These measures were performed in the JHU ICTR Core Laboratory.
Brain histopathology
After brain autopsies were performed as previously described [29], aggregated amyloid-β proteins and paired helical filaments of tau tangles in slices of brain regions were identified using 10D5 (dilution 1:300; courtesy of Elan Pharmaceuticals, South San Francisco, CA). Pathologic diagnoses of AD were made using NIA-Reagan and modified CERAD criteria. Staging of neurofibrillary pathology was made by Braak Staging in the Rush MAP [29]. Amyloid-β and tangle burden were calculated using the average percent density in brain slices for eight relevant brain regions, namely, the entorhinal cortex, calcarine cortex, cingulate cortex, inferior parietal gyrus, inferior temporal gyrus, hippocampus, middle frontal gyrus (MFG), and superior frontal cortex (SFC). Finally, overall amyloid-β and tangle scores for the entire brain were obtained using the mean of those from all brain regions.
Cognitive performance
The cognitive performance of participants was measured by administrating a mini-mental state examination (MMSE) and cognitive performance tests each year, 19 of which assessed five cognitive domains, including episodic memory, semantic memory, working memory, perceptual speed, and visuospatial ability, as previously described [30]. Raw scores from each test were first standardized to z scores and averaged to yield a participant’s global cognitive function (GCF) score. Then, cognitive score slopes were analyzed with adjustment both demographic (sex, age, and education level) and pathologies of the brain such as amyloid-β, PHF tau tangles, gross chronic cerebral infarctions, chronic microinfarctions, Lewy body disease, TDP-43, hippocampal sclerosis, cerebral amyloid angiopathy, cerebral atherosclerosis, and arteriolosclerosis as described previously [31, 32].
Physical function
Gait speed and grip strength were assessed as previously reported [29, 33, 34].
Statistical analysis
SPSS 23 and GraphPad Prism 9 were used for statistical analysis. Normally distributed values are expressed as a mean ± standard deviation (SD), while non-normally distributed values are expressed as a median (interquartile range (IQR)). A p value of ≤ 0.05 was used as the threshold of statistical significance. An unpaired Student’s t test for normally distributed variables and Mann–Whitney U tests for non-normally distributed variables were performed to test for the significance of differences. A chi-square test was used to compare binary variables. Correlation analysis was performed with Spearman’s correlation test. Four group analysis were done with either one- or two-way ANOVA (with disease state (NCI vs AD) and ARB use as factors).
Results
Characteristics of study participants are given in Table 1. Body mass index (BMI) and indicators of functional outcomes, such as gait speed and grip strength, were not significantly different between the groups. NCI and AD participants who took ARB had fewer years of education than those who did not take ARB. We also observed a lower number of participants with hypertension diagnosis in the NCI group (p < 0.001) compared to the other groups. However, there were no significant difference in average systolic and diastolic blood pressure between the groups (p = 0.056, p = 0.558, respectively).
Table 1.
Characteristics of not cognitively impaired and Alzheimer’s dementia participants
| NCI (n = 30) | NCI + ARB (n = 30) | AD (n = 30) | AD + ARB (n = 30) | p value | |
|---|---|---|---|---|---|
| Age, mean (SD) | 90.24 (5.1) | 90.04 (3.5) | 90.11 (5.7) | 90.20 (5.5) | 0.999 |
| Sex, male, n (%) | 5 (16.7) | 5 (16.7) | 5 (16.7) | 5 (16.7) | 1 |
| Education, years, median (IQR) | 18 (21–16) | 15 (17–12) | 18 (21–18) | 16 (18–12) | < 0.001* |
| BMI, median (IQR) | 24.0 (25.2–22.6) | 25.6 (28.6–23.0) | 24.8 (29.0–23.4) | 26.2 (30–24.6) | 0.093 |
| Walking speed, m/s, mean (SD) | 0.63 (0.24) | 0.58 (0.17) | 0.56 (0.19) | 0.52 (0.20) | 0.373 |
| Grip strength, kg, mean (SD) | 33.5 (15.1) | 36.1 (11.0) | 30.1 (10.4) | 37.6 (16.2) | 0.402 |
| Coronary heart disease, n (%) | 7 (23.3) | 7 (23.3) | 7 (23.3) | 10 (33.3) | 0.766 |
| Stroke, n (%) | 6 (20) | 8 (26.7) | 6 (20) | 6 (20) | 0.902 |
| Cancer, n (%) | 12 (40) | 14 (46.7) | 9 (30) | 10 (33.3) | 0.561 |
| Hypertension, n (%) | 16 (53.3) | 29 (96.7) | 25 (83.3) | 24 (80) | < 0.001* |
| Thyroid disease, n (%) | 5 (16.7) | 12 (40) | 11 (36.7) | 14 (46.7) | 0.086 |
| Claudication, n (%) | 5 (16.7) | 7 (23.3) | 11 (36.7) | 7 (23.3) | 0.344 |
| Diabetes, n (%) | 3 (10) | 6 (20) | 7 (23.3) | 8 (26.7) | 0.413 |
| Average systolic BP, mean (SD) | 124 (18) | 136 (18) | 135 (15) | 138 (19) | 0.056 |
| Average diastolic BP, mean (SD) | 69 (10) | 71 (10) | 73 (11) | 72 (10) | 0.558 |
| PMI, hours, median (IQR) | 5.63 (15.3–4.2) | 5.33 (6.5–4.5) | 6.93 (10.58–4.25) | 5.43 (13.2–4.4) | 0.841 |
AD, Alzheimer’s dementia; ARB, angiotensin receptor blockers; BMI, body mass index; BP, blood pressure; IQR, interquartile range; NCI, not cognitively impaired; PMI, post-mortem interval; SD, standard deviation; *p ≤ 0.05
Treatment with angiotensin receptor blockers was associated with higher levels of brain AT4R only in participants without cognitive impairment
Angiotensin II, which is a bioactive product of angiotensinogen, acts mainly through two angiotensin receptor subtypes: AT1R and AT2R. Ang II can also be converted to angiotensin IV by glutamyl aminopeptidase A (AP-A) and alanyl aminopeptidase N (AP-N) [35]. Ang IV acts through AT4R. To elucidate the effects of ARB on brain angiotensin receptors, we measured protein levels of AT1R, AT2R, and AT4R and compared them between ARB treated and untreated groups. Our results (Fig. 1) show that in the not cognitively impaired group (without MCI or dementia), there is no significant difference in AT1R protein levels (median (IQR) 0.47 (0.63–0.19) vs. 0.46 (0.68–0.30), NCI vs. NCI + ARB, p = 0.243) (Fig. 1A). Similarly, no significant difference is observed in AT2R protein levels (median (IQR) 0.45 (1.08–0.26) vs. 0.39 (1.11–0.27), NCI vs. NCI + ARB, p = 0.848) with the ARB treatment (Fig. 1B). Nevertheless, ARB treatment was associated with higher AT4R protein levels in NCI participants (median (IQR), 0.17 (0.76–0.01) vs. 0.69 (0.96–0.23), NCI vs. NCI + ARB, p = 0.018) (Fig. 1C).
Fig. 1.

Protein levels of angiotensin receptors in not cognitively impaired groups. (A) Protein levels of angiotensin II type 1 receptor (AT1R). (B) Angiotensin II type 2 receptor (AT2R). (C) Angiotensin type 4 receptor (AT4R) in not cognitively impaired (NCI) group treated (n = 30) and untreated (n = 30) with angiotensin receptor blockers (ARB). Data are presented with median and interquartile range. Comparisons were conducted using Mann–Whitney U test. (*p ≤ 0.05). au, arbitrary unit. (D) Western blot images of the AT1R, AT2R, and AT4R
In contrast, in the AD group, AT1R protein levels were significantly lower in ARB treatment group (0.59 (0.76–0.45) vs. 0.47 (0.63–0.21), AD vs. AD + ARB, p = 0.023) (Fig. 2A). However, there was no significant difference in either AT2R (0.46 (0.79–0.32) vs. 0.52 (0.67–0.34), AD vs. AD + ARB, p = 0.965) or AT4R protein levels (0.13 (0.51–0.03) vs. 0.06 (0.22–0.01), AD vs. AD + ARB, p = 0.117) (Fig. 2B and C). Representative western blot images were added as a supplementary file.
Fig. 2.

Protein levels of angiotensin receptors in Alzheimer’s dementia groups. (A) Protein levels of angiotensin II type 1 receptor (AT1R). (B) Angiotensin II type 2 receptor (AT2R). (C) Angiotensin type 4 receptor (AT4R) in Alzheimer’s dementia (AD) group treated (n = 30) and untreated (n = 30) with angiotensin receptor blockers (ARB). Data are presented with median and interquartile range. Comparisons were conducted using Mann–Whitney U test (*p ≤ 0.05). au, arbitrary unit. (D) Western blot images of the AT1R, AT2R, and AT4R
ARB treatment was associated with lower brain oxidative stress levels in the not cognitively impaired participants
Oxidative stress is highlighted as a primary theory of aging and plays a prominent role in cognitive decline in older individuals [36]. While AT1R is known for its detrimental pro-oxidative stress effects, AT2R and AT4R possess anti-inflammatory and anti-oxidative stress downstream signaling effects. The balance among angiotensin receptors is essential for homeostasis [5]. Levels of brain protein carbonyl (PC), a biomarker of oxidative stress, were quantified and compared between the groups.
PC levels were lower in the NCI + ARB group compared to the NCI group (median and (IQR) 10.6 (11.9–9.5) vs. 11.3 (13.8–10.5), NCI + ARB vs. NCI, p = 0.035) (Fig. 3A). In contrast, no significant difference in brain PC level was observed in the AD group treated with ARB (10.6 (13.3–9.6) vs. 10.9 (13.9–9.9), AD vs. AD + ARB, p = 0.478) (Fig. 3B).
Fig. 3.
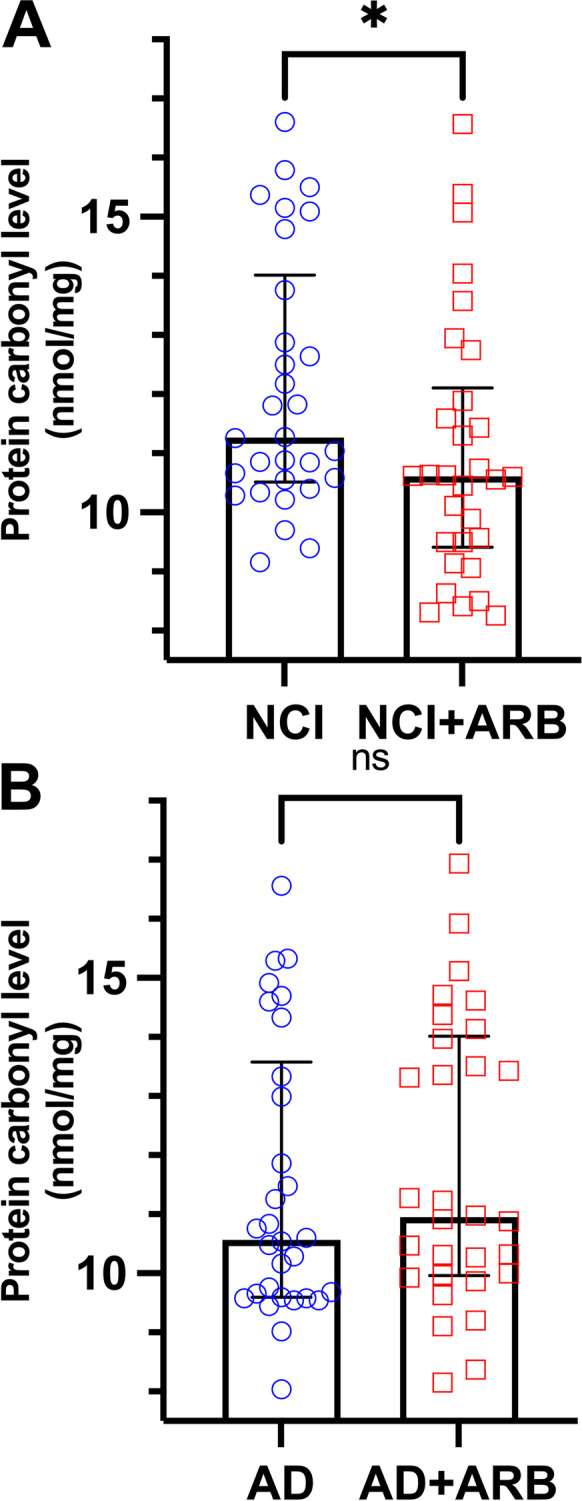
Brain protein carbonyl (oxidative stress marker) differences between angiotensin receptor blockers treated and untreated groups. Levels of brain protein carbonyls (PC) in (A) not cognitively impaired (NCI) group treated (n = 30) and untreated (n = 30) with angiotensin receptor blockers (ARB) and (B) Alzheimer’s dementia (AD) group treated (n = 30) and untreated (n = 30) with ARB. Data are presented with median and interquartile range. Comparisons were conducted using Mann–Whitney U test (ns, non-significant; *p ≤ 0.05)
Given the tight connection between chronic inflammation and cognitive decline, we examined the impact of ARB on the brain and systemic (circulating) inflammatory markers (IL-6, TNF-α, IL-1β, and IFN-γ). Our data show no significant difference in brain and serum cytokine levels between the NCI and AD groups, treated and not treated with ARB. However, brain AT4R protein levels were negatively correlated with brain IL-6 levels (r = − 0.523 p = 0.003) (Fig. 4B) and serum IL-1β (r = − 0.460 p = 0.020) in AD + ARB group (Fig. 4D). There was no significant correlation in other groups (Fig. 4A and C).
Fig. 4.

Illustration of correlation between AT4R and cytokine levels. Scatter plot showing the correlation (and 95% confidence interval of the regression line) between levels of (A) AT4R and brain IL-6 (n = 30) (p = 0.871) in Alzheimer’s dementia group. (B) AT4R and brain IL-6 (n = 30) (r = − 0.523 p = 0.003) in Alzheimer’s dementia group treated with angiotensin receptor blockers. (C) AT4R and serum IL-1β (n = 26) (p = 0.898) in Alzheimer’s dementia group. (D) AT4R and serum IL-1β (n = 26) (r = − 0.460 p = 0.020) in Alzheimer’s dementia group treated with angiotensin receptor blockers. Spearman’s correlation analysis was used
Treatment with angiotensin receptor blockers was associated with lower amyloid-β burden in the NCI participants but no effect in AD participants
Next, we examined if the higher brain AT4R levels combined with lower oxidative stress in the ARB-treated NCI group or the lower brain AT1R levels in the ARB-treated AD group impacted brain amyloid-β or tau pathologies. There were significantly lower calcarine cortex amyloid-β and inferior temporal amyloid-β scores in ARB-treated NCI group compared to the control untreated NCI group (1.92 (4.44–1.17) vs. 0.43 (2.39–0), p = 0.011, 3.47 (8.04–1.61) vs. 1.09 (3.83–0), p = 0.030, NCI vs. NCI + ARB, respectively) (Fig. 5A and B). There were no significant differences in amyloid-β in other regions of the brain, including hippocampus and entorhinal cortex compared to the control untreated NCI group (with 1.15 (2.04–0) vs. 0 (0.56–0), p = 0.057, 3.30 (6.43–1.27) vs. 0.60 (3.58–0), p = 0.051, NCI vs. NCI + ARB, respectively). Additionally, there were no significant difference in amyloid-β in the AD groups, treated and untreated.
Fig. 5.

Amyloid-β scores in not cognitively impaired groups and Alzheimer’s dementia groups. (A) Calcarine cortex amyloid-β scores in not cognitively impaired (NCI) group treated (n = 30) and untreated (n = 26) with angiotensin receptor blockers (ARB). (B) Inferior temporal cortex amyloid-β scores in not cognitively impaired (NCI) group treated (n = 30) and untreated (n = 24) with angiotensin receptor blockers (ARB). (C) Calcarine cortex amyloid-β scores in Alzheimer’s dementia (AD) group treated (n = 30) and untreated (n = 30) with ARB. (D) Inferior temporal cortex amyloid-β scores in Alzheimer’s dementia (AD) group treated (n = 30) and untreated (n = 30) with ARB. Data are presented with median and interquartile range. Comparisons were conducted using Mann–Whitney U test (ns, non-significant; *p ≤ 0.05)
There were no significant differences in tangle density between treated and untreated participants in any group.
Despite the absence of significant difference in brain inflammation, oxidative stress, or amyloid-β burden, our data shows that AD participants treated with ARB had higher semantic memory scores (mean ± SD) − 2.5 ± 1.8 vs. − 1.3 ± 0.9, AD vs. AD + ARB, p = 0.013) (Fig. 6A). Furthermore, the slope of semantic memory trajectory, adjusted for demographics (age, sex, and education level) as well as AD brain pathologies, was higher with ARB treatment (− 0.06 ± 0.13 vs. 0.007 ± 0.06), AD vs. AD + ARB, p = 0.017) (Fig. 6B).
Fig. 6.

Semantic memory scores and slope of the semantic memory in Alzheimer’s dementia group treated with angiotensin receptor blockers. (A) Semantic memory score in Alzheimer’s dementia (AD) group treated (n = 25) and untreated (n = 26) with ARB. (B) Slope of the semantic memory in Alzheimer’s dementia (AD) group treated (n = 30) and untreated (n = 29) with ARB. Data are presented with mean and standard deviation. Comparisons were conducted using Student’s t test (*p < 0.05)
Angiotensin receptor blockers act differently on brain angiotensin receptor levels in NCI and AD participants
Finally, to determine if ARB treatment had differential effects in NCI group as compared to AD group, we performed a two-way ANOVA analysis for all four groups. There was a significant interaction effect between ARB use and disease status (AD vs. NCI) on angiotensin type 4 receptor level (F (1116) = 9.349, p = 0.003, ηp2 = 0.075). The significance of the interaction term showed that the effect of ARB use on AT4R level is different between the AD and NCI groups. In the follow-up simple effect analysis, while ARB use had a statistically significant effect on AT4R level (p = 0.014) in NCI group, there was no significant effect in AD group (p = 0.093). Similarly, there was a significant interaction effect between ARB use and disease group on angiotensin type 1 receptor level (F (1116) = 5.351, p = 0.022, ηp2 = 0.044). In the follow-up simple effect analysis, ARB use had a significant effect on AT1R level (p = 0.017) in AD group, with no significant changes in the NCI group (p = 0.260). There were no interaction or main effect of ARB treatment and disease state (AD vs NCI) on angiotensin type 2 receptors.
Discussion
This study investigated b-RAS differences associated with ARB use in the postmortem brains of not cognitively impaired individuals and Alzheimer’s dementia participants. In the NCI individuals who had used ARB, higher AT4R protein levels, lower oxidative stress, and lower amyloid-β scores were seen. However, we did not see any of these differences in comparing AD participants who had used and not used ARB. The only significant difference in angiotensin receptor expression that we saw in AD participants who had used ARB was lower AT1R protein levels. Also, semantic memory scores were higher in AD participants using ARB. There was no difference in inflammation between both NCI and AD participants who had used and not used ARB (Fig. 7).
Fig. 7.

Summary of the changes with angiotensin receptor blocker (ARB) treatment in both not cognitively impaired and Alzheimer’s dementia participants. ACE, angiotensin-converting enzyme; Ang, angiotensin; ARB, angiotensin receptor blocker; AP-A, aminopeptidase A; AP-N, aminopeptidase N; AT1R, angiotensin II type 1 receptor; AT2R, angiotensin II type 2 receptor; AT4R, angiotensin type 4 receptor
Few human studies examined the association of ARB use with changes in brain renin-angiotensin system receptors to the best of our knowledge. Our results demonstrate discordance between the impact of ARB on the brains of participants with normal cognition as compared to those with AD and a possible role for AT4R in the NCI group. AT4R is one of the recent members of the functional angiotensin receptor family, primarily found in cholinergic neurons of the cortex and hippocampus [9]. The AT4R subtype is known for its beneficial effects on cerebral blood flow, neuroprotection, long-term potentiation, and memory consolidation and retrieval [14]. Brazsko et al. suggested that Ang II is converted in the brain to Ang IV and that this conversion is responsible for the cognitive protective effects of Ang II in mice [37]. Our results are also buttressed by a prior study reporting that the beneficial effects of losartan, an angiotensin receptor blocker, are abolished by the concomitant blockade of AT4R in mice [38].
Furthermore, our finding that ARB use was associated with lower oxidative stress levels in not cognitively impaired individuals is consistent with previous Alzheimer’s disease animal models and in vitro studies [39–41]. It was recently shown that higher Ang IV levels are associated with lower oxidative stress [42].
Finally, our result that ARB use is associated with lower amyloid-β levels in not cognitively impaired participants is supported by preclinical evidence suggesting the upregulation of enzymes responsible for the catabolism and clearance of amyloid-β such as neprilysin and transthyretin with ARB use [43–47]. A direct association between ARB use and reduced amyloid-β levels was shown in several animal studies [39, 44, 48, 49] and a human postmortem study [50]. A recent study reported an association between the use of ARB and ACEI and lower amyloid-β accumulation, with superiority of ARB compared to ACEI [47]. The lack of beneficial effects in AD in our study is difficult to assess. It is also likely confounded by several factors, including the severity of AD, the time between diagnosis and starting ARB medication, and duration of treatment.
In contrast to previous literature, there were no anti-inflammatory effects of ARB in our data, although such anti-inflammatory properties have been shown in in vitro and animal studies for both Alzheimer’s and non-Alzheimer’s models [48, 51–53]. Small sample size, assay sensitivity, and protein degradation in postmortem brains may have contributed to this negative result. Our data shows a negative correlation between AT4R and brain IL-6 and serum IL-1β only in ARB-treated AD participants, in agreement with a recent study that demonstrated lower levels of these cytokines with angiotensin IV infusion and reversal of this effects by divalinal-Ang IV (AT4R antagonist) in chronic cerebral hypoperfusion rat models [54].
There are several limitations to our study. First, we do not know the type of ARB used by study participants. This is particularly important since different ARB currently used in clinical setting have different blood–brain penetrability, affinity to the AT1R, and any additional non-angiotensin effects (for example activation levels of peroxisome proliferator-activated receptor gamma with losartan treatment) [5, 40, 55]. Second, the duration for which participants have used ARB is unknown, which precludes us from accounting for their time-dependent effects. Another limitation to our study is the low/lack of specificity of commercially available antibodies against AT1R and AT2R [56–58]. Until the development of more sensitive technology, western blot techniques using validated antibodies remain the cornerstone for measuring angiotensin receptors. As another limitation, in regard to western blot technique, we have not verified linearity of the protein band densities, because of the high variability between different human samples. Another limitation of our study is using protein carbonyl levels as a single marker of oxidative stress. We decided to use protein carbonyl levels as an oxidative stress marker in our study based on their common use, stability, parallelism with protein nitration in AD, and previous studies of the brain angiotensin system and neurodegenerative diseases [59–67]. However, there is a need for more comprehensive analysis of oxidative stress pathways that link to the development of AD. Finally, the small size limits our ability to control important confounding factors such as age, sex, hypertension, education level, and PMI. High blood pressure is a significant contributor to the development and progression of AD. Previous research has shown that higher levels of blood pressure are associated with higher levels of amyloid-β [68–70]. Interestingly, our group analysis shows lower amyloid-β levels in the NCI group treated with ARBs compared to the untreated NCI group, despite higher percentage of hypertension in the treated group.
As of today, there is no cure or effective prevention strategy for Alzheimer’s dementia. AT1R-blockers have attracted the attention of the AD research community after epidemiological evidence favoring the use of ARB against AD for prevention and treatment [71, 72] and after the discovery of brain RAS, which has several effects on the central nervous system beyond well-known endocrine RAS hypertensive effects. Our results collectively suggest a role for AT4R in mediating protective effects of ARB in not-cognitively impaired individuals. Further research is needed to understand better the impact of onset and duration of treatment with ARB in AD individuals.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Figure 7 was created with BioRender.com.
Funding
This work was supported by the Bright Focus Foundation Research Award (PMA), the Johns Hopkins University Claude D. Pepper Older Americans Independence Center, which is funded by the National Institute on Aging of the National Institutes of Health under award number P30AG021334 and NIH Grants R01AG046441 and K23 AG035005, and R01AG17917; and the Nathan W. and Margaret T. Shock Aging Research Foundation, Nathan Shock Scholar in Aging (PMA). This research was supported in part by the Intramural Research Program at the National Institute on Aging. Its contents are solely the authors’ responsibility and do not necessarily represent the official view of the NIH.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
This manuscript does not contain material previously published and is not under consideration for publication elsewhere, in whole or in part. Every author listed on our manuscript meets the qualification for authorship and has had the opportunity to read and comment upon the submitted manuscript.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.James BD, Bennett DA. Annu Rev Public Health. 2019;40:65–84. doi: 10.1146/annurev-publhealth-040218-043758. [DOI] [PubMed] [Google Scholar]
- 2.K.B. Rajan, J. Weuve, L.L. Barnes, E.A. McAninch, R.S. Wilson, D.A. Evans, Alzheimers Dement. J. Alzheimers Assoc. (2021).
- 3.Albers DS, Beal MF. J Neural Transm Suppl. 2000;59:133–154. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- 4.Yin F, Sancheti H, Patil I, Cadenas E. Free Radic Biol Med. 2016;100:108. doi: 10.1016/j.freeradbiomed.2016.04.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.C. Cosarderelioglu, L. Nidadavolu, C. George, E. Oh, D.A. Bennett, J.D. Walston, P. Abadir, Front. Neurosci. 14 (2020). [DOI] [PMC free article] [PubMed]
- 6.J.L. Labandeira-Garcia, A.I. Rodríguez-Perez, P. Garrido-Gil, J. Rodriguez-Pallares, J.L. Lanciego, M.J. Guerra, Front. Aging Neurosci. 9 (2017). [DOI] [PMC free article] [PubMed]
- 7.Wright JW, Harding JW. J Alzheimers Dis. 2019;67:469–480. doi: 10.3233/JAD-181035. [DOI] [PubMed] [Google Scholar]
- 8.Zhu D, Shi J, Zhang Y, Wang B, Liu W, Chen Z, Tong Q. PLoS ONE. 2011;6:e16037. doi: 10.1371/journal.pone.0016037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson L, Eldahshan W, Fagan S, Ergul A. Int J Mol Sci. 2018;19:876. doi: 10.3390/ijms19030876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abadir PM, Carey RM, Siragy HM. Hypertens Dallas Tex. 2003;1979(42):600–604. doi: 10.1161/01.HYP.0000090323.58122.5C. [DOI] [PubMed] [Google Scholar]
- 11.Wright JW, Harding JW. J Renin Angiotensin Aldosterone Syst. 2008;9:226–237. doi: 10.1177/1470320308099084. [DOI] [PubMed] [Google Scholar]
- 12.R. Vajapey, D. Rini, J. Walston, P. Abadir, Front. Physiol. 5 (2014). [DOI] [PMC free article] [PubMed]
- 13.T.M. De Silva, F.M. Faraci, Front. Physiol. 3 (2013). [DOI] [PMC free article] [PubMed]
- 14.Wright JW, Harding JW. J Alzheimers Dis JAD. 2019;67:469–480. doi: 10.3233/JAD-181035. [DOI] [PubMed] [Google Scholar]
- 15.C. Cosarderelioglu, L.S. Nidadavolu, C.J. George, R. Marx, L. Powell, Q.-L. Xue, J. Tian, J. Salib, E. Oh, L. Ferrucci, P. Dincer, D.A. Bennett, J.D. Walston, P.M. Abadir, J. Gerontol. Ser. A (2021) glab376.
- 16.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. J Alzheimers Dis JAD. 2018;64:S161–S189. doi: 10.3233/JAD-179939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett DA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Curr Alzheimer Res. 2012;9:646–663. doi: 10.2174/156720512801322663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colurso GJ, Nilson JE, Vervoort LG. Life Sci. 2003;73:1795–1803. doi: 10.1016/s0024-3205(03)00512-5. [DOI] [PubMed] [Google Scholar]
- 19.Diaz-Ruiz C, Villar-Cheda B, Dominguez-Meijide A, Garrido-Gil P, Guerra MJ, Labandeira-Garcia JL. J Gerontol Ser A. 2020;75:416–424. doi: 10.1093/gerona/gly259. [DOI] [PubMed] [Google Scholar]
- 20.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O’Rourke B, Walston JD. Proc Natl Acad Sci. 2011;108:14849–14854. doi: 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Garrote M, Perez-Villalba A, Garrido-Gil P, Belenguer G, Parga JA, Perez-Sanchez F, Labandeira-Garcia JL, Fariñas I, Rodriguez-Pallares J. Cells. 2019;8:1551. doi: 10.3390/cells8121551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drobin K, Assadi G, Hong M-G, Andersson E, Fredolini C, Forsström B, Reznichenko A, Akhter T, Ek WE, Bonfiglio F, Hansen MB, Sandberg K, Greco D, Repsilber D, Schwenk JM, D’Amato M, Halfvarson J. Inflamm Bowel Dis. 2019;25:306–316. doi: 10.1093/ibd/izy326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valenzuela R, Costa-Besada MA, Iglesias-Gonzalez J, Perez-Costas E, Villar-Cheda B, Garrido-Gil P, Melendez-Ferro M, Soto-Otero R, Lanciego JL, Henrion D, Franco R, Labandeira-Garcia JL. Cell Death Dis. 2016;7:e2427. doi: 10.1038/cddis.2016.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franco R, Lillo A, Rivas-Santisteban R, Rodríguez-Pérez AI, Reyes-Resina I, Labandeira-García JL, Navarro G. Int J Mol Sci. 2020;21:E9602. doi: 10.3390/ijms21249602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernando RN, Larm J, Albiston AL, Chai SY. J Comp Neurol. 2005;487:372–390. doi: 10.1002/cne.20585. [DOI] [PubMed] [Google Scholar]
- 26.Keller SR, Scott HM, Mastick CC, Aebersold R, Lienhard GE. J Biol Chem. 1995;270:23612–23618. doi: 10.1074/jbc.270.40.23612. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J-H, Hanesworth JM, Sardinia MF, Alt JA, Wright JW, Harding JW. J Pharmacol Exp Ther. 1999;289:1075–1083. [PubMed] [Google Scholar]
- 28.Maity-Kumar G, Thal DR, Baumann B, Scharffetter-Kochanek K, Wirth T. FASEB J. 2015;29:2843–2858. doi: 10.1096/fj.14-265157. [DOI] [PubMed] [Google Scholar]
- 29.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Curr Alzheimer Res. 2012;9:646–663. doi: 10.2174/156720512801322663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson RS, Boyle PA, Yu L, Barnes LL, Sytsma J, Buchman AS, Bennett DA, Schneider JA. Neurology. 2015;85:984–991. doi: 10.1212/WNL.0000000000001935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.P.L. De Jager, J.M. Shulman, L.B. Chibnik, B.T. Keenan, T. Raj, R.S. Wilson, L. Yu, S.E. Leurgans, D. Tran, C. Aubin, C.D. Anderson, A. Biffi, J.J. Corneveaux, M.J. Huentelman, Alzheimer’s disease neuroimaging initiative, J. Rosand, M.J. Daly, A.J. Myers, E.M. Reiman, D.A. Bennett, D.A. Evans, Neurobiol. Aging 33 (2012) 1017.e1–15. [DOI] [PMC free article] [PubMed]
- 32.Jansen WJ, Wilson RS, Visser PJ, Nag S, Schneider JA, James BD, Leurgans SE, Capuano AW, Bennett DA, Boyle PA. Neurobiol Aging. 2018;61:138–145. doi: 10.1016/j.neurobiolaging.2017.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buchman AS, Boyle PA, Leurgans SE, Evans DA, Bennett DA. Proc Am Thorac Soc. 2009;6:581–587. doi: 10.1513/pats.200905-030RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchman AS, Shah RC, Leurgans SE, Boyle PA, Wilson RS, Bennett DA. Arthritis Care Res. 2010;62:1287–1293. doi: 10.1002/acr.20200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright JW, Kawas LH, Harding JW. Prog Neurobiol. 2015;125:26–46. doi: 10.1016/j.pneurobio.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Baierle M, Nascimento SN, Moro AM, Brucker N, Freitas F, Gauer B, Durgante J, Bordignon S, Zibetti M, Trentini CM, Duarte MMMF, Grune T, Breusing N, Garcia SC. Oxid Med Cell Longev. 2015;2015:e804198. doi: 10.1155/2015/804198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braszko JJ, Walesiuk A, Wielgat P. J Renin Angiotensin Aldosterone Syst. 2006;7:168–174. doi: 10.3317/jraas.2006.027. [DOI] [PubMed] [Google Scholar]
- 38.Royea J, Zhang L, Tong X-K, Hamel E. J Neurosci. 2017;37:5562–5573. doi: 10.1523/JNEUROSCI.0329-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeda S, Sato N, Takeuchi D, Kurinami H, Shinohara M, Niisato K, Kano M, Ogihara T, Rakugi H, Morishita R. Hypertens Dallas Tex. 2009;1979(54):1345–1352. doi: 10.1161/HYPERTENSIONAHA.109.138586. [DOI] [PubMed] [Google Scholar]
- 40.Saavedra JM. Cell Mol Neurobiol. 2016;36:259–279. doi: 10.1007/s10571-015-0327-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ongali B, Nicolakakis N, Tong X-K, Aboulkassim T, Papadopoulos P, Rosa-Neto P, Lecrux C, Imboden H, Hamel E. Neurobiol Dis. 2014;68:126–136. doi: 10.1016/j.nbd.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 42.Royea J, Martinot P, Hamel E. Neurobiol Dis. 2020;134:104644. doi: 10.1016/j.nbd.2019.104644. [DOI] [PubMed] [Google Scholar]
- 43.Drews HJ, Yenkoyan K, Lourhmati A, Buadze M, Kabisch D, Verleysdonk S, Petschak S, Beer-Hammer S, Davtyan T, Frey WH, Gleiter CH, Schwab M, Danielyan L. Neurotherapeutics. 2019;16:725–740. doi: 10.1007/s13311-019-00723-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti GM. J Clin Invest. 2007;117:3393–3402. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benson Stephen C., Pershadsingh Harrihar A., Ho Christopher I., Chittiboyina Amar, Desai Prashant, Pravenec Michal, Qi Nianning, Wang Jiaming, Avery Mitchell A., Kurtz Theodore W., Hypertension 43 (2004) 993–1002. [DOI] [PubMed]
- 46.M.O. Grimm, J. Mett, C.P. Stahlmann, V.J. Haupenthal, V.C. Zimmer, T. Hartmann, Front. Aging Neurosci. 5 (2013). [DOI] [PMC free article] [PubMed]
- 47.Ouk M, Wu C-Y, Rabin JS, Edwards JD, Ramirez J, Masellis M, Swartz RH, Herrmann N, Lanctôt KL, Black SE, Swardfager W. Neurobiol Aging. 2021;100:22–31. doi: 10.1016/j.neurobiolaging.2020.12.011. [DOI] [PubMed] [Google Scholar]
- 48.Danielyan L, Klein R, Hanson LR, Buadze M, Schwab M, Gleiter CH, Frey WH. Rejuvenation Res. 2010;13:195–201. doi: 10.1089/rej.2009.0944. [DOI] [PubMed] [Google Scholar]
- 49.Mogi M, Li J-M, Tsukuda K, Iwanami J, Min L-J, Sakata A, Fujita T, Iwai M, Horiuchi M. Biochem Biophys Res Commun. 2008;375:446–449. doi: 10.1016/j.bbrc.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 50.Hajjar I, Brown L, Mack WJ, Chui H. Arch Neurol. 2012;69:1632–1638. doi: 10.1001/archneurol.2012.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pang T, Benicky J, Wang J, Orecna M, Sanchez-Lemus E, Saavedra JM. J Hypertens. 2012;30:87–96. doi: 10.1097/HJH.0b013e32834dde5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benicky J, Sánchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, Leng Y, Chuang D-M, Saavedra JM. Neuropsychopharmacology. 2011;36:857–870. doi: 10.1038/npp.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu Y, Xu Y, Wang Y, Wang Y, He L, Jiang Z, Huang Z, Liao H, Li J, Saavedra JM, Zhang L, Pang T. Brain Behav Immun. 2015;50:298–313. doi: 10.1016/j.bbi.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 54.Wang Q-G, Xue X, Yang Y, Gong P-Y, Jiang T, Zhang Y-D. J Renin Angiotensin Aldosterone Syst. 2018;19:1470320318799587. doi: 10.1177/1470320318799587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.J.L. Labandeira-Garcia, R. Valenzuela, M.A. Costa-Besada, B. Villar-Cheda, A.I. Rodriguez-Perez, Prog. Neurobiol. (2020). [DOI] [PMC free article] [PubMed]
- 56.Benicky J, Hafko R, Sanchez-Lemus E, Aguilera G, Saavedra JM. Cell Mol Neurobiol. 2012;32:1353–1365. doi: 10.1007/s10571-012-9862-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Hypertension. 2013;61:253–258. doi: 10.1161/HYPERTENSIONAHA.112.203679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hafko R, Villapol S, Nostramo R, Symes A, Sabban EL, Inagami T, Saavedra JM. PLoS ONE. 2013;8:e69234. doi: 10.1371/journal.pone.0069234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.R.L. Levine, J.A. Williams, E.P. Stadtman, E. Shacter, in: Methods Enzymol., Academic Press, 1994, pp. 346–357. [DOI] [PubMed]
- 60.Grune T, Jung T, Merker K, Davies KJA. Int J Biochem Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 61.Brioschi M, Polvani G, Fratto P, Parolari A, Agostoni P, Tremoli E, Banfi C. PLoS ONE. 2012;7:e35841. doi: 10.1371/journal.pone.0035841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weber D, Stuetz W, Toussaint O, Debacq-Chainiaux F, Dollé MET, Jansen E, Gonos ES, Franceschi C, Sikora E, Hervonen A, Breusing N, Sindlinger T, Moreno-Villanueva M, Bürkle A, Grune T. Oxid Med Cell Longev. 2017;2017:e1401452. doi: 10.1155/2017/1401452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Domenico F, Coccia R, Butterfield DA, Perluigi M. Biochim. Biophys Acta BBA - Proteins Proteomics. 1814;2011:1785–1795. doi: 10.1016/j.bbapap.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 64.Jung T, Engels M, Klotz L-O, Kröncke K-D, Grune T. Free Radic Biol Med. 2007;42:773–786. doi: 10.1016/j.freeradbiomed.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 65.Lopez-Real A, Rey P, Soto-Otero R, Mendez-Alvarez E, Labandeira-Garcia JL. J Neurosci Res. 2005;81:865–873. doi: 10.1002/jnr.20598. [DOI] [PubMed] [Google Scholar]
- 66.Ding Q, Shults NV, Gychka SG, Harris BT, Suzuki YJ. Int J Mol Sci. 2021;22:1687. doi: 10.3390/ijms22041687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muñoz A, Rey P, Guerra MJ, Mendez-Alvarez E, Soto-Otero R, Labandeira-Garcia JL. Neuropharmacology. 2006;51:112–120. doi: 10.1016/j.neuropharm.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 68.Sparks DL, Scheff SW, Liu H, Landers TM, Coyne CM, Hunsaker JC. J Neurol Sci. 1995;131:162–169. doi: 10.1016/0022-510x(95)00105-b. [DOI] [PubMed] [Google Scholar]
- 69.Ashby EL, Miners JS, Kehoe PG, Love S. J Alzheimers Dis. 2016;50:1191–1203. doi: 10.3233/JAD-150831. [DOI] [PubMed] [Google Scholar]
- 70.Walker KA, Power MC, Gottesman RF. Curr Hypertens Rep. 2017;19:24. doi: 10.1007/s11906-017-0724-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.T.J. Oscanoa, J. Amado, X. Vidal, R. Romero-Ortuno, Curr. Clin. Pharmacol. (2020). [DOI] [PubMed]
- 72.Scotti L, Bassi L, Soranna D, Verde F, Silani V, Torsello A, Parati G, Zambon A. Pharmacol Res. 2021;166:105515. doi: 10.1016/j.phrs.2021.105515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.