

Summary
Genome-wide association studies (GWAS) on diverse ancestry groups are lacking, resulting in deficits of genetic discoveries and polygenic scores. We conducted GWAS for 76 phenotypes in Korean biobank data, namely the Korean Genome and Epidemiology Study (KoGES) (n = 72,298). Our analysis discovered 2,242 associated loci, including 122 novel associations, many of which were replicated in Biobank Japan (BBJ) GWAS. We also applied several up-to-date methods for genetic association tests to increase the power, discovering additional associations that are not identified in simple case-control GWAS. We evaluated genetic pleiotropy to investigate genes associated with multiple traits. Following meta-analysis of 32 phenotypes between KoGES and BBJ, we further identified 379 novel associations and demonstrated the improved predictive performance of polygenic risk scores by using the meta-analysis results. The summary statistics of 76 KoGES GWAS phenotypes are publicly available, contributing to a better comprehension of the genetic architecture of the East Asian population.
Keywords: genome-wide association study (GWAS), Korean Genome and Epidemiology Study (KoGES), meta-analysis, Polygenic risk score (PRS), pleiotropy
Graphical abstract
Highlights
-
•
GWAS on individuals in Korean biobank identifies 122 novel associations
-
•
Investigation of pleiotropic genes and genetic correlation across traits
-
•
A meta-analysis including Korean GWAS improves risk prediction accuracy in East Asians
Genome-wide association studies (GWAS) on diverse ancestry groups are lacking, resulting in deficits of genetic discoveries. Nam et al. report a GWAS of 76 traits on Korean individuals, identifying 122 novel associations and demonstrating the improvement of the predictive performance of polygenic risk scores by the meta-analysis.
Introduction
Population-based biobanks, such as UK Biobank2,3 and FinnGen,4 facilitate genome-wide association studies (GWAS) in tens of thousands or even millions of samples across a large number of traits. These extensive resources helped identify numerous genetic associations and elucidate genetic components of complex traits.5,6 Using the analysis results, genome-based prediction models have been built and successfully identified individuals with a high risk of disease. Despite the success, a major limitation of the current status of GWAS is the relative lack of non-European samples.7 As rare variants in Europeans can have high minor allele frequencies (MAFs) in other ancestry groups, the lack of non-European samples can limit further discoveries. In addition, it can cause health disparities if the use of genetic discovery in clinical practice is limited to individuals of European ancestry.8
We report a GWAS of 76 phenotypes in 72,298 Korean individuals from the Korean Genome and Epidemiology Study (KoGES), a large biobank conducted by the National Biobank of Korea. Previously several GWAS were performed using KoGES data, including GWAS for anthropometric traits and some metabolites.9, 10, 11 However, these studies mainly focused on one or a few traits of interest. Recently, significant efforts have been made to catalog genetic associations in East Asians by analyzing a large number of phenotypes, including phenome-wide analysis of the Biobank Japan (BBJ)12, 13, 14 and the Taiwan Biobank15 data. By increasing the sample size and demographic diversity of East Asian GWAS samples, our analysis contributes to novel discoveries. Through the analysis of 14 binary diseases endpoints, 31 biomarkers, 23 dietary information, and 8 alcohol consumption phenotypes, we identified 2,242 associated loci for 47 phenotypes at the genome-wide significance level (p < 5 × 10−8). To fully use the information in the KoGES data, in addition to the mixed effect model for continuous, binary, and categorical phenotypes, we applied up-to-date analysis methods for survival GWAS16 and methods to incorporate family disease history,4 and identified 19 additional significant associations. Among associations, 122 were novel, and more than 70% of novel associations with corresponding phenotypes and genetic variants in BBJ were replicated at a nominal p value of 0.05. Many of the novel loci had very low MAFs in Europeans, demonstrating power increment by utilizing samples from diverse ancestry groups.
To find East Asian-specific genetic associations, we conducted meta-analyses for 32 traits using KoGES and BBJ (n = 251,000) GWAS results. We identified 379 novel loci for 25 traits, mostly in clinical biomarkers, and 85% of these loci were not identified in individual studies. We also constructed polygenic risk scores (PRSs) with the meta-analyzed GWAS summary and showed that the PRS trained with the meta-analyzed KoGES and BBJ could have 20% larger R2 in the prediction of the trait values in East Asian samples in UK Biobank compared with PRSs trained from BBJ GWAS only, demonstrating one potential utility of our analysis results. We publicly provide all GWAS summary statistics to broaden the understanding of the genetic basis of the East Asian population.
Results
KoGES
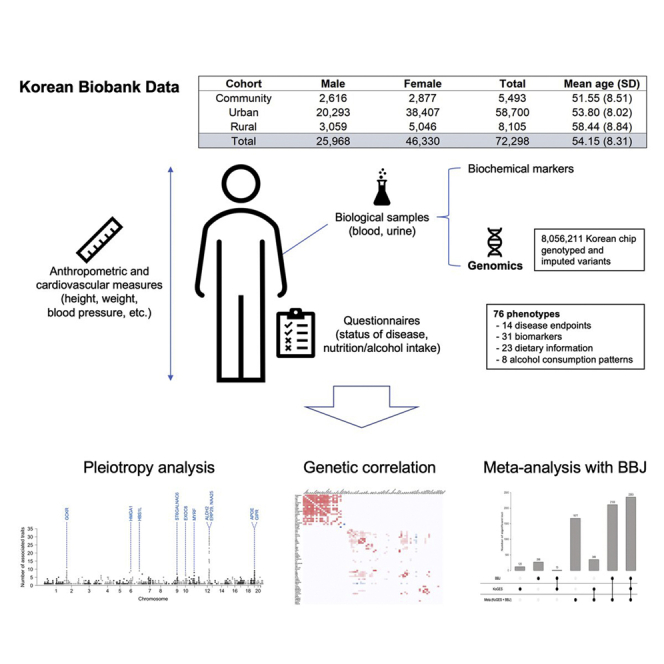
KoGES, part of the National Biobank of Korea, is a prospective cohort study with a comprehensive range of phenotypic measures and biological samples, such as DNA, serum, plasma, and urine, collected on approximately 210,000 individuals. KoGES includes the community-based Ansan and Ansung study, the urban community-based health examinee study, and the rural community-based cardiovascular disease association study. Each cohort has the baseline assessment and follow-up measurement thereafter, and we used the baseline measures only in this study (see STAR Methods for details). The table in Figure 1 describes the sample size and the mean age of KoGES data by cohort. A total of 72,000 samples with KoreanChip17 genotyped and imputed were used in our analysis (see STAR Methods).
Figure 1.

Overview of the KoGES data and our study
KoGES data consist of three cohorts: community-based, urban, and rural cohort. Participants in KoGES are recruited from the national health examinee registry, age ≥40 at baseline. The sample size in the figure indicates the number of samples for which both genotype (after QC) and baseline assessment data exist. See also Table S1.
GWAS of 76 traits
An overview of our analysis is shown in Figure 1, and the studied traits are described in Table S1. We analyzed 14 binary disease endpoints, 31 biomarkers, 23 dietary information, and 8 traits about alcohol consumption patterns. A total of 8,056,211 genotyped and imputed variants were used in our analysis. For continuous and binary traits, SAIGE18 was used to maximize power while controlling type I error. For ordinal categorical phenotypes, we applied a proportional odds logistic mixed effect model.19 A total of 2,223 loci for 47 traits satisfying a genome-wide significance threshold (p < 5 × 10−8) where significant clumped variants are identified using a window width of 5 Mb and a linkage disequilibrium threshold of R2 = 0.1(Table S2). The estimated false discovery rate (FDR) is 0.0017 (FDR = 76 [number of traits] × 106 [number of independent loci] × 5 × 10−8 [genome-wide significance threshold]/2,223 [number of significant loci]), with assuming 1 million independent loci that correspond to genome-wide α = 5 × 10−8.20, 21, 22, 23 When a more stringent criterion adjusted for the number of phenotypes at the top of the genome-wide significant level (p < 5 × 10−8/76 = 6.58 × 10−10) was used, the number of significant loci was 1,455 for 42 phenotypes.
We performed linkage disequilibrium score regression (LDSC)24,25 to estimate heritability and genetic correlation (Table S3). There were no significant confounding biases as the mean LDSC intercept values were 1.0212 for all 76 traits. As expected, height had the largest heritability (h2 = 0.400), followed by weight (h2 = 0.270) and blood platelet count (h2 = 0.265). The estimated heritabilities were similar to those of UKBB and BBJ (h2 = 20.402 and 0.386 for height, respectively). We also computed pairwise genetic correlations to discover the genetic relationship between phenotypes and represented them as a heatmap (Figure S1). To avoid false-positive findings, a genetic correlation was treated as zero when the p value was greater than each threshold. We observed several phenotype clusters with high genetic correlations. Twenty-one phenotypes related to nutrition intake form the largest cluster. In addition, we identified sets of closely related phenotypes, such as (1) liver-related biochemical markers (aspartate transaminase, gamma-glutamyl transpeptidase, and alanine aminotransferase), (2) cardiovascular phenotypes (systolic blood pressure [SBP], diastolic blood pressure [DBP], and hypertension), and (3) hematological traits (hemoglobin, hematocrit, white blood cell count, and red blood cell count).
Using the first onset age, we conducted survival analysis for 14 disease endpoints using SPACox, a method using Cox proportional hazards regression model. We replicated 15 significant associations for 3 traits that were not significant in case-control phenotype analysis. Incidence plots show that these loci influence the disease prevalence (Figure S2). In addition, the association signals identified in case-control GWAS became more significant when applying survival analysis (Table S4).
By incorporating the family disease history, the association test power can be improved. We used the family disease history information of disease endpoints with TAPE.26 In two types of cancer: gastric cancer and gallbladder cancer, we identified an additional four independent association signals that were not detected in the analysis without family disease history. We calculated the mean value of the TAPE-adjusted phenotypes by the genotype of the top SNP of each locus (Table S5). As the number of minor alleles increases, we observe that the TAPE-adjusted phenotypes monotonically increase or decrease.
Genetic pleiotropy analysis
Since our analysis results show numerous associations, we investigated pleiotropy. To evaluate it at a gene level, we first mapped the most significant variant in each associated locus to a gene using FUMA27 and then counted the number of phenotypes associated (Table S6). Overall, 826 genes had more than 2 associations. Many genes in chromosome 12 showed high levels of pleiotropy. Two neighboring genes in chromosome 12, ERP29 (12:112,451,230-112,461,253; GRCh37) and NAA25 (12:112,464,493-112,546,587; GRCh37), were the most pleiotropic genes with 30 associated phenotypes, and then 8 genes, including ALDH2, with 29 associated phenotypes (Figure 2). Except for genes in chromosome 12, GCKR in chromosome 2, associated with 11 phenotypes, was the most pleiotropic. GCKR encodes glucokinase regulatory protein and is related to many phenotypes affected by glucose metabolisms, such as fasting glucose and insulin measurement.28 This result is consistent with the studies on other biobank data such as BBJ and UKB.14
Figure 2.

Genetic pleiotropy analysis results
Manhattan plot with the y axis being the number of significantly associated (p < 5 × 10−8) traits per gene for 76 traits in KoGES data. To avoid double-counting the associations in the same phenotypes, results from SPACox and TAPE were excluded when counting the number of significant associations. See also Table S6.
The number of associated traits per variant is also used to quantify the degree of pleiotropy. Two hundred and sixty-two variants in chromosome 12 were associated with more than 10 traits. Nine variants, including rs671, a missense variant in the ALDH2 region, were the most pleiotropic variants (27 traits). Except for variants in chromosome 12, rs1260326, a missense variant at the GCKR locus (and 12 variants nearby), was the most pleiotropic variant (11 traits).
Novel associations and replications
We identified 122 novel associations for 32 traits among the significant associations (Table S7). We defined the association as a novel if the association is not reported in the GWAS catalog and the p value is not genome-wide significant (p < 5 × 10−8) in the BBJ GWAS (see STAR Methods). Among 122 novel associations, 53 top SNPs for 18 phenotypes were present in BBJ, and 38 SNPs (71.7%) out of 53 had BBJ p < 0.05. With a more stringent threshold for replication by Bonferroni correction (p < 0.05/53 = 9.43 × 10−4), 25 top SNPs (47.2%) were replicated. Many of the corresponding variants have low MAFs in the European population (Figure S3). For example, rs939955, an intergenic variant between CYP3A4-CYP3A7, was associated with triglyceride (TG) level (p = 2.47 × 10−9; BBJ p = 1.2 × 10−4). The MAF of rs939955 in KoGES was 0.22, but it was very rare among Europeans (MAFEUR = 0.002). Both CYP3A4 and CYP3A7 belong to the cytochrome P450 (CYP) superfamily and are well known for drug metabolism. A variant rs1314013, at the ZEB1 locus, was associated with weight (p = 7.19 × 10−11; BBJ p = 2.7 × 10−4, MAFKoGES = 0.17, MAFEUR = 0.04). In an experiment on mice, the zinc finger E-box binding homeobox (ZEB1) transcription factor was a repressor of adiposity.29 A variant rs118190473, at ANXA3, was associated with HDL cholesterol level (p = 4.49×10−8; BBJ p = 7.0 × 10−6, MAFKoGES = 0.155, MAFEUR = 0.005). Since adipocyte differentiation and lipid accumulation is the potential function of Annexin A3 (ANXA3),30,31 our result may provide a link between HDL level and the ANXA3 locus. ANXA3 encodes a member of the annexin family and is predicted to be involved in phospholipase A2 (PLA2s) inhibitor activity. Secretory PLA2s are known to be associated with HDL, and a mouse study has shown that overexpression of secretory PLA2 caused the decrease in serum HDL.32,33 We identified rs9921399, an intron variant at CES1, was associated with LDL cholesterol (p = 1.37 × 10−9; BBJ p = 5.5 × 10−5, MAFKoGES = 0.45, MAFEUR = 0.23). Although this locus has not been demonstrated in GWAS, it is known that LDL cholesterol levels were reduced in carboxylesterase 1-deficient mice.34 We compared effect sizes and 95% confidence interval for those four loci and drew locus zoom plots (Figure S4).
Meta-analysis with BBJ
To identify genetic associations in the East Asian population, we conducted a meta-analysis for 9 disease endpoints and 23 biomarkers for KoGES together with BBJ and identified 289 genome-wide significant associations for disease endpoints and 6,197 significant associations for biomarkers (Table S8). Figure 3 represents the number of associations identified in the meta-analysis across KoGES and BBJ. As expected, meta-analysis substantially increased the number of significant associations. Of the total 6,486 associated locus-trait pairs, 1,677 (25.8%) were newly identified by the meta-analysis. For example, alanine aminotransferase GWAS identified 26 loci in KoGES and 52 in BBJ, yet 124 loci were significant in the meta-analysis. Among the identified associations, 379 (2 disease associated and 377 biomarker associated) were novel and 321 novel associations were not significant in individual GWAS of KoGES or BBJ.
Figure 3.

The number of significant associations identified in the KoGES, BBJ, and meta-analysis
Black dots indicate significance in the analysis, and a line connected between dots represents simultaneous significance in multiple cohorts. The number of loci is counted based on the meta-analysis summary statistics after clumping for the variants with p values less than 5 × 10−8, window size of 5 Mb, and linkage disequilibrium threshold R2 of 0.1. See also Table S8.
PRS improvement
We calculated PRSs based on the East Asian meta-analysis GWAS results across KoGES and BBJ and compared them with the BBJ-based and UK-Biobank European-based PRS models. Using PRS-CS,35 we trained the PRS model for SBP, DBP, high-density lipoprotein cholesterol (HDLC), low-density lipoprotein cholesterol, and TGs. To estimate unbiased prediction performance, we used East Asian samples in the UK Biobank as the test samples.
For the five phenotypes we tested, PRS based on the East Asian meta-analysis (PRSEAS-Meta) provided better predictive performance, in terms of R2, compared with BBJ-based PRS (PRSBBJ) in all models (Table S9A). Interestingly, the European-based PRS model (PRSEUR) performed better than two East Asian-based PRS models (i.e., PRSEAS-Meta and PRSBBJ) for two blood pressure traits (SBP and DBP). We also conducted a multi-ethnic PRS analysis,36 which linearly combines PRSs from Europeans and East Asians (Table S9B). For all five phenotypes, the multi-ethnic PRS model based on PRSEUR and PRSEAS-Meta performed better than the model constructed by PRSEUR and PRSBBJ. To evaluate whether the improvement of the use of PRSEAS-Meta over PRSBBJ is significant, we fitted the models with PRSEAS-Meta and PRSBBJ (Table S9C). The first two models included each PRS only, and the third model had both PRSEAS-Meta over PRSBBJ. When these two PRSs were included in the model, only PRSEAS-Meta was statistically significant for all five phenotypes we tested. In addition, the R2 values of the model with two PRSs were not substantially different from the R2 values of the model with PRSEAS-Meta. This suggests that PRS based on meta-analysis explains the phenotype better than PRSBBJ. Overall, our analysis result demonstrates that the meta-analysis, including the KoGES GWAS, can contribute to the improvement of risk prediction for East Asians.
Discussion
In this paper, we carried out GWAS for 76 phenotypes in 72,000 Korean samples and identified a large number of significant associations. Our analysis found 122 novel associations previously unknown, and many of them were replicated in BBJ. We also performed pleiotropy analysis and illustrated that the most pleiotropic regions, such as a region of chromosome 12, including ERP29, NAA25, and ALDH2, and a chromosome 2 region, including GCKR. Through the meta-analysis with BBJ, we further identified a large number of significant associations. Since most of the novel associations in the meta-analysis with BBJ were not identified in individual GWAS, our study contributes to increasing the effective sample size. We also compared the prediction models based on the meta-analysis results and BBJ summary statistics. We demonstrated that a model based on the meta-analysis across KoGES and BBJ has better prediction performance when predicting trait values for East Asian samples in UK Biobank.
Biobanks collect additional disease information from either surveys or electronic records, such as time to disease onset and family disease history. In addition to genetic association analysis of continuous, binary, and categorical phenotypes, we applied survival analysis (SPACox) and model with family history (TAPE) to utilize time-to-onset age and family history. These methods found 19 more significant loci in 5 traits; all of them were previously known, validating the approach. In addition, many of the significant loci p values were improved. Our analysis demonstrates that time-to-onset and family history are valuable data and can help to identify true associations.
In our pleiotropy analysis, many genes in chromosome 12 were highly pleiotropic and many of the associated phenotypes were related to alcohol consumption. Based on this, it is reasonable to assume that ALDH2 association with alcohol drives the signals. The variant rs671 in ALDH2 had a significant negative effect on alcohol consumption and, hence, affected many alcohol-consumption phenotypes and alcohol-related phenotypes, such as blood pressure and cholesterol level. Interestingly, ERP29 and NAA25 had an additional association with thyroid disease. NAA25 is known for its association with hypothyroidism,37 and both ERP29 and NAA25 genes are linked to several traits, including blood pressure and alcohol-related traits.38
We acknowledge that the validation rate of novel variants in BBJ was lower than expected, and it did not substantially change even when we applied the Bonferroni corrected threshold. For variants not replicated in BBJ, allele frequencies in KoGES and BBJ were not substantially different. The low validation is probably due to the difference between these two biobanks, such as cohort characteristics, phenotype definition, and measurement.
Although we highlighted the novel loci with low MAF among Europeans, there exist many novel loci that are not rare among Europeans. For example, rs1314013 (MAFEUR = 0.0492) for body weight and rs9921399 (MAFEUR = 0.2646) for LDL cholesterol did not show a signal for association among Europeans (EUR p = 0.75 and 0.17, respectively). This supports the necessity for further investigation of the genetic difference between ancestry groups.
It is not surprising that most dietary-related traits showed a high genetic correlation. In the UK Biobank study,39 there were several clusters with strong correlations among food-liking phenotypes. Interestingly, we found a negatively strong correlation between sugar intake and retinol (vitamin A1) intake (rg = −0.90, p = 3.4 × 10−7), both adjusted for overall energy intake. Since there were no genome-wide significant loci for both phenotypes, we could not identify the set of variants or genes to explain this correlation.
We further estimated the heritability of (single) top variants to show the proportion of variance explained by those variants. The single variant heritability can be calculated as , where β is the estimated effect size of the variant and MAF is the minor allele frequencies.40 For rs939955, a variant associated with TG, ; and for rs118190473, a variant associated with HDLC. These variants only explain the modest amount of heritability compared with the most significant SNPs ( for rs74368849 with TG and for rs72786786 with HDLC). However, it appears to be comparable with other known variants. For example, rs56156922 is associated with TG in both KoGES and BBJ (KoGES p = 9.5 × 10−9; and BBJ p = 1.5 × 10−13), and the heritability explained by it was 4.0 × 10−4 and 4.5 × 10−4 in KoGES and BBJ, respectively.
For SBP and DBP, European-based PRS showed better prediction performance than East Asian-based PRS. There may be two possible reasons. As the UK Biobank data were used for constructing EUR-based PRS, the phenotype definition and genotyping platform were identical to the test set (East Asians in UK Biobank data), while KoGES and BBJ were not. It is also possible that the genetics of blood pressure may be less varying across ancestry groups than lipid phenotypes. In this case, predictive performance can be more affected by the sample sizes. The EUR-based PRS models were built using GWAS of 400K samples, while sample sizes of BBJ and meta-analysis were 140K and 210K samples, respectively.
To better predict and prevent complex diseases, we need large GWAS in diverse populations. Ideally, GWAS results should be publicly available for meta-analysis and downstream analysis, including novel association identification and replication, PRS calculation, and Mendelian randomization. All our GWAS and meta-analysis results are publicly available on a PheWeb41 website with interactive visualization of Manhattan, Q-Q, and locus zoom plots. By providing East Asian GWAS on many phenotypes, our results will contribute to elucidating the genetic architecture of complex traits.
Limitations of the study
There are several limitations to our study. First, the disease status phenotypes in KoGES are collected through a self-reported survey and have not been verified by an expert diagnosis. Second, the nutrition intake data in KoGES are derived from a food frequency questionnaire involving 103 foods (see STAR Methods), which can have errors. Third, since there is no information on medication in KoGES data, calibration of several continuous phenotypes, such as blood pressure, was not feasible. Despite the limitations, our study is the only existing research that analyzed a large number of phenotypes in the Korean population.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| KoGES summary statistics and plots | This paper |
https://koges.leelabsg.org https://zenodo.org/record/7042518 |
| Individual-level genotype and phenotype data in KoGES | Moon et al., 2019 | 4851-302 |
| Biobank Japan summary statistics | Sakaue et al., 2021 | https://pheweb.jp/ |
| UK Biobank summary statistics | Pan-UKB team, 2020 | https://pan.ukbb.broadinstitute.org/ |
| Software and algorithms | ||
| SAIGE | Zhou et al., 2018 | https://github.com/weizhouUMICH/SAIGE |
| POLMM | Bi et al., 2021 | https://github.com/WenjianBI/POLMM |
| SPACox | Bi et al., 2020 | https://github.com/WenjianBI/SPACox |
| TAPE | Zhuang et al., 2022 | https://github.com/styvon/TAPE |
| LDSC | Bulik-Sullivan et al., 2015 | https://github.com/bulik/ldsc |
| FUMA | Watanabe et al., 2017 | https://fuma.ctglab.nl/ |
| PRS-CS | Ge et al., 2015 | https://github.com/getian107/PRScs |
| PLINK | Chang et al., 2015 | https://www.cog-genomics.org/plink/2.0/ |
| gwasrapidd | Magno et al., 2020 | https://github.com/ramiromagno/gwasrapidd |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Seunggeun Lee (lee7801@snu.ac.kr).
Materials availability
This study did not generate new unique reagents.
Method details
KoGES data
All samples in the analysis were genotyped with KoreanChip. KoreanChip is a customized array optimized for the Korean population. It has 833K variants selected using 2,576 Korean sequencing data (397 WGS and 2,179 WES). Among them, 600K variants are tagging variants for genome-wide coverage. The details of the KoreanChip can be found elsewhere.17 We used measures of the baseline recruitment, and only genotyped samples that met the following exclusion criteria were used: low call rate (<97%), excessive heterozygosity, excessive singletons, gender discrepancy, and cryptic first-degree relatives. SNPs with low HWE p value (<10−6) or low call rate (<95%) were excluded. After quality control, data were phased using Eagle v2.3 and imputed using IMPUTE4 with 1000 Genomes Project Phase 3 data and the Korean reference genome as a reference panel. Variants with imputation quality score (IQS) < 0.8 and MAF <1% were excluded after imputation. We analyzed 8,056,211 variants in total after these processes.
Anthropometric and clinical measurements in KoGES were obtained by physical examinations and clinical investigations, and the disease status of the participants and family members was collected by the interview. Nutritional intake data were calculated based on a categorical food frequency questionnaire (FFQ) involving 103 foods. The exact sample size and unit of measurement for each phenotype are described in Table S1. Detailed methods for the measurements, interviews, and questionnaires are described elsewhere.42,43
GWAS of KoGES data
For both binary and quantitative traits, we conducted GWAS using a linear mixed model implemented in SAIGE (version 0.44.5) in order to maximize power while controlling type I error for case-control imbalance. In step 1, we used 327,540 variants with Imputation Quality Score (IQS) = 1 to obtain the genetic relationship matrix (GRM). We used the top 10 principal components (PC), age, sex, and adjustment of assessment details such as cohort and year of examination, as covariates in step 1. For quantitative traits, we applied rank-based inverse normal transformation and leave-one-chromosome-out (LOCO) scheme to remove the proximal contamination. For nutrition intake phenotypes, we additionally adjusted for the total energy intake since most nutrients are closely correlated with caloric intake.44 For ordinal categorical traits, we used a proportional odds logistic mixed model (POLMM) to model the nature of ordinal categorical phenotypes. POLMM is also known to be robust for imbalanced phenotypic distributions, while linear mixed models do not control type I error rate well. After GWAS, we conducted clumping analysis using PLINK245 for the variants with p values less than 5 × 10−8, a window size of 5Mb, and linkage disequilibrium threshold R2 of 0.1 to count independent genome-wide significant loci. We carried out LD score regression using ldsc (version 1.0.1) to estimate SNP-based heritability, confounding bias, and genetic correlation. All statistical tests from SAIGE and POLMM are two-sided. We reported the results for all 76 studied traits (Table S2).
Survival analysis and incorporating family history
We performed survival analysis for 14 disease endpoints with SPACox (version 0.1.2), a Cox proportional hazards regression model with saddlepoint approximation, using the first onset age in KoGES. We used the same samples and covariates as normal GWAS (top 10 PCs, age, sex, and batch effect). The exact sample size and unit of measurement for each phenotype are described in Table S1.
In KoGES, family disease history data collected by the survey are available. This data indicates whether a family member (separated into paternal, maternal, and siblings) has ever been diagnosed with the disease. With these family disease histories in our data, we conducted an association analysis using TAPE (version 0.2.1). We first adjusted phenotypes (disease status) using three types of family history of disease: paternal, maternal, and siblings for 12 disease endpoints. In TAPE, the adjusted phenotype for individual i (Zi) is defined as:
where 1 denotes indicator function, ρ is a pre-specified constant indicating the increase in latent disease risk and . In this study, we adjusted phenotypes assuming ρ is equal to 0.5. After adjusting phenotypes, we used the same samples and covariates as normal GWAS (top 10 PCs, age, sex, and batch effect). All statistical tests from SPACox and TAPE are two-sided.
Gene-level genetic pleiotropy analysis
We measured the degree of pleiotropy by counting the number of associated traits (out of 76 traits) per gene and per variant for KoGES GWASs. We excluded the results from survival analysis (SPACox) and model with family history (TAPE) when counting the number of associated traits. For gene-level pleiotropy, we used the SNP2GENE function of FUMA27 to map SNPs in GWAS results to a gene with 1000 Genome Phase 3 EAS as a reference panel. FUMA is a bioinformatic tool that uses multiple sources of information, including LD structure, functional score, and chromatin interaction, to link associated variants to relevant genes. FUMA first characterizes independent significant variants and surrounding genomic loci based on LD structure. Next, those variants are annotated using various tools and databases such as ANNOVAR,46 CADD,47 RegulomeDB,48 and Hi-C data.49 Then annotated variants are mapped to genes using position, eQTL association, and chromatin interaction. All parameters used in gene mapping are default values provided by FUMA. For variant-level pleiotropy, we counted the number of associated traits satisfying a genome-wide significance threshold (p < 5 × 10−8).
Novel association identification
We searched for existing associations using the GWAS catalog50 within ±500 kb from the lead variant to regard an associated locus as novel. In order to screen for variants that have been previously reported, we first used gwasrapidd51 R package to access the representational state transfer (REST) application programming interface (API) of the GWAS catalog. To map traits reported under different names of traits, we used experimental factor ontology (EFO) traits, and we again exhaustively searched for existing reports of association in the same or similar phenotypes. For example, variants that have been reported for blood-pressure-related traits were excluded from the novel loci for hypertension. Since the results by BBJ14 were not listed in the GWAS catalog at the time of evaluation for the novel association, we additionally excluded variants that were genome-wide significant in BBJ.
Meta-analysis with biobank Japan
Summary statistics of BBJ were downloaded from the Biobank Japan PheWeb website (https://pheweb.jp/). 9 disease endpoints and 23 biomarker phenotypes were presented in both KoGES and BBJ, and a total of 6,907,490 variants were overlapped in these two studies. The z-score-based meta-analysis method was used to calculate p values and we used the inverse variance method to obtain the effect sizes for risk prediction. To identify the novel loci, we applied the same criteria as mentioned in the previous section.
PRS evaluation
We calculated polygenic risk scores (PRS) using PRS-CS (version released on June 4, 2021). We used East Asian (EAS) in 1000 Genomes Project phase 3 samples for BBJ-based and meta-analysis-based models and European (EUR) samples for European-based models as the LD reference panel. All parameters used in our analysis are default values provided by PRS-CS software, and we did not specify the global shrinkage parameter phi to use a fully Bayesian approach. The method uses a gamma-gamma prior with a = 1 and b = 0.5. For Markov Chain Monte Carlo (MCMC) in PRS-CS, the total number of iterations is 1,000, the number of burn-in iteration is 500, and thinning factor is equal to 5. When training the PRS models, we used summary statistics after filtering variants that exist in all UK Biobank, KoGES, and BBJ. We additionally restricted variants in HapMap3 as in Prive et al.,52 so a total of 900,746 variants were used. The effect sizes of the meta-analysis were calculated from inverse variance methods described in the meta-analysis part.
In addition, we conducted a multi-ethnic PRS analysis,36 which combines PRS from Europeans and East Asians. Multi-ethnic PRS is defined as the linear combination of two PRS: . We used half of the East Asian samples in UK Biobank to estimate w1 and w2, and the other half was used as a test set. To evaluate the improvement, we compared the significance of PRS in three linear regression models: (1) , (2) , and (3) .
Acknowledgments
This research was supported by the Brain Pool Plus (BP+, Brain Pool+) Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2020H1D3A2A03100666). Data in this study were from the Korean Genome and Epidemiology Study (KoGES; 4851-302), the National Research Institute of Health, Centers for Disease Control and Prevention, and Ministry for Health and Welfare, Republic of Korea. UK Biobank data were accessed under the accession number UKB: 45227. We thank Dr. Cristen Willer for the constructive comments and suggestions.
Author contributions
K.N. and S.L. designed the experiments. K.N., J.K., and S.L. analyzed the KoGES data. K.N. and S.L. wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: October 5, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100189.
Supplemental information
We report basic information on studied phenotypes, including sample size, mean, median, and unit of measurement.
We report summary statistics of genome-wide significant loci identified in KoGES. Beta indicates the genetic effect size calculated from SAIGE/POLMM.
We report LD score regression results for 76 studied phenotypes.
We report gene mapping results obtained by using the SNP2GENE function of FUMA.
We report information, including GWAS results and minor allele frequencies of the novel loci identified in KoGES.
We report (1) summary statistics of genome-wide significant loci identified by meta-analysis, (2) gene mapping of meta-analysis results using FUMA.
We report (1) predictive performance (R2) in single-ethnic PRS, (2) predictive performance (R2) in multi-ethnic PRS, and (3) statistical significance of PRS.
Data and code availability
The summary statistics generated during this study are submitted to Zenodo: https://zenodo.org/record/7042518. Manhattan plots and quantile-quantile plots generated by the summary statistics are publicly available at https://koges.leelabsg.org. BBJ summary statistics used in this study were downloaded from the Biobank Japan PheWeb: https://pheweb.jp/ and summary statistics for Europeans in UK Biobank were downloaded from Pan-UK Biobank1: https://pan.ukbb.broadinstitute.org/. This paper does not report custom code or software. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Pan-UKB team 2020. https://pan.ukbb.broadinstitute.org
- 2.Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M., et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O'Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurki M.I., Karjalainen J., Palta P., Sipilä T.P., Kristiansson K., Donner K., Reeve M.P., Laivuori H., Aavikko M., Kaunisto M.A., et al. FinnGen: unique genetic insights from combining isolated population and national health register data. medRxiv. 2022 doi: 10.1101/2022.03.03.22271360. Preprint at. [DOI] [Google Scholar]
- 5.Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J. 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017;101:5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tam V., Patel N., Turcotte M., Bossé Y., Paré G., Meyre D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019;20:467–484. doi: 10.1038/s41576-019-0127-1. [DOI] [PubMed] [Google Scholar]
- 7.Peterson R.E., Kuchenbaecker K., Walters R.K., Chen C.Y., Popejoy A.B., Periyasamy S., Lam M., Iyegbe C., Strawbridge R.J., Brick L., et al. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell. 2019;179:589–603. doi: 10.1016/j.cell.2019.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin A.R., Kanai M., Kamatani Y., Okada Y., Neale B.M., Daly M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho H.W., Jin H.S., Eom Y.B. A genome-wide association study of novel genetic variants associated with anthropometric traits in Koreans. Front. Genet. 2021;12:669215. doi: 10.3389/fgene.2021.669215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park J.S., Kim Y., Kang J. Genome-wide meta-analysis revealed several genetic loci associated with serum uric acid levels in Korean population: an analysis of Korea Biobank data. J. Hum. Genet. 2022;67:231–237. doi: 10.1038/s10038-021-00991-1. [DOI] [PubMed] [Google Scholar]
- 11.Kim T., Park A.Y., Baek Y., Cha S. Genome-wide association study reveals four loci for lipid ratios in the Korean population and the constitutional subgroup. PLoS One. 2017;12:e0168137. doi: 10.1371/journal.pone.0168137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akiyama M., Okada Y., Kanai M., Takahashi A., Momozawa Y., Ikeda M., Iwata N., Ikegawa S., Hirata M., Matsuda K., et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet. 2017;49:1458–1467. doi: 10.1038/ng.3951. [DOI] [PubMed] [Google Scholar]
- 13.Kanai M., Akiyama M., Takahashi A., Matoba N., Momozawa Y., Ikeda M., Iwata N., Ikegawa S., Hirata M., Matsuda K., et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 2018;50:390–400. doi: 10.1038/s41588-018-0047-6. [DOI] [PubMed] [Google Scholar]
- 14.Sakaue S., Kanai M., Tanigawa Y., Karjalainen J., Kurki M., Koshiba S., Narita A., Konuma T., Yamamoto K., Akiyama M., et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021;53:1415–1424. doi: 10.1038/s41588-021-00931-x. [DOI] [PubMed] [Google Scholar]
- 15.Chen C.-Y., Chen T.-T., Anne Feng Y.-C., Longchamps R.J., Lin S.-C., Wang S.-H., Hsu Y.-H., Yang H.-I., Kuo P.-H., Daly M.J., et al. Analysis across Taiwan Biobank, Biobank Japan and UK Biobank identifies hundreds of novel loci for 36 quantitative traits. medRxiv. 2021 doi: 10.1101/2021.04.12.21255236. Preprint at. [DOI] [PubMed] [Google Scholar]
- 16.Bi W., Fritsche L.G., Mukherjee B., Kim S., Lee S. A fast and accurate method for genome-wide time-to-event data analysis and its application to UK biobank. Am. J. Hum. Genet. 2020;107:222–233. doi: 10.1016/j.ajhg.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moon S., Kim Y.J., Han S., Hwang M.Y., Shin D.M., Park M.Y., Lu Y., Yoon K., Jang H.M., Kim Y.K., et al. The Korea biobank array: design and identification of coding variants associated with blood biochemical traits. Sci. Rep. 2019;9:1382. doi: 10.1038/s41598-018-37832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou W., Nielsen J.B., Fritsche L.G., Dey R., Gabrielsen M.E., Wolford B.N., LeFaive J., VandeHaar P., Gagliano S.A., Gifford A., et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018;50:1335–1341. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bi W., Zhou W., Dey R., Mukherjee B., Sampson J.N., Lee S. Efficient mixed model approach for large-scale genome-wide association studies of ordinal categorical phenotypes. Am. J. Hum. Genet. 2021;108:825–839. doi: 10.1016/j.ajhg.2021.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pe'er I., Yelensky R., Altshuler D., Daly M.J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 21.Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sobota R.S., Shriner D., Kodaman N., Goodloe R., Zheng W., Gao Y.T., Edwards T.L., Amos C.I., Williams S.M. Addressing population-specific multiple testing burdens in genetic association studies. Ann. Hum. Genet. 2015;79:136–147. doi: 10.1111/ahg.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanai M., Tanaka T., Okada Y. Empirical estimation of genome-wide significance thresholds based on the 1000 Genomes Project data set. J. Hum. Genet. 2016;61:861–866. doi: 10.1038/jhg.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics, C. Patterson N., Daly M.J., Price A.L., Neale B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulik-Sullivan B., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.R., ReproGen Consortium. Robinson E.B., et al. Psychiatric Genomics Consortium. Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3 An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015;47:1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhuang Y., Wolford B.N., Nam K., Bi W., Zhou W., Willer C.J., Mukherjee B., Lee S. Incorporating family disease history and controlling case–control imbalance for population-based genetic association studies. Bioinformatics. 2022:btac459. doi: 10.1093/bioinformatics/btac459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe K., Taskesen E., van Bochoven A., Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017;8:1826. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masotti M., Guo B., Wu B. Pleiotropy informed adaptive association test of multiple traits using genome-wide association study summary data. Biometrics. 2019;75:1076–1085. doi: 10.1111/biom.13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saykally J.N., Dogan S., Cleary M.P., Sanders M.M. The ZEB1 transcription factor is a novel repressor of adiposity in female mice. PLoS One. 2009;4:e8460. doi: 10.1371/journal.pone.0008460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grewal T., Enrich C., Rentero C., Buechler C. Annexins in adipose tissue: novel players in obesity. Int. J. Mol. Sci. 2019;20:E3449. doi: 10.3390/ijms20143449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe T., Ito Y., Sato A., Hosono T., Niimi S., Ariga T., Seki T. Annexin A3 as a negative regulator of adipocyte differentiation. J. Biochem. 2012;152:355–363. doi: 10.1093/jb/mvs084. [DOI] [PubMed] [Google Scholar]
- 32.Tietge U.J.F., Maugeais C., Lund-Katz S., Grass D., deBeer F.C., Rader D.J. Human secretory phospholipase A2 mediates decreased plasma levels of HDL cholesterol and apoA-I in response to inflammation in human apoA-I transgenic mice. Arterioscler. Thromb. Vasc. Biol. 2002;22:1213–1218. doi: 10.1161/01.atv.0000023228.90866.29. [DOI] [PubMed] [Google Scholar]
- 33.Curcic S., Holzer M., Pasterk L., Knuplez E., Eichmann T.O., Frank S., Zimmermann R., Schicho R., Heinemann A., Marsche G. Secretory phospholipase A2 modified HDL rapidly and potently suppresses platelet activation. Sci. Rep. 2017;7:8030. doi: 10.1038/s41598-017-08136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J., Xu Y., Xu Y., Yin L., Zhang Y. Global inactivation of carboxylesterase 1 (Ces1/Ces1g) protects against atherosclerosis in Ldlr (-/-) mice. Sci. Rep. 2017;7:17845. doi: 10.1038/s41598-017-18232-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ge T., Chen C.Y., Ni Y., Feng Y.C.A., Smoller J.W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 2019;10:1776. doi: 10.1038/s41467-019-09718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Márquez-Luna C., Loh P.R., South Asian Type 2 Diabetes SAT2D Consortium. SIGMA Type 2 Diabetes Consortium. Price A.L. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 2017;41:811–823. doi: 10.1002/gepi.22083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kichaev G., Bhatia G., Loh P.R., Gazal S., Burch K., Freund M.K., Schoech A., Pasaniuc B., Price A.L. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feitosa M.F., Kraja A.T., Chasman D.I., Sung Y.J., Winkler T.W., Ntalla I., Guo X., Franceschini N., Cheng C.Y., Sim X., et al. Novel genetic associations for blood pressure identified via gene-alcohol interaction in up to 570K individuals across multiple ancestries. PLoS One. 2018;13:e0198166. doi: 10.1371/journal.pone.0198166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.May-Wilson S., Matoba N., Wade K.H., Hottenga J.J., Concas M.P., Mangino M., Grzeszkowiak E.J., Menni C., Gasparini P., Timpson N.J., et al. Large-scale GWAS of food liking reveals genetic determinants and genetic correlations with distinct neurophysiological traits. Nat. Commun. 2022;13:2743. doi: 10.1038/s41467-022-30187-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shim H., Chasman D.I., Smith J.D., Mora S., Ridker P.M., Nickerson D.A., Krauss R.M., Stephens M. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One. 2015;10:e0120758. doi: 10.1371/journal.pone.0120758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gagliano Taliun S.A., VandeHaar P., Boughton A.P., Welch R.P., Taliun D., Schmidt E.M., Zhou W., Nielsen J.B., Willer C.J., Lee S., et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat. Genet. 2020;52:550–552. doi: 10.1038/s41588-020-0622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim Y., Han B.G., KoGES group Cohort profile: the Korean genome and Epidemiology study (KoGES) consortium. Int. J. Epidemiol. 2017;46:1350. doi: 10.1093/ije/dyx105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahn Y., Kwon E., Shim J.E., Park M.K., Joo Y., Kimm K., Park C., Kim D.H. Validation and reproducibility of food frequency questionnaire for Korean genome epidemiologic study. Eur. J. Clin. Nutr. 2007;61:1435–1441. doi: 10.1038/sj.ejcn.1602657. [DOI] [PubMed] [Google Scholar]
- 44.Willett W.C., Howe G.R., Kushi L.H. Adjustment for total energy intake in epidemiologic studies. Am. J. Clin. Nutr. 1997;65:1220S–1228S. doi: 10.1093/ajcn/65.4.1220S. [DOI] [PubMed] [Google Scholar]
- 45.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S., et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmitt A.D., Hu M., Jung I., Xu Z., Qiu Y., Tan C.L., Li Y., Lin S., Lin Y., Barr C.L., Ren B. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016;17:2042–2059. doi: 10.1016/j.celrep.2016.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Magno R., Maia A.T. gwasrapidd: an R package to query, download and wrangle GWAS catalog data. Bioinformatics. 2020;36:649–650. doi: 10.1093/bioinformatics/btz605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Privé F., Arbel J., Vilhjálmsson B.J. LDpred2: better, faster, stronger. Bioinformatics. 2020;36:5424–5431. doi: 10.1093/bioinformatics/btaa1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
We report basic information on studied phenotypes, including sample size, mean, median, and unit of measurement.
We report summary statistics of genome-wide significant loci identified in KoGES. Beta indicates the genetic effect size calculated from SAIGE/POLMM.
We report LD score regression results for 76 studied phenotypes.
We report gene mapping results obtained by using the SNP2GENE function of FUMA.
We report information, including GWAS results and minor allele frequencies of the novel loci identified in KoGES.
We report (1) summary statistics of genome-wide significant loci identified by meta-analysis, (2) gene mapping of meta-analysis results using FUMA.
We report (1) predictive performance (R2) in single-ethnic PRS, (2) predictive performance (R2) in multi-ethnic PRS, and (3) statistical significance of PRS.
Data Availability Statement
The summary statistics generated during this study are submitted to Zenodo: https://zenodo.org/record/7042518. Manhattan plots and quantile-quantile plots generated by the summary statistics are publicly available at https://koges.leelabsg.org. BBJ summary statistics used in this study were downloaded from the Biobank Japan PheWeb: https://pheweb.jp/ and summary statistics for Europeans in UK Biobank were downloaded from Pan-UK Biobank1: https://pan.ukbb.broadinstitute.org/. This paper does not report custom code or software. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.