

Summary
Gene expression is controlled by transcription factors (TFs) that bind cognate DNA motif sequences in cis-regulatory elements (CREs). The combinations of DNA motifs acting within homeostasis and disease, however, are unclear. Gene expression, chromatin accessibility, TF footprinting, and H3K27ac-dependent DNA looping data were generated and a random-forest-based model was applied to identify 7,531 cell-type-specific cis-regulatory modules (CRMs) across 15 diploid human cell types. A co-enrichment framework within CRMs nominated 838 cell-type-specific, recurrent heterotypic DNA motif combinations (DMCs), which were functionally validated using massively parallel reporter assays. Cancer cells engaged DMCs linked to neoplasia-enabling processes operative in normal cells while also activating new DMCs only seen in the neoplastic state. This integrative approach identifies cell-type-specific cis-regulatory combinatorial DNA motifs in diverse normal and diseased human cells and represents a general framework for deciphering cis-regulatory sequence logic in gene regulation.
Keywords: multi-omics, gene regulation, cis-regulatory logic, random-forest model, massively parallel reporter assay
Graphical abstract
Highlights
-
•
Profiling of 15 diploid human cell types via RNA-seq, ATAC-seq, and H3K27ac HiChIP
-
•
Identification of 838 cell-type-specific, recurrent heterotypic DNA motif combinations
-
•
Functional validation of regulatory DMCs via massively parallel reporter assays
-
•
Cancer-type-specific DMCs are linked to neoplasia-enabling processes
The cis-regulatory logic encoded within DNA sequences that mediate cell-type-specific gene expression is undefined. Here Donohue et al. generate multi-omics data across 15 diploid human cell types and present a new integrative framework for identifying regulatory DNA motif combinations (DMCs). Specifically, they identify cell-type- and -state-specific DMCs and anticipate broad applicability of the approach.
Introduction
The cis-regulatory logic encoded in the regulatory DNA sequences that control cell-type-specific gene expression is undefined. Deciphering this logic has been challenging because many cis-regulatory sequences1 reside in non-coding elements2 distant from the transcription start sites (TSSs) of their targets.3,4,5 Additionally, the human genome contains millions of potential enhancers,6,7 with a specific active subset in any given cell type.8 Gene dysregulation is a hallmark of disease,9,10 and whether diseased cells engage new regulatory logic as opposed to modulating the activity of normal logic is unknown. Integrating high-resolution epigenomic profiling with computational modeling and functional assays across diverse human cell types and disease states may help address current knowledge gaps.
One approach to genome-scale mapping of cis-regulatory DNA sequence logic involves identifying the recurrent DNA motifs present in non-coding CREs of specific cell types, including promoters (P) and enhancers (E) associated with cell-type-specific gene expression.11,12,13 Promoters lie ∼250 bp directly upstream of TSSs,14 and enhancers can directly contact promoters and other enhancers, forming E-E, E-P, and P-P loops in three-dimensional (3D) space.15 Active enhancers and promoters are marked by H3K27ac histones,16,17,18 which enables mapping of the 3D architecture of gene regulation. Transcription factors (TFs) act in a combinatorial fashion at CREs to modulate gene transcription by cooperatively binding specific DNA motifs.19 Cell-type-specific gene expression is hence believed to be dependent on cis-regulatory logic of TF motif combinations, referred to as the cis-regulatory lexicon.20,21 Computational efforts have attempted to predict this lexicon 22,23,24,25,26,27; however, these models rely on nearest gene annotations of the most proximal E to a given P along the linear DNA rather than known 3D E-P linkages. While genome-wide regulatory maps have been generated across a number of human cell types,28,29 identifying functional cell-type-specific DNA motif combinations (DMCs) across E-P linkages for the vast majority of normal human cell types is not fully defined, nor is it known how such combinations are altered in disease.
Here, we generate chromatin accessibility, 3D chromatin looping, and gene expression data across 15 diploid human cell types to define cell-type-specific open chromatin peaks within enhancers looped to open chromatin peaks at target gene promoters of expressed transcripts, or peak-loop-transcripts (PLTs). TF footprinting analysis extracted DNA sequence motifs directly bound by TFs within these PLT-associated CREs and a random-forest model was applied to derive cell-type-specific DMCs for each of these 15 cell types. Statistical co-enrichment analysis of TF footprint motifs produced activity predictions for cell-type-linked DMCs, which were validated by massively parallel reporter assays (MPRAs) in relevant cell types. Functionally, regulatory DMCs fell into four distinct classes: synergistic, buffering, redundant, and single driver. Applying this framework to parallel data generated in cancer cells demonstrated that malignant cells not only engage new DMCs but that they also differentially modulate normal lineage DMCs controlling cancer-relevant genes mediating proliferation, metabolism, and cell migration. This integrative approach uncovered a human DMC lexicon driving cell-type-specific gene transcription in a variety of normal cells and their malignant counterparts and provides a framework for future efforts to define the DNA sequence logic that enables cell-type-specific gene expression.
Results
Characterizing epigenomic landscapes in 15 diploid human cell types
To map gene regulatory elements and their putative target genes in diverse cell types, chromatin accessibility, H3K27ac chromatin looping, and RNA sequencing (RNA-seq) data were generated in 15 primary human cell types, including cells of epithelial origin from tissues in which 12 of the most common human cancers arise. These were lung airway, breast (human mammary epithelial cell [HMEC]), bladder, colon, esophageal, skin keratinocytes (KC), ovarian, pancreas, prostate, renal, thyroid, and uterine cells, as well as two cell types of neural origin, primary human astrocytes and melanocytes (MC), and the diploid human lymphoblastoid cell line, GM12878. Replicated 3′ mRNA-seq, ATAC-seq (assay for transposase-accessible chromatin followed by high-throughput sequencing), and H3K27ac HiChIP (Hi-C library preparation followed by a chromatin immunoprecipitation) data were generated for each cell type (Figure 1A). Principal-component analysis (PCA) showed high consistency between biological replicates (Figures S1A–S1C), although differences in read depth likely contributed to variance (Table S2). Publicly available data11,30,31,32,33 cover a portion of cell types studied here; however, primary human melanocytes and airway, bladder, esophageal, ovarian, thyroid, and uterine epithelial cells have been largely unprofiled. These data provide a resource to begin to decode the regulatory logic of active CREs in primary cells from distinct human tissues.
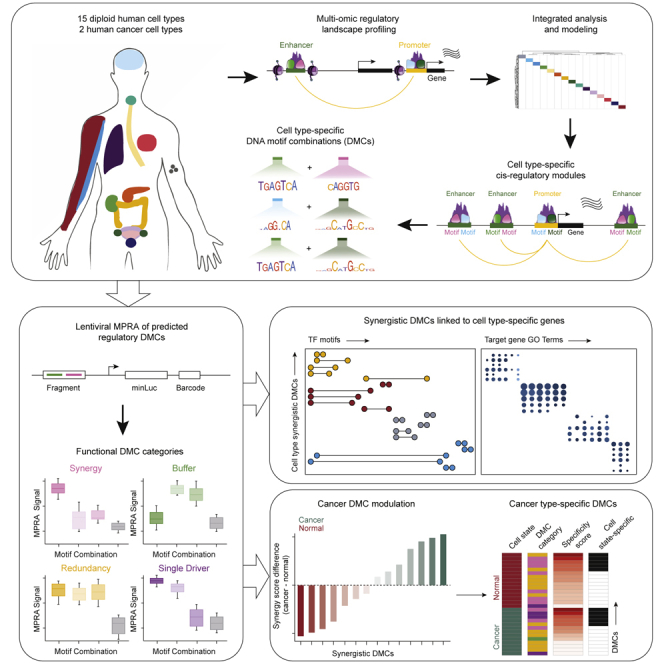
Figure 1.

An integrated multi-omic resource in 15 diploid human cell types
(A) Workflow for cell-type-specific ATAC peaks, HiChIP loops, and target gene transcripts (PLTs) across 15 diploid human cell types.
(B) Schematic of transcription factor (TF) footprinting analysis within PLTs to identify inputs for a random-forest model to derive cell type CRMs. Co-enrichment analysis within CRMs extracted DMCs.
(C) Native genomic instances of putative intra-enhancer and intra-promoter DMCs were tested via MPRA. Combinatorial mutations were used to assess cooperativity of DMCs in a lentiviral setup.
(D) Schematic of MPRA-validated functional categories of DMC interactions.
(E) Schematic bar plot comparing synergistic DMC MPRA activity of normal and cancer-derived DMCs in corresponding cell types.
Epigenomic landscapes and molecular subtypes of diploid human cells
RNA-seq, ATAC-seq, and H3K27ac HiChIP data across these 15 human cell types were integrated to assess cell-type-specific features in regulatory DNA. RNA-seq identified 14,098 total expressed genes, 7,531 of which were differentially expressed (Figure 2A). Similar to PCA analysis, these differential RNA transcripts clustered into four distinct groups, including two epithelial cell groups: (1) Epithelial Cluster 1 (EC1), including colon, esophageal, ovarian, pancreas, renal, and thyroid epithelial cells; (2) Epithelial Cluster 2 (EC2), including airway, bladder, KC, HMEC, prostate, and uterine epithelial cells; (3) neuroendocrine/neural crest lineage (N) astrocytes and MC; and (4) hematopoietic lymphoblastoid GM12878 cells. Relevant expected genes for cell-lineage-specific expression programs were associated with these differential clusters, such as IRF4 in GM12878,34 RUNX2 in astrocytes,35 WT1 in EC1,36,37 and TP63 in EC238,39 (Figures 2A and S1A). ATAC-seq identified 2,342,155 total accessible regions, of which 30,519 (1.3%) exhibited significant variation across all 15 cell types. Chromatin accessibility separated the cell types into the same four clusters found by differential RNA transcripts, EC1, EC2, N, and GM12878 (Figures 2B and S1B). H3K27ac HiChIP data identified 2,822,181 loop anchors, 46,540 (1.6%) of which were differential across all 15 diploid human cell types. Differential regulatory loops clustered into EC1, EC2, MC, astrocyte, and GM12878 (Figures 2C and S2C). Further characterization of these differential regulatory loops revealed expected putative target genes such as CD22 in GM12878,40 SYNDIG1 in astrocytes,41 MLANA in MC,42 TFF1 in EC1,43 and KRT1 in EC244 (Figure 2C). Hierarchical clustering of differential regulatory loops revealed cell type relatedness, with broad clustering of the hematopoietic GM12878 B cells of mesoderm origin, endocrine and gastrointestinal system-related EC1 cells of mesoderm and endoderm origin, and the neuroendocrine astrocytes and MCs of neuroectoderm origin clustering more closely to exocrine-system-related EC2 cells, including keratinocytes and HMECs (Figure 2D). These data identified tens of thousands of putative enhancers physically linked to thousands of expressed genes.
Figure 2.

Epigenomic landscape reveals distinct molecular subtypes of human cells
(A) RNA transcripts (rows) versus cell types (columns) of differential gene expression (log2 fold change >0.1, t test, FDR-adjusted p value <0.05).
(B) Heatmap of accessible peaks (rows) versus cell types (columns) indicating differential ATAC peaks. ATAC peaks with the highest inter-group SD shown.
(C) Heatmap of H3K27ac HiChIP loops (rows) versus cell types (columns) indicating differential loops. Differential loops with the highest inter-group SD shown.
(D) Hierarchical clustering of differential H3K27ac HiChIP loops.
(E) Bar plot depicting cell-type-specific 3D chromatin architecture and overlap between the 15 different cell types.
(F) Bar plot depicting distribution of P-P, E-P, and E-E interactions by cell type.
(G) Bar plot depicting putative enhancers and target genes identified in different E-P interaction types.
(H) Regulatory loop module functional enrichment using GO biological processes. EC1 and EC2 are grouped together. Dot color corresponds to the p value of the GO enrichment (hypergeometric test).
(I) Virtual 4C visualization at 5-kb resolution and RNA and ATAC-seq tracks centered at the ZNF750 TSS. > and < denote gene orientation on plus and minus DNA strand respectively.
(J) Virtual 4C visualization for IL10.
(K) Virtual 4C visualization for TYRP1. Related to Figures S1, S2, and Table S2.
To characterize 3D genomic architecture across these cell types, significant looping interactions identified by H3K27ac HiChIP were investigated, and 10,117 common anchors were shared across all cell types. These linked to 453 commonly expressed target genes, 36 of which are housekeeping genes45 and 112 are essential genes.46 Between 2.5% and 45% of HiChIP interactions detected in a given cell type were unique to that cell type (Figure 2E), and 80% of all HiChIP interactions occurred between DNA regions within 180 kb of each other (Figure S1D). Significant loop anchors were classified into putative enhancers and promoters. Through integration of HiChIP and ATAC-seq data, a putative enhancer was defined as a promoter-interacting region (PIR) containing accessible chromatin peaks within matched datasets by cell type. Promoters were defined as regions containing accessible chromatin peaks and the TSS of a gene. Of the 1,175,428 total looping interactions, 58.4% were between putative enhancer loci (E-E), 33.6% were E-P, and 8.0% were P-P (Figure 2F). A single promoter was assigned a median of two putative enhancers. Promoters linked to expressed genes had a greater number of E-P linkages than non-expressed genes (Mann-Whitney U test, p value = 1 × 10−41). Within E-P interactions, 198,896 cell-type-unique putative enhancers were identified, of which 24,557 directly contact the promoter of the single nearest target gene only, 38,524 putative enhancers directly contact both the nearest target gene and distal gene(s), while 135,815 putative enhancers contact only distal genes (Figure 2G). P-P interactions have been identified at clusters of co-regulated genes,47,48 and recently promoters have also been shown to function in vivo as long-range enhancers.5,49 In 32.0% of P-P interactions, both genes were expressed, in 45.8% one gene but not the other was expressed, and in 22.2% neither gene was expressed (Figure S1E), suggesting some promoters serve enhancer functions and highlighting the 3D complexity of CREs across human cell types.
Relevant biological process terms were enriched in cell-type-specific putative regulatory loops, such as B cell activation, differentiation, and proliferation in GM12878 cells, synapse organization and neuron axonogenesis in astrocytes, pigmentation and melanocyte differentiation in MC, maintenance of gastrointestinal epithelium and epithelial cell morphogenesis in EC1, and epidermis development in EC2 (Figure 2H). The association of cluster and cell-type-specific processes suggests that CREs harbor lineage-specific regulatory roles. Indeed, ZNF750, a known regulator of epidermal differentiation in KC,50,51 was found to be an EC2-specific expressed gene contacted by two EC2-specific putative enhancers (Figures 2I and S1F). Two GM12878-specific putative enhancers were found to directly contact the cytokine IL10, important for B cell regulation52 (Figure 2J). TYRP1, which enables melanin biosynthesis,53 similarly displayed contact with an MC-specific putative enhancer in concert with MC-specific expression (Figure 2K). Integrated HiChIP, ATAC-seq, and RNA-seq data provide a putative map of physically linked regulatory elements to their biologically relevant target genes across diverse normal human cell types.
Consistent with prior work,54,55,56 cell-type-specific CREs identified contained risk-associated variants for diseases of their corresponding tissues. Cell-type-specific distal CREs were intersected with disease-linked variants from the genome-wide association studies (GWAS) catalog.57 HaploReg v4 58 was then used to identify linked single nucleotide variants (SNVs) above a linkage disequilibrium (LD) threshold of 0.8 for 55,202 SNVs linked to risk of developing 15 cancer types arising from the cell types profiled. Additionally, a total of 31,276 SNVS in LD with nine inflammatory diseases were also assessed, including systemic sclerosis, inflammatory bowel disease, and ulcerative colitis, and 82,610 unique SNVs at 5% FDR were significantly enriched across all traits at identified CREs in a disease- and cell-type-specific manner (Figures S2A and S2B). For example, SNVs linked to risk for endometrial cancer and lymphoma were found to reside within putative enhancers that loop to the BUB1B mitotic checkpoint kinase gene, known to be important in cancer growth,58,59 in uterine cells and GM12878 cells, respectively (Figure S2C); these lymphoma-associated SNVs also score as BUB1B tissue-selective eQTLs (expression quantitative trait loci) in GTEx whole-blood data and created motifs for several B cell-relevant TFs (Figure S2D). Additionally, the prostate cancer-linked SNV, rs6983267,60 and the renal carcinoma-linked SNV, rs35252396,61 were found to reside in CRE loci that loop to the MYC oncogene in their respective cell types (Figure S2E) and lymphoma- and pancreatic cancer-linked SNVs were also enriched in GM12878 and pancreas distal CREs, respectively. E-P-linked cell-type-specific CREs thus contain disease-relevant variants with putative functional effects on target gene regulation, potentially through disruption of relevant TF motifs.
cis-Regulatory modules identify a lexicon across human cells
We next searched for cell-type-specific DMCs in cell-type-specific CREs. First, the HINT-ATAC62 package performed TF footprinting to identify putative DNA bases bound by proteins in ATAC-seq data. TF position-weight matrices from HOCOMOCO v1163 were then used to match putative TFs to TF footprints (Figure S3A). Putative TF motif footprints were linked to CRE-based transcriptional regulation if the motif footprint’s corresponding putative TF was expressed in the relevant cell type. Next, we built a package called Pan-omics to identify the TF motif footprints present in proximal and distally looped CREs to a cell-type-specific target gene’s TSS to nominate cis-regulatory modules (CRMs) (Figure S3B). In addition to the identity and number of expressed TF motifs, CRM attributes include the number of unique and total loops contained within the CRM, the number of ATAC peaks present in the CRM, and the identity and transcripts per million (TPM) expression of the target gene. Between 290,391 and 1,786,988 motif footprints were found per cell type. Expressed genes were found to have an increased number of footprints per CRE, with an average of 9.0 footprints for expressed genes (TPM > 1), 5.4 for lowly expressed genes (0 < TPM ≤ 1), and 1.6 for non-expressed genes (TPM = 0). Such gene-centric CRMs captured cell-type-specific 3D contact information, chromatin accessibility, and transcription machinery that may contribute to cell-type-specific transcription.
Consistent with this premise, using a random-forest, tree-based algorithm, the model successfully determined the cell type of a CRM (Figure 3A). When the CRM model features were selected based on assay of origin, it was found that the combination of RNA, ATAC, HiChIP, and TF footprinting was necessary to achieve the highest cell-type performance. Using one-dimensional (1D) RNA and ATAC information as a baseline (purple line), the addition of 3D HiChIP alone (orange line) contributed to a 0.17 increase in model performance (area under the receiver operating characteristic curve [auROC]), 1D putative TF motifs alone (red line) contributed to a 0.32 increase, yet the addition of distally located putative TF motifs (blue line) performed best, contributing to a 0.46 increase in model performance (Figures 3B and S3C–S3F). Thus, models lacking looping and TF motif data performed poorly on the cell-type prediction task, indicating the importance of distal enhancers and TF motif identity in cell-type-specific CRMs. Interestingly, TF motifs in putative enhancers contributed the most to cell-type-specific prediction accuracy. While including putative enhancers decreases the sparsity of CRM motif matrix representations and thus augments model performance, models where the distal enhancer was linked to the HiChIP-identified gene promoter performed 24% better than models where enhancers were linked to the nearest gene and 10% better than models where enhancers were linked to random intrachromosomal genes (Figure S3F). This suggests that DNA looping data capture distal enhancers that mediate cell-type-selective gene expression.
Figure 3.

TF motif enrichment via footprinting cell-type CRMs
(A) Confusion matrix depicting the positive predictive value (PPV) for the cell type prediction model.
(B) Scatterplot showing auROC versus percentage of CRMs learned in the random-forest-based cell-type prediction model. Lines are fitted to the points using logistic regression.
(C) Virtual 4C visualization along with the POU2F2 position-weight matrix (PWM), TF footprint sequence, and surrounding ATAC peak centered at FLG.
(D) Virtual 4C visualization along with the POU2F2 PWM, TF footprint sequence, and surrounding ATAC peak centered at UGDH.
(E) Heatmap (left) depicts normalized log2(TPM) values for nominated TFs corresponding to motifs derived from TF footprinting analysis (rows) in the 15 cell types (columns). TFs are ordered by expression similarity. Dot plot (right) depicts GO enrichment for target genes (x axis) proximal or distally looped to TF footprint motifs in cell-type-specific CRMs (y axis). Dots are colored by cell type. Size corresponds to the −log10(p value) of the GO enrichment (hypergeometric test). Related to Figure S3.
Cell-type-specific CRMs may underlie transcriptional differences between cell types. For example, Gene Ontology (GO) enrichment analysis revealed cell-type-relevant biological terms, such as tumor necrosis factor signaling, linked to recurrent CRM motifs for known B cell TFs, IRF4 and IRF8,34 and IKZF164 in GM12878 cells (Figure 3E). In addition to enrichment for distinct putative TF motifs regulating the same target genes within a single cell type, the same motifs in distinct putative enhancers were looped to genes involved in specific cellular processes. (Figure 3E). For example, the POU2F2 motif lies within a putative enhancer looped to the KC differentiation gene, FLG65 (Figure 3C). The POU2F2 motif was also found in a unique prostate CRM looped to UGDH, a regulator of androgen activity in prostate cells66 (Figure 3D). These results suggest that TF motifs in CRMs link to regulation of target gene expression programs important for establishing relevant cell-type-specific biological processes.
A cell-type-specific cis-regulatory logic of heterotypic motif combinations
The enrichment of commonly shared TF motifs across cell-type-specific expression programs suggested that specific combinations of motifs, distinct from cell-type-unique motifs, contribute to cell-type-specific transcription. To determine potential synergistic relationships between TF DNA motifs in cell-type-specific gene regulation, a co-enrichment test (Fisher’s exact, Bonferroni-corrected p <0.05) was done on all pairwise hetero motif-motif combinations in the CRMs associated with each cell type. This analysis identified 838 total DMCs, ranging from 12 to 106 per cell type, with an average of 55.9 (Figures 4A–4C; Table S4). These DMCs identify known co-regulators, such as keratinocyte differentiation cooperative TFs KLF4 and TP63, MAF and MAFB, among others in KC DMCs67,68,69,70,71 (Figure 4A). This suggests that significantly co-occurring TF motifs are linked to distinct processes in cellular contexts.
Figure 4.

Co-enrichment analysis reveals DMCs
(A) Co-enrichment dot plot of TF motifs within KC CRMs depicting putative cooperativity (Fisher’s exact, Bonferroni-corrected p < 0.05). Dots are colored by −log10(p value). Size corresponds to normalized number of shared genes. Red outlined dots indicate known cooperative KC TFs.
(B) Bar plot depicting the distribution of DMCs based on CRM epigenomic interactions for MPRA-tested KC DMCs.
(C) Bar plot of number of cell-type-specific DMCs in the 15 cell types.
(D) Genomic instance of intra-promoter KC DMC HMGA1+KLF5 at the SCNN1A TSS.
(E) Genomic instance of putative intra-enhancer KC DMC HMGA1+KLF5 looping to PPARD. RC, reverse complement.
(F) Genomic instance of putative inter-enhancer-promoter KC DMC HMGA1+KLF5 proximal to FNBP1L. Related to Figure S4 and Table S4.
Next, all genomic instances of the nominated TF regulatory DMCs within each cell type were identified and the genomic locations of the motifs within the pairwise combination determined. Interestingly, while some DMCs have a strong bias toward 1D intra-promoter interactions such as KLF4-SALL4 and EGR2-KLF4, others have a strong bias toward 1D putative intra-enhancer interactions, such as HBP1-RORA and EGR2-JUNB, and nearly all DMCs occur across a 3D putative inter-enhancer-promoter interaction (Figures 1B, 4B, and S4A). The statistical co-enrichment of TF motifs across these distinct epigenomic interactions suggest that identified DMC cis-regulatory logic acts at local proximal promoters, distal putative enhancers, and across 3D E-P contacts to control cell-type-specific activity.
All three DMC interaction types were associated with cell-type-relevant target gene biological processes. For example, a DMC harboring the KLF5 KC differentiation motif,72 KLF5-HMGA1, resides in an intra-promoter genomic instance in KC at the SCNN1A gene, a subunit of the epithelial sodium channel important for body temperature regulation in skin73 (Figure 4D). Also in KC, an intra-enhancer genomic instance of KLF5-HMGA1 looped to PPARD, which stimulates keratinocyte differentiation and improves skin barrier74,75 (Figure 4E). Finally, a 3D E-P genomic instance of KLF5-HMGA1 in KC links to FNBP1L, a regulator of another known skin barrier gene, N-WASP76,77 (Figure 4F). These findings suggest that the identified DMC cis-regulatory logic operates both distally and proximally to control cell-type-relevant gene expression.
Functional assessment of cell-type-enriched DMCs via MPRA
If the DMCs identified above capture the sequence logic of cell-type-specific gene regulation, then motif combinations should synergistically drive cell-type-specific transcription when tested in diverse cell types. To assess this quantitatively at scale, MPRAs were performed on a subset of cell-type DMCs derived from cells representative of each major cluster studied, including primary human colon (EC1), KC (EC2), MC (N), and GM12878 lymphoblastoid cells. Ten native genomic instances of 42 colon, 49 KC, 26 MC, and 39 GM12878 heterotypic cis DMCs were selected, along with matched sequences in which one or both DNA motif nucleotide sequences were iteratively scrambled. These were cloned into a lentiviral MPRA library containing 62,400 sequences, including controls (Figure 5A). MPRA was performed in primary human colon, KC, MC, and diploid GM12878 cells; sample clustering demonstrated high reproducibility and clear separation between cell types (Figures S5A–S5E). Each native DMC genomic instance was compared with its corresponding scrambled controls (individual motifA, individual motifB, and jointly scrambled motifA and motifB). An expected additive change in MPRA signal, computed from individually scrambled motifs, was compared with the observed MPRA signal with both motifs present to assess the cell-type-specific activity of each DMC and the interaction between its constituent motifs.
Figure 5.

MPRAs validate TF DMCs in human cells
(A) Schematic representation of MPRA design and validated functional categories of DMC interactions.
(B) Box-and-whisker plot showing the normalized log2 MPRA signal for the different motifA-motifB combinations in the synergy DMC MITF + ZNF589 in MC. Each point on the plot represents the signal value in one genomic instance in one replicate. ∗p < 0.05 (Mann-Whitney U test).
(C) Pie chart depicting percentage of DMCs by functional category.
(D) Top left: heatmap shows log2(TPM + 1) values for TFs involved in the functional synergistic DMC combinatorial motifs (columns) by cell type (rows). Left: combinatorial TFs of DMCs (rows). Motifs (columns) that make up the DMC are circles connected by a black line. Circles are colored based on DMC cell type. Right: dot plot shows the GO terms enriched for target genes (x axis) that utilize the DMC (y axis). Dots are colored by log2(target gene count). Dot sizes are the −log10(p value) of the GO enrichment. GO terms are colored by cell-type biological processes (hypergeometric test). Related to Figure S5 and Table S5.
Similar to previously identified TF interaction categories,78 four major patterns of motif interactions were observed within studied DMCs (Figure 5A). The expected pattern of synergy was observed for the MITF-ZNF589 DMC in MC where scrambling the motif for both MITF, a known MC master regulator,79 and ZNF589 had a greater combined negative impact than scrambling of each motif alone (Figure 5B). A pattern of redundancy was observed for an IRF4-PLAG1 DMC in GM12878 cells where scrambling either IRF4 or PLAG1 alone failed to alter transcription-directing activity (Figure S5F). A buffer pattern was observed for an ETV2-SP2 DMC in MC where mutation of either motif boosted expression over native DMC sequences (Figure S5G). Finally, a single-driver pattern was also observed in which only one DNA motif was necessary to achieve similar expression to that obtained with the native recurrent heterotypic motif combination. Examples of this included the EGR2-KLF4 DMC in KC (Figure S5H) and the FOXM1-PATZ1 DMC in colon cells (Figure S5I). While all tested DMCs included TF motifs where the associated TF had a TPM > 1 in the relevant cell type, it remains a possibility for single-driver DMCs that one of the TFs is lowly expressed, such as FOXM1 compared with PATZ1 in colon (Figure 3E), or that the annotated TF is not relevant for the given cell type. The frequencies of these four patterns of motif interactions within DMCs were significant against expected pattern simulations (Figures S5J–K), with synergy (32%) being the most common (Figure 5C; Table S5).
Combinatorial TF motifs within synergistic DMCs may be linked to cell-type-specific gene expression. Indeed, cell-type-specific synergistic DMCs correspond to known important lineage TFs and linked target genes are enriched in GO terms related to relevant cellular processes, such as colon synergistic DMCs harboring the KLF5 motif co-regulating terms related to transforming growth factor β (TGF-β) and Wnt signaling.80,81 Furthermore, the RORA motif was found to co-regulate keratinization and cornification terms in KC,82 EBF1 and IZKF1 motifs co-regulate lymphocyte activation and proliferation in GM12878s,64 and the MITF motif co-regulates intracellular transport in MC83 (Figure 5D). Previous studies modeled the configuration of TF binding motifs,22,84 thus we further investigated the spacing of combinatorial TF motifs within the 10 tested genomic instances of validated synergistic DMCs. While no global spacing-to-MPRA signal relationship was observed (Figure S5L), some DMCs did display recurrent spacing features. For example, some DMCs drove signal when <25 bp apart, such as HBP1-IRF8 in GM12878s (Figure S5M), while others drove signal when ∼40 bp apart, such as PRDM1-RORA in KCs (Figure S5N). These results indicate that heterotypic TF motif pairs synergistically regulate transcription through a cis-regulatory logic and that this regulation occurs in the absence of a strict global pattern of TF motif spacing.
Synergistic KC and MC DMCs are differentially modulated in malignancy
Do diseased cells modulate the activity of normal lineage DMCs or do they engage entirely new DMCs? To explore this, the present framework was applied to malignant counterparts of two of the primary cell types studied here that give rise to some of the most common cancers in humans: keratinocytes and melanocytes. Replicate RNA-seq, ATAC-seq, and HiChIP data were generated across independent human malignant melanoma (MM) (WM-266-4 [WM] and COLO 829 [COLO]) and cutaneous squamous cell carcinoma (cSCC) (CAL27, SCC13, and A431) cell lines (Jung et al., in preparation) then DMCs identified as above. Fifty-three putative heterotypic DMC pairs were nominated in MM and 155 in cSCC (Figure 6A). These findings suggest that DMC cis-regulatory logic is not only cell-type-specific but potentially cell-state specific, such as between a disease cell and its healthy cell of origin.
Figure 6.

MPRAs identify regulatory DMCs in cancer
(A) Upset plot depicting number of DMCs determined from MM and cSCC cell lines and the size of their overlapping sets.
(B) Pie chart depicting percentage of functional DMC categories by MPRA in cSCC and MM cells.
(C) Bar plot showing the synergy score difference for KC- and cSCC-identified DMCs; p value based on a rank-sum Wilcoxon test.
(D) Left to right: panel colored by cell type/state; panel colored by functional DMC category; heatmap panel of −log10(p value) cell-type-/state-specificity score (STAR Methods); panel colored by cell-type-/state-specific expression (Wilcoxon rank-sum test p value <0.10).
(E) Top left: heatmap shows log2(TPM + 1) values for TFs in synergistic DMCs (columns) by normal KC- and cSCC-specific cell state (rows). Left: combinatorial TFs of the DMC (rows). Motifs that make up the DMC (columns) are circles with a black line connecting them. Circles are colored based on DMC cell state. Right: dot plot shows GO terms enriched for target genes (x axis) that utilize the DMC (y axis). Dots are colored by log2(target gene count). Dot sizes are the −log10(p value) of the GO enrichment. GO terms are colored by cell state biological processes.
(F) Genomic instance of putative inter-enhancer-promoter cSCC-specific synergistic DMC SP1+ARNT at ADAP1. Related to Figure S6 and Table S5.
To address disease DMC commonalities and differences with their normal counterparts, wild-type native DMC genomic instances were selected from 40 MM and 43 cSCC heterotypic motif pairs, for a total of 33,200 sequences then lentiviral MPRA performed in COLO and A431 cells. MPRA readouts for the entire library were obtained and sample clustering demonstrated high reproducibility and clear separation between the two cancer types and normal primary human cells (Figure S5E). Of the MPRA-validated cancer DMCs, 36.4% were determined to be functionally synergistic (Figure 6B; Table S5). The spacing patterns between TF motifs of these synergistic cancer DMCs were then assessed. Similar to healthy cells, some DMCs drove MPRA signal when <25 bp apart, such as SOX10-SOX13 in MM cells (Figure S6A). However, it was also observed that the EGR2 motif drove MPRA signal in KCs when 12–22 bp apart from the RARG motif (Figure S6B), while driving signal in MM cells when <10 bp or >27 bp apart from the NFE2L1 motif (Figure S6C), suggesting the spacing of several DMCs may be important for regulatory function in these cancer cells. Interestingly, comparing synergistic DMCs between cancer cells and their normal cell type of origin (MM versus MC and cSCC versus KC) revealed that, for the same target gene, DMCs were co-enriched for different combinatorial TF pairs, such as the 1P validated synergistic ETV-PRRX1 DMC in MC versus the 1P ZBTB49-NF2L1 DMC in MM at the MLANA locus, a gene involved in melanosome biogenesis85 (Figure S6D). These findings suggest that normal and disease-state cells might display a differential cis-regulatory lexicon to regulate gene expression programs that are biologically relevant for their given cell type of origin.
To investigate whether different cell states mediate gene expression through shared DMCs or through cell-state-specific DMCs, the relationship between synergistic regulatory DMCs in normal human cells versus their malignant counterparts was explored. The difference in synergy scores between the A431 cSCC line and primary KCs was calculated. A significant distributional shift in DMC synergy scores was observed in cSCC cells (Wilcoxon rank-sum test, p = 5.16 × 10−5) (Figures 6C, S6G, and S6H), where cSCC DMCs had higher synergistic scores in the A431 cSCC line than in KCs and KC DMCs had higher synergistic scores in KCs than in the A431 cSCC cells. Due to the differential modulation of synergistic DMCs in a healthy cell type of origin versus a disease cell state, all functional DMCs were assessed by their global expression across all six cell types assayed by MPRA to compare DMC activity. Five synergistic DMCs were found to be cSCC specific and three were MM specific (Wilcoxon signed rank test, p value <0.10), suggesting different mechanisms may control this process in normal versus malignant cells (Figures 6D and S6F). These cSCC- and MM-specific synergistic DMCs implicate known cancer-associated TFs such as SP1 in cSCC86,87,88 and SOX10 in MM.89,90,91 Hence, cSCC- and MM-specific DMCs determined by the CRM model drive cSCC- and MM-specific expression.
To investigate whether validated cell-state-specific synergistic cancer DMCs regulate the same gene modules as their healthy cell type of origin or regulate new biological processes relevant to a disease state, GO enrichment analysis was performed on the putative target genes of normal and cSCC- and MM-specific DMCs. While synergistic DMCs in both cSCC and MM linked to processes identified in normal cell function, such as epidermis development and pigment metabolic processing, respectively, there is also a de-enrichment of terminal differentiation processes in cSCC-specific DMCs, such as keratinization and cornification, compared with their enrichment in KC-specific DMCs (Figures 6E and S6E). Furthermore, cSCC- and MM-specific processes were also enriched, such as regulation of nuclear division and stem cell population maintenance, respectively. Indeed, the cooperative SP1-ARNT cancer-associated DMC92 was found to be cSCC specific, and a 3EP genomic instance is linked to the ADAP1 gene, a mediator of TGF-β-induced invasion in cSCC93 (Figure 6F). These results suggest that malignant-cell-type DMCs synergistically regulate both normal cell types of origin as well as cancer-relevant target gene expression in MM and cSCC through distinctive patterns of disease-specific cis-regulatory logic.
Discussion
Here, we describe a framework for identifying cell-type-specific DMCs that regulate cell-type-specific transcription built on a newly generated resource of RNA-seq, ATAC-seq, and H3K27ac HiChIP in 15 diploid human cell types. This resource was designed to serve as a repository for further studies of cell-type cis-regulatory control. Modeling these data suggested a combinatorial lexicon of cooperative DNA motifs encoded in cell-type-specific putative enhancers and promoters of actively expressed target genes to derive 7,531 cell-type-specific CRMs. MPRA experiments validated predicted cell-type-specific cis-regulatory logic, helping account for regulation of cell-type-specific transcripts between primary human cell types and selected cancer cell counterparts. The functional synergy of regulatory DMCs was found to shift between normal cells and a disease state, namely cancer, identifying cell-type- and state-enriched DMCs. This suggested that pathogenic gene dysregulation engages disease-type-unique motif combinations while also modulating existing cell lineage lexicons relevant to pathogenesis. The present work thus suggests that cell-type-specific gene expression is mediated by a code of TF DMCs in regulatory DNA whose activity is modulated in disease.
Previous studies provided high-throughput chromatin looping datasets linking distal CREs to annotate functional target gene promoters across a diverse array of cell types.54,55,56,94 The current integrative framework using cell-type-specific ATAC, HiChIP, and RNA-seq data extends these efforts to imply patterns of combinatorial, motif-based, cis-regulatory control. Including the looping-data-inferred location, proximal or distal, of CREs relative to the promoter enhanced model accuracy for cell-type-specificity in cis-regulatory logic. Moreover, cell-type-specificity predictions perform best when all features derived from ATAC, HiChIP, and RNA-seq of a CRM are present, demonstrating the value of epigenomic and transcriptomic data in relevant cellular contexts. This genome-wide map provides a resource of testable hypotheses on cell-type-specific transcription.
Additionally, while this work echoes previous efforts in model interpretation methods to catalog cis-regulatory logic and discover active motif instances and co-occurrence patterns, the present framework provides validation of extracted CRE logic. The epigenomic and transcriptomic data provided a genome-wide map of CREs for 15 human cell types and enabled resolution of CRE logic to the level of individual genes. Furthermore, cell-type-specific, regulatory DMCs were validated functionally via MPRA. These DMCs could then be categorized into classes based on their cooperative patterns. Motifs for established transcriptional regulators, such as MITF in melanocytes,79 IKZF1 in lymphocytes,64 and KLF5 in colon epithelial cells,80,81 are found in concert with other cell-type-specific as well as more globally expressed TFs to drive cell-type-specific processes. These cooperative patterns of DMCs suggest that discrete logic patterns guide transcription regulatory mechanisms to achieve differential cell-type-specific gene transcription.
The present work also compared functional cis-regulatory logic between diseased cells and healthy human cells from which they arise. Specifically, MPRA enabled direct comparison of the transcription-driving activity of synergistic TF motif combinations between normal human skin cells and their malignant counterparts; the latter, namely cSCC and MM, represent two of the most common cancers in humans. This demonstrated that CRM-predicted cell-state-specific TF regulatory DMCs are functionally synergistic in normal and cancer cells. These cell-state-derived synergistic regulatory DMCs were enriched for processes specific to cell state and cell type of origin, such as pigment metabolism, response to oxygen levels, regulation of amide metabolism in MM and epidermis development, Ras signal transduction, and mitotic nuclear division in cSCC. MM- and cSCC-specific processes were also linked to TFs with established roles in their corresponding tumors, such as SOX10 in MM and SP1 in cSCC, each associated with several paired TF motif co-regulators. Cell-state-specific synergistic TF regulatory DMCs were further functionally validated and found to be differentially modulated between healthy versus disease cell states. Finally, spacing of TF motifs within functional DMCs failed to exhibit strict global spatial patterns that could identify synergy, buffering, redundancy, and single driver patterns, but several spatial patterns were identified for specific synergistic DMCs in both healthy and disease cells. This suggests that, while a normal cell and a disease cell may retain a shared cis-regulatory logic linked to the originating cell lineage, cells in the pathogenic state shift toward using a disease-state-selective lexicon, thus altering the homeostatic balance of transcriptional regulation toward pathophysiological processes.
Limitations of the study
The information about DNA sequence lexicons underlying cell-type-specific gene expression provided here raises issues for future exploration. For example, due to the 3D nature of the CRMs, synthesizing cooperative patterns across proximal and distal elements into a relatively high-throughput, unbiased, testable manner represents a future, albeit technically challenging, goal. The chosen MPRA of combinatorial DNA motifs was limited to two cooperative motifs found in pre-existing 145-bp genomic instances, likewise limiting the assay to 3D DMCs also found in a 1D context. Moreover, MPRA does not test candidate DMCs in their native context and does not functionally validate target gene expression. Additionally, MPRA tested heterotypic motifs only with a pre-existing spacing and orientation based on the genomic instances used. Future modeling efforts, however, could be designed in synthetic sequences to explore the CRE logic with finer granularity and for a wider range of combinatorial lexical patterns. Finally, the epigenomic and transcriptomic data are derived from a static snapshot of different human cell types, and extending these efforts to model dynamic developmental processes influenced by CRMs is a future extension of these efforts. High-throughput perturbational experimental assays, such as SPEAR-ATAC95 and Perturb-seq,96 may offer orthogonal means of identifying functionally active DMCs to help decipher additional patterns of cell-type-specific cis-regulatory logic in development, homeostasis, and disease progression.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3K27ac | Abcam | Cat#Ab4729; RRID:AB_2118291 |
| Bacterial and virus strains | ||
| Stellar Competent Cells | Takara | Cat#636766 |
| Biological samples | ||
| Primary Keratinocytes | Stanford University School of Medicine | De-identified |
| Primary Melanocytes | Stanford University School of Medicine | De-identified |
| Human Bronchial/Tracheal Epithelial Cells (Airway) | ATCC | Cat#PCS-300-010 |
| Human Astrocytes (astrocytes) | Lonza | Cat#N7805-100 |
| Primary Human Bladder Epithelial Cells (Bladder) | ATCC | Cat#PCS-420-010 |
| Human Primary Colonic Epithelial Cells (Colon) | CellBiologics | Cat#H-6047 |
| Human Primary Esophageal Epithelial Cells (Esophageal) | CellBiologics | Cat#H-6046 |
| Human Mammary Epithelial Cells (HMECs) | Lonza | Cat#CC-2551 |
| Human Primary Ovarian Epithelial Cells (Ovarian) | CellBiologics | Cat#H-6036 |
| Human Primary Pancreatic Epithelial Cells (Pancreas) | CellBiologics | Cat#H-6037 |
| Human Prostate Epithelial Cells (Prostate) | Lonza | Cat#CC-2555 |
| Human Primary Proximal Tubular Epithelial Cells (Renal) | CellBiologics | Cat#H-6015 |
| Human Primary Thyroid Epithelial Cells (Thyroid) | CellBiologics | Cat#H-6040 |
| Endometrial Epithelial Cells (Uterine) | Lifeline Cell Technology | Cat#FC-0078 |
| Chemicals, peptides, and recombinant proteins | ||
| PrimeSTAR® Max DNA Polymerase | Takara | Cat#R045B |
| EcoRI-HF | NEB | Cat#R3101L |
| BamHI-HF | NEB | Cat#R3136L |
| XhoI | NEB | Cat#R0146L |
| NheI-HF | NEB | Cat#R3131L |
| rSAP: shrimp alkaline phosphatase | NEB | Cat#M0371L |
| T4 DNA ligase | NEB | Cat#M0202L |
| SuperScript™ IV Reverse Transcriptase | Thermo Fisher | Cat#18090200 |
| Thermolabile Exonuclease I | NEB | Cat#M0568L |
| Lenti-X Concentrator | Takara | Cat#631231 |
| Turbo DNase | Thermo Fisher | Cat#AM2239 |
| Optimem | Thermo Fisher | Cat#31985062 |
| Lipofectamine 3000 | Thermo Fisher | Cat#L3000015 |
| SYBR™ Green I Nucleic Acid Gel Stain, 10,000X concentrate in DMSO | Thermo Fisher | Cat#S7567 |
| Polybrene | Sigma-Aldrich | Cat#H9268-5G |
| Keratinocyte-SFM | Thermo Fisher | Cat#17005042 |
| Medium 154 | Thermo Fisher | Cat#M-154-500 |
| HKGS Supplement | Thermo Fisher | Cat#s-002-5 |
| Medium 254 | Thermo Fisher | Cat#M-254-500 |
| HMGS Supplement | Thermo Fisher | Cat#s-002-5 |
| Airway Epithelial Cell Basal Medium | ATCC | Cat#PCS-300-030 |
| Airway Epithelial Cell Basal Medium - supplement | ATCC | Cat#PCS-300-040 |
| Gibco Astrocyte Medium | ThermoFisher | Cat#A1261301 |
| CComplete Human Epithelial Cell Medium supplemented with Human Epithelial Cell Medium Supplement Kit | CellBiologics | Cat#H6621 |
| MEGM Mammary Epithelial Cell Growth Medium BulletKit | Lonza | Cat#CC-3150 |
| Prostate Epithelial Basal Medium | Lonza | Cat#CC-3165 |
| Renal Epithelial Cell Basal Medium | ATCC | Cat#PCS-400-030 |
| Renal Epithelial Cell Growth Kit | ATCC | Cat#PCS-400-040 |
| ReproLife™ Reproductive Medium Complete Kit | Lifeline Cell Technology | Cat#LL-0068 |
| RPMI-1640 | A1049101 | Cat#A1049101 |
| L-glutamine | Sigma-Aldrich | Cat#G8540-25G |
| DMEM F:12 | Thermo Fisher | Cat#11995-065 |
| Digitonin | Promega | Cat#G9441 |
| MboI | NEB | Cat#R0147 |
| Large Klenow Fragment | NEB | Cat#M0210 |
| Proteinase K | Thermo Fisher | Cat#AM2526 |
| BlueJuice loading buffer | Thermo Fisher | Cat#10816015 |
| Critical commercial assays | ||
| Miseq Reagent kit v3 (150-cycle) | Illumina | Cat# MS-102-3001 |
| Lexogen Quant-seq 3′ mRNA-seq Library Prep Kit | Lexogen | Cat#015.96 |
| BioAnalyzer High Sensitivity DNA Kit | Agilent | Cat#5067-4626 |
| Zymo DNA Clean and Concentrator-5 Kit | Zymo | Cat#D4014 |
| Turbo DNA-free kit | Thermo Fisher | Cat#AM1907 |
| Dynabeads mRNA direct kit | Thermo Fisher | Cat#61012 |
| Kapa Library Quantification Kit | Roche | Cat#KK4854 |
| NucleoSpin Gel and PCR Clean-Up | Takara | Cat#740609.25 |
| Deposited data | ||
| RNA-seq | This paper | GEO: GSE186947 |
| ATAC-seq | This paper | GEO: GSE188398 |
| HiChIP | This paper | GEO: GSE188401 |
| MPRA | This paper | GEO: GSE188403 |
| HOCOMOCO PWMs v11 | (Kulakovskiy et al., 2018)63 | #0000FF; https://hocomoco11.autosome.ru/ |
| Housekeeping genes | (Hsiao et al., 2001)45 | #0000FF; https://www.gsea-msigdb.org/gsea/msigdb/cards/HSIAO_HOUSEKEEPING_GENES |
| Essential genes | (Wang et al., 2015)46 | #0000FF; https://doi.org/10.1126/science.aac7041 |
| Haploreg v4 | (Ward and Kellis, 2016)97 | #0000FF; https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php |
| GWAS Catalog | (Buniello et al., 2019)57 | #1155CC; https://www.ebi.ac.uk/gwas/home |
| Experimental models: Cell lines | ||
| Human: GM12878 | Coriel | Cat#GM12878 |
| Human: A-431 | ATCC | Cat#CRL-1555; RRID:CVCL_0037 |
| Human: CAL27 | ATCC | Cat#CRL-2095; RRID:CVCL_1107 |
| Human: SCC-13 | Harvard Human Skin Disease Resource Center, James Rheinwald Lab | RRID:CVCL_4029 |
| Human: WM-266-4 human malignant melanoma cell line | ATCC | Cat#CRL-1676 |
| Human: COLO-823 human malignant melanoma cell line | ATCC | Cat#CRL-1974 |
| Human: SK-MEL-5 human malignant melanoma cell line | ATCC | Cat#HTB-70 |
| Human: HEK293T Lenti-X | Takara | Cat#632180 |
| Oligonucleotides | ||
| Primers for RNAseq: Please see Table S1 | This paper | N/A |
| Primers for ATACseq: Please see Table S1 | This paper | N/A |
| Primers for HiChIP: Please see Table S1 | This paper | N/A |
| Primers for MPRA: Please see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pGreenFire1-mCMV (EF1α-puro) | System Biosciences | Cat# TR010PA-P |
| pD2-miniluc | This paper | AddGene:174105 |
| pCMV R d8.91 | (Stewart et al., 2003)98 | AddGene:2221 |
| pUC-MDG VSVG | EPFL Laboratory of Virology and Genetics, Didier Trono Lab | AddGene:12259 |
| Software and algorithms | ||
| CRM code | This paper | GitHub: https://github.com/mguo123/pan_omics; Zenodo: https://zenodo.org/record/6981951 |
| Samtools | (Li et al., 2009)99 | http://www.htslib.org/; RRID:SCR_002105 |
| Bedtools | (Quinlan and Hall, 2010)100 | http://bedtools.readthedocs.io/en/latest/; RRID:SCR_006646 |
| Pybedtools | (Dale et al., 2011)101 | #72C02C; https://daler.github.io/pybedtools/#; RRID:SCR_021018 |
| BWA | (Li and Durbin, 2009)102 | https://sourceforge.net/projects/bio-bwa/ |
| Bowtie2 | (Langmead and Salzberg, 2012)103 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml; RRID:SCR_016368 |
| Picard Tools | #1155CC; http://broadinstitute.github.io/picard/ |
RRID:SCR_006525 |
| ENCODE ATAC pipeline | (2008)33 | https://github.com/ENCODE-DCC/atac-seq-pipeline |
| ChIPSeeker | (Yu et al., 2015)104 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| ChIPpeakAnno | (Zhu et al., 2010)98 | https://bioconductor.org/packages/release/bioc/html/ChIPpeakAnno.html |
| Rsubread | (Liao et al., 2019)105 | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| GRanges | (Lawrence et al., 2013)106 | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| BiomaRt | (Durinck et al., 2009)107 | https://bioconductor.org/packages/3.8/bioc/html/biomaRt.html |
| ClusterProfiler | (Wu et al., 2021)108 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| MotifBreakR | (Coetzee et al., 2015)109 | https://bioconductor.org/packages/release/bioc/html/motifbreakR.html |
| RSEM | (Li and Dewey, 2011)110 | #0000FF; https://deweylab.github.io/RSEM/; RRID:SCR_013027 |
| STAR | (Dobin et al., 2013)111 | https://github.com/alexdobin/STAR |
| Limma | (Ritchie et al., 2015)112 | #0000FF; https://bioconductor.org/packages/release/bioc/html/limma.html; RRID:SCR_010943 |
| DEseq2 | (Love et al., 2014)113 | #0000FF; https://bioconductor.org/packages/release/bioc/html/DESeq2.html; RRID:SCR_015687 |
| MOODs | (Korhonen et al., 2009)114 | https://pypi.org/project/MOODS-python/ |
| MACS2 | (Zhang et al., 2008)115 | https://github.com/taoliu/MACS; RRID:SCR_013291 |
| HINT-ATAC | (Li et al., 2019)62 | #0000FF; http://www.regulatory-genomics.org/hint/introduction/ |
| HiC-Pro | (Servant et al., 2015)116 | #0000FF; https://github.com/nservant/HiC-Pro; RRID:SCR_017643 |
| Hichipper | (Lareau and Aryee, 2018a)117 | #0000FF; https://github.com/aryeelab/hichipper |
| FitHiChIP | (Bhattacharyya et al., 2019)118 | #0000FF; https://github.com/ay-lab/FitHiChIP |
| Diffloop | (Lareau and Aryee, 2018b)119 | #0000FF; http://bioconductor.org/packages/release/bioc/html/diffloop.html |
| Pandas | #0000FF; https://pandas.pydata.org/ |
RRID:SCR_018214 |
| Numpy | #0000FF; http://www.numpy.org/ |
RRID:SCR_008633 |
| Scipy | #0000FF; https://www.scipy.org/ |
RRID:SCR_008058 |
| scikit-learn | #0000FF; http://scikit-learn.org/ |
RRID:SCR_002577 |
| Statsmodel | #0000FF; http://www.statsmodels.org/ |
RRID:SCR_016074 |
| Matplotlib | #0000FF; http://matplotlib.sourceforge.net/ |
RRID:SCR_008624 |
| Seaborn | #0000FF; https://seaborn.pydata.org/ |
RRID:SCR_018132 |
| ggplot2 | #0000FF; https://cran.r-project.org/web/packages/ggplot2/index.html |
RRID:SCR_014601 |
| RColorBrewer | #0000FF; https://cran.r-project.org/web/packages/RColorBrewer/index.html |
RRID:SCR_016697 |
| Pheatmap | #0000FF; https://www.rdocumentation.org/packages/pheatmap/versions/0.2/topics/pheatmap |
RRID:SCR_016418 |
| Viridis | #0000FF; https://cran.r-project.org/web/packages/viridis/vignettes/intro-to-viridis.html |
RRID:SCR_016696 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Paul A. Khavari (khavari@stanford.edu).
Material availability
Plasmid pD2-miniluc generated in this study has been deposited to Addgene, catalog number 174105.
Experimental model and subject details
Human tissue samples
Primary human keratinocytes and melanocytes were isolated and cultured from fresh, surgically discarded neonatal foreskin. All human cells were collected and analyzed by protocols approved by the Stanford Human Subjects Institutional Review Board and in accordance with the NIH genomic data sharing policy. Keratinocytes were maintained in a 1:1 mixture of Keratinocyte-SFM (ThermoFisher, 17005042) and Medium 154 (ThermoFisher, M-154-500) supplemented with HKGS (ThermoFisher, S-001-5). Keratinocyte differentiation was induced by the addition of 1.2 mM calcium for 3 or 6 days at full confluence. Melanocytes were maintained in Medium 254 (ThermoFisher, m-254-500) and supplemented with HMGS supplement (ThermoFisher, s-002-5) and 1% anti-mycoplasma and 1% pen/strep.
Human cell culture
Bronchial/Tracheal Epithelial Cells (Airway) were obtained from ATCC (PCS-300-010) and grown in Airway Epithelial Cell Basal Medium (ATCC, PCS-300-030) supplemented with Bronchial Epithelial Cell Growth Kit (ATCC, PCS-300-040). Cells were grown in 15 cm dishes.
Human astrocytes (astrocytes) were obtained from Lonza (N7805-100) and grown in Gibco Astrocyte Medium (ThermoFisher, A1261301). Cells were grown in 15 cm dishes.
Primary Human Bladder Epithelial Cells (A/T/N) (Bladder) were obtained from ATCC (PCS-420-010) and grown in Bladder Epithelial Cell Basal Medium ((ATCC PCS-420-032) supplemented with Bladder Epithelial Cell Growth Kit (ATCC PCS-420-042). Cells were grown in 15 cm dishes.
Human Primary Colonic Epithelial Cells (Colon) were obtained from CellBiologics (H-6047) and grown in Complete Human Epithelial Cell Medium supplemented with Human Epithelial Cell Medium Supplement Kit (CellBiologics, H6621). Cells were grown in 15 cm dishes.
Human Primary Esophageal Epithelial Cells (Esophageal) were obtained from CellBiologics (H-6046) and grown in Complete Human Epithelial Cell Medium supplemented with Human Epithelial Cell Medium Supplement Kit (CellBiologics H6621). Cells were grown in 15 cm dishes.
Human Mammary Epithelial Cells (HMECs) were obtained from Lonza (CC-2551) and grown in MEGM Mammary Epithelial Cell Growth Medium BulletKit (Lonza, CC-3150). Cells were grown in 15 cm dishes.
Human Primary Ovarian Epithelial Cells (Ovarian) were obtained from CellBiologics (H-6036) and grown in grown in Complete Human Epithelial Cell Medium supplemented with Human Epithelial Cell Medium Supplement Kit (CellBiologics, H6621). Cells were grown in 15 cm dishes.
Human Primary Pancreatic Epithelial Cells (Pancreas) were obtained from CellBiologics (H-6037) and grown in Complete Human Epithelial Cell Medium supplemented with Human Epithelial Cell Medium Supplement Kit (CellBiologics, H6621). Cells were grown in 15 cm dishes.
Human Prostate Epithelial Cells (Prostate) were obtained from Lonza (CC-2555) and grown in Prostate epithelial basal medium (Lonza, CC-3165) and supplemented with PrEGM Prostate Epithelial Cell Growth Medium SingleQuots Supplements and Growth Factors (Lonza, CC-4177)
Human Primary Proximal Tubular Epithelial Cells (Renal) were obtained from CellBiologics (H-6015) and grown in Complete Human Epithelial Cell Medium supplemented with Human Epithelial Cell Medium Supplement Kit (CellBiologics, H6621). Cells were grown in 15 cm dishes.
Human Primary Thyroid Epithelial Cells (Thyroid) were obtained from CellBiologics (H-6040) and grown in Complete Human Epithelial Cell Medium supplemented with Human Epithelial Cell Medium Supplement Kit (CellBiologics, H6621). Cells were grown in 15 cm dishes.
Endometrial (Uterine) Primary Epithelial Cells were obtained from Lifeline Cell Technology (FC-0078) and grown in ReproLife™ Reproductive Medium Complete Kit (Lifeline Cell Technology, LL-0068). Cells were grown in 15 cm dishes.
Cell lines
GM12878 were obtained from Coriel (Catalog # GM12878) and grown in RPMI-1640 supplemented with 2mM L-glutamine (Thermo Fisher 25030149), 15% non-heat-activated FBS (HyClone, ThermoFisher) and 1% pen/strep. Cells were grown in T-25 or T-75 flasks in accordance with ENCODE guidelines.
Lenti-X 293T cell line was obtained from Takara (Catalog # 632180) and grown in DMEM F:12 (ThermoFisher, 11995-065) supplemented with 10% FBS (HyClone, ThermoFisher) and pen/strep.
SK-MEL-5 human malignant melanoma cell line was obtained from ATCC (HTB-70) and grown in DMEM F:12 (ThermoFisher, 11995-065) supplemented with 10% FBS and 1% Pen/Strep. Cells were grown in T-75 flasks.
WM-266-4 human malignant melanoma cell line was obtained from ATCC (CRL-1676) and grown in DMEM F:12 (ThermoFisher, 11995-065) supplemented with 10% FBS. Cells were grown in T-75 flasks.
COLO 829 human melanoma cell line was obtained from ATCC (CRL-1974) and grown in RPMI 1640 Media (ThermoFisher, A1049101) supplemented with 10% FBS. Cells were grown in T-75 flasks.
A-431 human epidermoid carcinoma cell line was obtained from ATCC (CRL-1555) and grown in DMEM F:12 (ThermoFisher, 11995-065) supplemented with 10% FBS. Cells were grown in 15 cm dishes.
CAL27 human squamous cell carcinoma cell line was obtained from ATCC (CRL-2095) and grown in DMEM F:12 (ThermoFisher, 11995-065) supplemented with 10% FBS. Cells were grown in 15 cm dishes.
SCC-13 human squamous cell carcinoma cell line was a generous gift from J.G. Rheinwald, Dana-Farber/Harvard Cancer Center and grown in Keratinocyte-SFM (Thermo Fisher, 17005042) supplemented with HKGS (ThermoFisher, S-001-5). Cells were grown in 15 cm dishes.
All cells were grown at 37°C in a humidified chamber with 5% CO2. All cell lines were negative for mycoplasma with MycoAlert (Lonza, Basel, Switzerland) immediately before use.
Method details
RNA-seq library preparation and sequencing
RNA-seq was performed on biological replicates using the Lexogen Quant-seq 3′ mRNA-seq Library Prep Kit FWD for Illumina protocol (Lexogen, 015.96). Briefly, total RNA was extracted from cells using the RNeasy Mini Kit (QIAGEN, 74104). 1 ug of total RNA was hybridized with an oligo-dT primer containing an Illumina-compatible sequence at its 5′ end and reverse transcription is performed. Following first strand cDNA synthesis, RNA is removed. Double-stranded cDNA was synthesized followed by a purification step. qPCR was performed to determine optimal PCR cycle number using the PCR Add-on Kit for Illumina (Lexogen, 020.96). i7 adapters for Illumina sequencing were added during PCR amplification (see Table S1, Primers and oligos, for RNA-seq adapter sequences). Following purification, RNA-seq libraries were quantified using the BioAnalyzer High Sensitivity DNA Kit (Agilent, 5067-4626) prior to sequencing using 1 × 150 bp single-end reads on an Illumina HiSeq 4000 instrument at a depth of 50 million reads per sample (see Table S2, Sequencing QC, for RNA-seq read depth information).
ATAC-seq library preparation and sequencing
Fast-ATAC sequencing on biological replicates was performed as previously described.120 Briefly, 55,000 viable cells were pelleted and resuspended in 50 uL of ATAC resuspension buffer (RSB) with 0.1% Igepal CA-630 (NP-40), 0.1% Tween 20, and 0.01% digitonin (Promega, G9441). After 3 min on ice, 1 mL of ATAC RSB with 0.1% Tween 20 was added, tubes were inverted, and nuclei pelleted by centrifugation at 500 RCF for 10 min at 4C. Supernatant was carefully removed and the nuclei pellet was resuspended in 50 uL of transposition mixture (25 uL TD buffer, 2.5 uL of TDE1 (Illumina, 20034197), 16.5 uL PBS, 0.5 uL 1% digitonin, 0.5 uL 10% Tween 20, 5 uL nuclease-free water). Transposition reactions were incubated at 37°C for 30 min in an Eppendorf ThermoMixer with agitation at 1000 RPM. Transposed DNA was purified using a Zymo DNA Clean and Concentrator-5 Kit (Zymo, D4014) and purified DNA was eluted in 20 ul elution buffer (10 mM Tris-HCl, pH 8). Transposed fragments were amplified and purified as described previously121 (briefly, transposed fragments were amplified for 5 cycles, then 5 uL of the pre-amplified mixture was run in a 15 uL qPCR and the amplification profiles assessed manually to determine the required number of additional cycles to amplify the remainder of the pre-amplified DNA.) with modified primers122 (see Table S1 for ATAC-seq adapter sequences). Libraries were quantified using qPCR (Kapa Library Quantification Kit for Illumina, Roche, #KK4854) prior to sequencing. All Fast-ATAC libraries were sequenced using paired-end 2 × 75 bp, dual-index sequencing on an Illumina HiSeq 4000 at a depth of 50 million reads per sample (see Table S2 for ATAC-seq read depth information).
HiChIP library preparation and sequencing
The HiChIP protocol was performed as previously described.123 Briefly, 5 million live cells were crosslinked using freshly prepared 1% formaldehyde. The reaction was quenched using 125 mM glycine and cells were stored in −80C prior to performing the HiChIP protocol. Crosslinked cells were resuspended in 500 uL Hi-C Lysis Buffer and rotated at 4C for 30 min. Cells were spun down at 2500 rcf for 5 min at 4C. Supernatant was removed and the pelleted nuclei were resuspended in 500 uL Hi-C Lysis Buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% NP-40, 1X protease inhibitors, water). Spin down and wash steps were performed twice. The pellet was resuspended in 100 uL of 0.5% SDS, split in half, and incubated at 62 for 10 min. SDS was quenched using 285 uL H20 and 50 uL 10% Triton X-100 with rotation at 37C for 15 min. Chromatin was digested using 50 uL NEBuffer 2 (NEB, B7002S) and 8 uL of 375 U of MboI restriction enzyme (NEB, R0147) with rotation at 37C for 15 min. Digested chromatin was spun down for 5 min at 2500 rcf, supernatant was removed, and the pellet was resuspended in 500 uL 1X NEBuffer 2. This step was repeated twice. Restriction fragment overhangs were filled in and DNA ends were marked with biotin through addition of 52 uL of fill-in master mix (37.5 uL 0.4mM biotin-ATP (Jena Bioscience, NU-835-BIO14-L), 1.5 uL 10 mM dCTP, 1.5 uL 10 mM dGTP, 1.5 uL 10 mM dTTP (ThermoFisher, 10297018), and 10 uL 5U/uL DNA Polymerase I, Large Klenow Fragment (NEB, M0210) and rotation at 37C for 1 h. 948 uL ligation master mix (150 uL 10X NEB TF DNA ligase buffer with 10 mM ATP (NEB, B0202), 125 uL 10% Triton X-100 (Sigma, T8787-100ML), 3 uL 50 mg/mL BSA (ThermoFisher, AM2616), 10 uL 400 U/uL T4 DNA Ligase (NEB, M0202) and 660 uL water) was added and chromatin was resuspended before incubation at RT for 4 h with rotation. Nuclei was pelleted at 2500 rcf for 5 min, supernatant was removed, 880 uL Nuclease Lysis Buffer (50 mM Tris-HCl pH7.5, 10 mM EDTA, 1% SDS, 1X protease inhibitors, water) was added, and nuclei were moved to 1 mL Covaris tubes (milliTUBE 1 mL AFA Fiber(100), Covaris). Samples were sheared using a Covaris E220 using the following parameters: Fill Level = 10, Duty Cycle = 5, PIP = 140, Cycles/Burst = 200, Time = 4 min and then clarified by centrifugation for 15 min at 16100 rcf at 4°C. 10X volume of ChIP Dilution Buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 7.5, 167 mM NaCl, water) was added to achieve an SDS concentration of 0.1%. 4 ug of H3K27ac antibody was added (Abcam, ab4729) and chromatin was incubated overnight at 4C with rotation. We captured the chromatin-antibody complex with 34 uL Protein A beads (Thermo Fisher, 10001D) and rotation at 4C for 2 h. Beads were washed three times each with 500 uL Low Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-Hcl, 150 mM NaCl, water), High Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 500 mM NaCl, water), and LiCl Wash Buffer (10 mM Tris pH 7.5, 250 mM LiCl, 1% NP-40, 1% Na-DOC, 1 mM EDTA, water) at RT using magnet swishing and removing the supernatant. Split samples were recombined when adding the first Low Salt Wash Buffer.
ChIP samples were resuspended in 100 uL Elution Buffer (50 mM NaHCO3, 1% SDS, water) and incubated for 10 min at RT with rotation, followed by 3 min at 37C shaking. Samples were placed on a magnet and the supernatant was moved to a new tube. This step was repeated twice for a final volume of 200 uL ChIP DNA. 10 uL Proteinase K (ThermoFisher, AM2546) was added and samples were incubated at 55C for 45 min. The temperature was then increased to 67C for 1.5 h with shaking. Samples were purified using Zymo ChIP DNA Clean & Concentrator (Capped Columns) (Zymo, D5205) and eluted in 10 uL of water. Qubit quantification following ChIP ranged from 125-150 ng. (ThermoFisher, Q32851) Up to 150 ng DNA was resuspended with 5 uL Streptavidin C-1 (ThermoFisher, 65001) beads resuspended in 10 uL Binding Buffer (10 mM Tris-HCl pH 7.5, 1mM EDTA, 2M NaCl, water) and incubated at RT for 15 min with rotation. Beads were separated on a magnet and the supernatant removed. Beads were washed twice with 500 uL Tween Wash Buffer (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20, water) and incubated at 55C for 2 min shaking. Beads were washed with 100 uL 1X TD Buffer. Beads were resuspended in 25 uL of 2X TD Buffer (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 20% Dimethylformamide, water), the appropriate amount of Tn5 used and number of PCR cycles performed were based on the post-ChIP Qubit amounts, as previously described123 (briefly, a maximum of 4 uL Tn5 was used for samples with 125 ng of DNA transpose and then amplified in 5 cycles), and water up to 50 uL. Samples were incubated at 55C with interval shaking for 10 min, placed on a magnet, and the supernatant removed. 300uL of 50 mM EDTA (ThermoFisher, 15575020) was added and samples were incubated at 50C for 30 min. Samples were placed on a magnet and the supernatant removed, washed twice with 300 uL 50 mM EDTA, and incubated at 50 for 3 min with interval shaking and the supernatant removed. Samples were then washed twice with 500 uL Tween Wash Buffer and incubated at 55C for 2 min with interval shaking, removing the supernatant on the magnet. Samples were washed once with 500 uL 10 mM Tris. Beads were resuspended in 50 uL PCR master mix in a strip tube (25 uL Phusion HF 2X (NEB, M0531S), 1 uL 12.5 uM Ad 1.x, 1 uL 12.5 uM Ad2.x100 (see Table S1 for H3K27AC HiChIP adapter sequences), and 23 uL water) and run at 72C for 5 min, 98C for 1 min, and for 5 cycles of 98C for 15 s, 63C for 30 s, and 72C for 1 min. Supernatant was transferred to fresh tubes. Samples were purified using the Zymo kit (Zymo, D4013) and eluted in 10 uL of water. 1 uL of 10X BlueJuice loading buffer (ThermoFisher, 10816015) was added and samples were run on a 6% PAGE gel (ThermoFisher, EC6265BOX) for 30 min at 160V. The gel was soaked in SYBR Safe (ThermoFisher, S33102) and TBE buffer (ThermoFisher, LC6675)) for 5 min. HiChIP samples were size selected by PAGE purification (300-700 bp) for effective paired-end tag mapping and were therefore removed of all primer contamination. Gel slices were placed in doubled tubes with a hole in the smaller one and tubes were centrifuged for 3 min at max speed. 300 uL Crushed Salt Buffer (500 mM NaCl, 1 mL EDTA, 0.05% SDS, water) was added to each tube and incubated at 55C overnight. CSB buffer and gel slurry were transferred to Spin-X columns (Sigma, CLS8162-24EA) and spun down at max speed for 2 min. Samples were Zymo purified using the DNA Clean & Concentrator-5 kit (Zymo, D4013) and eluted in 10 uL elution buffer. Libraries were quantified using qPCR (Kapa Library Quantification Kit for Illumina, Roche) prior to sequencing. All libraries were sequenced using 2 × 150 bp reads on the Illumina NovaSeq 6000 instrument to an average read depth of 300 million total reads (see Table S2 for H3K27ac HiChIP read depth information).
MPRA
Oligo design and selection
TF DMCs were selected from the co-enrichment analysis. A total of 49 keratinocyte, 42 colon, 39 GM12878, and 26 melanocyte normal DMCs were curated based on literature search, which prioritized TF’s with cell type-specific function in the corresponding cell type. Additionally, 43 squamous cell carcinoma and 40 melanoma DMCs were curated, for a total of 239 DMCs to be tested. For each DMC, 10 genomic instances of the DMCs in a 135 bp segment were selected. Segments where the DMCs were closer than 50 bp apart were prioritized. Positive and negative control sequences were added. Positive controls were genomic sequences of the 150 bp upstream of the TSS of the 72 highest expressed genes in selected cell type DMCs. Negative controls were genomic sequences of the 150 bp upstream of the TSS of 89 genes that were not expressed, had no looping, and had no accessible sites in any of the selected cell types. A list of the 239 selected DMCs can be found in Table S5, MPRA DMC categories. All genomic sequences were extracted via API querying of hg19 version of the UCSC browser.124 Scrambling of the motif instances of the two motifs within the DMC were done so that four configurations existed for each DMC: both motifs scrambled, motif A scrambled, motif B scrambled, and no motifs scrambled. The scrambling was done iteratively. After each iteration, the sequences were scanned for via the MOODs python package,114 to ensure that the scrambled version did not contain the motif of interest and to minimize the possibility that other possible motifs were introduced. To design the MPRA sequences for synthesis and subsequence cloning, sequences first were filtered to ensure the restriction sites for EcoRI, BamHI, XhoI, and NheI were not present. Each MPRA library oligo included, in order: a forward PCR primer binding site (5′-ACTGGCCGCTTCACTG-3′), the 145 bp genomic instance sequence, a XhoI restriction site, a 10bp randomly generated spacer sequence, a NheI restriction site, a 20bp barcode, and a reverse PCR primer binding site (5′-AGATCGGAAGAGCGTCG-3′) (see Table S1 for complete MPRA oligo and primer information). The 10bp spacer sequence was included to improve restriction enzyme cutting efficiency and to reduce template switching in the initial PCR amplification cloning step, and is later removed following digestion with NheI and XhoI. The 20bp barcodes are all a minimum Hamming distance of 3 apart. Each unique genomic instance is barcoded 10 times. The smaller number of barcodes may reduce sensitivity,125 but 10 barcodes were sufficient to identify active time-dependent combinatorial DNA motifs.20 This yields a 97,210-221bp oligo library that was synthesized by the Agilent oligo synthesis process.
Cloning
Cloning proceeded in 2 steps. Agilent oligo library was resuspended in Ultra-Pure H2O then diluted to make a 10 pg/ul stock. Resuspended oligo pool was amplified 24 cycles using PrimeSTAR Max DNA Polymerase (Takara, R045B) with a forward primer 5′-GCTAAGGAATTCACTGGCCGCTTCACTG-3′ and reverse primer 5′-GCTAAGGGATCCCGACGCTCTTCCGATC-3′ to introduce the EcoRI and BamHI restriction sites upstream and downstream of the oligo, respectively. Product was gel purified using a 2% agarose gel then using an MN nucleospin kit (Takara, 740609.250). A BamHI site was added to the pGreenFire1-mCMV (EF1α-puro) plasmid (System Biosciences, TR010PA-P) between the luciferase and WPRE element by mutagenesis with the following primer: GAGGTTGATTGTCGAGTCGAGGATCCTTACAATTTGGACTTTCCGCCC. This is referred to as the pGreenFire-MPRA plasmid. 64ug of pGreenFire-MPRA plasmid (32 reactions) and the purified PCR library product (6 reactions) were digested with EcoRI-HF (NEB, R3101L) and BamHI-HF (NEB, R3136L) for 1 h at 37C. The pGreenFire plasmid was also simultaneously rSAP (NEB, M0371L) treated. The pGreenFire-MPRA plasmid gel purified using a 0.7% agarose gel, while the digested oligo library was PCR purified using the same MN kit. Digested library and pGreenFire-MPRA vector were ligated using T4 Ligase (NEB, #M0202L) at a 2:1 insert:vector ratio for 2 h at room temperature (10-20ul reactions). All of the ligation product was transformed into Stellar Competent cells (Takara, 636766) with 2 ul of ligation mix per 50 ul of cells and a total of 80 transformation reactions. Reactions were done in two sets of 40, with a test transformation followed by Sanger sequencing to confirm the ligation was a success. Full scale transformation was done in large volume liquid cultures, allowed to recover for 1 h-post heat shock, and incubated at 37C for 8 h in ampicillin-treated LB. The expanded library was isolated by Qiagen Plasmid Plus Max Kit (Qiagen, 12963). This pGreenFire-library vector was then digested with XhoI (NEB, R0146L) and NheI-HF (NEB, R3131L) and rSAP treated for 90 min at 37C. The pMPRA-d2-miniluciferase plasmid was simultaneously PCR amplified with 5′-TTGTAAAACGACGGCCAGTGAATTCG-3′ and 5′-ACATCATGGTCGCTAGCGGGCGTAGCGCTTCATGGCT-3′ for 34 cycles and then subsequently digested with XhoI and NheI-HF for 90 min at 37C. Both the digested pGreenFire-library vector and the minluc insert products were gel purified using 0.5 and 1.6% agarose gel respectively. Purified miniluc and pGreenFire-library vector were ligated using T4 Ligase at a 3:1 insert:vector ratio for 2 h at room temperature (5-20ul reactions). A similar round of 40 transformations using Stellar Competent cells was performed, as done in the previous cloning step with an 8 h incubation of liquid cultures. The final plasmid library was isolated by Qiagen Plasmid Plus Maxi Kit (Qiagen, 12963). Plasmid library was sequenced to ensure adequate coverage of the designed oligo library pool, according to manufacturer instructions. Briefly, Plasmid was amplified on a qPCR Stratagene MX3005P machine using the PrimeSTAR MAX polymerase and SYBR green (ThermoFisher, S7567) until the linear phase (around cycles 10-15) on the qPCR machine was achieved. PCR product was PCR purified then gel purified with a 549 bp band using the MN Nucleospin kit (Takara, 740609.250). Library concentration was determined using Kapa Library Quantification Kit (Roche Diagnostics Corporation, KK4854) and Bioanalyzer. Sequencing was done using an Illumina Miseq (Illumina, MS-102-3001), and the number of barcodes present in the library was determined. Multiple iterations of the cloning process were done and pooled to form the final plasmid library to obtain near complete coverage of the library.
Virus generation
LentiX cells (passage < P8) were grown in 15cm plates until ∼80% confluent. Plasmids pCMV R d8.91126 (25 ug/plate), pUC-MDG VSVG (Addgene, 12259) (10 ug/plate), and the plasmid library (25 ug/plate) were transfected using Lipofectamine 3000 (ThermoFisher, L3000015) (2ul/ug DNA) in Optimem (ThermoFisher, 31985062) (7mL/plate). Supernatant was harvested 48 and 72 h post transfection. Supernatant was concentrated using Lenti-X concentrator (Takara, 631232) at a 3:1 vol:vol ratio of supernatant: concentrator, then aliquoted and frozen down to −80C.
Infection and cell collection
In each cell type, optimal puromycin concentration was determined, and the virus was titrated using CellTiterBlue (Promega, G8080) assays to minimize virus toxicity and maximize infection efficiency for each cell type. Additionally, average integrants per cell was determined for infected cells. Briefly, gDNA was extracted from infected cells post-selection via Qiagen tissue extraction kits (Qiagen, 69504). Serial dilutions of the original plasmid library and the gDNA were made. qPCR was performed on all serial dilutions using primers designed for the oligo library sequences to determine the number of copies of the integrants present in each gDNA sample, using the formula: log10(copies) = PLASMID_INTERCEPT ∗ Cq + PLASMID_SLOPE. Cell number for each gDNA sample was approximated based on the assumption that there is roughly 6.6 pg of gDNA per cell. The average integrants per cell was calculated by dividing the number of copies present in a gDNA sample by the number of estimated cells. Average number of integrants per cell greater than 4 were desired.
For non-GM12878 cell types, cells were infected by trypsinizing then counting cells. At least 12.2 million cells per replicate were desired for infection. A virus-polybrene-cell-media mix of 8 ul/mL of polybrene (Sigma, H9268-5G) and 100,000 cells/mL of media was made to seed the cells in 6 15-cm plates. The amount of virus/200,000 cells was previously determined using a CellTiterBlue (Promega, G8080) toxicity screen (see Table S3, MPRA cell culture conditions, for concentrations for each cell type). 2 wells of a 6-well plate were also seeded, one with virus and the other without, for monitoring the antibiotic selection. Plates were returned to the 37C incubator and allowed to recover for 24 h. For GM12878 cells, virus was not concentrated and fresh virus suspended in RPMI-1640 media was used. Cells were counted and at least 26 millions cells per replicated were desired for infection. A virus-polybrene-cell-media mix of 4 ul/mL polybrene was plated onto 6-well plates and spun at 731g (2000 rpm) at room temp for 2 h. Plates were incubated at 37C for 6 h before the cells were pelleted and resuspended into the T-25 flasks in normal media. Media was changed 24 h post-infection. 48 h post-infection, cells were selected using puromycin (1.0 ug/mL for keratinocytes) for 48-72 h. CellTiterBlue (Promega, G8080) assays were used to ensure all noninfected, antibiotic-treated cells were dead. Cells were changed to normal media and allowed to divide until the desired number of cells was achieved (usually 2-4 days post-selection). Cells were lysed on plate using the Lysis/binding buffer from the Dynabeads mRNA direct kit (ThermoFisher, 61012). Cells were homogenized using a 2gg needle and syringe and stored at −80C for sequencing library preparation.
Sequencing library preparation
mRNA from the cells was extracted using the Dynabeads mRNA direct kit (ThermoFisher, 61012) per manufacturer’s instructions and eluted to 30 ul per replicated. Extracted mRNA was Turbo DNase treated (ThermoFisher, AM1907) for 1 h at 37C and then subsequently purified using Ampure XP beads (Beckman Coulter, A63881) at a 1:1.9 sample:Ampure bead ratio. cDNA was generated using Super-Script IV Reverse Transcriptase (ThermoFisher, 18090050) in 500 ng RNA reactions using a P5 primer (see Supplemental Information Table S1 for full MPRA primer sequences), per manufacturer’s instructions. No RT enzyme conditions were included as a control. Reaction were treated with 1ul of thermolabile exonuclease I (NEB, M0293) and incubated at 37C for 10 min then heat inactivated. A second Ampure XP purification step was done, using a 1:1.1 sample to beads ratio. Test qPCR assays using PrimeStar Max Polymerase, the Illumina P7 primer and 5′-AATGATACGGCGACCACCGAGATCTAC-3′ (see Table S1 for full MPRA primer sequences) and were run to determine the optimal number of cycles with a 50C annealing temperature for 15 s and a 72C extension temperature for 20 s. Samples were removed when qPCR reached a linear phase (typically cycle 20-25). Care was taken to ensure the cycle stopped prior to when the NRT negative control began to rise. qPCR amplification product was gel purified on a 2% agarose gel and the resultant amplicon was 277 bp. Library concentration of the cDNA libraries was determined using Kapa Library Quantification Kit (Roche Diagnostics Corporation, KK4854) and BioAnalyzer prior to pooling for sequencing.
Sequencing
cDNA libraries were sequenced using 2 × 150 bp reads on the Illumina NovaSeq 6000 instrument to an average read depth of 200 million reads per sample.
Quantification and statistical analysis
Coding platform
Statistical analyses were performed with R version 3.6.1 and Python 3.7.4 in Jupyter Notebook. Parameters such as number of replicates, the number of independent experiments, measures of center, dispersion, and precision (mean ± SD or SEM), statistical test and significance, are reported in Figures and Figure Legends. Raw sequencing data were processed on Stanford’s Sherlock cluster. RNA-seq (GSE186947), ATAC-seq (GSE188398), HiChIP (GSE188401) and MPRA (GSE188403) data have been deposited at GEO (GSE188405) and are publicly available as of the date of publication. Raw sequencing files for primary keratinocyte and melanocyte data are restricted and access is in accordance with NIH genomic data sharing policy. Python packages pandas, numpy, scipy, statsmodel, and matplotlib were also used for modeling. The seaborn Python package was used for visualization using viridis color palettes. Plots in R were made with tidyverse, ggplot2, RColorBrewer, and pheatmap packages.
Computational pipeline for RNA-seq
To quantify gene expression, single end reads were mapped to the hg19 reference genome with GRCh37 Ensembl annotations using STAR aligner (version 2.5.4b)111 using default parameters and the Gencode V19 gene annotation gtf. Sample expression counts and transcripts per million (TPM) values were generated using RSEM (version 1.3.0)110 and default parameters. Conversion between Ensemble IDs and HGNC symbols was performed using the biothings api client python package. Cell type-specific genes were defined as genes expressed at a TPM>1 across both biological replicates in a single cell type and at a TPM <1 in all other cell types.
Computational pipeline for ATAC-seq
ATAC-seq read alignment, quality filtering, duplicate removal, transposase shifting, peak calling, and signal generation were all performed through the ENCODE ATAC-seq pipeline (https://github.com/ENCODE-DCC/atac-seq-pipeline). Briefly, adapter sequences were trimmed, sequences were mapped to the hg19 reference genome using Bowtie2 (-X2000),103 poor quality reads were removed (params), PCR duplicates were removed (Picard Tools MarkDauplicates, http://broadinstitute.github.io/picard/), chrM reads were removed, and read ends were shifted +4 on the positive strand or −5 on the negative strand to produce a set of filtered high-quality reads. These reads were put through MACS2115 to get peak calls and signal files. Finally, IDR analysis was run on the two replicate peak files to produce an IDR peak file that is the reproducible set of peaks across both replicates. Cell type-specific ATAC-seq peaks were identified using the IDR peak files across both biological replicates that were unique to a single cell type. The full pipeline can be found on the ENCODE portal. ATAC Footprinting was performed using the HINT-ATAC62 package. Transcription factor motif position-weight matrices (PWMs) from the HOCOMOCO v1163 database were processed to remove non-informative bases. Additionally, motifs were matched to transcription factors using the annotations provided. Ends of the PWMs were trimmed by an information content (IC) threshold at the end of IC > 0.4. Overall 770 PWMs remained post-processing. Footprinting using the “rgt-hint footprinting” and “rgt-motifanalysis matching” commands were done using default parameters over the filtered ATAC bam file and the IDR peak file derived from ATAC processing. A TF motif footprint was considered present within a cell type-specific regulatory region if the associated TF had a TPM >1.
Computational pipeline for HiChIP
HiChIP paired-end reads were aligned to the hg19 genome using the HiC-Pro pipeline.116 Default settings were used to remove duplicate reads, assign reads to MboI restriction fragments, filter for valid interactions, and generate binned interaction matrices. HiC-Pro filtered reads were then processed using hichipper117 using the {EACH, ALL} settings to call HiChIP peaks to MboI restriction fragments. HiC-Pro valid interaction pairs and hichipper HiChIP peaks were then processed using FitHiChIP118 to call significant chromatin contacts using the default settings except for the following: MappSize = 500, Int-Type = 3, BINSIZE = 5000, QVALUE = 0.01, UseP2PBackgrnd = 0, Draw = 1, TimeProf = 1. Significant HiChIP interactions from either biological replicate were used to identify unique interactions to a single cell type. Cell type-specific HiChIP data were analyzed. The distribution of loop width was determined. The number of unique, total, common across cell types loops and anchors (anchors are defined as one of two 5 kb regions a loop is connecting) was found. The distribution of loop types (promoter:promoter, promoter:promoter-interacting, promoter-interacting:promoter-interacting) was determined. An “enhancer” was determined as a promoter-interacting region, or HiChIP anchor, that contained at least one ATAC peak in the match cell type data. Results were compared with HiChIP data from previous studies.
Differential “omics” analysis
Differential RNA-seq analysis
Differential RNA-seq was performed using the limma112 package using cell type as the grouping variable and an absolute log fold change >0.1 and an FDR-adjusted p value < 0.05 as thresholds. Hierarchical clustering was used to determine the four main clusters (Epithelial C1 (Airway, Bladder, Keratinocytes, HMEC, Prostate, and Uterine), Epithelial C2 (Colon, Esophageal, Ovarian, Pancreas, Renal, and Thyroid), neurogenic (Melanocytes and astrocytes), and immune (GM12878)). biomaRt107 was used to obtain gene identifiers. Differential expression by cluster (Epithelial C1, Epithelial C2, neurogenic, and immune) revealed 7531 differentially expressed genes which was further filtered down to 2952 prioritized differential genes. Results were plotted as a heatmap using R package pheatmap and the R function scale to Z score by column (cell type). ClusterProfiler R package108 was used to functionally characterize the different genesets using GO Term enrichment.
Differential ATAC-seq analysis
Differential ATAC-seq analysis was performed on the IDR filtered ATAC peak files for each tissue and in accordance with the RNA-seq extracted groups. Consensus peak regions were established using the R package Granges.106 Counts of reads within consensus regions were determined using the R package Rsubread.105 The R package DESeq2113 was used to determine differentially accessible peak regions between the groups for a total of 34253 peak regions, and results were plotted using R package pheatmap and the R function scale to Z score by column (cell type).
Differential HiChIP analysis
Differential loop regions were determined by first creating a loop Object in R using package diffloop.119 Loop objects are matrix-like with rows as loops, columns as cell type samples and values as the number of read counts part of the loop. Differential loops are called using limma,112 similar to how differential RNAseq analysis is done with 46,540 unique loops. Heatmap of results is plotted using R package pheatmap and the R function scale to Z score by column (cell type).
Integrative pairwise comparison
Differential analyses from the different “omics” datasets were compared by determining the Jaccard similarity and odds ratio (via Fisher exact test) for the genesets extracted from differential analysis of different methods. Genesets for the different groups for RNAseq are trivially determined. Genesets for the differential ATAC peaks and the differential HiChIP loops are determined by annotating peaks with the R packages ChIPseeker104 and ChIPpeakAnno98
Tracks visualization
HiChIP HiC-Pro interaction matrices were generated as described above. v4C visualization plots were generated from HiChIP interaction matrices by filtering the matrix for all bin pairs in which one bin matched a single anchor bin. The interaction profile of a specific 5-kb bin containing the anchor of a loci of interest was then plotted in R and smoothed with the rollmean function of the zoo package. Depth normalization was achieved by scaling counts by the total number of filtered reads in each sample. High-confidence FitHiChIP loop calls were loaded into the WashU Epigenome Browser (http://epigenomegateway.wustl.edu/browser/) along with corresponding RNA and ATAC-seq profiles. Browser shots from WashU track sessions were included in v4C and interaction map anecdotes. RefSeq gene track locations were also shown. Samtools99 and bedtools100 were used for formatting gene track locations and sequencing profiles. Pybedtools101 was used for processing bedfiles in python environment.
Cis-regulatory module analysis
Generation
cis-Regulatory Modules (CRMs) are defined as the transcription factors motif footprints present in proximal or distally looped regions to a target gene’s transcription start site (TSS). The proximal region, or promoter region, is defined as 500 bp downstream and 2 kb upstream of the TSS, or the entire H3K27ac HiChIP anchor region, if it includes the promoter region, if one is present. CRMs attributes include the number of unique and total loops contained within the CRM, the number of ATAC peaks present in the CRM, and the identity and TPM expression of the target gene. Since each RNA, ATAC, and HiChIP information is cell type-specific, the CRM for a target gene is also cell type specific. CRMs were extracted from the processed ATAC, HiChIP, and RNA data using a package we built pan-omics. In addition to the actual CRMs for a target gene, randomly looped CRMs were generated as a proxy for model testing, by randomly scrambling HiChIP anchor regions with a fixed number of total and unique loop counts. HiChIP anchor regions were also linked to the nearest TSS promoter. Original code to generate CRMs is available on Github at https://github.com/mguo123/pan_omics and Zenodo at https://zenodo.org/record/6981951 (https://doi.org/10.5281/zenodo.6981951).
Cell type specificity prediction
Various combinations of CRM attributes, pending on whether the attribute was derived from ATAC, ATAC footprinting, and/or HiChIP data were used to predict cell type using various machine learning architectures. Random forest classifiers from Python package scikit-learn with minimal hyperparameter tuning (number of estimators = 200, features are square-rooted) was found to have the best performance and confusion matrices showing positive predictive values were created. Additional performance metrics such as auROC, auPRC, sensitivity, specific, and accuracy were determined. Additionally, the % of CRM’s needed to achieve a given level of performance was determined.
Cooperativity of TF DMC analysis
Pairwise enrichment from the CRM setup between motifs was calculated using a Fisher Exact test and a Bonferroni-corrected p value < 0.05 was used to determine putatively cooperative motif pairs. Genesets connected to DMCs from the different configurations (promoter-promoter, promoter-enhancer, enhancer-enhancer) were determined, and distribution of configurations across motif pairs was determined. DMCs were determined based on the genomic presence in cis of the motif pairs occurring on more than 20 genomic instances for the differentially expressed genes in the matching RNA-seq cell type data. A total of 838 DMCs across the 15 normal cell types, 155 DMCs from the 3 squamous cell carcinoma cell lines, and 53 DMCs from the 2 melanoma cell lines were extracted (see Table S4, MPRA DMC categories, for a full list of the 838 extracted DMCs). This list was further curated down to 239 DMCs to be tested for 6 of the different cell types in an MPRA setup (see Table S5, MPRA DMC categories, for a list of the 239 DMCs tested via MPRA).
GWAS enrichment method
GWAS SNV information was downloaded from GWAS catalog57 in January 2019. SNVs associated with major cancer types (esophageal squamous cell carcinoma, cutaneous squamous cell carcinoma, ovarian cancer, lymphoma, renal cell carcinoma, pancreatic cancer, breast cancer, lung cancer, colon cancer, prostate cancer, bladder cancer, endometrial cancer, thyroid cancer, basal cell carcinoma, melanoma), autoimmune diseases (dermatomyositis, eosinophilic esophagitis, systemic sclerosis, systemic lupus erythematosus, rheumatoid arthritis, asthma, inflammatory bowel disease, ulcerative colitis, and type 1 diabetes mellitus), and skin diseases (cutaneous inflammation, acne, psoriasis, basal cell carcinoma, cutaneous squamous cell carcinoma, alopecia areata, atopic dermatitis, and rosacea) were retrieved and defined as index SNVs. The SNV list was then expanded by adding SNVs within a linkage disequilibrium (LD) block of r2 > 0.8 to the GWAS SNVs. The LD information was obtained from Haploreg v458 (http://archive.broadinstitute.org/mammals/haploreg/data/). The SNVs located in exon or UTR regions were removed, to yield 254960 SNVs for this analysis. To perform the enrichment, regions of cell type-specific HiChIP anchors overlapped with ATAC peaks were defined as cell type-specific regulatory regions. bwa102 was used to preformat sam files. “bedtools shuffle” was used to create 1000 permutations of the cell type specific regulatory regions, excluding blacklisted regions on ENCODE. The number of SNVs that fall in the permuted regions were recorded and used to construct a null distribution. Empirical p values were calculated by counting the times where the number of SNVs in the original regulatory regions were less than the number of SNVs in a given permutation, then dividing by 1000. 5% FDR cutoff was used as the significance threshold (Benjamin-Hochberg method). Fold changes were determined by finding the ratio of the number of SNVs in the original regulatory regions to the mean of the number of SNVs in the permuted data. Motifs present at SNP loci with the reference and alternate allele were determined using R package motifBreakR.109
MPRA analysis
RNA/DNA read analysis
DNA plasmid library and MPRA library reads were sequenced and analyzed using the same approach. Fastq read files from MPRA sequencing were pre-processed as such: reads were trimmed to only include the 20 bp barcode section in the correct orientation. The UMI from index 2 was processed and joined to the barcode. The barcode + UMI sequences were collapsed to remove PCR duplicates. The 20 bp barcode sequences were saved, and aligned to the MPRA library using bowtie2.110 Counts for each barcode were determined and summed across all sequencing lanes performed through Novogene. RNA counts were normalized to plasmid fractions as done previously.20 Briefly, MPRA counts were multiplied by the plasmid fractions, converted to fractions, then multiplied by the total count across the MPRA library for the sample. These processed counts were then run through the regularized log transformation (rlog) from DESeq2113 to get a normalized MPRA signal for each barcode in each cell type. Normalized counts were used in further downstream analysis and visualization.
DMC GO terms
GO biological process enrichment for the DMCs was performed by extracting target genes for each DMC in the corresponding cell type-specific CRMs. GO analysis was performed using ClusterProfiler108 and a hypergeometric test p value cut off of p < 0.05 was used. Panels were plotted using R ggplot2 and pheatmap packages.
DMC class determination
For each DMC genomic instance in the corresponding cell type, a Mann-Whitney U test was used, with an fdr-corrected significance threshold of 0.05, to determine the significance for the following DMC mutation configurations:
-
1.
A_B > scrA_scrB.
-
2.
scrA_B > scrA_scrB.
-
3.
A_scrB > scrA_scrB.
Each DMC instance was graded as synergy, redundancy, buffer, or driver motif A or driver motif B (the driver motif A and B were merged into a single category in subsequent analysis), using the following rules:
-
•
Synergy (AND) = (A_B > scrA_scrB) AND NOT(scrA_B > scrA_scrB) AND NOT(A_scrB > scrA_scrB)
-
•
Redundancy (OR) = (A_B > scrA_scrB) AND (scrA_B > scrA_scrB) AND (A_scrB > scrA_scrB)
-
•
Buffer (XOR) = NOT(A_B > scrA_scrB) AND (scrA_B > scrA_scrB) AND (A_scrB > scrA_scrB)
-
•
Single Driver A = (A_B > scrA_scrB) AND NOT(scrA_B > scrA_scrB) AND (A_scrB > scrA_scrB)
-
•
Single Driver B = (A_B > scrA_scrB) AND (scrA_B > scrA_scrB) AND NOT(A_scrB > scrA_scrB)
If a DMC instance failed to meet any of the above classifications, then it was labeled as “other.” Simulations were then run for a given category from a particular cell type. First, we randomly assigned the actual MPRA value to a DMC configuration (A_B, scrA_B, A_scrB, or scrA_scrB). Then we classified each simulated DMC interaction into the defined groupings (Synergy, Redundancy, Buffer, Single Driver, or No deterministic relationship (‘other’). Pie charts of simulations versus observed DMC genomic instance categories were plotted using R. The mode class for each DMC across all instances was used to determine DMC class determination, where ties were broken by manual inspection to ensure subthreshold significant trends were observed. See Table S5 for the annotated consensus classification for the 239 DMCs tested via MPRA.
DMC cancer versus normal synergy score
Normalized MPRA RNA counts for each genomic instance from corresponding cancer versus normal cell types were used to calculate a synergy score difference. First, synergy scores for cancer (Sc) and normal (Sn) cell types were determined by subtracting the MPRA signal from the configuration of the native genomic sequence (A_B) to the motif configuration where both motifs were scrambled (scrA_scrB). The difference between normal cell type and cancer synergy scores (Sdiff = Sc - Sn) was calculated and represents the putative modulation of DMC activity due to disease. A one-sided Wilcoxon signed rank test (p < 0.10) was used to determine if there is a significant difference in sign and magnitude of Sdiff for cancer versus normal DMCs.
DMC cell type and state-specificity determination
Normalized MPRA RNA counts for each genomic instance, averaged across the 10 respective barcodes, was calculated for each cell type in the MPRA assay (colon, MC, KC, GM12878, A431, and COLO). These MPRA signals for each genomic instance were used to calculate a cell type-specificity score. The MPRA signal from the cell type from which the DMC was originally identified was compared against the mean MPRA signal from the other five cell types via a one-sided Wilcoxon signed rank test (p < 0.10) to determine if there is a significant difference in MPRA signal between cell types.
Acknowledgments
We thank M.P. Snyder, W.J. Greenleaf, A.E. Oro, A. Kundaje, and H.Y. Chang for pre-submission review. We thank members of the Khavari lab for helpful discussions. We thank G. Rayyant and K. Fields for helpful discussions and generous support. L.K.H.D. was supported by the National Science Foundation (NSF) Graduate Research Fellowship Program (GRFP). This work was supported by USVA Office of Research and Development and by NHGRI/NIH U24HG010856, NIAMS/NIH AR045192, and NIAMS/NIH AR076965 to P.A.K.
Author contributions
L.K.H.D., M.G.G., D.S.K., and P.A.K. conceived this project. L.K.H.D., M.G.G., Y.Z., N.J., R.T.B., P.H.N., L.N.K., O.S.G., and R.M.M. performed experiments and analyzed data. P.A.K. guided experiments and data analysis. L.K.H.D., M.G.G., and P.A.K. wrote the manuscript with input from all authors.
Declaration of interests
The authors declare no competing interests.
Published: October 5, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100191.
Supplemental information
Oligo and primer information for RNA, ATAC, H3K27ac HiChIP, and MPRA assays.
QC information for RNA, ATAC, and H3K27ac HiChIP assays.
Cell culture conditions and infection parameters associated with the different cell types assayed in the MPRA setup.
DMCs extracted from the cell-type-specific CRMs for the 15 normal cell types and two different cancer types.
DMCs tested via MPRA assay (n = 239), including instances and sequences chosen, as well as their annotated consensus classification.
Data and code availability
-
•
RNA-seq (GEO: GSE186947), ATAC-seq (GEO: GSE188398), HiChIP (GEO: GSE188401) and MPRA (GEO: GSE188403) data have been deposited at GEO: GSE188405 and are publicly available as of the date of publication. Raw sequencing files for primary keratinocyte and melanocyte data are restricted and access is in accordance with NIH genomic data sharing policy. Accession numbers are listed in the key resources table.
-
•
Original code to generate CRMs is available on Github: https://github.com/mguo123/pan_omics and Zenodo: https://zenodo.org/record/6981951 (https://doi.org/10.5281/zenodo.6981951). Analysis scripts are available as a series of jupyter notebooks used for generating figures for this paper. All code is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Istrail S., Davidson E.H. Logic functions of the genomic cis-regulatory code. Proc. Natl. Acad. Sci. USA. 2005;102:4954–4959. doi: 10.1073/pnas.0409624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banerji J., Rusconi S., Schaffner W. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 3.Calo E., Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol. Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ong C.-T., Corces V.G. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dao L.T.M., Galindo-Albarrán A.O., Castro-Mondragon J.A., Andrieu-Soler C., Medina-Rivera A., Souaid C., Charbonnier G., Griffon A., Vanhille L., Stephen T., et al. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nat. Genet. 2017;49:1073–1081. doi: 10.1038/ng.3884. [DOI] [PubMed] [Google Scholar]
- 6.Coppola C.J., C Ramaker R., Mendenhall E.M. Identification and function of enhancers in the human genome. Hum. Mol. Genet. 2016;25:R190–R197. doi: 10.1093/hmg/ddw216. [DOI] [PubMed] [Google Scholar]
- 7.Gasperini M., Tome J.M., Shendure J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 2020;21:292–310. doi: 10.1038/s41576-019-0209-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinz S., Romanoski C.E., Benner C., Glass C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatia S., Kleinjan D.A. Disruption of long-range gene regulation in human genetic disease: a kaleidoscope of general principles, diverse mechanisms and unique phenotypic consequences. Hum. Genet. 2014;133:815–845. doi: 10.1007/s00439-014-1424-6. [DOI] [PubMed] [Google Scholar]
- 10.Kleinjan D.A., van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roadmap Epigenomics Consortium. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiter F., Wienerroither S., Stark A. Combinatorial function of transcription factors and cofactors. Curr. Opin. Genet. Dev. 2017;43:73–81. doi: 10.1016/j.gde.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Rubin A.J., Barajas B.C., Furlan-Magaril M., Lopez-Pajares V., Mumbach M.R., Howard I., Kim D.S., Boxer L.D., Cairns J., Spivakov M., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat. Genet. 2017;49:1522–1528. doi: 10.1038/ng.3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choudhuri S. Academic Press; 2014. Bioinformatics for Beginners: Genes, Genomes, Molecular Evolution, Databases and Analytical Tools. [Google Scholar]
- 15.Pennacchio L.A., Bickmore W., Dean A., Nobrega M.A., Bejerano G. Enhancers: five essential questions. Nat. Rev. Genet. 2013;14:288–295. doi: 10.1038/nrg3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chai X., Nagarajan S., Kim K., Lee K., Choi J.K. Regulation of the boundaries of accessible chromatin. PLoS Genet. 2013;9:e1003778. doi: 10.1371/journal.pgen.1003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siggens L., Ekwall K. Epigenetics, chromatin and genome organization: recent advances from the ENCODE project. J. Intern. Med. 2014;276:201–214. doi: 10.1111/joim.12231. [DOI] [PubMed] [Google Scholar]
- 18.Collings C.K., Anderson J.N. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenet. Chromatin. 2017;10:18. doi: 10.1186/s13072-017-0125-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T. The human transcription factors. Cell. 2018;172:650–665. doi: 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 20.Kim D.S., Risca V.I., Reynolds D.L., Chappell J., Rubin A.J., Jung N., Donohue L.K.H., Lopez-Pajares V., Kathiria A., Shi M., et al. The dynamic, combinatorial cis-regulatory lexicon of epidermal differentiation. Nat. Genet. 2021;53:1564–1576. doi: 10.1038/s41588-021-00947-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levo M., Segal E. In pursuit of design principles of regulatory sequences. Nat. Rev. Genet. 2014;15:453–468. doi: 10.1038/nrg3684. [DOI] [PubMed] [Google Scholar]
- 22.Avsec Ž., Weilert M., Shrikumar A., Krueger S., Alexandari A., Dalal K., Fropf R., McAnany C., Gagneur J., Kundaje A., Zeitlinger J. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 2021;53:354–366. doi: 10.1038/s41588-021-00782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Boer C.G., Vaishnav E.D., Sadeh R., Abeyta E.L., Friedman N., Regev A. Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol. 2020;38:56–65. doi: 10.1038/s41587-019-0315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fulco C.P., Nasser J., Jones T.R., Munson G., Bergman D.T., Subramanian V., Grossman S.R., Anyoha R., Doughty B.R., Patwardhan T.A., et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019;51:1664–1669. doi: 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daily K., Patel V.R., Rigor P., Xie X., Baldi P. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics. 2011;12:495. doi: 10.1186/1471-2105-12-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelley D.R., Snoek J., Rinn J.L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016;26:990–999. doi: 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaishnav E.D., de Boer C.G., Molinet J., Yassour M., Fan L., Adiconis X., Thompson D.A., Levin J.Z., Cubillos F.A., Regev A. The evolution, evolvability and engineering of gene regulatory DNA. Nature. 2022;603:455–463. doi: 10.1038/s41586-022-04506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nasser J., Bergman D.T., Fulco C.P., Guckelberger P., Doughty B.R., Patwardhan T.A., Jones T.R., Nguyen T.H., Ulirsch J.C., Lekschas F., et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593:238–243. doi: 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agrawal P., Heimbruch K.E., Rao S. Genome-wide maps of transcription regulatory elements and transcription enhancers in development and disease. Compr. Physiol. 2018;9:439–455. doi: 10.1002/cphy.c180028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duan A., Wang H., Zhu Y., Wang Q., Zhang J., Hou Q., Xing Y., Shi J., Hou J., Qin Z., et al. Chromatin architecture reveals cell type-specific target genes for kidney disease risk variants. BMC Biol. 2021;19:38. doi: 10.1186/s12915-021-00977-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giambartolomei C., Seo J.-H., Schwarz T., Freund M.K., Johnson R.D., Spisak S., Baca S.C., Gusev A., Mancuso N., Pasaniuc B., Freedman M.L. H3K27ac HiChIP in prostate cell lines identifies risk genes for prostate cancer susceptibility. Am. J. Hum. Genet. 2021;108:2284–2300. doi: 10.1016/j.ajhg.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren B., Yang J., Wang C., Yang G., Wang H., Chen Y., Xu R., Fan X., You L., Zhang T., Zhao Y. High-resolution Hi-C maps highlight multiscale 3D epigenome reprogramming during pancreatic cancer metastasis. J. Hematol. Oncol. 2021;14:120. doi: 10.1186/s13045-021-01131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shukla V., Lu R. IRF4 and IRF8: governing the virtues of B lymphocytes. Front. Biol. 2014;9:269–282. doi: 10.1007/s11515-014-1318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tiwari N., Pataskar A., Péron S., Thakurela S., Sahu S.K., Figueres-Oñate M., Marichal N., López-Mascaraque L., Tiwari V.K., Berninger B. Stage-specific transcription factors drive astrogliogenesis by remodeling gene regulatory landscapes. Cell Stem Cell. 2018;23:557–571.e8. doi: 10.1016/j.stem.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosono S., Luo X., Hyink D.P., Schnapp L.M., Wilson P.D., Burrow C.R., Reddy J.C., Atweh G.F., Licht J.D. WT1 expression induces features of renal epithelial differentiation in mesenchymal fibroblasts. Oncogene. 1999;18:417–427. doi: 10.1038/sj.onc.1202311. [DOI] [PubMed] [Google Scholar]
- 37.Hsu S.Y., Kubo M., Chun S.Y., Haluska F.G., Housman D.E., Hsueh A.J. Wilms’ tumor protein WT1 as an ovarian transcription factor: decreases in expression during follicle development and repression of inhibin-alpha gene promoter. Mol. Endocrinol. 1995;9:1356–1366. doi: 10.1210/mend.9.10.8544844. [DOI] [PubMed] [Google Scholar]
- 38.Rock J.R., Onaitis M.W., Rawlins E.L., Lu Y., Clark C.P., Xue Y., Randell S.H., Hogan B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA. 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang A., Kaghad M., Wang Y., Gillett E., Fleming M.D., Dötsch V., Andrews N.C., Caput D., McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 40.Tedder T.F., Tuscano J., Sato S., Kehrl J.H. CD22, a B lymphocyte–specific adhesion molecule that regulates antigen receptor signaling. Annu. Rev. Immunol. 1997;15:481–504. doi: 10.1146/annurev.immunol.15.1.481. [DOI] [PubMed] [Google Scholar]
- 41.Crowe E.P., Tuzer F., Gregory B.D., Donahue G., Gosai S.J., Cohen J., Leung Y.Y., Yetkin E., Nativio R., Wang L.-S., et al. Changes in the transcriptome of human astrocytes accompanying oxidative stress-induced senescence. Front. Aging Neurosci. 2016;8:208. doi: 10.3389/fnagi.2016.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du J., Miller A.J., Widlund H.R., Horstmann M.A., Ramaswamy S., Fisher D.E. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am. J. Pathol. 2003;163:333–343. doi: 10.1016/S0002-9440(10)63657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aihara E., Engevik K.A., Montrose M.H. Trefoil factor peptides and gastrointestinal function. Annu. Rev. Physiol. 2017;79:357–380. doi: 10.1146/annurev-physiol-021115-105447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roth W., Kumar V., Beer H.-D., Richter M., Wohlenberg C., Reuter U., Thiering S., Staratschek-Jox A., Hofmann A., Kreusch F., et al. Keratin 1 maintains skin integrity and participates in an inflammatory network in skin through interleukin-18. J. Cell Sci. 2012;125:5269–5279. doi: 10.1242/jcs.116574. [DOI] [PubMed] [Google Scholar]
- 45.Hsiao L.L., Dangond F., Yoshida T., Hong R., Jensen R.V., Misra J., Dillon W., Lee K.F., Clark K.E., Haverty P., et al. A compendium of gene expression in normal human tissues. Physiol. Genomics. 2001;7:97–104. doi: 10.1152/physiolgenomics.00040.2001. [DOI] [PubMed] [Google Scholar]
- 46.Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li G., Ruan X., Auerbach R.K., Sandhu K.S., Zheng M., Wang P., Poh H.M., Goh Y., Lim J., Zhang J., et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mifsud B., Tavares-Cadete F., Young A.N., Sugar R., Schoenfelder S., Ferreira L., Wingett S.W., Andrews S., Grey W., Ewels P.A., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015;47:598–606. doi: 10.1038/ng.3286. [DOI] [PubMed] [Google Scholar]
- 49.Diao Y., Fang R., Li B., Meng Z., Yu J., Qiu Y., Lin K.C., Huang H., Liu T., Marina R.J., et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods. 2017;14:629–635. doi: 10.1038/nmeth.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cohen I., Birnbaum R.Y., Leibson K., Taube R., Sivan S., Birk O.S. ZNF750 is expressed in differentiated keratinocytes and regulates epidermal late differentiation genes. PLoS One. 2012;7:e42628. doi: 10.1371/journal.pone.0042628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sen G.L., Boxer L.D., Webster D.E., Bussat R.T., Qu K., Zarnegar B.J., Johnston D., Siprashvili Z., Khavari P.A. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev. Cell. 2012;22:669–677. doi: 10.1016/j.devcel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vazquez M.I., Catalan-Dibene J., Zlotnik A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine. 2015;74:318–326. doi: 10.1016/j.cyto.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghanem G., Fabrice J. Tyrosinase related protein 1 (TYRP1/gp75) in human cutaneous melanoma. Mol. Oncol. 2011;5:150–155. doi: 10.1016/j.molonc.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jung I., Schmitt A., Diao Y., Lee A.J., Liu T., Yang D., Tan C., Eom J., Chan M., Chee S., et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet. 2019;51:1442–1449. doi: 10.1038/s41588-019-0494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nott A., Holtman I.R., Coufal N.G., Schlachetzki J.C.M., Yu M., Hu R., Han C.Z., Pena M., Xiao J., Wu Y., et al. Brain cell type-specific enhancer-promoter interactome maps and diseaserisk association. Science. 2019;366:1134–1139. doi: 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song M., Yang X., Ren X., Maliskova L., Li B., Jones I.R., Wang C., Jacob F., Wu K., Traglia M., et al. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet. 2019;51:1252–1262. doi: 10.1038/s41588-019-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanks S., Coleman K., Reid S., Plaja A., Firth H., Fitzpatrick D., Kidd A., Méhes K., Nash R., Robin N., et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004;36:1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 59.Jiao C.Y., Feng Q.C., Li C.X., Wang D., Han S., Zhang Y.D., Jiang W.J., Chang J., Wang X., Li X.C. BUB1B promotes extrahepatic cholangiocarcinoma progression via JNK/c-Jun pathways. Cell Death Dis. 2021;12:63. doi: 10.1038/s41419-020-03234-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wasserman N.F., Aneas I., Nobrega M.A. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010;20:1191–1197. doi: 10.1101/gr.105361.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grampp S., Platt J.L., Lauer V., Salama R., Kranz F., Neumann V.K., Wach S., Stöhr C., Hartmann A., Eckardt K.-U., et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat. Commun. 2016;7:13183. doi: 10.1038/ncomms13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Z., Schulz M.H., Look T., Begemann M., Zenke M., Costa I.G. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019;20:45. doi: 10.1186/s13059-019-1642-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A., et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018;46:D252–D259. doi: 10.1093/nar/gkx1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nutt S.L., Kee B.L. The transcriptional regulation of B cell lineage commitment. Immunity. 2007;27:361. doi: 10.1016/j.immuni.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 65.Sandilands A., Sutherland C., Irvine A.D., McLean W.H.I. Filaggrin in the frontline: role in skin barrier function and disease. J. Cell Sci. 2009;122:1285–1294. doi: 10.1242/jcs.033969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei Q., Galbenus R., Raza A., Cerny R.L., Simpson M.A. Androgen-stimulated UDP-glucose dehydrogenase expression limits prostate androgen availability without impacting hyaluronan levels. Cancer Res. 2009;69:2332–2339. doi: 10.1158/0008-5472.CAN-08-3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin-Shiao E., Lan Y., Welzenbach J., Alexander K.A., Zhang Z., Knapp M., Mangold E., Sammons M., Ludwig K.U., Berger S.L. p63 establishes epithelial enhancers at critical craniofacial development genes. Sci. Adv. 2019;5:eaaw0946. doi: 10.1126/sciadv.aaw0946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lopez-Pajares V., Qu K., Zhang J., Webster D.E., Barajas B.C., Siprashvili Z., Zarnegar B.J., Boxer L.D., Rios E.J., Tao S., et al. A LncRNA-MAF:MAFB transcription factor network regulates epidermal differentiation. Dev. Cell. 2015;32:693–706. doi: 10.1016/j.devcel.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rossi A., Jang S.I., Ceci R., Steinert P.M., Markova N.G. Effect of AP1 transcription factors on the regulation of transcription in normal human epidermal keratinocytes. J. Invest. Dermatol. 1998;110:34–40. doi: 10.1046/j.1523-1747.1998.00071.x. [DOI] [PubMed] [Google Scholar]
- 70.Rozenberg J.M., Bhattacharya P., Chatterjee R., Glass K., Vinson C. Combinatorial recruitment of CREB, C/EBPβ and c-Jun determines activation of promoters upon keratinocyte differentiation. PLoS One. 2013;8:e78179. doi: 10.1371/journal.pone.0078179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liberati N.T., Datto M.B., Frederick J.P., Shen X., Wong C., Rougier-Chapman E.M., Wang X.-F. Smads bind directly to the Jun family of AP-1 transcription factors. Proc. Natl. Acad. Sci. USA. 1999;96:4844–4849. doi: 10.1073/pnas.96.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cavazza A., Miccio A., Romano O., Petiti L., Malagoli Tagliazucchi G., Peano C., Severgnini M., Rizzi E., De Bellis G., Bicciato S., Mavilio F. Dynamic transcriptional and epigenetic regulation of human epidermal keratinocyte differentiation. Stem Cell Rep. 2016;6:618–632. doi: 10.1016/j.stemcr.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hanukoglu I., Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016;579:95–132. doi: 10.1016/j.gene.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Westergaard M., Henningsen J., Svendsen M.L., Johansen C., Jensen U.B., Schrøder H.D., Kratchmarova I., Berge R.K., Iversen L., Bolund L., et al. Modulation of keratinocyte gene expression and differentiation by PPAR-selective ligands and tetradecylthioacetic acid. J. Invest. Dermatol. 2001;116:702–712. doi: 10.1046/j.1523-1747.2001.01329.x. [DOI] [PubMed] [Google Scholar]
- 75.Schmuth M., Haqq C.M., Cairns W.J., Holder J.C., Dorsam S., Chang S., Lau P., Fowler A.J., Chuang G., Moser A.H., et al. Peroxisome proliferator-activated receptor (PPAR)-beta/delta stimulates differentiation and lipid accumulation in keratinocytes. J. Invest. Dermatol. 2004;122:971–983. doi: 10.1111/j.0022-202X.2004.22412.x. [DOI] [PubMed] [Google Scholar]
- 76.Kalailingam P., Tan H.B., Jain N., Sng M.K., Chan J.S.K., Tan N.S., Thanabalu T. Conditional knock out of N-WASP in keratinocytes causes skin barrier defects and atopic dermatitis-like inflammation. Sci. Rep. 2017;7:7311. doi: 10.1038/s41598-017-07125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ho H.-Y.H., Rohatgi R., Lebensohn A.M., Ma L., Li J., Gygi S.P., Kirschner M.W. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 78.Rubin A.J., Parker K.R., Satpathy A.T., Qi Y., Wu B., Ong A.J., Mumbach M.R., Ji A.L., Kim D.S., Cho S.W., et al. Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell. 2019;176:361–376.e17. doi: 10.1016/j.cell.2018.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Levy C., Khaled M., Fisher D.E. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006;12:406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 80.Ghaleb A.M., Nandan M.O., Chanchevalap S., Dalton W.B., Hisamuddin I.M., Yang V.W. Krüppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res. 2005;15:92–96. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McConnell B.B., Ghaleb A.M., Nandan M.O., Yang V.W. The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays. 2007;29:549–557. doi: 10.1002/bies.20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dai J., Brooks Y., Lefort K., Getsios S., Dotto G.P. The retinoid-related orphan receptor RORα promotes keratinocyte differentiation via FOXN1. PLoS One. 2013;8:e70392. doi: 10.1371/journal.pone.0070392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu X., Bowers B., Rao K., Wei Q., Hammer J.A., 3rd Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function in vivo. J. Cell Biol. 1998;143:1899–1918. doi: 10.1083/jcb.143.7.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Farley E.K., Olson K.M., Zhang W., Rokhsar D.S., Levine M.S. Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proc. Natl. Acad. Sci. USA. 2016;113:6508–6513. doi: 10.1073/pnas.1605085113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoashi T., Watabe H., Muller J., Yamaguchi Y., Vieira W.D., Hearing V.J. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J. Biol. Chem. 2005;280:14006–14016. doi: 10.1074/jbc.M413692200. [DOI] [PubMed] [Google Scholar]
- 86.Tuong Z.K., Lewandowski A., Bridge J.A., Cruz J.L.G., Yamada M., Lambie D., Lewandowski R., Steptoe R.J., Leggatt G.R., Simpson F., et al. Cytokine/chemokine profiles in squamous cell carcinoma correlate with precancerous and cancerous disease stage. Sci. Rep. 2019;9:17754. doi: 10.1038/s41598-019-54435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khou S., Popa A., Luci C., Bihl F., Meghraoui-Kheddar A., Bourdely P., Salavagione E., Cosson E., Rubod A., Cazareth J., et al. Tumor-associated neutrophils dampen adaptive immunity and promote cutaneous squamous cell carcinoma development. Cancers. 2020;12:1860. doi: 10.3390/cancers12071860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Das Mahapatra K., Pasquali L., Søndergaard J.N., Lapins J., Nemeth I.B., Baltás E., Kemény L., Homey B., Moldovan L.-I., Kjems J., et al. A comprehensive analysis of coding and non-coding transcriptomic changes in cutaneous squamous cell carcinoma. Sci. Rep. 2020;10:3637. doi: 10.1038/s41598-020-59660-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.D’Arcangelo D., Scatozza F., Giampietri C., Marchetti P., Facchiano F., Facchiano A. Ion channel expression in human melanoma samples: in silico identification and experimental validation of molecular targets. Cancers. 2019;11:E446. doi: 10.3390/cancers11040446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Graf S.A., Busch C., Bosserhoff A.-K., Besch R., Berking C. SOX10 promotes melanoma cell invasion by regulating melanoma inhibitory activity. J. Invest. Dermatol. 2014;134:2212–2220. doi: 10.1038/jid.2014.128. [DOI] [PubMed] [Google Scholar]
- 91.Bedogni B., Powell M.B. Hypoxia, melanocytes and melanoma - survival and tumor development in the permissive microenvironment of the skin. Pigment Cell Melanoma Res. 2009;22:166–174. doi: 10.1111/j.1755-148X.2009.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsuchiya Y., Nakajima M., Yokoi T. Critical enhancer region to which AhR/ARNT and Sp1 bind in the human CYP1B1 gene. J. Biochem. 2003;133:583–592. doi: 10.1093/jb/mvg075. [DOI] [PubMed] [Google Scholar]
- 93.Van Duzer A., Taniguchi S., Elhance A., Tsujikawa T., Oshimori N. ADAP1 promotes invasive squamous cell carcinoma progression and predicts patient survival. Life Sci. Alliance. 2019;2:e201900582. doi: 10.26508/lsa.201900582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mumbach M.R., Satpathy A.T., Boyle E.A., Dai C., Gowen B.G., Cho S.W., Nguyen M.L., Rubin A.J., Granja J.M., Kazane K.R., et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017;49:1602–1612. doi: 10.1038/ng.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pierce S.E., Granja J.M., Greenleaf W.J. High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat. Commun. 2021;12:2969. doi: 10.1038/s41467-021-23213-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dixit A., Parnas O., Li B., Chen J., Fulco C.P., Jerby-Arnon L., Marjanovic N.D., Dionne D., Burks T., Raychowdhury R., et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167:1853–1866.e17. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ward L.D., Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44:D877–D881. doi: 10.1093/nar/gkv1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu L.J., Gazin C., Lawson N.D., Pagès H., Lin S.M., Lapointe D.S., Green M.R. ChIPpeakAnno: a bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dale R.K., Pedersen B.S., Quinlan A.R. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics. 2011;27:3423–3424. doi: 10.1093/bioinformatics/btr539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu G., Wang L.-G., He Q.-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 105.Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Durinck S., Spellman P.T., Birney E., Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. The Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Coetzee S.G., Coetzee G.A., Hazelett D.J. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015 doi: 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Korhonen J., Martinmäki P., Pizzi C., Rastas P., Ukkonen E. MOODS: fast search for position weight matrix matches in DNA sequences. Bioinformatics. 2009;25:3181–3182. doi: 10.1093/bioinformatics/btp554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.-J., Vert J.-P., Heard E., Dekker J., Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lareau C.A., Aryee M.J. hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat. Methods. 2018;15:155–156. doi: 10.1038/nmeth.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bhattacharyya S., Chandra V., Vijayanand P., Ay F. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 2019;10:4221. doi: 10.1038/s41467-019-11950-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lareau C.A., Aryee M.J. diffloop: a computational framework for identifying and analyzing differential DNA loops from sequencing data. Bioinformatics. 2018;34:672–674. doi: 10.1093/bioinformatics/btx623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Buenrostro J.D., Wu B., Litzenburger U.M., Ruff D., Gonzales M.L., Snyder M.P., Chang H.Y., Greenleaf W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mumbach M.R., Rubin A.J., Flynn R.A., Dai C., Khavari P.A., Greenleaf W.J., Chang H.Y. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods. 2016;13:919–922. doi: 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gordon M.G., Inoue F., Martin B., Schubach M., Agarwal V., Whalen S., Feng S., Zhao J., Ashuach T., Ziffra R., et al. Author Correction: lentiMPRA and MPRAflow for high-throughput functional characterization of gene regulatory elements. Nat. Protoc. 2021;16:3736. doi: 10.1038/s41596-020-00422-z. [DOI] [PubMed] [Google Scholar]
- 126.Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S.Y., Hahn W.C., Sharp P.A., et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligo and primer information for RNA, ATAC, H3K27ac HiChIP, and MPRA assays.
QC information for RNA, ATAC, and H3K27ac HiChIP assays.
Cell culture conditions and infection parameters associated with the different cell types assayed in the MPRA setup.
DMCs extracted from the cell-type-specific CRMs for the 15 normal cell types and two different cancer types.
DMCs tested via MPRA assay (n = 239), including instances and sequences chosen, as well as their annotated consensus classification.
Data Availability Statement
-
•
RNA-seq (GEO: GSE186947), ATAC-seq (GEO: GSE188398), HiChIP (GEO: GSE188401) and MPRA (GEO: GSE188403) data have been deposited at GEO: GSE188405 and are publicly available as of the date of publication. Raw sequencing files for primary keratinocyte and melanocyte data are restricted and access is in accordance with NIH genomic data sharing policy. Accession numbers are listed in the key resources table.
-
•
Original code to generate CRMs is available on Github: https://github.com/mguo123/pan_omics and Zenodo: https://zenodo.org/record/6981951 (https://doi.org/10.5281/zenodo.6981951). Analysis scripts are available as a series of jupyter notebooks used for generating figures for this paper. All code is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.