

Abstract
The consumption of fructose as sugar and high-fructose corn syrup has markedly increased during the past several decades. This trend coincides with the exponential rise of metabolic diseases, including obesity, nonalcoholic fatty liver disease, cardiovascular disease, and diabetes. While the biochemical pathways of fructose metabolism were elucidated in the early 1990s, organismal-level fructose metabolism and its whole-body pathophysiological impacts have been only recently investigated. In this review, we discuss the history of fructose consumption, biochemical and molecular pathways involved in fructose metabolism in different organs and gut microbiota, the role of fructose in the pathogenesis of metabolic diseases, and the remaining questions to treat such diseases.
Keywords: fructose, ketohexokinase, fatty liver, lipogenesis, intestine, gut microbiota
1. HISTORY OF FRUCTOSE CONSUMPTION
1.1. Fructose History
In all living creatures, glucose is a major carbohydrate in the animal kingdom, whereas sucrose, a disaccharide form of glucose and fructose, is a major carbohydrate in the plant kingdom. In some plants, a plentiful fructose monomer also exists. When animals consume plants, their digestion system quickly hydrolyzes sucrose into glucose and fructose monomers via the sucrase enzyme in the small intestine (56). Early Homo sapiens had consumed fructose in various plants and fruits, especially in the fall, to store energy for survival during the winter. The overall total energy intake from fructose was efficiently consumed without causing excessive energy accumulation. However, in modern society with readily available sugar-containing processed foods, fructose has become the major dietary carbohydrate (45) and contributes to diverse systemic diseases and health burdens.
Historically, it is believed that sucrose was first isolated from sugarcane and sugar beet in the form of juice around 800 BC in India (145) (Figure 1). In the early centuries AD, the Indians successfully produced granulated sucrose crystals from juice, promoting efficient trade to the Islamic world. Such sucrose was used for medicinal purposes as well as for an expensive spice called sweet salt. In the seventeenth century, widespread cultivation and advanced processing of cane sugar in Europe and the United States made sucrose more affordable. Since then, sucrose has become a semi-essential ingredient widely used in a vast variety of beverages and food.
Figure 1.
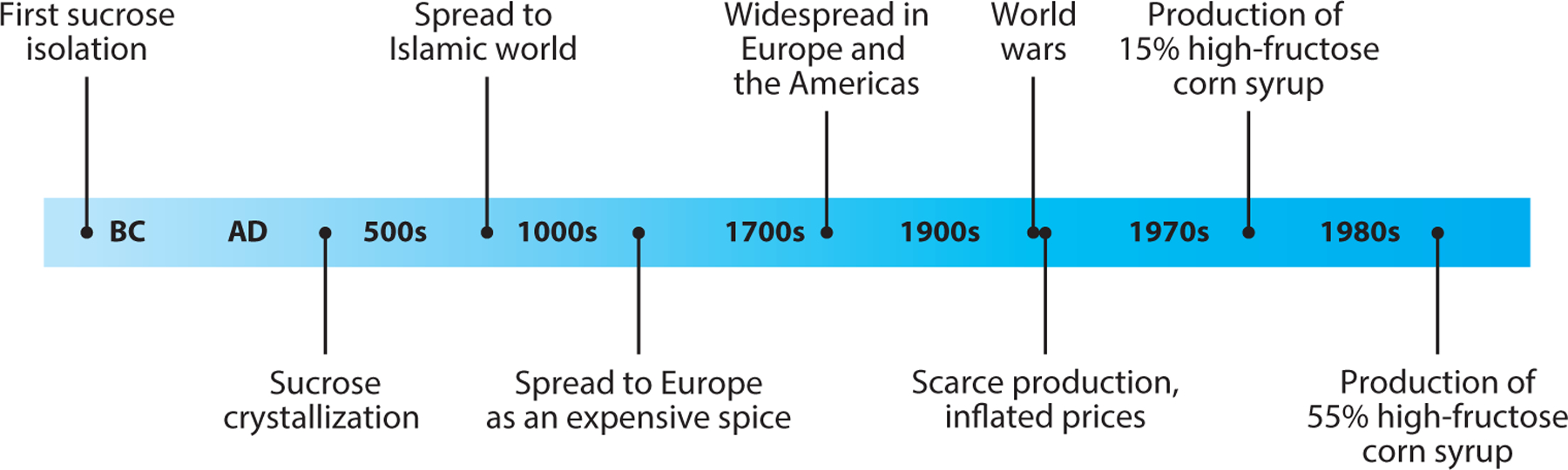
Fructose history: from the isolation of plant-based sucrose to the industrial production of 55% high-fructose corn syrup, a common sweetener in modern society.
In the mid-twentieth century, because of the destruction of the sugar industry and political instability during the two world wars, sucrose became scarce, and prices inflated. This caused producers to seek new ingredients for sweetness and led to the development of corn starch as an alternative. Corn starch, which was a plentiful and dependable agricultural raw material, was used to produce corn syrup. However, glucose, which is the main component of corn syrup, is not as sweet as sucrose. In the 1970s, a manufacturing breakthrough occurred in the sugar industry. High-fructose corn syrup (HFCS) containing 15% fructose was developed through an enzyme reaction in which glucose isomerase converts glucose into fructose (134). Then, manufacturers focused on increasing the fructose content in HFCS. In the 1980s, HFCS containing 55% fructose, which is a similar ratio of glucose and fructose to that in sucrose, was produced and has been widely used for beverages (56). It has replaced sucrose as a cheaper and sweeter substitute and has been used for almost all popular processed foods, including chocolate, jelly, ice cream, and sweetened drinks.
1.2. Increased Fructose Consumption in Modern Society
Since the 1980s, obesity and overweight have increased remarkably. Currently, approximately 35% of Americans are classified as obese or overweight (45). Such a fast rise is attributed to excess energy intake relative to energy expenditure because genetic backgrounds and other intrinsic biological processes cannot be changed in such a short time. In this respect, one of the most significant changes in food consumption in modern society was the increased intake of fructose.
In particular, fructose intake as HFCS has substantially increased in the United States over the past few decades (113). From the 1970s to 2000s, the average American’s annual intake of HFCS increased tremendously from 0.23 kg to 28.4 kg, while intake of sucrose moderately decreased from 46.4 kg to 30.5 kg. Daily fructose consumption has also increased by 26%, from 64 g/day in the 1970s to 81 g/day in the 2000s (48, 100). Thus, fructose has become a significant proportion of energy intake in the typical American diet.
2. FRUCTOSE METABOLISM
2.1. Fructose Uptake in the Intestine, Liver, and Other Organs
Like other dietary nutrients, fructose is absorbed by the small intestine, regardless of pure fructose, sucrose, or HFCS ingestion. Fructose is transported through specific fructose transporters, glucose transporter 5 (GLUT5) and GLUT2 (53, 114) (Figure 2). GLUT5 is highly expressed in the small intestine, kidneys, adipose tissue, skeletal muscle, and brain, but barely expressed in the liver. GLUT5 is located on the apical side of the enterocyte luminal pole, facilitating fructose transport from the intestinal lumen into the epithelial cells with a high affinity for fructose (Km = 6.0 mM) (12, 114). Contrary to glucose, this process does not require adenosine triphosphate (ATP) hydrolysis and is independent of sodium absorption. GLUT2, which has a low fructose affinity (Km = 66.0 mM), diffuses fructose out of the enterocyte into the portal circulation (18, 53). GLUT5 immunoreactivity was also reported in the basolateral membrane of human enterocytes (9), but the release of fructose into the portal circulation through GLUT5 remains unclear. In the liver, fructose transport is primarily mediated by GLUT2 (37), which is also highly expressed in the kidneys. GLUT8 highly expressed in the liver and heart was reported to play a role in fructose transport in mice (27).
Figure 2.
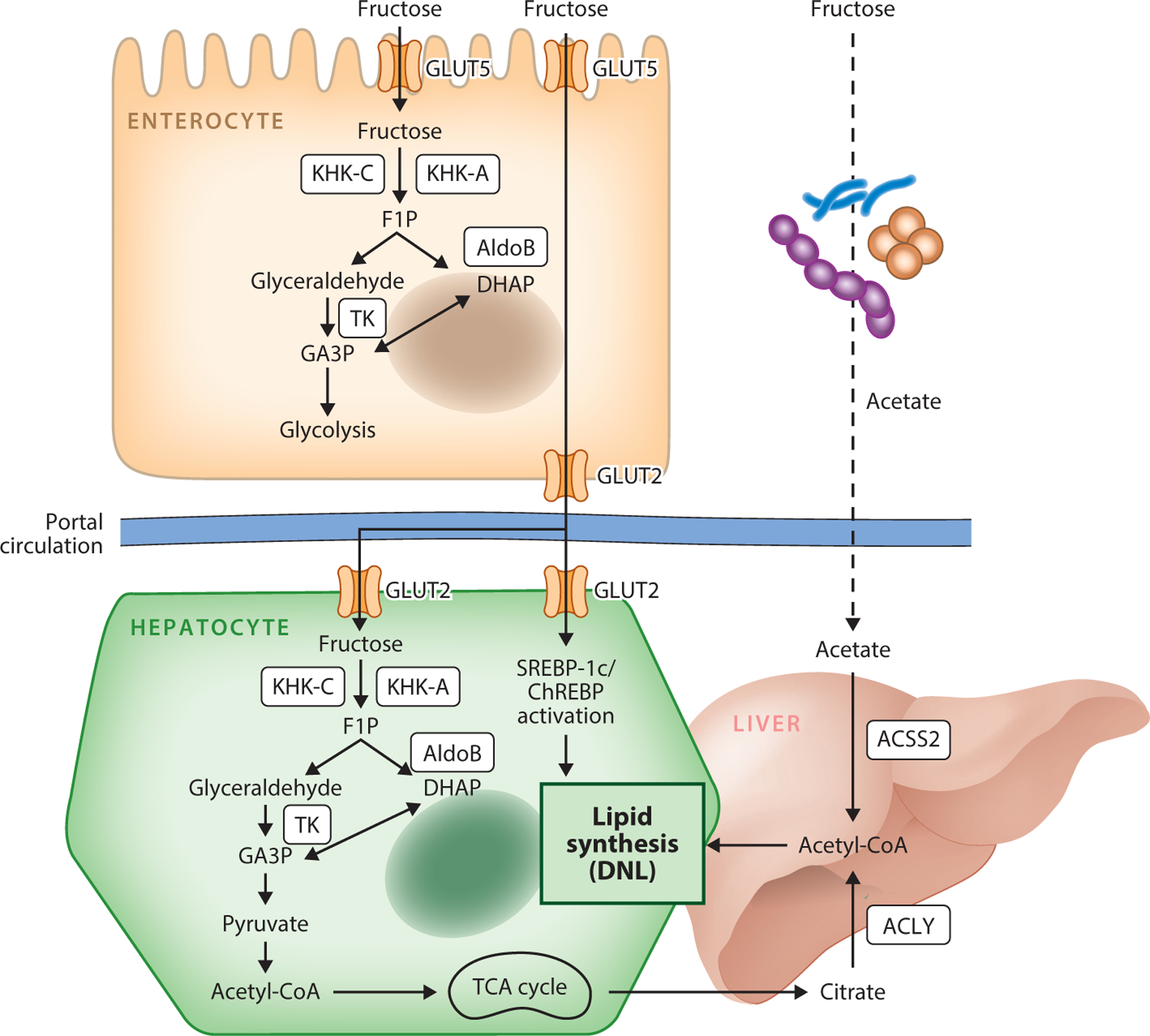
Fructose metabolism. Fructose is taken up by GLUT5 or GLUT2 in enterocytes or hepatocytes. Fructose is subsequently phosphorylated by KHK-C/A into F1P, which is cleaved by AldoB into DHAP and glyceraldehyde. Both then enter the glycolysis and TCA cycle. In liver, some citrate is converted to cytosolic acetyl-CoA via the ACLY enzyme. Alternatively, acetate, which is catabolized from fructose by gut microbiota, is converted into cytosolic acetyl-CoA by ACSS2 and is used for hepatic lipid synthesis. Fructose catabolism also activates DNL signaling pathways via the transcription factors SREBP-1c and ChREBP. Abbreviations: acetyl-CoA, acetyl coenzyme A; ACLY, ATP citrate lyase; ACSS2, acetyl-CoA synthetase 2; AldoB, aldolase B; ChREBP, carbohydrate-responsive element-binding protein; DHAP, dihydroxyacetone phosphate; DNL, de novo lipogenesis; F1P, fructose-1-phosphate; GA3P, glyceraldehyde-3-phosphate; GLUT2, glucose transporter 2; GLUT5, glucose transporter 5; KHK-A, ketohexokinase-A; KHK-C, ketohexokinase-C; SREBP-1c, sterol-responsive element-binding protein; TCA, tricarboxylic acid; TK, triose kinase.
The importance of GLUT5 as a major intestinal fructose transporter has been demonstrated by genetically modified mice. GLUT5 whole-body knockout (KO) mice do not show any defects under a typical chow diet but exhibit lethal phenotypes under fructose feeding (114). In contrast, Glut2 whole-body KO mice develop only mildly decreased fructose absorption (53). In humans, hereditary fructose intolerance, potentially involving GLUT5 deficiency or other mutations in fructose catabolic enzymes, manifests a colonic dilation and increased intestinal permeability (5). GLUT5 expression can be stimulated by increased fructose consumption (23), largely through carbohydrate-responsive element-binding proteins (ChREBPs) (78), and the thioredoxin-interacting protein (35).
2.2. Fructolysis in the Intestine and Liver
Although glucose and fructose are chemically very similar (both are C6H12O6), fructose has a keto group, whereas glucose has an aldehyde group. This relatively small structural difference results in dramatically distinct cellular metabolisms of glucose and fructose. Ketohexokinase (KHK) initiates the fructose mechanism through phosphorylation of fructose to fructose-1-phosphate (F1P). This reaction can lead to ATP depletion when excessive fructose is catabolized (31, 59). KHK has two alternatively spliced isoforms: KHK-A and KHK-C. KHK-A is ubiquitously expressed in most tissues (31) but has a very low affinity (Km = 1 mM). KHK-C, with a high affinity for fructose (Km = 20 μM), is expressed primarily in the liver, intestine, and kidney, making it of primary importance in fructolysis (31, 66). Although KHK-A has a low fructose affinity, KHK-C isoform-specific KO mice showed a high residual (~30–50%) intestinal fructose catabolic activity (69), suggesting that KHK-A significantly contributes to fructose metabolism, at least in the intestine where the luminal fructose level is high enough to activate KHK-A. Importantly, Khk-A/C whole-body double-KO mice showed resistance to metabolic syndromes induced by fructose consumption, with most fructose excreted by urine (66). This key finding led to the development of KHK inhibitors for the treatment of fructose-related pathology (see below).
Aldolase B (AldoB), the next enzyme that breaks down F1P, is also crucial for fructose catabolism. AldoB splits F1P into glyceraldehyde and dihydroxyacetone phosphate, the latter of which enters glycolysis (10, 51). Glyceraldehyde is converted to glyceraldehyde-3-phosphate via triose kinase, which is an important step that affects fructose-dependent fat synthesis and pathologies (93). AldoB whole-body KO mice showed high hepatic F1P accumulation with lethal fat accumulation and fibrosis in the liver, which in part mimics human patients with fructose intolerance (110). Importantly, these pathological phenotypes caused by AldoB depletion were largely rescued by KHK inhibition (86), demonstrating that F1P is the major cause of hepatic detrimental effects. AldoB deficiency in humans also impairs renal function upon fructose ingestion (83). KHK inhibitors are likely to suppress this fructose-induced kidney disorder, which requires more clinical investigations.
2.3. Fructose-Induced Lipogenesis
Compared with glucose, fructose is a more potent inducer of hepatic de novo lipogenesis (DNL), which converts excess carbons into lipids. Fructose-derived carbons first enter the tricarboxylic acid (TCA) cycle and generate citrate to provide cytosolic acetyl coenzyme A (acetyl-CoA) for DNL. ATP citrate lyase (ACLY) is the essential enzyme for generating such lipogenic cytosolic acetyl-CoA (74). Recently, an alternative pathway of DNL, especially under high-fructose feeding, was shown to be activated through acetyl-CoA synthetase 2 (ACSS2). When a high amount of fructose is consumed, unabsorbed fructose reaches the colon and is converted into short-chain fatty acids (SCFAs) (primarily acetate) by the gut microbiota (68). Acetate is then transported through the portal blood and feeds hepatic acetyl-CoA via ACSS2. To initiate DNL, cytosolic acetyl-CoA should be converted to malonyl-CoA by acetyl-CoA carboxylase, which is the rate-limiting step of DNL. After palmitate is synthesized via fatty acid synthase, elongation and desaturation of palmitate subsequently occur (54, 131). Importantly, fructose not only provides carbons for DNL but also activates lipogenic transcription machinery. Such a signaling effect of fructose is mainly mediated by the transcription factors ChREBP (especially ChREBP-β) and sterol-responsive element-binding protein (SREBP-1c). Although the signaling molecule in the fructolysis pathway that activates ChRBEPs and SREBP-1c remains controversial, the key role of ChREBP in fructose catabolism and consequent lipogenesis has been demonstrated (79).
3. FRUCTOSE-RELATED DISEASES AND UNDERLYING MECHANISMS
Epidemiological and experimental feeding studies have indicated the causal relationship between excessive fructose intake and metabolic diseases including obesity, nonalcoholic fatty liver disease (NAFLD), cardiovascular disease (CVD), and type 2 diabetes (Figure 3). For example, Olsen & Heitmann (109) found a positive correlation between the intake of sweetened beverages and obesity in a meta-analysis of 19 clinical studies. In the NAFLD study, 80% of patients had a soft drink more than once per day, while only 17% of healthy controls had a soft drink more than once per day for the 6-month observation period. Fructose consumption through soft drinks also had an association with higher CVD risk factors such as waist circumference, fasting blood glucose, blood pressure, serum triglycerides, and cholesterol. These factors had a 48% higher prevalence in individuals who consumed soft drinks more than once per day compared with individuals who consumed soft drinks less than once per day (30). In addition, individuals drinking more than one sweetened beverage per day showed an 83% higher risk of type 2 diabetes compared with individuals drinking less than one beverage per day in an 8-year prospective cohort study (128). In this section, we discuss the link between fructose and each disease and the potential molecular mechanisms.
Figure 3.
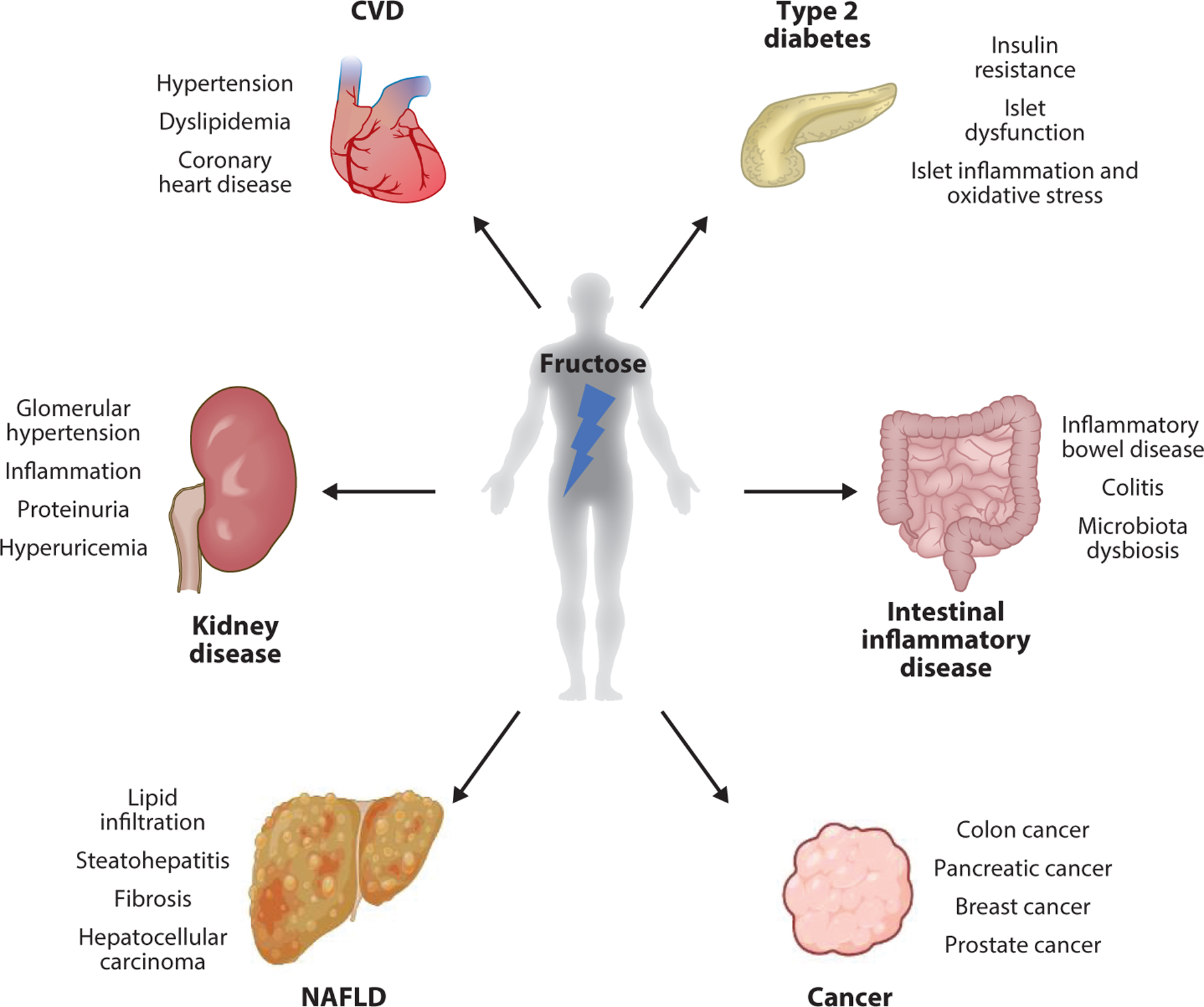
Fructose-related pathologies. Excessive fructose metabolism and consequent metabolic products contribute to diverse metabolic and inflammatory diseases, including nonalcoholic fatty liver disease (NAFLD), type 2 diabetes, dyslipidemia, colitis, cardiovascular disease (CVD), and renal disease. Fructose consumption is also linked to many different types of cancers.
3.1. Nonalcoholic Fatty Liver Disease
Among the many diseases related to fructose, NAFLD has emerged as the most prevalent disease associated with chronic fructose intake. NAFLD is diagnosed on the basis of the presence of hepatocytes with lipid infiltration but no evidence of infection, inborn metabolic disorder, or steatogenic drug or alcohol consumption (146). NAFLD can progress to nonalcoholic steatohepatitis, which is histologically characterized by the presence of steatosis and lobular inflammation in hepatocytes (16). NAFLD has the potential to evolve into cirrhosis, end-stage fibrotic liver disease, and occasionally hepatocellular carcinoma (16). Fructose intake has a dose-dependent correlation with NAFLD development and progression (1). Meanwhile, lifestyle changes along with restricting dietary fructose intake have reduced hepatic triglyceride accumulation (39). While some human studies did not find a clear association between fructose intake and NAFLD development due to many variable factors (genetic and environmental) (118), mounting evidence indicates that fructose consumption, especially in a liquid form, is linked to the development of NAFLD.
Fructose can induce NAFLD in many different ways. Due to its delightful taste, fructose increases food consumption, elevating total energy intake. Studies have also shown that fructose directly affects neuronal and hormonal signaling that controls appetite (96). In addition, the unique biochemistry and organ metabolism of fructose may mediate fructose-induced NAFLD. Unlike glucose, fructose is mainly catabolized by the small intestine followed by the liver (114). High-dose fructose, however, overwhelms the small intestinal fructose clearance, causing excess fructose to reach the liver (68).
Fructose catabolism by hepatocytes can deplete ATP and activate the adenosine monophosphate (AMP) deaminase pathway, inducing excessive uric acid production (70). Accumulation of uric acid in the hepatocytes inhibits CoA hydratase activity with consequent decrease of fatty acid β-oxidation, which leads to hepatic lipid accumulation (20). Interestingly, a recent paper shows an alternative relationship between fructose and metabolites in the AMP deaminase pathway in NAFLD development. The authors showed that inosine monophosphate can induce NAFLD via the induction of AMP deaminase 2 and purine degradation (3). While the epidemiological evidence between high circulating uric acid and NAFLD is ample, their causal relationship is less clear, as animal models with hyperuricemia do not develop NAFLD (97).
Another important effect of fructose intake on the liver is the induction of lipogenesis. In patients with NAFLD, DNL was shown to be threefold greater than in healthy individuals (84). It has long been believed that fructose itself delivers excessive carbons for lipogenesis to the hepatocytes. However, a recent study using liver-specific KO mice of Acly, an essential enzyme for lipogenesis, challenged this notion (148). While these mice were not able to use cytosolic citrate for lipogenesis, they still developed NAFLD under a high-fructose diet. Using isotope tracing, the group discovered that copious amounts of fructose feed hepatic lipogenesis via gut microbiota–derived acetate, which bypasses ACLY. Antibiotics treatment or liver-specific knock out of Acss2, the essential enzyme for acetate catabolism, sufficiently reduced fructose-dependent hepatic lipogenesis.
In addition to lipogenic acetate production, fructose that reaches the large intestine also has other detrimental effects that contribute to NAFLD, such as increased intestinal epithelial permeability (47). This causes the delivery of toxic microbial metabolites from the gut lumen to the liver, activating inflammatory signals in liver cells (hepatocytes and immune cells) to trigger NAFLD (105). Chronic fructose intake can also induce microbiota dysbiosis, which augments inflammatory signals (105). Mice lacking Toll-like receptor 4 (TLR4), the inflammatory signal receptor, showed reduced onset of NAFLD (44), while TLR4 agonists trigger lipogenesis and NAFLD (92). On the other hand, a recent systematic genetics approach using the progeny of fructose-sensitive and -resistant mouse strains found no connection between NAFLD and TLR4 (34). Therefore, the role of hepatic inflammation via TLR4 signaling in NAFLD development is likely context dependent.
Altogether, these findings highlight the complex interactions between the liver, intestine, and gut microbiota in NAFLD development. Several recent studies further support this organ cross talk by generating various mouse models, including liver- or intestine-specific Khk-A/C KO mice, intestine-specific Khk-C (the active isoform) KO mice, and intestine-specific Khk-C overexpressing transgenic mice (69, 148). Two recent reviews nicely summarized the various phenotypes and implications of these mouse models (43, 61). Overall, these data showed that intestinal fructose catabolism shields the liver from excess fructose exposure while hepatic fructose metabolism is the major cause of NAFLD. Given that Pfizer’s orally available KHK inhibitor is now in phase II clinical trials, understanding the drug’s distribution and action in different organs will be crucial to maximizing its therapeutic effects.
3.2. Type 2 Diabetes
The incidence of type 2 diabetes has increased at an epidemic rate, and this increase is also linked to changes in diet and reduced physical activity. Type 2 diabetes prevalence is 20% higher in countries with higher availability of HFCS compared with countries that have low availability of HFCS, independently of obesity prevalence (52). Accumulative human studies suggest that fructose-induced liver fat accumulation contributes to hepatic lipotoxicity and the development of insulin resistance (139). Such hepatic insulin resistance leads to hyperlipidemia and consequent lipid accumulation and lipotoxicity in other organs including the skeletal muscle (137).
Type 2 diabetes is characterized by defective insulin responses and eventual failure of insulin secretion, which is related to pancreatic islet cell dysfunction or diminished β-cell mass. In pancreatic β cells, fructose treatment alone did not cause insulin secretion, yet constant exposure led to enhanced reactivity of pancreatic β cells to glucose (6). In a rat model of type 2 diabetes, fructose consumption accelerated islet dysfunction via induction of islet inflammation and oxidative stress (24). While these studies suggest that fructose may act directly on the pancreas, further investigations are needed to demonstrate whether circulating fructose after intestinal and hepatic clearance is sufficiently high to affect the pancreas in vivo.
Despite the strong epidemiological evidence, direct fructose feeding to human subjects has shown variable effects on insulin sensitivity and metabolic phenotypes. For example, fructose feeding in healthy subjects showed only a mild effect on insulin sensitivity (22), which contrasts with fructose-induced deleterious effects in obese and diabetic patients (98). Different kinetics and efficiency of intestinal and hepatic fructose absorption/catabolism between healthy and obese/diabetic individuals may be a potential explanation. Fructose effects are also potentiated by glucose, whose circulating levels are high in obese and diabetic patients (122). Therefore, future studies are required to identify genetic and environmental factors that confer interindividual variations in sensitivity to fructose-induced diabetes.
3.3. Kidney Disease
Fructose intake may also have a direct or indirect role in the development of chronic kidney disease (CKD), given the epidemiological relationship between fructose, hypertension, and diabetes. A cross-sectional analysis from the National Health and Nutrition Examination Survey (1999–2004) reported that the intake of two or more sugar-containing beverages per day was associated with an increased risk of albuminuria (129). In addition, epidemiological studies have linked dietary fructose with an increased risk for kidney stones (135). People who drink less than one 24-oz. can of soft drinks per week showed a 10% lower risk for kidney stones (130). Therefore, epidemiological findings provide a correlation between fructose intake and the risk for kidney diseases.
Experimental studies also support fructose intake being a mechanism for CKDs such as glomerular hypertension, renal inflammation, and tubulointerstitial injury. In fact, feeding fructose, but not glucose, accelerated the CKD progression by exacerbating proteinuria, renal dysfunction, and glomerulosclerosis in a rat remnant kidney model (49). Furthermore, the role of endogenous fructose in diabetic nephropathy has been suggested (87). Importantly, the lack of KHK activity protects against aging-associated renal disease in both mice and humans (121), indicating that fructose catabolism, not fructose itself, is crucial for pathogenesis. Fructose metabolism by renal KHK increases sodium hydrogen exchanger activity in renal proximal tubular cells by decreasing intracellular cyclic AMP. This results in increased renal sodium reabsorption and blood pressure, which were not observed in Khk KO mice (58). Finally, studies in rats documented that fructose intake results in glomerular hypertension and reduced renal blood flow, leading to the development of kidney vascular disease (40).
Interestingly, the renal injury associated with fructose intake mimics what is observed in subjects with an abnormally high level of blood uric acid. In a longitudinal study with 627 patients at the initial stage of CKD, serum levels of more than 7.5 mg/dL in uric acid were a risk factor for a renal malfunction (120). One potential source of such uric acid is fructose catabolism in kidneys. Khk KO mice showed protection from fructose-induced CKD phenotypes with decreased uric acid, oxidative stress, and inflammation (4). Taken together, these findings suggest that more clinical and mechanistic studies are needed to determine if limiting fructose intake to suppress uric acid production may benefit subjects with kidney disease.
3.4. Cardiovascular Disease
CVD comprises a group of disorders in the heart and blood vessels and is the number one cause of death worldwide. There is increasing evidence that higher fructose consumption increases CVD risk by contributing to the development of hypertension, dyslipidemia, inflammation, and coronary heart disease (103). The risk of CVD is increased by 26% by a high intake of fructose-sweetened beverages (99). Although increased CVD risk may be partially attributed to fructose-induced obese or insulin-resistant states, cardiac-specific fructose toxicity is also possible. Consistent with this notion, the relationship between fructose intake and increased risk for CVD is independent of body mass index (99).
One potential mechanism is related to direct fructose catabolism in the heart because both GLUT5 and KHK are expressed in cardiomyocytes (102). In normal, healthy individuals, intestinal and hepatic fructose catabolism efficiently clears fructose. Circulating fructose levels are thus unlikely high enough to trigger ATP depletion in the heart. Moreover, the heart normally expresses the KHK-A isoform, which further suggests that cardiac fructose metabolism is likely insignificant. However, a recent study showed that cardiac activation of hypoxia-inducible factor 1α drives the ectopic expression of KHK-C through the induction of the splice factor SF3B1 (106). This KHK-C expression may trigger ATP depletion when an excessive amount of fructose reaches the heart due to reduced intestinal and hepatic fructose clearance. Another suggested mechanism of fructose-induced CVD is the effect of fructose on the glycation of cardiac proteins. Fructose can directly impact the structure and function of cardiac proteins through posttranslational modifications such as glycation and O-GlcNAcylation (101), which are now regarded as novel targets for CVD interventions. Further studies are required to determine the causal relationships between these suggested mechanisms and CVD in humans.
3.5. Intestinal Inflammatory Disease
Given the fact that the intestine is the first organ that is exposed to high levels of dietary fructose and its catabolism, it is not surprising that excessive fructose intake is linked to intestinal diseases. Fructose is linked to intestinal inflammatory disease, including inflammatory bowel disease (IBD) and colitis, and its incidence has increased recently worldwide. In 2015, an estimated 1.3% of US adults had received a diagnosis of intestinal inflammatory disease, a number that had almost doubled since 1999 (25). Previous research in animal models has found that a high-fructose diet can cause intestinal inflammatory disease (75). However, human epidemiologic studies have not always shown a correlation between refined sugar intake and intestinal inflammatory disease (60). Intriguingly, a diet abundant in HFCS increases the risk of intestinal inflammatory disease only when dietary fiber intake is low (62). Similarly, the increased consumption of fructose along with ultraprocessed foods, which contain little dietary fiber (55), parallels the increasing incidence of intestinal inflammatory disease. It is crucial to investigate the mechanism behind fructose-induced intestinal inflammatory disease and whether restricting fructose in the diet can decrease this risk.
Fructose consumption was shown to worsen colonic inflammation with effects on the gut microbiome, including changes in their compositions, metabolism, and localization within the colon (107). Interestingly, the detrimental effects of high-fructose feeding on colitis severity are completely reversed by switching back to a nonfructose diet (108). Meanwhile, a recent study showed that fructose consumption increases the survival of intestinal epithelial cells in hypoxia conditions (136). This induces the elongation of the intestinal villi, enhancing the absorption of various nutrients such as fat and causing excessive calorie intake and obesity.
The intestinal immune system also can be a critical factor that mediates fructose-induced intestinal inflammatory disease. Recent findings showed altered activity of immune cells, such as dendritic cells and macrophages, after exposure to fructose. In dendritic cells, the critical antigen-presenting cells that initiate an immune response, acute exposure to high amounts of fructose, but not glucose, induced an increase in proinflammatory cytokines (67). Similarly, in human monocytes and mouse macrophages, the key cell types of the innate immune system, fructose increases inflammatory cytokine production (73). While these studies in cultured cells suggest a potential involvement of the gut immune system in fructose-induced intestinal pathologies, more in vivo studies are required.
3.6. Cancer
Fructose consumption is linked to many different types of cancers. The observation of GLUT5 expression in several types of tumors led to the idea that tumor cells may directly utilize fructose. For example, GLUT5 expression was observed in human epithelial colorectal adenocarcinoma (57). Expression of GLUT5 in pancreatic tumor cells (91) led to a cohort study involving 88,802 patients, which revealed that fructose consumption was the strongest risk factor for pancreatic tumors in subjects with obesity and little physical activity (104) and in women (126). Moreover, a 7.2-year follow-up study also showed that high sugar consumption results in a greater risk of pancreatic cancer (88). Importantly, in pancreatic cancer patients, the serum concentration of fructose was higher than that in normal individuals (65). In addition to pancreatic tumors, GLUT5 is also overexpressed in brain cancer, liver cancer, and prostate cancer (17, 36). While these findings suggest direct fructose utilization by cancer cells, the question remains whether a sufficient amount of fructose is present in the circulation or in the tumor microenvironment.
Mounting experimental evidence indicates that increased fructose consumption can trigger tumor formation, progression, and metastasis. In lung adenocarcinoma, depletion of GLUT5 decreased tumor cell proliferation and invasion and increased cell death (144). On the other hand, overexpression of GLUT5 increased cell proliferation, migration, invasion, and tumorigenesis (144). In acute myeloid leukemia, fructose uptake increased cell proliferation, colony growth, migration, and invasion (19). In prostate cancer, dietary fructose increased the growth of patient-derived xenografts (15). In breast cancer, KHK-A overexpression induced metastasis under fructose-fed conditions (77). In intestinal cancer, daily gavage of high-dose fructose promoted tumor growth in the tumor-prone genetic mouse model (50).
The observation of ectopic expression of GLUT5 and increased fructose uptake by certain types of cancers raises the exciting potential of using positron emission tomography (PET)-fructose imaging as a specific tumor-detection tool, in conjunction with conventional glucose-PET imaging (133). Another important aspect to consider is whether blocking fructose usage by tumor cells can be a therapeutic strategy. While a fructose-restricted diet would be an approach, patients’ compliance can be an issue given the fact that most processed foods contain high amounts of fructose. In addition, since fructose can be endogenously generated, KHK blockade may serve as a better approach.
4. THE NEXT FRONTIER: MICROBIAL FRUCTOSE METABOLISM
Recent studies found that high consumption of dietary fructose contributes to the development of diabetes, NAFLD, and IBD via gut microbial dysbiosis (85). The genetic contents of the human gut microbiome are over 100 times more diverse than those of human cells, indicating that the gut microbiome can produce an enormous diversity of exogenic molecules from fructose (90). Therefore, fructose-mediated gut microbial changes and their products can affect distinct host physiology (26) and pathology (41). In this section, we discuss how a high-fructose diet changes gut microbial activities and impacts host health.
4.1. Fructose-Induced Gut Microbiome Changes
Various human gut microbiota species encode fructose uptake and metabolizing genes (115). Among the 13 representative human gut bacterial species used in the study, 10 can grow on fructose as a sole carbohydrate source (29). However, because the human gut microbiota is a complex microbial community with competitive and mutualistic relationships for nutrient utilization (21), it may not be appropriate to focus on the fructose utilization of single bacterial species for an understanding of whole gut microbial changes during high fructose consumption.
It has been shown that high fructose consumption contributes to gut microbial dysbiosis and reduction of diversity in the mammalian intestine (132). To identify the causal microbiota species in host pathologies, recent studies intended to associate the host diseases induced by high fructose consumption with specific levels of gut microbiota. For example, high fructose intake causes a reduction of butyrate-producing bacteria (7). Faecalibacterium and Ruminococcus, which are known as the representative butyrate-producing bacteria, were decreased during a high-fructose diet feeding in healthy adult humans (7). In addition, the abundance of Ruminococcaceae and Lachnospiraceae, the major butyrate producers, was decreased in rats fed HFCS (132). Reduced levels of butyrate-producing bacteria in the gut correlated with diabetes and IBD (62).
Even though various mechanisms were reported concerning butyrate-producing bacteria and host diseases, the most dominant theory is that butyrate-producing bacteria are required to enhance intestinal barrier function and mucosal immunity (46). Butyrate improves the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase (AMPK) in colonocytes (116). Moreover, butyrate reduces inflammation by inhibiting nuclear factor κB activity (81). For these reasons, the reduction of butyrate-producing bacteria is linked with the host pathologies in high fructose consumption, but the exact causal relationships are still unclear. Therefore, further research is required to reveal the reason why high fructose intake reduces the levels of butyrate-producing bacteria and leads to the subsequent biological mechanisms in host pathologies.
During high fructose consumption, the other interesting feature in gut microbial changes is the increase in the composition of the phyla Bacteroidetes or Proteobacteria (7, 33). While high-fructose diet–fed mice showed overall decreased gut microbial diversity, the proportion of Proteobacteria markedly increased (33). These mice also showed increased gut permeability and inflammation. In another study, 10% fructose feeding led to significantly elevated Bacteroidetes and Proteobacteria in the murine fecal microbiome (132). Both Bacteroidetes and Proteobacteria are major phyla constituting the gram-negative bacteria of the human gut microbiome (123). Bacterial endotoxin or lipopolysaccharide (LPS), the cell wall components of gram-negative bacteria, are recognized by the innate immune system and induce inflammation (138). Therefore, high fructose intake increases gut permeability due to alterations to tight junction proteins caused by gut microbial dysbiosis, leading to LPS translocation to the portal vein (143). Evidence of LPS-induced liver injury has been reported in hepatitis and primary biliary cirrhosis patients (72). In this respect, the increases of Bacteroidetes and Proteobacteria in high fructose intake are presumed to be key causes for host pathologies.
Some studies showed gut microbial changes by high fructose consumption at the bacterial species level. Montrose et al. (107) found that elevated dietary fructose promotes the notable growth of Citrobacter rodentium in mice. C. rodentium, an enteric bacterial pathogen of the mouse intestinal tract, triggers severe inflammatory responses such as colitis (94). Furthermore, Akkermansia spp. was increased sevenfold after 8% fructose water feeding in C57/BL6 mice but not in FVB or DBA mouse strains (2). Interestingly, fecal transplantation of C57/BL6 mice or colonization of Akkermansia muciniphila suppressed fructose-induced weight gain and improved glycemic responses in the other strains. A. muciniphila is well known to strengthen intestinal barrier function and improve metabolism in obese and diabetic mice (14). In particular, Amuc_1100, a specific protein isolated from the outer membrane of A. muciniphila, interacts with Toll-like receptor 2 and modulates host immune response with the gut barrier (111). Supplementation with A. muciniphila in overweight and obese humans improved insulin sensitivity and reduced insulinemia and plasma total cholesterol (28). Because of these characteristics, there are attempts to develop A. muciniphila as a next-generation probiotic bacterium (28).
4.2. Metabolic Changes in Gut Microbial Environments from High Fructose Intake
As outlined above, various gut bacteria species can readily grow with fructose and produce a variety of metabolic products that influence host physiology and pathology (Figure 4). Among the gut microbial metabolites, SCFAs are the most studied metabolites whose levels are changed during high fructose intake (64). Gut microbiota produce SCFAs (acetate, propionate, and butyrate) from fructose via their unique metabolic pathways (i.e., Wood-Ljungdahl pathway, butyryl-CoA:acetate CoA-transferase route) (95). Fructose-derived SCFAs can directly serve as energy sources or activate G-protein-coupled receptors as signaling molecules. They can also affect epigenetics by providing carbon sources for histone acetylation (through acetate) or by inhibiting histone deacetylases (through butyrate) (82). Fructose-derived SCFAs thus affect various physiological processes and contribute to host health and diseases (140).
Figure 4.
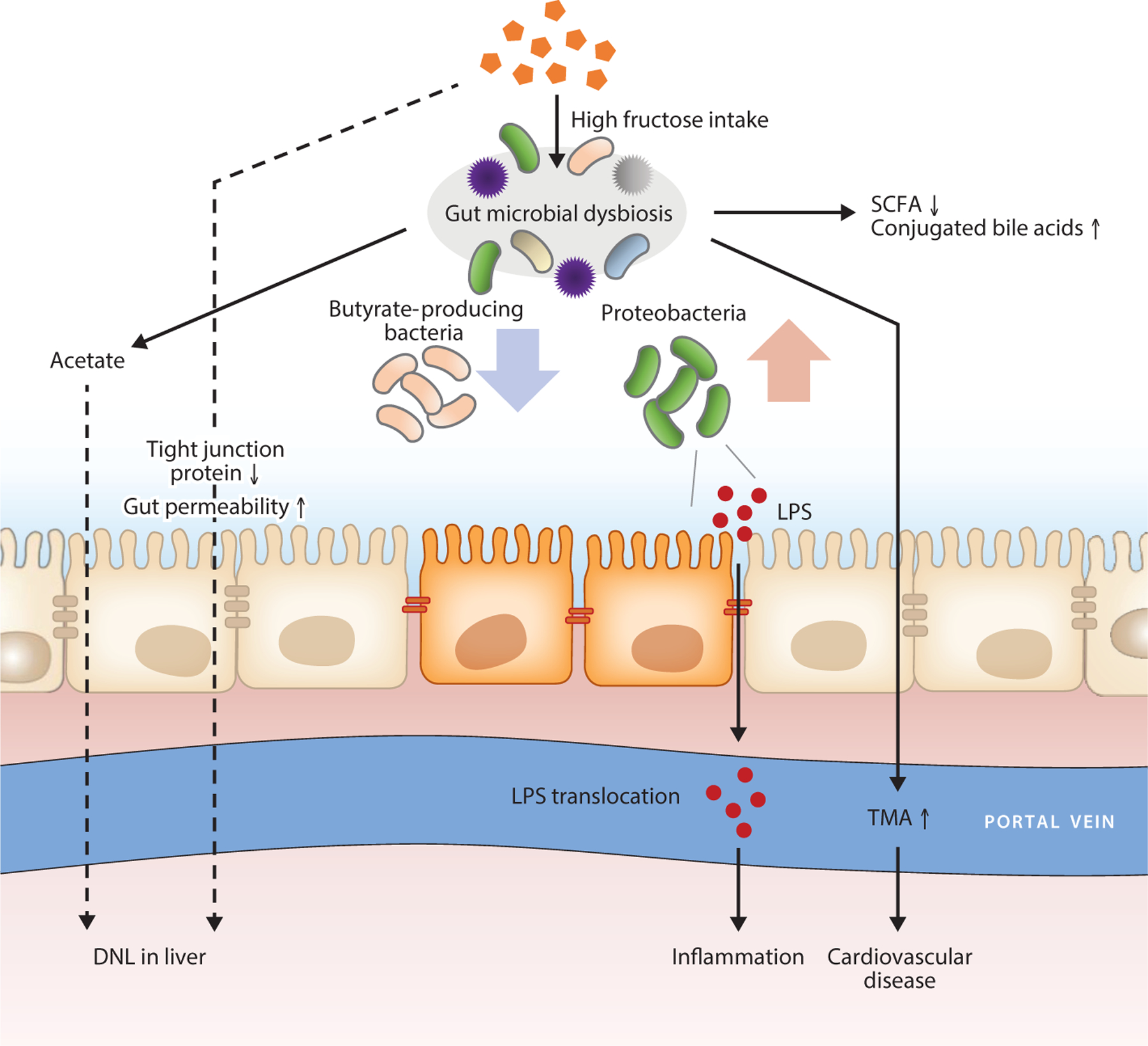
Role of gut microbiota in fructose-related pathologies. Chronic overconsumption of fructose induces gut dysbiosis, with decreased butyrate-producing bacteria and increased gram-negative Proteobacteria, which induces changes in microbial metabolites including SCFAs, TMA, LPS, and bile acids. Fructose also induces leaky gut, facilitating translocation of microbial toxic chemicals to the host organs. Abbreviations: DNL, de novo lipogenesis; LPS, lipopolysaccharide; SCFA, short-chain fatty acid; TMA, trimethylamine.
However, changes in SCFA levels during high fructose consumption remain controversial. Li et al. (89) showed that the fecal concentrations of all three SCFAs were significantly lower in fructose-fed mice. The cause of this reduction can be an altered and unhealthy status of the gut microbiota (89). Some studies have shown a connection between the reduction of SCFA levels and host pathologies such as obesity and colonic diseases (80). Supporting this notion, SCFA supplementation improved high-fructose-induced diseases through the increase of SCFA receptors in the kidney or amelioration of intestinal epithelial barrier impairment (64, 89). On the contrary, others reported that a high-fructose diet increased plasma levels of SCFAs (11, 63). The increase of SCFA levels in plasma is associated with the reduction of SCFA receptor expression in kidney and CVDs (8). This discrepancy may be due to the use of variable mouse strains, the amounts and methods of fructose feeding (liquid versus solid forms), and other unknown factors. Therefore, to clarify the exact relationships between high fructose intake, gut dysbiosis, and SCFA levels, a more systematic analysis of various biological samples and control of variables will be needed.
Recently, other gut microbial metabolite changes have gained significant attention (107). For example, a high-fructose diet increases plasma levels of trimethylamine (TMA) (64). TMA is generated from choline, betaine, and carnitine by gut microbiota, and it can be metabolized into trimethylamine-N-oxide (TMAO) (64). Importantly, a high blood TMAO level is strongly associated with increased CVDs (127). This increased TMA production by fructose is at least in part because dietary fructose increases the abundance of Escherichia, which encodes genes required for the formation of TMA (11). On the other hand, a high-fructose diet reduced bile salt hydrolase–expressing microbes and increased luminal conjugated bile acids (107). Elevated levels of fecal conjugated bile acids disrupt the gut barrier, contributing to IBD and worsening chemically induced colitis in mice (38). In addition, supplementation of nonconjugated bile acids prevents fructose-induced hepatic steatosis in mice through mechanisms involving protection against the fructose-induced translocation of intestinal bacterial endotoxin (141). Together, these studies strongly suggest that several gut microbial metabolite levels are changed with high fructose consumption. It will be valuable to perform comprehensive mapping of microbiota metabolites altered by fructose with advanced metabolomics and isotope tracing techniques.
4.3. Probiotic and Prebiotic Strategies for Treating Fructose-Related Diseases
Due to the association between fructose-induced gut microbial dysbiosis and host pathologies, recent studies tested the effectiveness of probiotics and prebiotics as microbiota-management tools for improving health (125). Probiotics are defined as live microorganisms that, when administered in adequate amounts, confer a health benefit on the host. The main advantages of probiotics are the effect on the interference with potential pathogens, improvement of gut barrier function, immunomodulation, and production of beneficial metabolites (124). On the basis of these findings, studies showed that oral administration of probiotics mitigates the pathological features associated with high fructose consumption (112, 149). The administration of Lactobacillus kefiri to mice fed a fructose-rich diet prevented weight gain, elevations of plasma triglycerides and leptin, and glucose intolerance (149). Moreover, the probiotic administration inhibited local inflammation in epidydimal adipose tissue and increased the abundance of Bacteroidetes in the gut. In addition, the administration of Lactobacillus curvatus and Lactobacillus plantarum lowered plasma glucose, insulin, triglycerides, and oxidative stress levels in high-fructose diet–fed rats (112). It also reduced liver mass, cholesterol, and lipogenesis while increasing FA β-oxidation.
Probiotics also prevent steatosis and hepatic inflammation through the decrease of reactive oxygen species production and activation of hepatic AMPK (147). In addition, probiotic administration increases the expression of the intestinal tight junction proteins such as claudin-1, ZO-1, and occludin, which leads to reduced translocation of pathogenic bacteria and their products, endotoxin (mainly LPS), into the portal circulation (142). While the probiotic effects on high-fructose conditions are generally limited to liver and intestinal pathologies, they also have a beneficial effect on CVD risk factors and CKDs (32, 71).
On the other hand, prebiotics are predominantly types of fiber that promote the growth of beneficial bacteria in the gut. Studies evaluated the effect of prebiotics on improving health during high fructose consumption (42). Busserolles et al. (13) showed that insulin plasma concentrations were elevated in rats fed a high-fructose diet but not in those supplemented with fructooligosaccharide (FOS). Moreover, FOS lowered plasma leptin levels and triglyceride accumulation in the liver. FOS also increased the growth and functionality of specific bacteria with the enhancement of epithelial integrity, the elaboration of beneficial bacteria-derived antimicrobial agents, and the reduction of pathogenic bacteria (42). Besides FOS, other oligosaccharides have recently gained attention as potential prebiotics (76). For instance, arabinoxylan oligosaccharides increase the SCFA pool size in the large intestine and decrease the risk of various diseases (117, 119). Therefore, identifying the best prebiotics to treat pathological features associated with fructose consumption will be an important future research direction.
5. CONCLUDING REMARKS
Due to the alarmingly increased incidence of metabolic diseases and associated fructose consumption, recent research has focused on the pathophysiological impacts of fructose on various organs and gut microbiota. Such efforts have greatly expanded our understanding of the complex interactions between dietary fructose, organ functions, and disease outcomes. While several molecular and biochemical mechanisms and their influence and pathophysiology have been elucidated using animal models, more clinical studies are required to pinpoint therapeutically targetable pathways for preventing and treating various human diseases associated with fructose consumption. The KHK inhibitors that target host organs hold promise, while more patient-specific personalized medicines that account for interindividual variations in gut microbiota can be another future direction.
ACKNOWLEDGMENTS
We thank all the members of the Jang laboratory for their discussions. This work was funded by grants from the National Research Foundation of Korea to S.J. (2021R1A6A3A-14039681) and H.B. (2021R1A6A3A14039132); the Korea Health Technology R&D Project through the Korea Health Industry Development Institute funded by the Ministry of Health & Welfare of Korea to W.S.S. (HI19C1352); and the AASLD Foundation Pinnacle Research Award in Liver Disease, the Edward Mallinckrodt, Jr. Foundation Award, and grant NIH/NIAAA R01 AA029124 to C.J.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, et al. 2010. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 51:1961–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn IS, Lang JM, Olson CA, Diamante G, Zhang G, et al. 2020. Host genetic background and gut micro-biota contribute to differential metabolic responses to fructose consumption in mice. J. Nutr 150:2716–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andres-Hernando A, Cicerchi C, Kuwabara M, Orlicky DJ, Sanchez-Lozada LG, et al. 2021. Umami-induced obesity and metabolic syndrome is mediated by nucleotide degradation and uric acid generation. Nat. Metab 3:1189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andres-Hernando A, Li N, Cicerchi C, Inaba S, Chen W, et al. 2017. Protective role of fructokinase blockade in the pathogenesis of acute kidney injury in mice. Nat. Commun 8:14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barone S, Fussell SL, Singh AK, Lucas F, Xu J, et al. 2009. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem 284:5056–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartley C, Brun T, Oberhauser L, Grimaldi M, Molica F, et al. 2019. Chronic fructose renders pancreatic β-cells hyper-responsive to glucose-stimulated insulin secretion through extracellular ATP signaling. Am. J. Physiol. Endocrinol. Metab 317:E25–41 [DOI] [PubMed] [Google Scholar]
- 7.Beisner J, Gonzalez-Granda A, Basrai M, Damms-Machado A, Bischoff SC. 2020. Fructose-induced intestinal microbiota shift following two types of short-term high-fructose dietary phases. Nutrients 12:3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bier A, Braun T, Khasbab R, Di Segni A, Grossman E, et al. 2018. A high salt diet modulates the gut microbiota and short chain fatty acids production in a salt-sensitive hypertension rat model. Nutrients 10:1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blakemore SJ, Aledo JC, James J, Campbell FC, Lucocq JM, Hundal HS. 1995. The GLUT5 hexose transporter is also localized to the basolateral membrane of the human jejunum. Biochem. J 309(Part 1):7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanco ABG. 2017. Carbohydrate Metabolism. London: Medical Biochemistry Academic [Google Scholar]
- 11.Brütting C, Lara Bisch M, Brandsch C, Hirche F, Stangl GI. 2021. Impact of dietary propionate on fructose-induced changes in lipid metabolism, gut microbiota and short-chain fatty acids in mice. Int. J. Food Sci. Nutr 72:160–73 [DOI] [PubMed] [Google Scholar]
- 12.Burant CF, Takeda J, Brot-Laroche E, Bell GI, Davidson NO. 1992. Fructose transporter in human spermatozoa and small intestine is GLUT5. J. Biol. Chem 267:14523–26 [PubMed] [Google Scholar]
- 13.Busserolles J, Gueux E, Rock E, Demigné C, Mazur A, Rayssiguier Y. 2003. Oligofructose protects against the hypertriglyceridemic and pro-oxidative effects of a high fructose diet in rats. J. Nutr 133:1903–8 [DOI] [PubMed] [Google Scholar]
- 14.Cani PD, de Vos WM. 2017. Next-generation beneficial microbes: the case of Akkermansia muciniphila. Front. Microbiol 8:1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carreno DV, Corro NB, Cerda-Infante JF, Echeverria CE, Asencio-Barria CA, et al. 2021. Dietary fructose promotes prostate cancer growth. Cancer Res. 81:2824–32 [DOI] [PubMed] [Google Scholar]
- 16.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, et al. 2012. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55:2005–23 [DOI] [PubMed] [Google Scholar]
- 17.Charrez B, Qiao L, Hebbard L. 2015. The role of fructose in metabolism and cancer. Horm. Mol. Biol. Clin. Investig 22:79–89 [DOI] [PubMed] [Google Scholar]
- 18.Cheeseman CI. 1993. GLUT2 is the transporter for fructose across the rat intestinal basolateral membrane. Gastroenterology 105:1050–56 [DOI] [PubMed] [Google Scholar]
- 19.Chen WL, Wang YY, Zhao A, Xia L, Xie G, et al. 2016. Enhanced fructose utilization mediated by SLC2A5 is a unique metabolic feature of acute myeloid leukemia with therapeutic potential. Cancer Cell 30:779–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi YJ, Shin HS, Choi HS, Park JW, Jo I, et al. 2014. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab. Investig 94:1114–25 [DOI] [PubMed] [Google Scholar]
- 21.Coyte KZ, Rakoff-Nahoum S. 2019. Understanding competition and cooperation within the mammalian gut microbiome. Curr. Biol 29:R538–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cozma AI, Sievenpiper JL, de Souza RJ, Chiavaroli L, Ha V, et al. 2012. Effect of fructose on glycemic control in diabetes: a systematic review and meta-analysis of controlled feeding trials. Diabetes Care 35:1611–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui XL, Schlesier AM, Fisher EL, Cerqueira C, Ferraris RP. 2005. Fructose-induced increases in neonatal rat intestinal fructose transport involve the PI3-kinase/Akt signaling pathway. Am. J. Physiol. Gastroin-test. Liver Physiol 288:G1310–20 [DOI] [PubMed] [Google Scholar]
- 24.Cummings BP, Stanhope KL, Graham JL, Evans JL, Baskin DG, et al. 2010. Dietary fructose accelerates the development of diabetes in UCD-T2DM rats: amelioration by the antioxidant, α-lipoic acid. Am. J. Physiol. Regul. Integr. Comp. Physiol 298:R1343–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dahlhamer JM, Zammitti EP, Ward BW, Wheaton AG, Croft JB. 2016. Prevalence of inflammatory bowel disease among adults aged ≥18 years – United States, 2015. MMWR Morb. Mortal. Wkly. Rep 65:1166–69 [DOI] [PubMed] [Google Scholar]
- 26.De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, et al. 2014. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156:84–96 [DOI] [PubMed] [Google Scholar]
- 27.DeBosch BJ, Chen Z, Saben JL, Finck BN, Moley KH. 2014. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J. Biol. Chem 289:10989–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Depommier C, Everard A, Druart C, Plovier H, Van Hul M, et al. 2019. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat. Med 25:1096–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, et al. 2016. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167:1339–53.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhingra R, Sullivan L, Jacques PF, Wang TJ, Fox CS, et al. 2007. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation 116:480–88 [DOI] [PubMed] [Google Scholar]
- 31.Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, et al. 2009. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J. Histochem. Cytochem 57:763–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon A, Robertson K, Yung A, Que M, Randall H, et al. 2020. Efficacy of probiotics in patients of cardiovascular disease risk: a systematic review and meta-analysis. Curr. Hypertens. Rep 22:74. [DOI] [PubMed] [Google Scholar]
- 33.Do MH, Lee E, Oh M-J, Kim Y, Park H-Y. 2018. High-glucose or -fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients 10:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doridot L, Hannou SA, Krawczyk SA, Tong W, Kim M-S, et al. 2021. A systems approach dissociates fructose-induced liver triglyceride from hypertriglyceridemia and hyperinsulinemia in male mice. Nutrients 13:3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dotimas JR, Lee AW, Schmider AB, Carroll SH, Shah A, et al. 2016. Diabetes regulates fructose absorption through thioredoxin-interacting protein. eLife 5:e18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Douard V, Ferraris RP. 2008. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab 295:E227–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drozdowski LA, Thomson AB. 2006. Intestinal sugar transport. World J. Gastroenterol 12:1657–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert M-A, et al. 2013. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62:531–39 [DOI] [PubMed] [Google Scholar]
- 39.Egli L, Lecoultre V, Theytaz F, Campos V, Hodson L, et al. 2013. Exercise prevents fructose-induced hypertriglyceridemia in healthy young subjects. Diabetes 62:2259–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan L, Gao W, Nguyen BV, Jefferson JR, Liu Y, et al. 2020. Impaired renal hemodynamics and glomerular hyperfiltration contribute to hypertension-induced renal injury. Am. J. Physiol. Ren. Physiol 319:F624–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan Y, Pedersen O. 2021. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol 19:55–71 [DOI] [PubMed] [Google Scholar]
- 42.Farias DP, de Araújo FF, Neri-Numa IA, Pastore GM. 2019. Prebiotics: trends in food, health and technological applications. Trends Food Sci. Technol 93:23–35 [Google Scholar]
- 43.Febbraio MA, Karin M. 2021. “Sweet death”: fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. 33:2316–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferreira DF, Fiamoncini J, Prist IH, Ariga SK, de Souza HP, de Lima TM. 2015. Novel role of TLR4 in NAFLD development: modulation of metabolic enzymes expression. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1851:1353–59 [DOI] [PubMed] [Google Scholar]
- 45.Flegal KM, Carroll MD, Ogden CL, Curtin LR. 2010. Prevalence and trends in obesity among US adults, 1999–2008. JAMA 303:235–41 [DOI] [PubMed] [Google Scholar]
- 46.Fu X, Liu Z, Zhu C, Mou H, Kong Q. 2019. Nondigestible carbohydrates, butyrate, and butyrate-producing bacteria. Crit. Rev. Food Sci. Nutr 59:S130–52 [DOI] [PubMed] [Google Scholar]
- 47.Fukui H 2016. Increased intestinal permeability and decreased barrier function: Does it really influence the risk of inflammation? Inflamm. Intest. Dis 1:135–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaby AR. 2005. Adverse effects of dietary fructose. Altern. Med. Rev 10:294–306 [PubMed] [Google Scholar]
- 49.Gersch MS, Mu W, Cirillo P, Reungjui S, Zhang L, et al. 2007. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am. J. Physiol. Ren. Physiol 293:F1256–61 [DOI] [PubMed] [Google Scholar]
- 50.Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, et al. 2019. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363:1345–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gopher A, Vaisman N, Mandel H, Lapidot A. 1990. Determination of fructose metabolic pathways in normal and fructose-intolerant children: a 13C NMR study using [U-13C]fructose. PNAS 87:5449–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goran MI, Ulijaszek SJ, Ventura EE. 2013. High fructose corn syrup and diabetes prevalence: a global perspective. Glob. Public Health 8:55–64 [DOI] [PubMed] [Google Scholar]
- 53.Gouyon F, Caillaud L, Carriere V, Klein C, Dalet V, et al. 2003. Simple-sugar meals target GLUT2 at enterocyte apical membranes to improve sugar absorption: a study in GLUT2-null mice. J. Physiol 552:823–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guynn RW, Veloso D, Veech RL. 1972. The concentration of malonyl-coenzyme A and the control of fatty acid synthesis in vivo. J. Biol. Chem 247:7325–31 [PubMed] [Google Scholar]
- 55.Hall KD, Ayuketah A, Brychta R, Cai H, Cassimatis T, et al. 2019. Ultra-processed diets cause excess calorie intake and weight gain: an inpatient randomized controlled trial of ad libitum food intake. Cell Metab. 30:67–77.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanover LM, White JS. 1993. Manufacturing, composition, and applications of fructose. Am. J. Clin. Nutr 58:724S–32S [DOI] [PubMed] [Google Scholar]
- 57.Harris DS, Slot JW, Geuze HJ, James DE. 1992. Polarized distribution of glucose transporter isoforms in Caco-2 cells. PNAS 89:7556–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayasaki T, Ishimoto T, Doke T, Hirayama A, Soga T, et al. 2019. Fructose increases the activity of sodium hydrogen exchanger in renal proximal tubules that is dependent on ketohexokinase. J. Nutr. Biochem 71:54–62 [DOI] [PubMed] [Google Scholar]
- 59.Heinz F, Lamprecht W, Kirsch J. 1968. Enzymes of fructose metabolism in human liver. J. Clin. Investig 47:1826–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Helwig U, Koch AK, Reichel C, Jessen P, Buning J, et al. 2021. A prospective multicenter study on the prevalence of fructose malabsorption in patients with chronic inflammatory bowel disease. Digestion 102:397–403 [DOI] [PubMed] [Google Scholar]
- 61.Herman MA, Birnbaum MJ. 2021. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. 33:2329–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Higuchi BS, Rodrigues N, Gonzaga MI, Paiolo JCC, Stefanutto N, et al. 2018. Intestinal dysbiosis in autoimmune diabetes is correlated with poor glycemic control and increased interleukin-6: a pilot study. Front. Immunol 9:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsu C-N, Lin Y-J, Hou C-Y, Tain Y-L. 2018. Maternal administration of probiotic or prebiotic prevents male adult rat offspring against developmental programming of hypertension induced by high fructose consumption in pregnancy and lactation. Nutrients 10:1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsu CN, Chang-Chien GP, Lin S, Hou CY, Tain YL. 2019. Targeting on gut microbial metabolite trimethylamine-N-oxide and short-chain fatty acid to prevent maternal high-fructose-diet-induced developmental programming of hypertension in adult male offspring. Mol. Nutr. Food Res 63:e1900073. [DOI] [PubMed] [Google Scholar]
- 65.Hui H, Huang D, McArthur D, Nissen N, Boros LG, Heaney AP. 2009. Direct spectrophotometric determination of serum fructose in pancreatic cancer patients. Pancreas 38:706–12 [DOI] [PubMed] [Google Scholar]
- 66.Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, et al. 2012. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. PNAS 109:4320–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaiswal N, Agrawal S, Agrawal A. 2019. High fructose-induced metabolic changes enhance inflammation in human dendritic cells. Clin. Exp. Immunol 197:237–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, et al. 2018. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 27:351–61.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jang C, Wada S, Yang S, Gosis B, Zeng X, et al. 2020. The small intestine shields the liver from fructose-induced steatosis. Nat. Metab 2:586–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, et al. 2018. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J. Hepatol 68:1063–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jia L, Jia Q, Yang J, Jia R, Zhang H. 2018. Efficacy of probiotics supplementation on chronic kidney disease: a systematic review and meta-analysis. Kidney Blood Press. Res 43:1623–35 [DOI] [PubMed] [Google Scholar]
- 72.Jirillo E, Caccavo D, Magrone T, Piccigallo E, Amati L, et al. 2002. The role of the liver in the response to LPS: experimental and clinical findings. J. Endotoxin Res 8:319–27 [DOI] [PubMed] [Google Scholar]
- 73.Jones N, Blagih J, Zani F, Rees A, Hill DG, et al. 2021. Fructose reprogrammes glutamine-dependent oxidative metabolism to support LPS-induced inflammation. Nat. Commun 12:1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaplan RS, Mayor JA, Johnston N, Oliveira DL. 1990. Purification and characterization of the reconstitutively active tricarboxylate transporter from rat liver mitochondria. J. Biol. Chem 265:13379–85 [PubMed] [Google Scholar]
- 75.Khan S, Waliullah S, Godfrey V, Khan MAW, Ramachandran RA, et al. 2020. Dietary simple sugars alter microbial ecology in the gut and promote colitis in mice. Sci. Transl. Med 12:eaay6218. [DOI] [PubMed] [Google Scholar]
- 76.Khangwal I, Shukla P. 2019. Potential prebiotics and their transmission mechanisms: recent approaches. J. Food Drug Anal 27:649–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim J, Kang J, Kang YL, Woo J, Kim Y, et al. 2020. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat. Commun 11:5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim M, Astapova II, Flier SN, Hannou SA, Doridot L, et al. 2017. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2:e96703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim MS, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, et al. 2016. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig 126:4372–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, et al. 2013. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun 4:1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kinoshita M, Suzuki Y, Saito Y. 2002. Butyrate reduces colonic paracellular permeability by enhancing PPARγ activation. Biochem. Biophys. Res. Commun 293:827–31 [DOI] [PubMed] [Google Scholar]
- 82.Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. 2016. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165:1332–45 [DOI] [PubMed] [Google Scholar]
- 83.Kranhold JF, Loh D, Morris RC Jr. 1969. Renal fructose-metabolizing enzymes: significance in hereditary fructose intolerance. Science 165:402–3 [DOI] [PubMed] [Google Scholar]
- 84.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. 2014. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146:726–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lambertz J, Weiskirchen S, Landert S, Weiskirchen R. 2017. Fructose: a dietary sugar in crosstalk with microbiota contributing to the development and progression of non-alcoholic liver disease. Front. Immunol 8:1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lanaspa MA, Andres-Hernando A, Orlicky DJ, Cicerchi C, Jang C, et al. 2018. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Investig 128:2226–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, et al. 2014. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J. Am. Soc. Nephrol 25:2526–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larsson SC, Bergkvist L, Wolk A. 2006. Consumption of sugar and sugar-sweetened foods and the risk of pancreatic cancer in a prospective study. Am. J. Clin. Nutr 84:1171–76 [DOI] [PubMed] [Google Scholar]
- 89.Li J-M, Yu R, Zhang L-P, Wen S-Y, Wang S-J, et al. 2019. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: a benefit of short-chain fatty acids. Microbiome 7:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li J, Jia H, Cai X, Zhong H, Feng Q, et al. 2014. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol 32:834–41 [DOI] [PubMed] [Google Scholar]
- 91.Liu H, Huang D, McArthur DL, Boros LG, Nissen N, Heaney AP. 2010. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 70:6368–76 [DOI] [PubMed] [Google Scholar]
- 92.Liu J, Zhuang ZJ, Bian DX, Ma XJ, Xun YH, et al. 2014. Toll-like receptor-4 signalling in the progression of non-alcoholic fatty liver disease induced by high-fat and high-fructose diet in mice. Clin. Exp. Pharmacol. Physiol 41:482–88 [DOI] [PubMed] [Google Scholar]
- 93.Liu L, Li T, Liao Y, Wang Y, Gao Y, et al. 2020. Triose kinase controls the lipogenic potential of fructose and dietary tolerance. Cell Metab. 32:605–18.e7 [DOI] [PubMed] [Google Scholar]
- 94.Liu Z, Zaki MH, Vogel P, Gurung P, Finlay BB, et al. 2012. Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem 287:16955–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Louis P, Hold GL, Flint HJ. 2014. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol 12:661–72 [DOI] [PubMed] [Google Scholar]
- 96.Lowette K, Roosen L, Tack J, Vanden Berghe P. 2015. Effects of high-fructose diets on central appetite signaling and cognitive function. Front. Nutr 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu J, Hou X, Yuan X, Cui L, Liu Z, et al. 2018. Knockout of the urate oxidase gene provides a stable mouse model of hyperuricemia associated with metabolic disorders. Kidney Int. 93:69–80 [DOI] [PubMed] [Google Scholar]
- 98.Mai BH, Yan LJ. 2019. The negative and detrimental effects of high fructose on the liver, with special reference to metabolic disorders. Diabetes Metab. Syndr. Obes 12:821–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Malik VS, Popkin BM, Bray GA, Despres JP, Willett WC, Hu FB. 2010. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care 33:2477–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Marriott BP, Cole N, Lee E. 2009. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr 139:1228S–35S [DOI] [PubMed] [Google Scholar]
- 101.McLarty JL, Marsh SA, Chatham JC. 2013. Post-translational protein modification by O-linked N-acetyl-glucosamine: its role in mediating the adverse effects of diabetes on the heart. Life Sci. 92:621–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mellor KM, Bell JR, Wendt IR, Davidoff AJ, Ritchie RH, Delbridge LM. 2011. Fructose modulates cardiomyocyte excitation-contraction coupling and Ca2+ handling in vitro. PLOS ONE 6:e25204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mellor KM, Ritchie RH, Davidoff AJ, Delbridge LM. 2010. Elevated dietary sugar and the heart: experimental models and myocardial remodeling. Can. J. Physiol. Pharmacol 88:525–40 [DOI] [PubMed] [Google Scholar]
- 104.Michaud DS, Liu S, Giovannucci E, Willett WC, Colditz GA, Fuchs CS. 2002. Dietary sugar, glycemic load, and pancreatic cancer risk in a prospective study. J. Natl. Cancer Inst 94:1293–300 [DOI] [PubMed] [Google Scholar]
- 105.Mirmonsef P, Zariffard MR, Gilbert D, Makinde H, Landay AL, Spear GT. 2012. Short-chain fatty acids induce pro-inflammatory cytokine production alone and in combination with Toll-like receptor ligands. Am. J. Reprod. Immunol 67:391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mirtschink P, Krishnan J, Grimm F, Sarre A, Horl M, et al. 2015. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 522:444–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Montrose DC, Nishiguchi R, Basu S, Staab HA, Zhou XK, et al. 2021. Dietary fructose alters the composition, localization, and metabolism of gut microbiota in association with worsening colitis. Cell. Mol. Gastroenterol. Hepatol 11:525–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nishiguchi R, Basu S, Staab HA, Ito N, Zhou XK, et al. 2021. Dietary interventions to prevent high-fructose diet-associated worsening of colitis and colitis-associated tumorigenesis in mice. Carcinogenesis 42:842–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Olsen NJ, Heitmann BL. 2009. Intake of calorically sweetened beverages and obesity. Obes. Rev 10:68–75 [DOI] [PubMed] [Google Scholar]
- 110.Oppelt SA, Sennott EM, Tolan DR. 2015. Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Mol. Genet. Metab 114:445–50 [DOI] [PubMed] [Google Scholar]
- 111.Ottman N, Reunanen J, Meijerink M, Pietilä TE, Kainulainen V, et al. 2017. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLOS ONE 12:e0173004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Park D-Y, Ahn Y-T, Huh C-S, McGregor RA, Choi M-S. 2013. Dual probiotic strains suppress high fructose-induced metabolic syndrome. World J. Gastroenterol 19:274–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Park YK, Yetley EA. 1993. Intakes and food sources of fructose in the United States. Am. J. Clin. Nutr 58:737S–47S [DOI] [PubMed] [Google Scholar]
- 114.Patel C, Douard V, Yu S, Tharabenjasin P, Gao N, Ferraris RP. 2015. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am. J. Physiol. Regul. Integr. Comp. Physiol 309:R499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Payne AN, Chassard C, Lacroix C. 2012. Gut microbial adaptation to dietary consumption of fructose, artificial sweeteners and sugar alcohols: implications for host–microbe interactions contributing to obesity. Obes. Rev 13:799–809 [DOI] [PubMed] [Google Scholar]
- 116.Peng L, Li Z-R, Green RS, Holzman IR, Lin J. 2009. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr 139:1619–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pereira GV, Abdel-Hamid AM, Dutta S, D’Alessandro-Gabazza CN, Wefers D, et al. 2021. Degradation of complex arabinoxylans by human colonic Bacteroidetes. Nat. Commun 12:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Perrar I, Buyken AE, Penczynski KJ, Remer T, Kuhnle GG, et al. 2021. Relevance of fructose intake in adolescence for fatty liver indices in young adulthood. Eur. J. Nutr 60:3029–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rivière A, Gagnon M, Weckx S, Roy D, De Vuyst L. 2015. Mutual cross-feeding interactions between Bifidobacterium longum subsp. longum NCC2705 and Eubacterium rectale ATCC 33656 explain the bifidogenic and butyrogenic effects of arabinoxylan oligosaccharides. Appl. Environ. Microbiol 81:7767–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rodenbach KE, Schneider MF, Furth SL, Moxey-Mims MM, Mitsnefes MM, et al. 2015. Hyperuricemia and progression of CKD in children and adolescents: the Chronic Kidney Disease in Children (CKiD) cohort study. Am. J. Kidney Dis 66:984–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Roncal-Jimenez CA, Ishimoto T, Lanaspa MA, Milagres T, Hernando AA, et al. 2016. Aging-associated renal disease in mice is fructokinase dependent. Am. J. Physiol. Ren. Physiol 311:F722–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rumessen JJ, Gudmand-Hoyer E. 1986. Absorption capacity of fructose in healthy adults. Comparison with sucrose and its constituent monosaccharides. Gut 27:1161–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Salguero MV, Al-Obaide MAI, Singh R, Siepmann T, Vasylyeva TL. 2019. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med 18:3461–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sánchez B, Delgado S, Blanco-Míguez A, Lourenço A, Gueimonde M, Margolles A. 2017. Probiotics, gut microbiota, and their influence on host health and disease. Mol. Nutr. Food Res 61:1600240. [DOI] [PubMed] [Google Scholar]
- 125.Sanders ME, Merenstein DJ, Reid G, Gibson GR, Rastall RA. 2019. Probiotics and prebiotics in intestinal health and disease: from biology to the clinic. Nat. Rev. Gastroenterol. Hepatol 16:605–16 [DOI] [PubMed] [Google Scholar]
- 126.Schernhammer ES, Hu FB, Giovannucci E, Michaud DS, Colditz GA, et al. 2005. Sugar-sweetened soft drink consumption and risk of pancreatic cancer in two prospective cohorts. Cancer Epidemiol. Biomarkers Prev 14:2098–105 [DOI] [PubMed] [Google Scholar]
- 127.Schiattarella GG, Sannino A, Toscano E, Giugliano G, Gargiulo G, et al. 2017. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. Eur. Heart J 38:2948–56 [DOI] [PubMed] [Google Scholar]
- 128.Schulze MB, Manson JE, Ludwig DS, Colditz GA, Stampfer MJ, et al. 2004. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 292:927–34 [DOI] [PubMed] [Google Scholar]
- 129.Shoham DA, Durazo-Arvizu R, Kramer H, Luke A, Vupputuri S, et al. 2008. Sugary soda consumption and albuminuria: results from the National Health and Nutrition Examination Survey, 1999–2004. PLOS ONE 3:e3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shuster J, Jenkins A, Logan C, Barnett T, Riehle R, et al. 1992. Soft drink consumption and urinary stone recurrence: a randomized prevention trial. J. Clin. Epidemiol 45:911–16 [DOI] [PubMed] [Google Scholar]
- 131.Softic S, Cohen DE, Kahn CR. 2016. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci 61:1282–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Song M 2019. Dietary fructose induced gut microbiota dysbiosis is an early event in the onset of metabolic phenotype. FASEB J. 33(S1):723.2 [Google Scholar]
- 133.Sprinz C, Altmayer S, Zanon M, Watte G, Irion K, et al. 2018. Effects of blood glucose level on 18F-FDG uptake for PET/CT in normal organs: a systematic review. PLOS ONE 13:e0193140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Takasaki Y 1966. Studies on sugar-isomerizing enzyme. Agric. Biol. Chem 30:1247–53 [Google Scholar]
- 135.Taylor EN, Curhan GC. 2008. Fructose consumption and the risk of kidney stones. Kidney Int. 73:207–12 [DOI] [PubMed] [Google Scholar]
- 136.Taylor SR, Ramsamooj S, Liang RJ, Katti A, Pozovskiy R, et al. 2021. Dietary fructose improves intestinal cell survival and nutrient absorption. Nature 597:263–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thirunavukkarasu V, Anitha Nandhini AT, Anuradha CV. 2004. Effect of α-lipoic acid on lipid profile in rats fed a high-fructose diet. Exp. Diabesity Res 5:195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ulevitch RJ, Tobias PS. 1999. Recognition of Gram-negative bacteria and endotoxin by the innate immune system. Curr. Opin. Immunol 11:19–22 [DOI] [PubMed] [Google Scholar]
- 139.Utzschneider KM, Kahn SE. 2006. The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab 91:4753–61 [DOI] [PubMed] [Google Scholar]
- 140.van der Beek CM, Dejong CHC, Troost FJ, Masclee AAM, Lenaerts K. 2017. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr. Rev 75:286–305 [DOI] [PubMed] [Google Scholar]
- 141.Volynets V, Spruss A, Kanuri G, Wagnerberger S, Bischoff SC, Bergheim I. 2010. Protective effect of bile acids on the onset of fructose-induced hepatic steatosis in mice. J. Lipid Res 51:3414–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang Y, Kirpich I, Liu Y, Ma Z, Barve S, et al. 2011. Lactobacillus rhamnosus GG treatment potentiates intestinal hypoxia-inducible factor, promotes intestinal integrity and ameliorates alcohol-induced liver injury. Am. J. Pathol 179:2866–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Y, Qi W, Song G, Pang S, Peng Z, et al. 2020. High-fructose diet increases inflammatory cytokines and alters gut microbiota composition in rats. Mediat. Inflamm 2020:6672636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Weng Y, Fan X, Bai Y, Wang S, Huang H, et al. 2018. SLC2A5 promotes lung adenocarcinoma cell growth and metastasis by enhancing fructose utilization. Cell Death Discov. 4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.White JS. 2008. Straight talk about high-fructose corn syrup: what it is and what it ain’t. Am. J. Clin. Nutr 88:1716S–21S [DOI] [PubMed] [Google Scholar]
- 146.Yki-Jarvinen H, Luukkonen PK, Hodson L, Moore JB. 2021. Dietary carbohydrates and fats in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 18:770–86 [DOI] [PubMed] [Google Scholar]
- 147.Zhang M, Wang C, Wang C, Zhao H, Zhao C, et al. 2015. Enhanced AMPK phosphorylation contributes to the beneficial effects of Lactobacillus rhamnosus GG supernatant on chronic-alcohol-induced fatty liver disease. J. Nutr. Biochem 26:337–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhao S, Jang C, Liu J, Uehara K, Gilbert M, et al. 2020. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579:586–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zubiría MG, Gambaro SE, Rey MA, Carasi P, Serradell MdLÁ, Giovambattista A. 2017. Deleterious metabolic effects of high fructose intake: the preventive effect of Lactobacillus kefiri administration. Nutrients 9:470. [DOI] [PMC free article] [PubMed] [Google Scholar]