

SUMMARY
Tissue-resident memory CD8+ T (TRM) cells are a subset of memory T cells that play a critical role in limiting early pathogen spread and controlling infection. TRM cells exhibit differences across tissues, but their potential heterogeneity among distinct anatomic compartments within the small intestine and colon has not been well-recognized. Here, by analyzing TRM cells from the lamina propria and epithelial compartments of the small intestine and colon, we showed that intestinal TRM cells exhibited distinctive patterns of cytokine and granzyme expression, along with substantial transcriptional, epigenetic, and functional heterogeneity. The T-box transcription factor Eomes, which represses TRM cell formation in some tissues, exhibited unexpected context-specific regulatory roles in supporting the maintenance of established TRM cells in the small intestine, but not in the colon. Taken together, these data provide previously unappreciated insights into the heterogeneity and differential requirements for the formation vs. maintenance of intestinal TRM cells.
Keywords: Tissue-resident memory CD8+ T cells, small intestine, colon, single-cell RNA-sequencing, single-cell ATAC-sequencing, Eomes
eTOC Blurb
The tissue- and context-specific regulation of tissue-resident memory T (TRM) cells, which provide protection in organs and at barrier sites, is an emerging concept. Lin, Duong, Limary, Kim, et al. show that CD8+ TRM cells from SI vs. colon exhibit unique molecular and functional attributes along with distinct transcriptional requirements for their maintenance.
Graphical Abstract
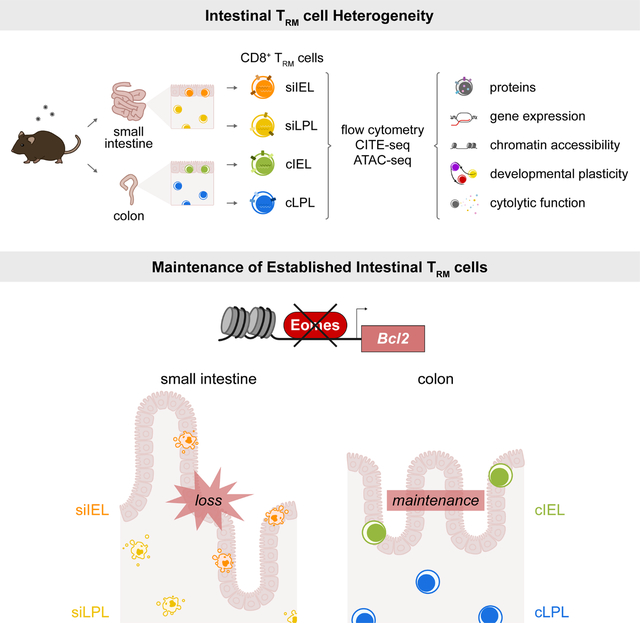
INTRODUCTION
During a microbial infection, CD8+ T cells give rise to short-lived effector cells that provide acute host defense and long-lived memory cells that provide sustained protection1. Memory CD8+ T cells can be broadly classified as circulating and tissue-resident memory (TRM) cells, with circulating memory T cells further subdivided into central memory (TCM), effector memory (TEM), terminal effector memory (t-TEM), and peripheral memory (TPM) cells, based on distinct phenotypic, homeostatic, functional, and migratory properties2–4. TRM cells are so named because they tend to ‘reside’ in the tissue in which they form, though it is now clear that TRM cells can egress into the circulation after reactivation5,6. TRM cells are a distinct component of the memory T cell compartment that play a crucial role in host defense against microbial pathogens, particularly infections at barrier sites, owing to their immediate effector capabilities and positioning in tissues7–10. Moreover, cells with TRM-like characteristics play crucial roles in mediating immune responses against tumors11–13. Conversely, T cells exhibiting a TRM-like phenotype can mediate pathology in autoimmune and inflammatory disorders14–17.
Early studies sought to identify shared features of TRM cells distinct from circulating memory cells, including increased expression of molecules necessary for trafficking to and retention in tissues, such as CD69 and αEβ7 integrin; decreased expression of molecules that promote egress to the circulation, such as the chemokine receptor CCR7, the receptor for sphingosine 1-phosphate (S1PR1), and the transcription factor KLF29,18,19; and induction of transcription factors that establish the tissue residency program, including Blimp1, Hobit, and Runx311,20. However, substantial transcriptional and functional heterogeneity exists among TRM cells derived from different tissues5,21–23, with TRM cells from disparate tissues exhibiting unique requirements for their formation. For example, Hobit is required for TRM cell formation in the skin, liver, and small intestine intraepithelial (siIEL) and lamina propria (siLPL) compartments20, but not in the lung24. IL-15 signaling is required for the formation of TRM cells in the skin, liver, salivary gland, and kidney, but not in the pancreas, female reproductive tract, or the siIEL and siLPL compartments18,25–27.
While the signals required for TRM cell formation are a focus of current research, the requirements for the maintenance of established TRM cells are less understood. The costimulatory molecule ICOS promotes the formation of TRM cells in the kidney, salivary gland, and siIEL and siLPL compartments, but is not required for their maintenance28. This illustrates the underappreciated concept that factors required for TRM cell formation may be distinct from those required for their maintenance. Moreover, the mechanisms underlying TRM cell formation and maintenance may be distinct across tissues. Lastly, it should be emphasized that conclusions based on siIEL TRM cells, which have been studied extensively, are typically generalized to all intestinal TRM cells, even though there is a paucity of work involving siLPL TRM cells20,28–30. Moreover, TRM cells from the colon IEL (cIEL) and LPL (cLPL) compartments have not been previously investigated in depth.
Here we performed an extensive comparative analysis of TRM cells from the siIEL, siLPL, cIEL and cLPL intestinal tissue compartments. Intestinal TRM cells exhibited heterogeneity in their expression of the widely used TRM cell markers CD69 and CD103, along with distinctive patterns of cytokine and granzyme expression. For example, siIEL TRM cells expressed the highest levels of GzmA and GzmB, whereas cIEL TRM cells exhibited the greatest capacity for IFNγ and TNF production. Furthermore, our studies revealed substantial transcriptional, epigenetic, and functional heterogeneity among TRM cells from the siIEL, siLPL, ciIEL, and cLPL compartments. We elucidated unexpected tissue- and context-specific regulatory roles for the T-box transcription factor Eomesodermin (Eomes), previously known to repress TRM cell formation in the skin, liver, and kidney31,32. We ascertained a role for Eomes in supporting the maintenance of established TRM cells in the small intestine (SI), but not in the colon, in part by inducing the anti-apoptotic molecule Bcl-2. Taken together, these data provide previously unappreciated insights into the heterogeneity and differential requirements for the formation vs. maintenance of TRM cells from the SI and colon.
RESULTS
Phenotypic and functional heterogeneity among CD8+ TRM cells in the SI and colon.
The current understanding of ‘intestinal’ CD8+ TRM cells is primarily focused on those that form in the siIEL compartment, even though TRM cells also form in the siLPL29,30, cIEL, and cLPL compartments33. Thus, in order to directly compare CD8+ TRM cells from each of these four intestinal tissue compartments under the same experimental conditions, CD8+CD45.1+ P14 T cells, which have transgenic expression of a T cell receptor (TCR) that recognizes an immunodominant epitope of lymphocytic choriomeningitis virus (LCMV), were adoptively transferred into congenic CD45.2+ recipients that were subsequently infected intraperitoneally (i.p.) with the Armstrong strain of LCMV (Figure 1A). We analyzed donor P14 T cells from the spleens and the four intestinal tissue compartments of recipient mice after 21 days post-infection. To exclude circulating cells, anti-CD8α antibodies were injected intravenously (i.v.) into recipient mice 3–5 minutes prior to sacrifice and only ‘i.v.-negative’ cells were considered tissue-resident for subsequent downstream analyses34. We observed that siIEL and siLPL TRM cells were the most abundant intestinal TRM cells, followed by cLPL TRM cells, with cIEL TRM cells representing the least abundant intestinal TRM cells (Figures 1B and S1A), consistent with previously published results using a microscopy-based quantification method33.
Figure 1. CD8+ TRM cells in SI and colon express distinct levels of CD69 and CD103.
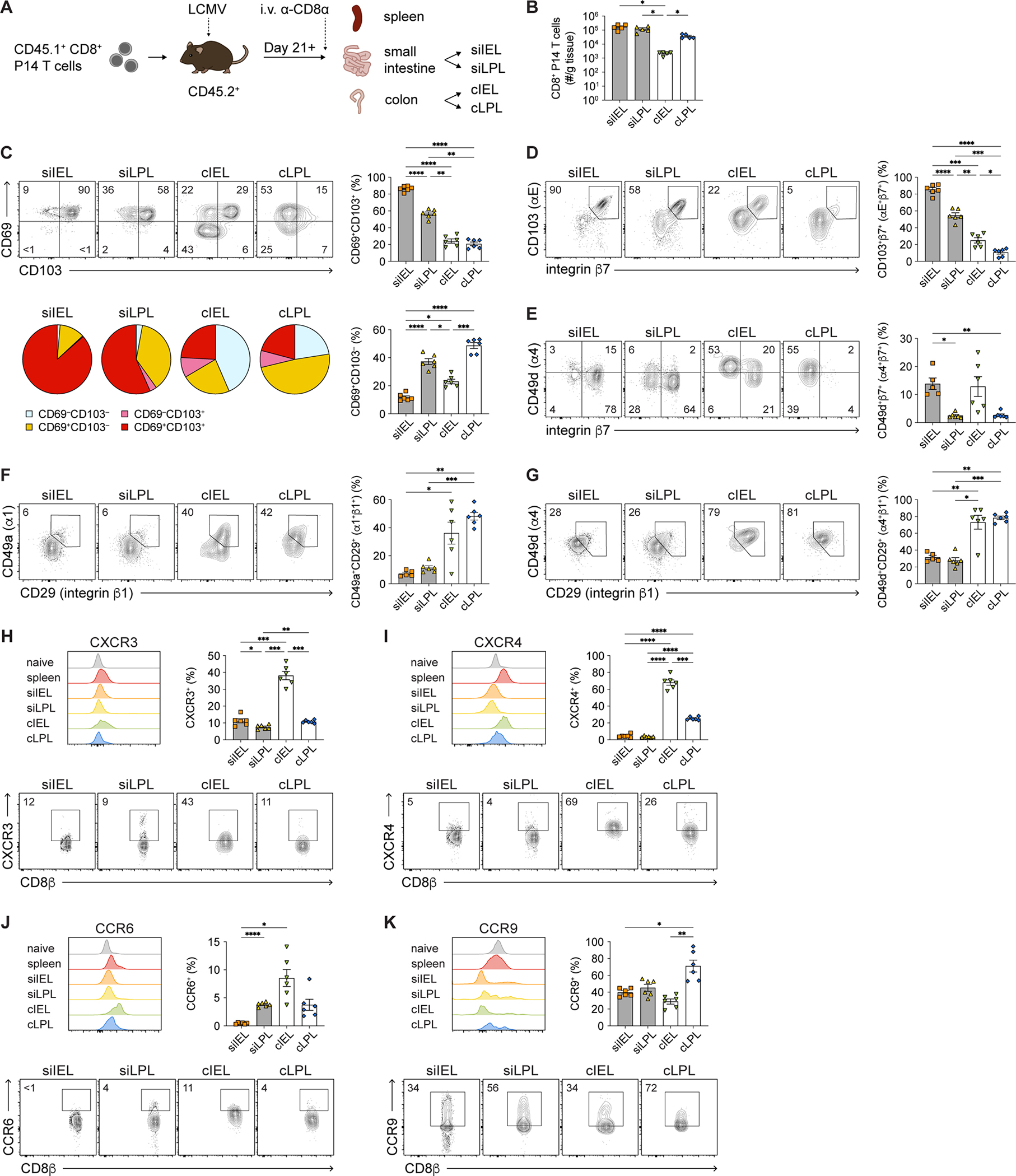
(A) Experimental design. Spleen and intestinal tissue compartments were isolated from CD45.2+ recipient mice ≥ 21 days after LCMV infection following adoptive transfer of donor CD8+CD45.1+ P14 T cells.
(B) Numbers of i.v.− intestinal P14 T cells, normalized to organ weights.
(C) Representative flow cytometry plots showing CD69 and CD103 expression (top left) by i.v.− intestinal P14 T cells. Frequencies (right) of intestinal CD69+CD103+ and CD69+CD103− P14 T cells. Distribution of intestinal P14 T cells expressing CD69 and/or CD103 (bottom left).
(D–G) Representative flow cytometry plots showing expression of CD103/integrin β7 (D), CD49d/integrin β7 (E), CD49a/CD29 (F), CD49d/CD29 (G) among i.v.− intestinal P14 T cells (left). Quantification of indicated integrin heterodimer expression among P14 T cells (right).
(H–K) Representative flow cytometry plots (bottom) showing expression of CXCR3 (H), CXCR4 (I), CCR6 (J), and CCR9 (K) among i.v.− intestinal P14 T cells. Frequencies of cells expressing each molecule (top right) and representative histograms (top left) indicating the distribution of expression for each molecule; expression by naïve (CD62LhiCD44lo) CD8+ T cells from a separate uninfected mouse is shown for comparison. Data are represented as mean ± SEM. Repeated measures one-way ANOVA. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001.
Data are representative of ≥3 independent experiments with n=5–6 mice per experiment.
See also Figure S1.
In addition to being a marker of early T cell activation, CD69 is often considered a marker for TRM cells in many tissues, whereas CD103 is expressed by TRM cells only in certain tissues with an epithelial component, such as in the skin and siIEL compartment7,9. In agreement with previously published data9, we observed that the vast majority of siIEL CD8+ TRM cells expressed high levels of both CD69 and CD103 (Figure 1C). By contrast, we observed that CD8+ TRM cells in the other three intestinal compartments were highly heterogeneous with respect to CD69 and CD103 expression. Approximately 60% of siLPL TRM cells, and far fewer ciIEL and cLPL TRM cells, expressed both CD69 and CD103 (Figure 1C); most siLPL and cLPL TRM cells expressed only CD69, and a proportion of cIEL and cLPL TRM cells expressed neither CD69 nor CD103 (Figure 1C). Since CD103 (integrin αE) can pair with different β integrins, we examined the expression of several integrin heterodimers and chemokine receptors. The expression patterns of integrin αEβ7 (CD103/β7) by TRM cells paralleled those observed for CD103 alone, with the highest levels in the siIEL compartment and progressively lower levels in the siLPL, cIEL, and cLPL compartments (Figure 1D), indicating that the levels of CD103 observed indeed reflected αEβ7 integrin expression. Few TRM cells expressed integrin α4β7 (CD49d/β7), but a greater proportion of IEL TRM cells expressed α4β7 integrin compared to LPL TRM cells (Figure 1E). A larger proportion of colon TRM cells expressed α1β1 (CD49a/β1) and α4β1 (CD49d/β1) integrins compared to SI TRM cells (Figures 1F and 1G). Compared to TRM cells in the other intestinal tissue compartments, a higher proportion of cIEL TRM cells tended to express CXCR3, CXCR4, and CCR6, and exhibited higher levels of these molecules on a per-cell basis (Figures 1H–1J). Compared to naïve and circulating memory T cells, intestinal TRM cells expressed lower levels of CCR9, but a greater proportion of cLPL TRM cells expressed CCR9 compared to TRM cells from the other intestinal compartments (Figure 1K).
Having established that TRM cells from the four intestinal tissue compartments exhibited phenotypic heterogeneity, we next sought to determine whether intestinal TRM cells demonstrated functional differences. Most siIEL TRM cells expressed the highest levels of GzmA and GzmB on a per-cell basis (Figures 2A and 2B). Small intestine LPL and cLPL TRM cells expressed low levels of GzmA but higher levels of GzmB, whereas cIEL TRM cells expressed relatively low levels of both granzymes. Few TRM cells were capable of IL-2 production, but TRM cells from all four intestinal compartments were capable of producing TNF, with the highest per-cell amounts produced by cIEL TRM cells (Figures 2C and 2D). TRM cells from the IEL compartments tended to produce higher levels of IFNγ compared to LPL TRM cells, with the highest levels of IFNγ produced by cIEL TRM cells on a per-cell basis; moreover, the vast majority of cIEL TRM cells were capable of producing IFNγ (Figure 2E). Colon IEL TRM cells were capable of the greatest polyfunctionality, followed by siIEL TRM cells (Figure 2F). This functional heterogeneity among intestinal TRM cells did not appear to be primarily driven by differences in CD69 and CD103 expression (Figures S1B–G). Lastly, we observed that siIEL and siLPL TRM cells exhibited greater longevity than cIEL and cLPL TRM cells, comparable to that of TCM cells (Figure 2G).
Figure 2. Small intestine IEL CD8+ TRM cells express high levels of granzymes whereas cIEL TRM cells exhibit high potential for cytokine production.

(A–E) Histograms (top left), bar graphs (top right), and representative flow cytometry plots (bottom) showing expression of GzmA (A), GzmB (B), IL-2 (C), TNF (D), and IFNγ (E) by i.v.− intestinal P14 T cells.
(F) Proportions of intestinal T cells expressing 0, 1, 2, or 3 cytokines.
(G) Relative change in numbers of intestinal P14 T cells between days 21 and 80 post-infection, normalized to TCM cells.
(H–J) Histograms (top left), bar graphs (top right), and representative flow cytometry plots (bottom) showing expression of CD127 (H), CD122 (I), and CD27 (J) by intestinal P14 T cells.
Data are represented as mean ± SEM. Repeated measures one-way ANOVA. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001. Data are representative of ≥3 independent experiments with n=5–6 mice per experiment.
See also Figure S1.
We next examined levels of cytokine and signaling receptors that have been previously associated with circulating and tissue-resident memory CD8+ T cells1. TRM cells from all four intestinal tissue compartments expressed similar levels of CD127 (IL-7Rα) (Figure 2H). By contrast, a higher proportion of colon TRM cells expressed CD122 (IL-2Rβ) compared to SI TRM cells; among all intestinal TRM cells, cIEL TRM cells exhibited the highest proportions expressing CD122 (Figure 2I). CD27, a costimulatory receptor that has been associated with circulating memory T cells, was expressed at lower levels by intestinal TRM cells compared to circulating memory cells; however, among intestinal TRM cells, a higher proportion of cIEL TRM cells expressed CD27 compared to cells from the other three compartments (Figure 2J). Taken together, these results indicated that TRM cells from the four intestinal compartments exhibit previously unappreciated phenotypic and functional heterogeneity.
Single-cell transcriptomic, epigenetic, and protein profiling reveal potential regulators of intestinal TRM cell heterogeneity.
To elucidate inter- and intra-intestinal TRM cell heterogeneity, we performed Cellular Indexing of Transcriptomes and Epitopes (CITE-seq), which enables measurement of proteins and the transcriptome at the single-cell level35. CD45.1+ P14 T cells were adoptively transferred into CD45.2+ recipients prior to infection with LCMV. After 30 days post-infection, P14 T cells from each intestinal tissue compartment were isolated by FACS and processed for CITE-seq using the 10x Genomics platform. Antibodies targeting 50 proteins, some of which have previously implicated in CD8+ T cell activation and differentiation, were selected for inclusion in the CITE-seq antibody panel (STAR Methods).
Uniform Manifold Approximation and Projection (UMAP) analyses revealed that while TRM cells from each intestinal tissue compartment clustered distinctly, siIEL and siLPL TRM cells clustered more closely together, whereas cIEL and cLPL TRM cells clustered more closely together (Figure S2A). We also observed additional heterogeneity within and among TRM cells from each intestinal tissue compartment; intestinal TRM cells separated into 15 clusters exhibiting disparate gene expression patterns (Figures S2A–S2C, Tables S1 and S2), with colon TRM cells predominantly contained within clusters 4, 5, 6, and 14, and SI TRM cells found within the remaining clusters. Pathway analyses36 of the clusters revealed that certain pathways, such as TCR signaling, were enriched in TRM cells across all clusters, while others, such as pathways regulating the cell cycle, were preferentially enriched in TRM cells from certain clusters (Figure S2D, Table S2).
Next we analyzed TRM cells from each of the four intestinal tissues as ‘pseudo-bulk’ samples in order to uncover the most substantial differences among TRM cells from each intestinal tissue compartment. Intestinal TRM cells exhibited numerous transcriptional disparities, with siIEL, siLPL, cIEL, and cLPL TRM cells exhibiting 605, 314, 754, and 723 differentially expressed genes, respectively (Figures 3A and 3B, Table S3). Focusing next on specific genes, we observed that SI TRM cells expressed higher levels of cytolytic molecules and certain surface receptors such as Cd160 (Figure 3B). By contrast, colon TRM cells expressed higher levels of transcripts encoding for killer lectin receptors and a number of surface receptors, including Il18r1, Ly6c, Il6ra, Ifngr1, and Slamf6 (Figure 3B). With respect to transcription factors, SI TRM cells expressed higher levels of factors previously implicated in siIEL TRM cell differentiation, such as Ahr, Prdm1 (Blimp1), Runx3, P2rx7, and Zfp683 (Hobit)11,20,37–39; conversely, colon TRM cells expressed higher levels of transcription factors previously associated with circulating memory T cells, including Tcf7 and Lef140,41. Compared to IEL TRM cells, LPL TRM cells tended to express higher levels of Areg and Rorc, while cIEL TRM cells expressed the highest levels of several transcription factors, including Bach2 (Figure 3B). Colon IEL TRM cells also expressed the highest levels of Eomes, an unexpected finding since the expression of Eomes has been previously shown to be extinguished during the differentiation of skin TRM cells31.
Figure 3. Colon CD8+ TRM cells express higher levels of Eomes than SI TRM cells.
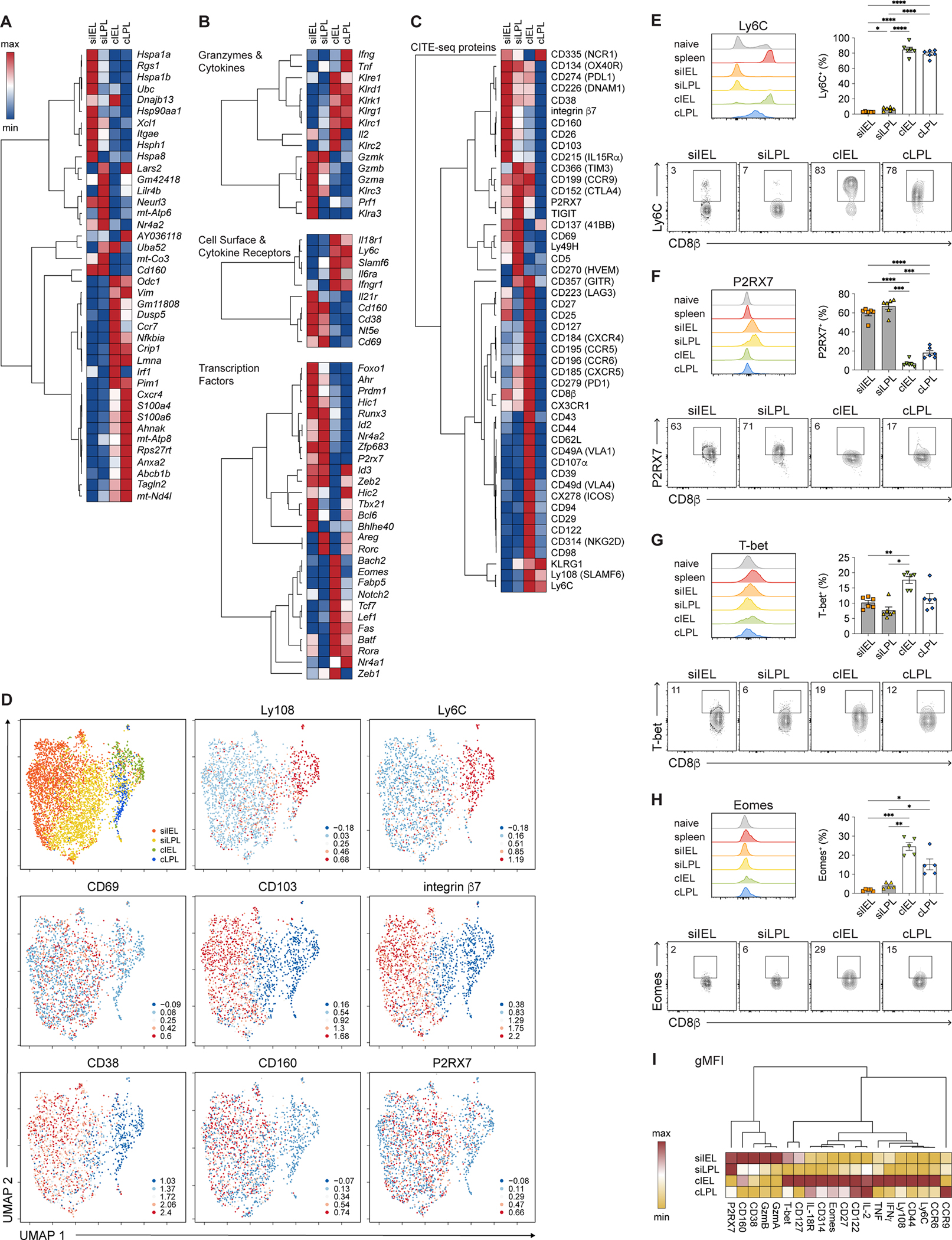
(A-C) Hierarchically clustered summary heatmaps, derived from CITE-seq data, representing top ten genes differentially expressed among intestinal TRM cells (A); relative expression of selected genes, divided by category (B); or relative expression of all proteins included in the CITE-seq antibody panel (C). Rows represent scaled expression of individual genes (A, B) or proteins (C); columns represent TRM cells from each of the 4 intestinal tissue compartments. Values are mapped to colors using the minimum and maximum of each row.
(D) UMAP plots colored by tissue compartment (top left) or expression of selected proteins superimposed onto individual cells.
(E–H) Histograms (top left), bar graphs (top right), and representative flow cytometry plots (bottom) showing expression of Ly6C (E), P2RX7 (F), T-bet (G), and Eomes (H) among intestinal P14 T cells.
(I) Geometric mean fluorescence intensity (gMFI) of expression of selected proteins by intestinal TRM cells, derived from flow cytometry analyses from Figures 2A–2E, 2H–2J, 3E–3H, and S3C–H, represented as a summary heatmap. Values are mapped to colors using the minimum and maximum of each row.
Data are represented as mean ± SEM. Repeated measures one-way ANOVA. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001. Data are representative of ≥3 independent experiments with n=5–6 mice per experiment.
See also Figures S2–S4 and Table S3.
Analyses of protein expression from the CITE-seq antibody panel (Figures 3C and 3D) along with independent flow cytometry experiments (Figures 3E–3I and S3C–S3H) confirmed some of the key findings observed at the transcriptional level. For example, colon TRM cells expressed higher levels of proteins such as Ly108 (SLAMF6) and Ly6C, whereas SI TRM cells expressed higher levels of proteins such as CD38, CD160, and P2RX7. Small intestine IEL TRM cells expressed the highest levels of CD103 and integrin β7; siLPL TRM cells expressed the highest levels of TIGIT, CD5, and CD270 (HVEM); and cIEL TRM cells expressed the highest levels of several proteins, such as CD314 (NKG2D) and CD44. The T-box transcription factor T-bet was detectable at low levels in intestinal TRM cells (Figure 3G). Eomes was detectable in all intestinal TRM cells (Figure 3H), but expressed by a higher proportion of colon TRM cells compared to SI TRM cells; among colon TRM cells, a higher proportion of cIEL TRM cells expressed Eomes protein compared to cLPL TRM cells, consistent with the transcriptional data (Figure 3B).
To investigate whether the transcriptional disparities among TRM cells from the four intestinal compartments were accompanied by epigenetic heterogeneity, we performed the assay for transposase-accessible chromatin with high-throughput sequencing at the single-cell level (scATAC-seq). UMAP analyses revealed that intestinal CD8+ TRM cells clustered distinctly based on differential chromatin accessibility (Figures S4A and S4B, Table S4). Additional analyses revealed 39, 23, 110, and 257 transcription factor binding motifs that were preferentially enriched within differentially accessible chromatin peaks in siIEL, siLPL, cIEL, and cLPL TRM cells, respectively (Figure S4C, Tables S5 and S6). For example, motifs for Ahr and basic helix-loop-helix family transcription factors, such as Hes1, Hes2, and Bhlhe40, were enriched in siIEL TRM cells (Figure S4D, Table S6), while motifs for IRF family members (Irf2, Irf3, Irf7, Irf8) were enriched in siLPL TRM cells (Figure S4E). Moreover, motifs for ROR/RAR family members, including Rora and Rarb, were enriched in cLPL TRM cells (Figure S4G), whereas motifs for a number of T-box transcription factor family members, including Tbx21 and Eomes, were enriched in ciIEL TRM cells (Figure S4F). We also identified transcription factor binding motifs that were shared between TRM cells from different intestinal tissue compartments, such as Runx3 for siIEL and cIEL TRM cells, Nfatc1 for siLPL and cLPL TRM cells, and Hic1 for cIEL and cLPL TRM cells (Figures S4H–S4J). Taken together, these findings reveal previously unappreciated phenotypic, transcriptional, and epigenetic heterogeneity among intestinal TRM cells.
Small intestine IEL and LPL TRM cells may exhibit differences in developmental plasticity.
We next sought to determine whether TRM cells from the intestinal tissue compartments exhibited disparities in developmental plasticity, as has been reported for TRM cells from other tissues21,22. Using the single-cell RNA-seq data, we first inferred the differentiation trajectories of siIEL and siLPL TRM cells by applying scVelo, a previously published framework to analyze transcriptional dynamics of splicing kinetics using a likelihood-based dynamical model42,43. These analyses predicted that although most siIEL and siLPL TRM cells followed similar differentiation paths, several subpopulations of siLPL TRM cells clustered distinctly from siIEL TRM cells (Figure 4A). These clusters of siLPL TRM cells were projected later in ‘latent time,’ an approximation of real time experienced by cells as they undergo differentiation (Figure 4B). These results raised the possibility that these siLPL TRM cell clusters may be more differentiated and exhibit less developmental plasticity than siIEL TRM cells, though it should be noted that trajectories, velocities, and pseudotime distributions inferred by scVelo and other single-cell trajectory inference methods may not always be aligned with real time. Thus, to test this prediction experimentally, CD45.1+ P14 T cells were adoptively transferred into CD45.2+ recipients subsequently infected with LCMV (Figure 4C). After day 21 post-infection, CD45.1+ P14 T cells from the siIEL or siLPL compartments (‘ex-siIEL’ or ‘ex-siLPL’) were FACS-purified and adoptively transferred into separate naïve CD45.2+ recipients subsequently infected with LCMV. Ten to 14 days later, we analyzed donor ‘ex-siIEL’ or ‘ex-siLPL’ P14 T cells from the spleens and the four intestinal tissue compartments of recipient mice. Compared to ex-siLPL TRM cells, ex-siIEL TRM cells appeared to be superior in their ability to give rise to secondary TRM cells in the siIEL and siLPL compartments. Moreover, compared to ex-siLPL TRM cells, ex-siIEL TRM cells tended to have an increased ability to give rise to secondary TRM cells in the colon and secondary memory cells in the spleen (Figures 4D–4F). Ex-siIEL TRM cells that gave rise to secondary TRM cells tended to adopt the phenotypic characteristics of their new environments. For example, ex-siIEL TRM cells that gave rise to secondary cLPL TRM cells no longer exhibited a CD69+CD103+ phenotype (Figure 4G) and instead expressed low levels of GzmA and high levels of Ly6C (Figures 4H and 4I), both distinctive features of primary cLPL TRM cells identified above (Figures 1C, 2A, 3C, 3D, and 3E). Lastly, ex-siIEL TRM cells gave rise to secondary TEM cells in the spleen, but not secondary TCM cells (Figure 4J), consistent with previously published data21. Taken together, these findings suggest that TRM cells from different anatomic regions within the same organ may exhibit disparate degrees of developmental plasticity.
Figure 4. Small intestine IEL TRM cells may exhibit higher developmental plasticity than siLPL TRM cells.
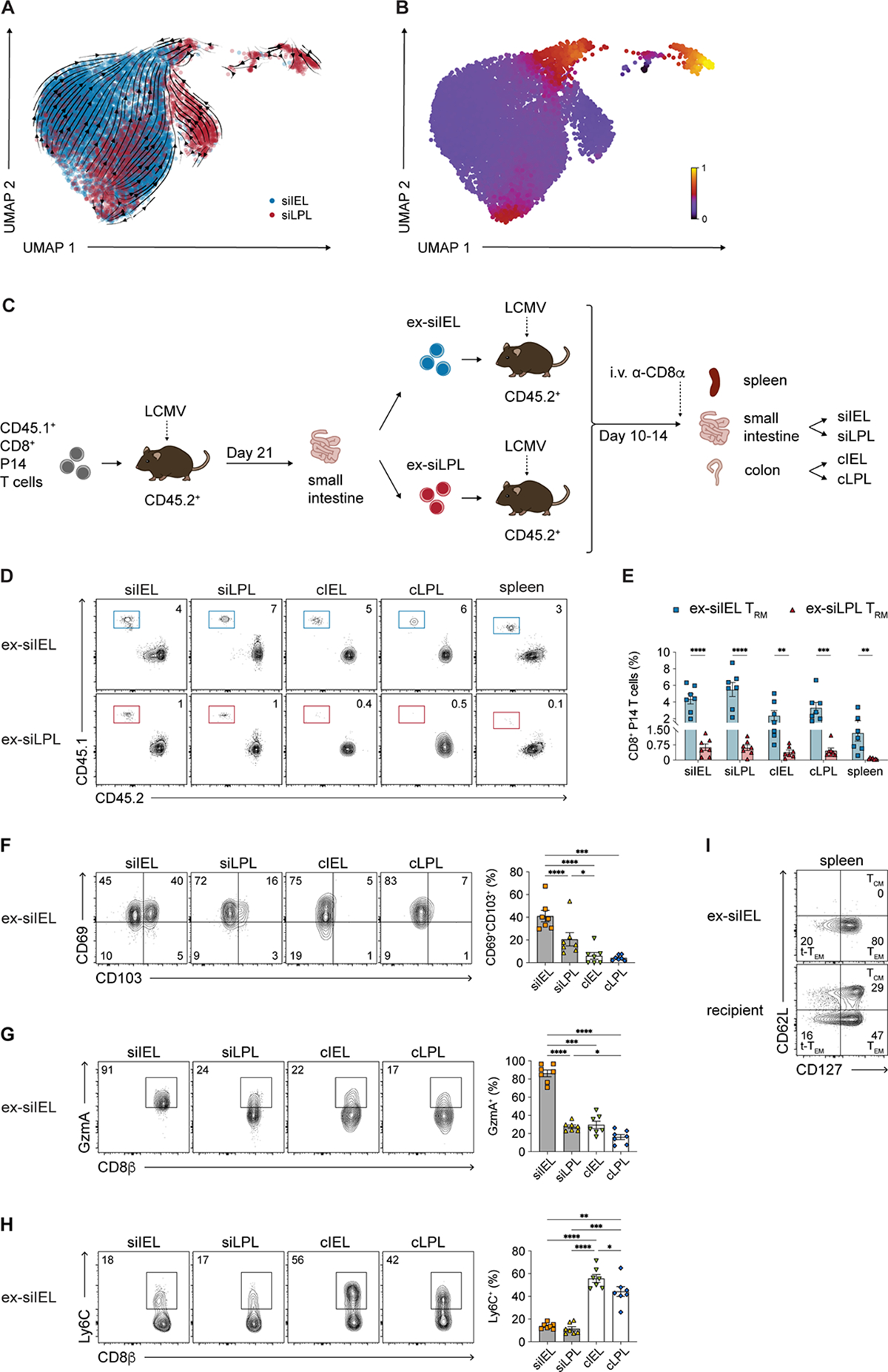
(A and B) Velocities (A) and latent time (B) of siIEL and siLPL TRM cells derived from scVelo projected onto a UMAP-based embedding.
(C) Experimental design. siIEL and siLPL TRM cells were FACS-purified from CD45.2+ recipient mice ≥21 days following adoptive transfer of donor CD45.1+ P14 T cells and LCMV infection. Cells were transferred into new, separate CD45.2+ recipients subsequently infected with LCMV and sacrificed 10–14 days later.
(D and E) Representative flow cytometry plots (D) and bar graphs (E) showing frequencies of transferred CD45.1+ ex-siIEL (left) or CD45.1+ ex-siLPL (right) TRM cells in the intestinal tissue compartments and spleen.
(F–H) Representative flow cytometry plots (left) and bar graphs (right) showing frequencies of cells expressing CD69 and CD103 (F), GzmA (G), or Ly6C (H) among ex-siIEL TRM cells in the intestinal tissue compartments.
(I) Representative flow cytometry plots showing expression of CD62L and CD127 among ex-siIEL CD8+CD45.1+ P14 T cells or recipient (non-P14) CD8+CD45.2+ T cells in the spleen.
Data are represented as mean ± SEM. Repeated measures one-way ANOVA. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001. Data are representative of ≥2 independent experiments with n=5–6 mice per experiment.
Eomes is dispensable for initial formation of intestinal CD8+ TRM cells, but plays a critical role in the maintenance of established SI CD8+ TRM cells.
The unexpected observation that Eomes was more highly expressed in colon TRM cells compared to those in the SI (Figures 3B and 3H) raised the possibility that Eomes might play a role in intestinal TRM cells that is distinct from its previously established role in skin, liver, and kidney TRM cells31,32. To evaluate whether Eomes plays a role in the initial formation of CD8+ TRM cells in each of the four intestinal tissue compartments, congenically distinct control and Eomesfl/flCd4Cre (Eomes conditional (c)KO) P14 T cells were adoptively transferred at a 1:1 ratio into recipient mice subsequently infected with LCMV (Figure 5A) and analyzed at day 7 and after 21 days post-infection. The proportions and absolute numbers of splenic and intestinal Eomes cKO P14 T cells were not substantially different than those of control cells at 7 days post-infection (Figures 5B, S5A–SC, and S6A). However, the proportions of Eomes cKO CD69+CD103+ P14 T cells were increased in each of the four intestinal tissue compartments (Figure 5C). In line with these observations, forced Eomes expression resulted in reduced proportions of CD69+CD103+ T cells in each of the four intestinal tissue compartments (Figure S6B). Compared to control counterparts, intestinal Eomes cKO CD8+ P14 T cells exhibited modest differences in the expression of cytolytic granules, markers of proliferation (Ki67), cytokines, and T-bet (Figures S6C–S6H). Splenic Eomes cKO CD8+ P14 T cells exhibited a decrease in the proportion of cells exhibiting a terminal effector phenotype (KLRG1hiCD127lo) and a slight increase in the proportion of cells exhibiting a memory precursor phenotype (KLRG1loCD127hi) phenotype, along with a modest decrease in GzmA expression (Figures S5D–S5I).
Figure 5. Eomes is dispensable for intestinal CD8+ TRM cell formation.
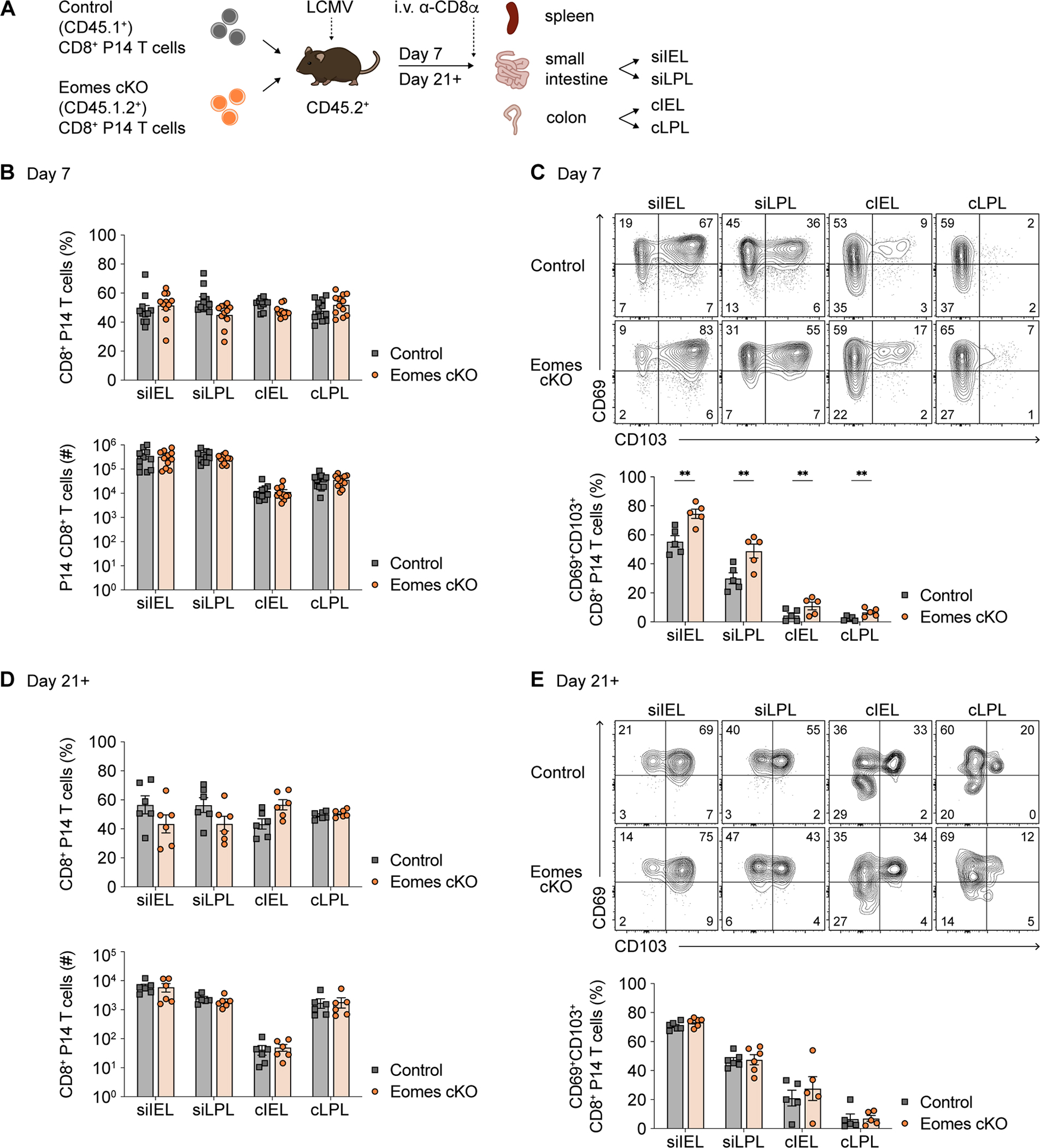
(A) Experimental design. CD8+ P14 T cells from congenic control and Eomesfl/flCd4-Cre+(Eomes cKO) mice were adoptively co-transferred at a 1:1 ratio into recipients subsequently infected with LCMV. Donor P14 T cells were isolated from spleen and intestinal tissue compartments as in Figure 1 at 7 days (B and C) or ≥21 days (D and E) post-infection.
(B and D) Quantification of the proportions (top) or absolute numbers (bottom) of control i.v.− control vs. Eomes cKO P14 T cells in each tissue compartment at 7 days (B) or ≥21 days (D) post-infection.
(C and E) Representative flow cytometry plots (top) and bar graphs (bottom) indicating frequencies of control vs. Eomes cKO intestinal P14 T cells expressing CD69 and CD103 at 7 days (C) or ≥21 days (E) post-infection.
Data are represented as mean ± SEM. Paired t-test. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001. Data are representative of ≥3 independent experiments with n=5–6 mice per experiment.
See also Figures S5 and S6.
Analyses performed after day 21 post-infection demonstrated similar proportions and absolute numbers of intestinal control and Eomes cKO P14 T cells (Figure 5D), along with similar proportions of CD69+CD103+ cells (Figure 5E) and expression patterns of granzymes and cytokines (Figures S6I and S6J). In the spleen, although the numbers of total circulating memory cells were unchanged by the absence of Eomes (Figure S5J), the proportion of TCM (CD62LhiCD127hi) cells was decreased with a corresponding increase in the proportion of TEM (CD62LloCD127hi) cells (Figure S5K). The reduced proportion of Eomes cKO TCM cells was associated with decreased expression of CD122, a known target of Eomes in circulating CD8+ T cells44, along with increased expression of T-bet by TCM cells (Figures S5L and S5M). Taken together, these findings indicate that Eomes is largely dispensable for CD8+ TRM cell formation in the four intestinal tissue compartments. Although Eomes does appear to play a role in repressing CD69 and CD103 expression early during intestinal TRM cell formation, as previously reported in skin TRM cells31, these effects did not result in sustained changes in the proportions or absolute numbers of Eomes cKO CD8+ TRM cells, compared to their control counterparts, in any of the four intestinal tissue compartments. Similar results were observed using P14 T cells in which Eomes was inducibly deleted prior to adoptive transfer and LCMV infection, arguing against the possibility of a compensatory adaptation by Eomes cKO CD8+ P14 T cells (Figures S6K and S6L).
We next sought to evaluate the role of Eomes in the maintenance of TRM cells, once established, in each of the four intestinal tissue compartments. Congenically distinct control and Eomesfl/flCre-ERT2 (Eomes inducible (i)KO) CD8+ P14 T cells were adoptively transferred at a 1:1 ratio into recipient mice subsequently infected with LCMV (Figure 6A). At 21 days post-infection, mice were treated with tamoxifen for 5 days and analyzed after 31 days post-infection. Compared to control cells, the proportions and absolute numbers of Eomes iKO CD8+ P14 T cells were markedly reduced in the siIEL and siLPL compartments, but not in the cIEL and cLPL compartments (Figures 6B and 6C). Eomes iKO P14 T cells exhibited a modest increase in the proportion of cells expressing both CD69 and CD103 (Figure 6D), along with minimal changes in GzmA and GzmB expression (Figures S7A and S7B). The reduction in proportions of Eomes iKO CD8+ P14 T cells in the siIEL and siLPL compartments appeared to affect both CD69+CD103+ and CD69+CD103− subpopulations equally (Figure 6E). In the spleen, the proportions and absolute numbers of total Eomes iKO circulating memory P14 T cells were decreased (Figure 6F), along with a reduced proportion of TCM cells and an increased proportion of t-TEM cells (Figure 6G), suggesting a role for Eomes in the maintenance of established TCM cells in addition to its known role in their formation45. Taken together, these findings reveal that Eomes plays a previously unappreciated role in the maintenance of established intestinal CD8+ TRM cells after their formation, with a greater role in the SI than in the colon.
Figure 6. Eomes plays a critical role in maintenance of established SI CD8+ TRM cells.
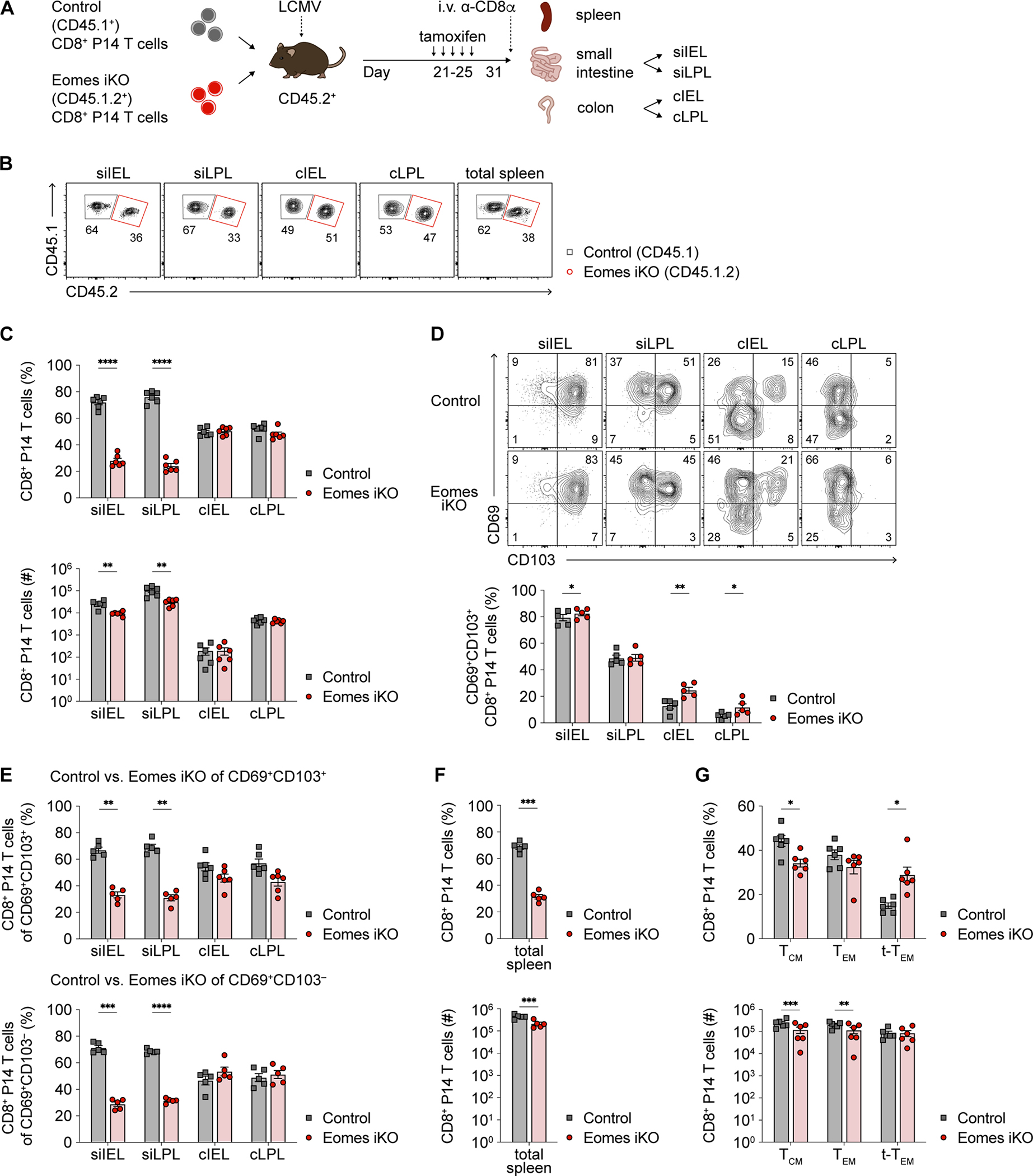
(A) Experimental design. CD8+ T cells from congenic control and Eomesfl/flErt2-Cre+ (Eomes iKO) P14 mice were adoptively co-transferred at 1:1 ratio into recipients subsequently infected with LCMV. Mice received tamoxifen i.p. once daily x 5 doses starting at day 21 post-infection. Spleen and intestinal P14 T cells were harvested >10 days later.
(B and C) Representative flow cytometry plots (B) and quantification (C) of the proportions (top) or absolute numbers (bottom) of i.v.− control vs. Eomes iKO P14 T cells in each intestinal tissue compartment.
(D) Representative flow cytometry plots (top) and bar graphs (bottom) indicating frequencies of control vs. Eomes iKO intestinal P14 T cells expressing CD69 and CD103.
(E) Proportions of control vs. Eomes iKO P14 TRM cells among CD69+CD103+ (top) or CD69+CD103− (bottom) subpopulations.
(F) Frequencies (top) or absolute numbers (bottom) of total control vs. Eomes iKO P14 T cells in the spleen.
(G) Bar graphs indicating the frequencies (top) or absolute numbers (bottom) of control vs. Eomes iKO TCM (CD62LhiCD127hi), TEM (CD62LloCD127hi), and t-TEM (CD62LloCD127lo) cells.
Data are represented as mean ± SEM. Paired t-test. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001. Data are representative of ≥3 independent experiments with n=5–6 mice per experiment.
See also Figure S7.
Eomes regulates the maintenance of SI TRM cells, in part, by inducing the anti-apoptotic regulator Bcl-2.
To elucidate potential mechanisms by which Eomes regulates the maintenance of SI TRM cells, we performed CITE-seq on Eomes iKO P14 CD8+ T cells harvested after day 31 post-infection following 5 daily doses of tamoxifen i.p. starting at day 21 post-infection. Pathway analyses of genes differentially expressed between control and Eomes iKO CD8+ TRM cells from each of the four intestinal tissue compartments revealed an enrichment of genes encoding components of pathways related to apoptosis, cell survival, TGFβ signaling, and cytokine signaling (Table S7). Indeed, transcripts encoding TGFβ signaling components such as Smad3 were reduced in Eomes iKO intestinal TRM cells compared to control cells, while expression of Il7r, the gene encoding for IL-7Rα (CD127), was reduced in Eomes iKO intestinal TRM cells compared to control cells (Figure 7A). Moreover, the anti-apoptotic genes Bcl2 and Mcl1 were more highly expressed in control SI TRM cells compared to Eomes iKO cells (Figure 7A; Table S7). At the protein level, Eomes iKO intestinal TRM cells expressed less CD127, both in terms of proportions and on a per-cell basis (Figure S7C). We also examined the levels of the purinergic receptor P2RX7, a sensor of extracellular ATP, in light of its reported role as a key regulator of TRM cell fitness and survival37,46. Eomes iKO intestinal TRM cells expressed higher levels of P2RX7 protein (Figure S7D), along with lower levels of Bcl-2 protein (Figure 7B). In line with these observations, forced expression of Eomes resulted in increased expression of Bcl-2 and reduced expression of P2RX7 (Figures 7C, S7E, and S7F). Taken together, these results raised the possibility that Eomes might support SI TRM cell maintenance by virtue of regulating genes and pathways involved in TGFβ signaling, responsiveness to homeostatic cytokines, and/or apoptosis.
Figure 7. Eomes promotes SI CD8+ TRM cell maintenance, in part, through effects on Bcl-2.
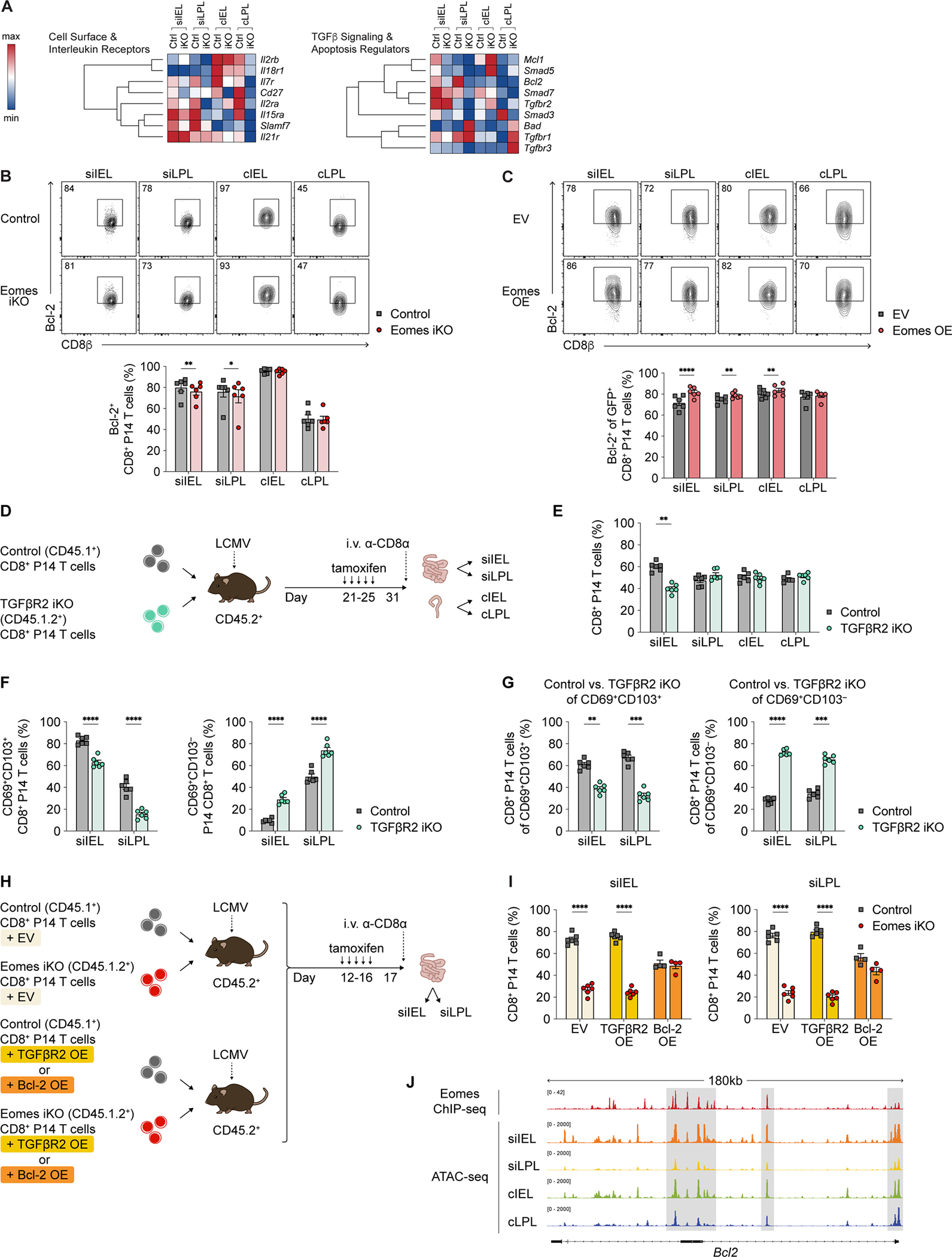
(A) Relative expression of selected genes derived using CITE-seq data from control vs. Eomesfl/flErt2-Cre+ (Eomes iKO) P14 T cells, represented as hierarchically clustered summary heatmaps; rows represent individual genes and columns represent P14 T cells from each of the 4 intestinal tissue compartments.
(B) Representative flow cytometry plots (top) and bar graphs displaying the frequencies of i.v.− control vs. Eomes iKO intestinal P14 T cells expressing Bcl-2.
(C) Representative flow cytometry plots (top) and bar graphs representing frequencies of adoptively co-transferred empty vector (EV)- vs. Eomes overexpression (OE)-transduced intestinal P14 T cells expressing Bcl-2 at day 7 following LCMV infection.
(D) Experimental design. CD8+ T cells from congenic control or Tgfbr2fl/flErt2-Cre+ (TGFβR2 iKO) P14 mice were adoptively co-transferred at a 1:1 ratio into recipients subsequently infected with LCMV, followed by treatment with tamoxifen as in Figure 6A.
(E and F) Bar graphs representing frequencies of total (E), CD69+CD103+ (F, left), or CD69+CD103− (F, right) control vs. TGFβR2 iKO i.v.− intestinal P14 T cells.
(H) Experimental design. CD8+ T cells from control (CD45.1+) or Eomes iKO (CD45.1.2+) P14 mice were activated and transduced with EV, TGFβR2 OE, or Bcl-2 OE constructs. Cells were mixed adoptively co-transferred at a 1:1 into CD45.2+ recipients subsequently infected with LCMV, followed by treatment with tamoxifen as in Figure 6A.
(I) Bar graphs representing frequencies of EV-transduced control vs. Eomes iKO P14 T cells (left); TGFβR2 OE-transduced control vs. Eomes iKO P14 T cells (middle); and Bcl-2 OE-transduced control vs. Eomes iKO P14 T cells (right) in the siIEL and siLPL tissue compartments.
(J) CD45.1+ intestinal P14 T cells were isolated from CD45.2+ recipient mice infected with LCMV 21 days prior, FACS-purified, and processed for scATAC-seq. Tracks representing Eomes ChIP-seq (top) and scATAC-seq peaks for each of the four intestinal tissue compartments (bottom) are shown for Bcl2.
Data are represented as mean ± SEM. Paired t-test. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001. Data are representative of ≥3 independent experiments with n=5–6 mice per experiment.
We next sought to test whether exogenous administration of IL-7 or shRNA-mediated deletion of P2RX7 might prevent the loss of SI TRM cells resulting from induced deletion of Eomes. Neither administration of IL-7-anti-IL-7 mAb complexes, which substantially increase the in vivo biological activity of IL-747, nor deletion of P2RX7 ameliorated the reduction of SI TRM cells resulting from the loss of Eomes (Figures S7G–S7J). We therefore asked whether induced deletion of TGFβR2 in established intestinal TRM cells might phenocopy the numerical deficiency of SI TRM cells resulting from the loss of Eomes. Accordingly, congenically distinct control and Tgfbr2fl/flCre-ERT2 (TGFβR2 inducible (i)KO) CD8+ P14 T cells were adoptively co-transferred at a 1:1 ratio into recipient mice subsequently infected with LCMV (Figure 7D). At 21 days after infection, mice were treated with tamoxifen daily for 5 days and then analyzed after 31 days post-infection. Compared to control cells, the proportions of TGFβR2 iKO CD8+ P14 T cells were reduced only in the siIEL compartment, but not in the other three intestinal tissue compartments (Figure 7E). However, in both the siEL and siLPL compartments, the loss of TGFβR2 led to decreased proportions of CD69+CD103+ TRM cells, but increased proportions of CD69+CD103− TRM cells (Figures 7F). Thus, CD69+CD103+ and CD69+CD103− TRM cells in the siIEL and siLPL compartments appeared to be discordantly impacted by the deletion of TGFβR2 (Figure 7G). This experimental finding was distinct from that observed from the loss of Eomes, which reduced both CD69+CD103+ and CD69+CD103− subpopulations among established Eomes iKO SI TRM cells (Figure 6E). In parallel, we observed that forced TGFβR2 overexpression failed to ameliorate the reduction of SI TRM cells resulting from the loss of Eomes (Figures 7H and 7I). Taken together, these results suggest that while TGFβ signaling plays a critical role in the maintenance of CD69+CD103+ SI TRM cells, the reduction in Eomes iKO SI TRM cells does not appear to be mechanistically linked to alterations in TGFβ signaling.
Lastly, as Eomes iKO intestinal TRM cells expressed lower levels of Bcl-2 (Figure 7B) whereas forced expression of Eomes resulted in increased expression of Bcl-2 (Figure 7C), we asked whether forced expression of Bcl-2 might be capable of preventing the loss of SI TRM cells resulting from the induced deletion of Eomes. Congenically distinct P14 T cells from control and Eomes iKO mice were transduced with empty vector (EV) or Bcl-2 retroviral constructs, mixed at a 1:1 ratio, and adoptively transferred into CD45.2+ recipients prior to infection with LCMV. Mice received 5 daily doses of tamoxifen starting at day 12 post-infection and intestinal tissue compartments were harvested on day 17 post-infection (Figure 7H). Eomes iKO cells transduced with the EV construct exhibited a competitive disadvantage in the SI compared to wild-type cells transduced with the EV construct (Figure 7I), recapitulating the phenotype observed in untransduced cells (Figure 6B, 6C). By contrast, Eomes iKO cells transduced with the Bcl-2 construct were much better able to compete with wild-type cells transduced with the Bcl-2 construct (Figure 7I). In order to distinguish whether the regulation of Bcl-2 by Eomes was direct or indirect, we analyzed our scATAC-seq intestinal TRM cell dataset in conjunction with a previously published Eomes ChIP-seq (chromatin immunoprecipitation sequencing) dataset32,48. These analyses revealed accessible regions of chromatin within the Bcl2 gene locus in intestinal TRM cells that corresponded to putative Eomes binding sites identified by ChIP-seq (Figure 7J). Taken together, these results support the hypothesis that Eomes promotes the survival and maintenance of SI TRM cells, in part, through potentially direct effects on Bcl-2.
DISCUSSION
Emerging data indicate that TRM cells from different tissues exhibit substantial heterogeneity5,21–23 and an increasing number of studies have highlighted heterogeneity even among TRM cells within the same organ. For example, CD103+ vs. CD103− TRM cells within the brain, salivary gland, and SI exhibit differences in transcriptional profiles10, developmental plasticity22, and responsiveness to secondary infection49,50, respectively. In the siIEL compartment, the IL-2Rαlo CD8+ T cell subpopulation contains putative TRM precursor cells, while the IL-2Rαhi T cell subpopulation may represent a transient, more terminally differentiated subpopulation51. Moreover, Blimp1hiId3lo and Blimp1loId3hi cells in the siIEL compartment are prominent at distinct phases of infection and exhibit distinct cytokine capabilities, secondary memory potential, and transcriptional programs11. Our data suggest differential requirements for the maintenance of TRM cell subpopulations within the same tissue, as TGFβ signaling was required to promote maintenance of CD103+, but not CD103− TRM cells, within the siIEL and siLPL compartments; by contrast, both CD103+ and CD103− TRM cells from the siIEL and siLPL compartments required Eomes for their continued maintenance.
Our study highlights the emerging concept that factors required for TRM cell formation may be distinct from those required for maintenance after their establishment. Factors that regulate TRM cell formation, but that have not been formally investigated in their maintenance, include Ahr (skin39); Blimp1 (skin, kidney, siIEL/siLPL20, and lung24); Hobit (skin, kidney, and siIEL/siLPL20,32); Bhlhe40 (lung52); Nr4a1 (siIEL, liver53), and Nr4a2 (siIEL51); and IL-15 signaling (salivary gland, kidney27, and skin18). Factors that play a critical role in both the initial formation and maintenance of TRM cells include Runx3 (siIEL, salivary gland, kidney, lung, skin11); TGFβ (skin18,54, siIEL18,23, and salivary gland23); and P2RX7 (siIEL37,38,46). Notably, however, a factor that plays a role in the formation of TRM cells need not be necessary for their maintenance, as previously demonstrated for ICOS in the kidney and siIEL compartment28. The data presented here indicate that, like ICOS, Eomes plays distinct roles in the formation vs. maintenance of intestinal TRM cells.
Eomes was first described as a regulator of effector CD8+ T cell responses owing to its effects in directly promoting GzmA expression55, and was subsequently shown to play an important role in the formation of circulating memory T cells, particularly TCM cells44,45. By contrast, TRM cells in the skin, lung, and siIEL compartment were observed to exhibit reduced expression of Eomes compared to circulating memory cells18. Moreover, forced expression of Eomes led to reduced TRM cell formation in the skin31, whereas deletion of endogenous Eomes led to increased TRM cell formation in the liver and kidney32. On the basis of these findings, Eomes has been generally regarded as a negative regulator of TRM cell formation, but its role in the maintenance of established TRM cells has not been previously investigated. Our data indicate that Eomes is dispensable for intestinal TRM cell formation, but plays a critical role in the maintenance of established TRM cells in the SI, but not in the colon, in spite of lower baseline expression of Eomes by SI TRM cells. This observation calls into question the seemingly intuitive notion that higher transcription factor expression levels imply greater functional importance. Taken together, these findings suggest that the role of Eomes in TRM cells may more nuanced than previously appreciated, with distinct functions that are dependent on the specific tissue compartment as well as the phase of differentiation.
Lastly, our study adds to the field’s understanding of TRM cell plasticity. It was initially thought that TRM cells are terminally differentiated, with limited proliferative capacity after rechallenge and low developmental plasticity. It is now understood that TRM cells are capable of proliferation after reactivation in situ56,57; moreover, reactivated TRM cells can leave tissues and egress to the circulation, giving rise to circulating memory T cells5,6. Progeny of intravenously transferred ex-siIEL TRM cells maintain a predilection for repopulating the siIEL and siLPL compartments, but not other tissue compartments such as the salivary gland or female reproductive tract5,9. Moreover, ex-siIEL TRM cells tend to acquire phenotypic characteristics of their new environment, while retaining some traces of their original tissue of residence5. Notably, the developmental plasticity of TRM cells has been reported to differ between some tissues, with ex-liver and ex-siIEL TRM cells exhibiting a greater degree of developmental plasticity than ex-skin TRM cells21,22; moreover, CD103− ex-salivary gland TRM cells exhibit a higher degree of developmental plasticity than CD103+ ex-salivary gland TRM cells22. Our data suggest that TRM cells located in different anatomic regions of the same organ may also exhibit distinct degrees of developmental plasticity, as ex-siIEL TRM cells were capable of giving rise to secondary TRM cells in all four intestinal tissue compartments and did so to a greater degree than ex-siLPL TRM cells. In addition, we observed that ex-siIEL TRM cells can contribute to the formation of secondary TEM cells in the spleen, but have limited capacity to form secondary TCM cells, in agreement with a prior study21. Furthermore, in light of a prior report showing that CD8+ TRM cells can migrate between the siIEL and siLPL compartments58, our data raise intriguing questions about the developmental relationships between TRM cells in the siIEL and siLPL compartments during their initial formation. Overall, our study reveals substantial phenotypic, transcriptional, epigenetic, and functional heterogeneity among TRM cells from the four intestinal tissue compartments, and provides a resource for the field to begin to investigate the differential molecular requirements of intestinal TRM cells.
Limitations of the study
In this study, we defined cells as TRM if they were ‘i.v.-negative’ following i.v. injection of anti-CD8α antibodies into recipient mice 3–5 minutes prior to sacrifice, a widely used approach that labels cells with access to the vasculature during the short pulse prior to sacrifice; it should be noted, however, that this technique does not directly identify TRM cells. Second, TRM cells were analyzed after 21 days post-infection, ~2 weeks after LCMV-Armstrong is cleared in the SI and colon59–61; however, it remains possible that antigen may persist in certain anatomical regions and could influence TRM cell differentiation and heterogeneity62. Third, although we identified a role for Eomes in the maintenance of established SI TRM cells, technical challenges precluded direct assessment of the consequences of Eomes-deficiency in intestinal TRM cells on host health. Lastly, it should be noted that differences in trafficking, proliferation, and/or survival between ex-siIEL and ex-siLPL TRM cells could contribute to apparent disparities in developmental plasticity.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for the resources and reagents should be directed to and will be fulfilled by the lead contact, John T. Chang (changj@ucsd.edu).
Materials Availability
All the mouse lines used in this study are available from Jackson Laboratories. This study did not generate new unique reagents.
Data and Code Availability
All data reported in this paper are available at Gene Expression Omnibus under accession number GSE205942.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were housed under specific pathogen-free conditions in an American Association of Laboratory Animal Care-approved facility at UCSD, and all procedures were approved by the UCSD Institutional Animal Care and Use Committee. C57BL/6J (CD45.2) and B6.SJL-PtprcaPepcb/BoyJ (CD45.1) mice were purchased from the Jackson Laboratory. Tgfbr2fl/fl, Eomesfl/fl, Cd4-Cre+, Ert2-Cre+ were purchased from Jackson Laboratory and bred together with P14 TCR transgenic mice to generate Tgfbr2fl/flErt2-Cre+ (TGFβR2 iKO), Eomesfl/flCd4-Cre+ (Eomes cKO) and Eomesfl/flErt2-Cre+ (Eomes iKO) P14 mice. All mice were used at 6–9 weeks of age.
METHOD DETAILS
Adoptive transfer and infection
CD45.1+ CD8+ P14 T cells were adoptively transferred into CD45.2+ recipient mice (1 × 105 cells/mouse). For competition experiments, TGFβR2 iKO, Eomes cKO, Eomes iKO CD8+ P14 T cells (1 × 105 cells/mouse) were co-transferred with control CD8+ P14 T cells (1 × 105 cells/mouse). Recipient mice were bled at 21 days post-infection to ensure successful adoptive transfer and to correct for the input ratios between control CD8+ P14 T cells vs. TGFβR2 iKO, Eomes cKO, or Eomes iKO CD8+ P14 T cells. Recipient mice were infected intraperitoneally (i.p.) with 2 × 105 plaque-forming units (p.f.u.) of LCMV-Armstrong 30 minutes following adoptive transfer.
Tamoxifen treatment
For inducible deletion of Eomes or Tgfbr2, tamoxifen was administered i.p. (1 mg/mouse daily × 5 days) at timepoints indicated in the figure legends. A 100 mg/mL tamoxifen stock solution prepared in 100% ethanol was subsequently diluted 1:10 in 100% sunflower seed oil; 100 μL of this was administered for a working dose of 1 mg/mouse/day.
Retroviral transduction
PLAT-E cells (Cell BioLabs) were transfected with empty vector (EV), overexpression vectors (Eomes, Bcl2, Tgfbr2), non-targeting shRNA construct, or shRNA constructs targeting P2rx7 (Transomic) with TransIT-LT1 Reagent (Mirus). The Eomes vector was provided by Dr. Steven Reiner, Columbia University; the Bcl2 vector was provided by Dr. Michael Croft, La Jolla Institute for Immunology; and the Tgfbr2 vector was provided by Dr. Wanjun Chen, National Institutes of Health. Retroviral supernatants were harvested 48h and 72h post-transfection. CD8+ T cells were isolated using the CD8+ T cell isolation kit (Miltenyi) and activated with plates coated with 100 μg/mL of goat anti-hamster IgG (H+L, Thermo Fisher Scientific), 10 μg/mL of anti-CD3 (3C11, BioXCell), and 10 μg/mL of anti-CD28 (37.51, BioXCell). After 18–22 hours of activation, the cells were ‘spinfected’ with retroviral supernatant supplemented with 8 g/mL of polybrene (Millipore) at 900g for 90 min at room temperature. The retroviral supernatant was replaced by culture media and the cells were incubated at 37°C. Transduction efficiency was measured based on ametrine or GFP signal using flow cytometry at 24h and 48h post-transduction.
Plasticity experiments
For experiments assessing TRM cell developmental plasticity, CD45.1+ CD8+ P14 T cells were adoptively transferred into congenic CD45.2+ recipients 30 minutes prior to infection with LCMV; siIEL and siLPL TRM cells were FACS-purified from recipient mice more than 21 days following infection, followed by transfer into new, separate CD45.2+ recipients 30 minutes prior to infection with LCMV and euthanized 10–14 days later.
Rescue experiments
Purified CD45.1+ control and CD45.1.2+ Eomes iKO CD8+ P14 T cells were isolated as described above; cells were activated in vitro and transduced with empty vector construct, overexpression constructs (Bcl2, Tgfbr2, P2rx7), non-targeting shRNA construct, or shRNA constructs targeting P2rx7 as described above. Retrovirally transduced cells were mixed at a 1:1 ratio and adoptively transferred into congenic recipients, followed by infection with LCMV. Recipient mice were bled at 10 days post-infection to ensure successful adoptive transfer and to correct for the input ratio between control and Eomes iKO CD8+ P14 T cells. For inducible deletion of Eomes, tamoxifen was administered i.p. daily × 5 days starting at 12 days post-infection. Recipient mice were euthanized at 17 days post-infection for analysis. For rescue experiments with IL-7-anti-IL-7 mAb complexes, CD45.1+ control (1 × 105 cells/mouse) and CD45.1.2+ Eomes iKO (1 × 105 cells/mouse) CD8+ P14 T cells were adoptively co-transferred into CD45.2+ recipient mice followed by i.p. infection with LCMV. Recipient mice were bled at 10 days post-infection to ensure successful adoptive transfer and to correct for the input ratio between control and Eomes iKO CD8+ P14 T cells. IL-7/anti-IL-7 mAb complexes were generating by mixing 1.5 μg of recombinant murine IL-7 (Peprotech) with 15 μg of anti-IL-7 mAb (M25, BioXCell), followed by incubation at 30 min at 37°C, as previously described47. IL-7/anti-IL-7 mAb complexes and tamoxifen were administered i.p. once daily × 5 days starting at day 12 post-infection. Recipient mice were euthanized at 17 days post-infection for analysis.
Lymphocyte isolation
3–5 minutes prior to sacrifice, mice were injected i.v. with anti-CD8α antibodies to label and exclude cells with access to the circulation. Lymphocyte isolation for spleen and the small intestine and colon IEL compartments was performed as previously reported51. To isolate lymphocytes from the small intestine and colon LPL compartment, the remaining tissues were incubated while shaking at 37°C for 15 minutes in IEL solution (1mM EDTA in 1x PBS). The tissues were washed with HBSS then cut thoroughly and resuspended in LPL solution (1640 RPMI, 10% FBS, 100 mg/mL DNase, and 0.02 g/L collagenase IV). Tissues were shaken and incubated at 37°C for 12 minutes. LPL supernatant was collected and filtered into a cell strainer to yield a single-cell suspension. Quantitation of TRM cell numbers reported in Figure 1B were derived by dividing the total number of i.v.-negative CD8 TRM cells quantitated in each intestinal compartment after digestion by the organ weight measured prior to digestion. The purpose of the organ weight normalization was to provide context for the reported numbers. The small intestine was typically larger (~0.7 g) than the colon (~0.2 g); hence, we aimed to show that the increased numbers of TRM cells observed in the small intestine were not simply due to increased size of that organ. Non-normalized absolute numbers are provided as Figure S1A.
Flow cytometry and sorting
For all analyses shown in this study, in order to exclude circulating cells, anti-CD8α antibodies were injected i.v. into recipient mice 3–5 minutes prior to sacrifice and only ‘i.v.-negative’ cells were considered to be tissue-resident for subsequent downstream analyses; this approach has been widely used to exclude circulating cells with access to the vasculature34. Spleen, siIEL, siLPL, cIEL, and cLPL intestinal tissue samples were isolated in a single-cell suspension and stained in Fixable Viability Dye eFluor780 (Thermo Fisher Scientific) at 1:1000 on ice for 10 minutes in the dark. Cells were then surface-stained for 30 minutes on ice with the following antibodies from Biolegend (CD103 (2E7), CD122 (TM-β1), CD127 (A7R34), CD160 (7H1), CCR6 (29-2L17), CCR9 (CW-1.2), CD27 (LG.3A10), CD29 (HMβ1 – 1), CD314 (CX5), CD38 (90), CD44 (IM7), CD45.1 (A20), CD45.2 (104), CD49a (HM1), CD49d (R1-2), CD62L (MEL-14), CD69 (H1.2F3), CD8a (53–6.7), CD8b (TS156.7.7), CXCR3 (CXCR3-173), CXCR4 (L276F12), IL-18R (A17071D), Integrin β7 (FIB27), Ki67 (11F6), KLRG1 (MAFA), Ly6c (HK1.4), P2RX7 (1F11) ); or R&D Systems (TGFR2). For experiments involving retrovirally transduced P14 T cells, cells were fixed in 2% paraformaldehyde (Electron Microscopy Services) at room temperature for 20 minutes. For intracellular and intranuclear staining, cells were fixed and permeabilized using the FoxP3/Transcription Factor Staining Buffer Kit (Thermo Fisher Scientific). For assessment of cytokine production, cells were cultured in the presence of LCMV gp33–41 peptide (GenScript) and Protein Transport Inhibitor Cocktail (Thermo Fisher Scientific) for 3h at 37°C, fixed and permeabilized with FoxP3/Transcription Factor Staining Buffer Kit for 30 minutes at room temperature, and then stained with the following antibodies from Biolegend (Bcl-2 (BCL/10C4), GzmB (QA16A02), IFN (XMG1.2), IL-2 (JES6-5H4), T-bet (4B10)); or Thermo Fisher (Eomes (Dan11mag), GzmA (GzA-3G8.5)) at room temperature for 30 minutes.
CITE-seq
Control and Eomes iKO CD8+ P14 T cells were harvested from the siIEL, siLPL, cIEL, and cLPL tissue compartments and spleens from 20 recipient mice infected more than 21 days prior, FACS-purified, and processed through the 10x Genomics pipeline. The single-cell library method used was the Chromium Next GEM Single Cell v3 with single-indexing. The RNA samples and ADT samples were pooled separately. RNA samples were sequenced to a depth of 20,000 reads/cell and ADT (protein) samples were sequenced to a depth of 5,000 reads/cell on a NovaSeq S4.
Single-cell ATAC-seq
CD8+ P14 T cells were harvested from the siIEL, siLPL, cIEL, and cLPL tissue compartments from recipient mice infected more than 21 days prior and purified by FACS. Nuclei were isolated and prepared for single-cell libraries using the Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1. The pooled libraries of each sample were sequenced on a NovaSeq S4.
QUANTIFICATION AND STATISTICAL ANALYSIS
Flow cytometry
For analysis, all samples were run on a LSRFortessa X-20 (BD Biosciences) or Novocyte 3000 (Agilent). Samples for FACS-purified with an Influx, FACSAria Fusion, or FACSAria2 (BD Biosciences). BD FACS DIVA (BD Biosciences) or NovoExpress (Agilent) software was used for data collection, and FlowJo software (BD Biosciences) was used for analysis of flow cytometry data. In general, when available, a biologic negative control not expressing the marker of interest was used to set gates; if not available, an isotype control or Fluorescence Minus One (FMO) control was used to set gates. A biologic negative control was used for the following antibodies: CCR6, CCR9, CD8β, CD27, CD29, CD38, CD44, CD45.1, CD45.2, CD49a, CD49d, CD62L, CD69, CD103, CD160, CD314, CXCR3, CXCR4, Eomes, GzmA, GzmB, IFNγ, IL-2, IL-18R, integrinβ7, Ki67, KLRG1, Ly108, Ly6C, P2RX7, T-bet, and TNF. An isotype negative control was used for the following antibodies: Bcl-2, CD122, and CD127. An FMO negative control was used for the following antibody: CD8α (i.v. label). Statistical analysis of flow cytometry data was performed using Prism software (GraphPad). P values of <0.05 were considered significant. Statistical details for each experiment are provided in the figure legends.
CITE-seq
The Cell Ranger 6.0.1 pipeline was used to align, filter, and quantify counts using a reference mouse genome file (mm10-3.0.0). The resulting UMI count matrices were read into Seurat 4.1.0.9001, and further filtered based on mitochondrial percentage. Cells with mitochondrial percentage greater than 5% were removed. The metrics from the Cellranger count pipeline were used to assess sample quality. All samples passed through the pipeline successfully. Sequencing saturation for samples was 88% and above. Valid barcodes and valid UMIs were all above the threshold of 75%, with values greater that 96% and 100% respectively. The fraction reads in cells for all samples were 85% and above, and all samples had reads mapped antisense to gene value of 2% or less, which passed the quality threshold of an ideal sample being less than 10%. There was no threshold for the number of cells collected during the Cellranger count pipeline. All samples had a %Q30 score of greater than 70, % perfect index reads of 85% and above, and raw cluster percentages of less than 7%.
After this pre-processing, cells from each sample were selected to be used for the downstream analysis with data from both the ‘Gene Expression’ and ‘Antibody Capture’ output matrices. Using the RNA assay, the Seurat object was log-normalized, scaled using variable features, and PCA-transformed. These steps were repeated on the ADT (antibody-derived tag) assay after removing features CD45.1 and CD45.2. After the removal of these two features, the Seurat object was normalized using centered log-ratio normalization, scaled, and PCA transformed. The command FindMultiModalNeighbors was used to construct the weighted nearest neighbor (WNN) graph, to find neighbors based on the weighted values of the RNA and ADT assays. The command FindClusters was used to identify clusters within the data base on the graph data from the WNN method. Non-linear dimensional reduction techniques tSNE and UMAP were utilized for visualization. Differentially expressed genes were found using command FindMarkers between clusters, tissue, and samples. Other visualization tools from the Seurat package such DoHeatmap, VlnPlot, and FeaturePlot were used to further explore the data. Pathway analyses were performed using Metascape36. It should be noted that the observed correlation between mRNA expression and protein abundance was suboptimal, consistent with prior studies35,63–65. There are likely to be many reasons for this lack of correlation, including biologic variations in mRNA stability and turnover as well as the technical limitation that only a small fraction of the transcriptome of the cell is captured (‘dropouts’) with the widely used 10x Genomics scRNA-seq platform. Thus, CITE-seq may not be the optimal approach to investigate mRNA – protein relationships. Ribosome profiling (termed ‘Ribo-seq’ or ‘ART-seq’ (active mRNA translation sequencing) based on sequencing of ribosome-protected mRNA fragments aims to identify transcripts undergoing active translation and may lead to better correlations between mRNA expression and protein abundance in future studies.
Single-cell ATAC-seq
Analysis of scATAC-seq data was completed using the Seurat extension Signac (v1.6.0)66. Cellranger outputs for each sample were individually imported into R as Seurat objects. The samples were randomly subsampled to 2000 cells per sample before being merged into a single Seurat object. Annotation, quality control, dimensional reduction, and UMAP creation were completed using the standard Signac workflow. Differentially accessible regions (DAR) between tissues were first obtained using the FindMarkers function in Signac. For each DAR, fragment counts per tissue were compiled using the CountsInRegion function and visualized with a heatmap using pheatmap (v1.0.8). The per-cell motif activity score was calculated by running Signac’s implementation of chromVAR. For each motif, differential activity scores between tissues, along with p values, were then determined by using the FindMarkers function on the chromVAR assay. Selected motifs that were differentially active in a single intestinal compartment (siIEL, siLPL, cIEL, cLPL) and their associated p values are shown in Figures S4D–G. A full list of all differentially active motifs (and associated p values) is also provided in Table S5. Selected motifs differentially active in two intestinal compartments and their associated p values are shown in Figures S4H (siIEL and cIEL), S4I (siLPL and cLPL), and S4J (cIEL and cLPL). For a single selected motif, violin plots in the lower right corner of each figure show the activity scores for individual cells from each of the four intestinal compartments. Statistics were not calculated specifically for the violin plots because the selected motifs shown were already identified as having statistically significant differential activity scores in CD8 TRM cells between the intestinal tissue compartments. Overlapping and unique enriched motifs between tissues were identified and visualized using the R package VennDiagram (v1.7.3). Signac’s MotifPlot function was used to generated Motif plots.
scVelo
Differentiation trajectories were inferred using scVelo (v0.2.4). The Cellranger output for each sample was converted into .loom files in command line for import into Python using Velocyto (v0.17.17). In Python, the samples were then merged into a single adata object using scVelo. Filtering, normalization, and moment computing for velocity estimation was completed using scVelo’s standard preprocessing steps. The samples were then analyzed using the Dynamical Modeling workflow to create velocity and latent time UMAPs.
Supplementary Material
Table S1. Differentially expressed genes for intestinal TRM cells separated by CITE-seq clusters, Related to Figures 3 and S2
Table S2. Gene and protein expression, and pathway analyses of intestinal TRM cells by CITE-seq clusters, Related to Figures 3 and S2
Table S3. Gene and protein expression of intestinal TRM cells, and differentially expressed genes of the same, Related to Figure 3
Table S4. Differentially accessible chromatin regions of intestinal TRM cells, Related to Figures 3 and S4
Table S5. Enriched transcription factor binding motifs within differentially accessible chromatin regions of intestinal TRM cells, Related to Figures 3 and S4
Table S6. Uniquely enriched or shared transcription factor binding motifs within differentially accessible chromatin regions of intestinal TRM cells, Related to Figures 3 and S4.
Table S7. Gene and protein expression, and pathway analysis of control vs. Eomes iKO intestinal TRM cells, Related to Figure 7.
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bcl-2 (BCL/10C4; FITC) | BioLegend | Cat#633504; RRID: AB_2028394 |
| Bcl-2 (BCL/10C4; AF647) | BioLegend | Cat#633510; RRID: AB_2274702 |
| CCR6 (29-2L17; PE/Dazzle 594) | BioLegend | Cat#129822; RRID: AB_2687019 |
| CCR9 (CW-1.2; FITC) | BioLegend | Cat#128706; RRID: AB_1186167 |
| CCR9 (CW-1.2; PE-Cy7) | BioLegend | Cat#128712; RRID: AB_10933082 |
| CD8α (clone: 53-6.7; BV510) | BioLegend | Cat#100752; RRID: AB_2563057 |
| CD8α (clone: 53-6.7; BV570) | BioLegend | Cat#100740; RRID: AB_2563055 |
| CD8β (clone TS156.7.7; PE/Dazzle 594) | BioLegend | Cat#126622; RRID: AB_2632630 |
| CD8β (clone TS156.7.7; PerCP/Cy5.5) | BioLegend | Cat#126610; RRID: AB_2260149 |
| CD8β (clone TS156.7.7; BV421) | BioLegend | Cat#126629; RRID: AB_2800620 |
| CD8β (clone TS156.7.7; BV510) | BioLegend | Cat#126631; RRID: AB_2800621 |
| CD27 (clone LG.3A10; BV510) | BioLegend | Cat#124229; RRID: AB_2565795 |
| CD27 (clone LG.3A10; BV605) | BioLegend | Cat#124249; RRID: AB_2860657 |
| CD27 (clone LG.3A10; BV785) | BioLegend | Cat#124241; RRID: AB_2800595 |
| CD29 (clone HMβ1-1; PerCP/Cy5.5) | BioLegend | Cat#102228; RRID: AB_2572079 |
| CD38 (clone 90; BV421) | BioLegend | Cat#102732; RRID: AB_2734153 |
| CD44 (clone IM7; BV421) | BioLegend | Cat#103040; RRID: AB_2616903 |
| CD44 (clone IM7; BV650) | BioLegend | Cat#103049; RRID: AB_2562600 |
| CD44 (clone IM7; BV785) | BioLegend | Cat#103059; RRID: AB_2571953 |
| CD45.1 (clone A20; FITC) | BioLegend | Cat#110706; RRID: AB_313495 |
| CD45.1 (clone A20; BV421) | BioLegend | Cat#110732; RRID: AB_2562563 |
| CD45.1 (clone A20; BV510) | BioLegend | Cat#110741; RRID: AB_2563378 |
| CD45.1 (clone A20; BV605) | BioLegend | Cat#110738; RRID: AB_2562565 |
| CD45.1 (clone A20; BV650) | BioLegend | Cat#110736; RRID: AB_2562564 |
| CD45.2 (clone 104; FITC) | BioLegend | Cat#109806; RRID: AB_313443 |
| CD45.2 (clone 104; BV421) | BioLegend | Cat#109832; RRID: AB_2565511 |
| CD45.2 (clone 104; BV510) | BioLegend | Cat#109838; RRID: AB_2650900 |
| CD45.2 (clone 104; BV605) | BioLegend | Cat#109841; RRID: AB_2563485 |
| CD45.2 (clone 104; BV650) | BioLegend | Cat#109836; RRID: AB_2563065 |
| CD49a (clone HMα1; APC) | BioLegend | Cat#142606; RRID: AB_2562253 |
| CD49d (clone R1-2; PE-Cy7) | BioLegend | Cat#103618; RRID: AB_2563700 |
| CD62L (clone MEL-14; APC) | BioLegend | Cat#104412; RRID: AB_313099 |
| CD62L (clone MEL-14; BV421) | BioLegend | Cat#104436; RRID: AB_2562560 |
| CD62L (clone MEL-14; BV605) | BioLegend | Cat#104438; RRID: AB_2563058 |
| CD62L (clone MEL-14; BV650) | BioLegend | Cat#104453; RRID: AB_2800559 |
| CD62L (clone MEL-14; BV785) | BioLegend | Cat#104440; RRID: AB_2629685 |
| CD69 (clone H1.2F3; PE/Dazzle 594) | BioLegend | Cat#104536; RRID: AB_2565583 |
| CD69 (clone H1.2F3; BV421) | BioLegend | Cat#104528; RRID: AB_2562328 |
| CD69 (clone H1.2F3; BV605) | BioLegend | Cat#104530; RRID: AB_2563062 |
| CD69 (clone H1.2F3; BV650) | BioLegend | Cat#104541; RRID: AB_2616934 |
| CD69 (clone H1.2F3; BV785) | BioLegend | Cat#104543; RRID: AB_2629640 |
| CD103 (clone 2E7; PE) | BioLegend | Cat#121406; RRID: AB_1133989 |
| CD103 (clone 2E7; BV421) | BioLegend | Cat#121422; RRID: AB_2562901 |
| CD103 (clone 2E7; BV605) | BioLegend | Cat#121433; RRID: AB_2629724 |
| CD103 (clone 2E7; BV785) | BioLegend | Cat#121439; RRID: AB_2800588 |
| CD122 (clone TM-β1; PE/Dazzle 594) | BioLegend | Cat#123218; RRID: AB_2572180 |
| CD127 (clone A7R34; FITC) | BioLegend | Cat#135008; RRID: AB_1937232 |
| CD127 (clone A7R34; PE) | BioLegend | Cat#135010; RRID: AB_1937251 |
| CD127 (clone A7R34; APC) | BioLegend | Cat#135012; RRID: AB_1937216 |
| CD127 (clone A7R34; BV421) | BioLegend | Cat#135024; RRID: AB_11218800 |
| CD127 (clone A7R34; BV605) | BioLegend | Cat#135041; RRID: AB_2572047 |
| CD127 (clone A7R34; BV650) | BioLegend | Cat#135043; RRID: AB_2629681 |
| CD127 (clone A7R34; BV785) | BioLegend | Cat#135037; RRID: AB_2565269 |
| CD160 (clone 7H1; PerCP/Cy5.5) | BioLegend | Cat#143008; RRID: AB_2562676 |
| CD160 (clone 7H1; PE-Cy7) | BioLegend | Cat#143010; RRID: AB_2562678 |
| CD314 (clone CX5; PE) | BioLegend | Cat#130208; RRID: AB_1227712 |
| CD314 (clone CX5; PE/Dazzle 594) | BioLegend | Cat#130214; RRID: AB_2728148 |
| CXCR3 (clone CXCR3-173; PE/Dazzle 594) | BioLegend | Cat#126534; RRID: AB_2566563 |
| CXCR3 (clone CXCR3-173; PerCP/Cy5.5) | BioLegend | Cat#126514; RRID: AB_1186015 |
| CXCR3 (clone CXCR3-173; BV605) | BioLegend | Cat#126523; RRID: AB_2561353 |
| CXCR3 (clone CXCR3-173; BV650) | BioLegend | Cat#126531; RRID: AB_2563160 |
| CXCR4 (clone L276F12; BV421) | BioLegend | Cat #146511; RRID: AB_2562788 |
| Eomes (clone Dan11mag; PE-Cy7) | ThermoFisher | Cat #25-4875-82; RRID: AB_2573454 |
| GzmA (clone GzA-3G8.5; PE) | ThermoFisher | Cat #12-5831-82; RRID: AB_2572631 |
| GzmA (clone GzA-3G8.5; PE-Cy7) | ThermoFisher | Cat #25-5831-82; RRID: AB_2573476 |
| GzmA (clone GzA-3G8.5; APC) | ThermoFisher | Cat #17-5831-82; RRID: AB_2573228 |
| GzmB (clone GB11; FITC) | BioLegend | Cat #515403; RRID: AB_2114575 |
| GzmB (clone QA16A02; PE/Dazzle 594) | BioLegend | Cat #372216; RRID: AB_2728383 |
| GzmB (clone GB11; AlexaFluor 647) | BioLegend | Cat #515406; RRID: AB_2566333 |
| GzmB (clone QA16A02; APC) | BioLegend | Cat #372204; RRID: AB_2687028 |
| GzmB (clone QA18A28; BV421) | BioLegend | Cat #396414; RRID: AB_2810603 |
| IFNγ (clone XMG1.2; PE/Dazzle 594) | BioLegend | Cat #505846; RRID: AB_2563980 |
| IFNγ (clone XMG1.2; BV785) | BioLegend | Cat #505838; RRID: AB_2629667 |
| IL-2 (clone JES6-5H4; BV421) | BioLegend | Cat #503826; RRID: AB_2650897 |
| IL-2 (clone JES6-5H4; BV605) | BioLegend | Cat #503829; RRID: AB_11204084 |
| IL-18R (clone A17071D; AlexaFluor 647) | BioLegend | Cat #157908; RRID: AB_2876539 |
| Integrin β7 (clone FIB504; FITC) | BioLegend | Cat #321213; RRID: AB_830857 |
| Integrin β7 (clone FIB504; PE/Dazzle 594) | BioLegend | Cat #321226; RRID: AB_2715983 |
| Ki67 (clone 16A8; PE) | BioLegend | Cat #652404; RRID: AB_2561525 |
| Ki67 (clone 16A8; PE/Dazzle 594) | BioLegend | Cat #652428; RRID: AB_2632696 |
| Ki67 (clone 16A8; PerCP/Cy5.5) | BioLegend | Cat #652424; RRID: AB_2629531 |
| Ki67 (clone 16A8; BV421) | BioLegend | Cat #652411; RRID: AB_2562663 |
| KLRG1 (clone 2F1/KLRG1; PE/Dazzle 594) | BioLegend | Cat #138424; RRID: AB_2564051 |
| KLRG1 (clone 2F1/KLRG1; PerCp/Cy5.5) | BioLegend | Cat #138418; RRID: AB_2563015 |
| KLRG1 (clone 2F1/KLRG1; BV421) | BioLegend | Cat #138414; RRID: AB_2565613 |
| KLRG1 (clone 2F1/KLRG1; BV605) | BioLegend | Cat #138419; RRID: AB_2563357 |
| KLRG1 (clone 2F1/KLRG1; BV785) | BioLegend | Cat #138429; RRID: AB_2629749 |
| Ly6C (clone HK1.4; BV605) | BioLegend | Cat #128036; RRID: AB_2562353 |
| Ly108 (clone 13G3; BV421) | BD Biosciences | Cat #740090; RRID: AB_2739850 |
| P2RX7 (clone 1F11; PerCP/Cy5.5) | BioLegend | Cat #148710; RRID: AB_2728183 |
| P2RX7 (clone 1F11; PE-Cy7) | BioLegend | Cat #148708; RRID: AB_2721686 |
| P2RX7 (clone 1F11; APC) | BioLegend | Cat #148706; RRID: AB_2650954 |
| T-bet (clone 4B10; BV785) | BioLegend | Cat #644835; RRID: AB_2721566 |
| TNFα (clone MP6-XT22; BV650) | BioLegend | Cat #506333; RRID: AB_2562450 |
| T-reg protector (anti-ARTC2 Nanobody) (clone S+16a) | BioLegend | Cat#149802; RRID: AB_2565494 |
| Goat anti-Hamster IgG (H+L) Secondary Antibody | ThermoFisher | Cat#31115; RRID: AB_228247 |
| InVivoMAb anti-mouse CD3 (clone 17A2) | BioXCell | Cat#BE0002; RRID: AB_1107630 |
| InVivoMAb anti-mouse CD28 (clone PV-1) | BioXCell | Cat#BE0015-5; RRID: AB_1107628 |
| InVivoMAb anti-mouse/human IL-7 (clone M25) | BioXCell | Cat #BE0048; RRID: AB_1107711 |
| TotalSeq™-A0214 anti-human/mouse integrin β7 Antibody | BioLegend | Cat#321227; RRID: AB_2750504 |
| TotalSeq™-A0201 anti-mouse CD103 Antibody | BioLegend | Cat#121437; RRID: AB_2750349 |
| TotalSeq™-A0198 anti-mouse CD127 (IL-7Rα) Antibody | BioLegend | Cat#135045; RRID: AB_2750009 |
| TotalSeq™-A0195 anti-mouse CD134 (OX-40) Antibody | BioLegend | Cat#119426; RRID: AB_2750376 |
| TotalSeq™-A0194 anti-mouse CD137 Antibody | BioLegend | Cat#106111; RRID: AB_2783048 |
| TotalSeq™-A1006 anti-mouse CD160 Antibody | BioLegend | Cat#143013; RRID: AB_2832512 |
| TotalSeq™-A0444 anti-mouse CD184 (CXCR4) Antibody | BioLegend | Cat#146520; RRID: AB_2800682 |
| TotalSeq™-A0846 anti-mouse CD185 (CXCR5) Antibody | BioLegend | Cat#145535; RRID: AB_2800681 |
| TotalSeq™-A0376 anti-mouse CD195 (CCR5) Antibody | BioLegend | Cat#107019; RRID: AB_2783049 |
| TotalSeq™-A0225 anti-mouse CD196 (CCR6) Antibody | BioLegend | Cat#129825; RRID: AB_2783083 |
| TotalSeq™-A0854 anti-mouse CD199 (CCR9) Antibody | BioLegend | Cat#128713; RRID: AB_2832466 |
| TotalSeq™-A0378 anti-mouse CD223 (LAG-3) Antibody | BioLegend | Cat#125229; RRID: AB_2783078 |
| TotalSeq™-A0852 anti-mouse CD226 (DNAM-1) Antibody | BioLegend | Cat#128823; RRID AB_2810393 |
| TotalSeq™-A0097 anti-mouse CD25 Antibody | BioLegend | Cat#102055; RRID: AB_2749982 |
| TotalSeq™-A0191 anti-mouse/rat/human CD27 Antibody | BioLegend | Cat#124235; RRID: AB_2750344 |
| TotalSeq™-A0190 anti-mouse CD274 (B7-H1, PD-L1) Antibody | BioLegend | Cat#153604; RRID: AB_2783125 |
| TotalSeq™-A0004 anti-mouse CD279 (PD-1) Antibody | BioLegend | Cat#109123; RRID: AB_2734169 |
| TotalSeq™-A0570 anti-mouse/rat CD29 Antibody | BioLegend | Cat#102233; RRID: AB_2783042 |
| TotalSeq™-A0184 anti-mouse CD335 (NKp46) Antibody | BioLegend | Cat#137633; RRID: AB_2734199 |
| TotalSeq™-A0193 anti-mouse CD357 (GITR) Antibody | BioLegend | Cat#126319; RRID: AB_2734195 |
| TotalSeq™-A0003 anti-mouse CD366 (Tim-3) Antibody | BioLegend | Cat#119729; RRID: AB_2734178 |
| TotalSeq™-A0110 anti-mouse CD43 Antibody | BioLegend | Cat#143211; RRID: AB_2750541 |
| TotalSeq™-A0073 anti-mouse/human CD44 Antibody | BioLegend | Cat#103045; RRID: AB_2734154 |
| TotalSeq™-A0850 anti-mouse CD49a Antibody | BioLegend | Cat#142613; RRID: AB_2800659 |
| TotalSeq™-A0078 anti-mouse CD49d Antibody | BioLegend | Cat#103623; RRID: AB_2734159 |
| TotalSeq™-A0112 anti-mouse CD62L Antibody | BioLegend | Cat#104451; RRID: AB_2750364 |
| TotalSeq™-A0197 anti-mouse CD69 Antibody | BioLegend | Cat#104546; RRID: AB_2750539 |
| TotalSeq™-A0230 anti-mouse CD8b (Ly-3) Antibody | BioLegend | Cat#126623; RRID: AB_2800615 |
| TotalSeq™-A1009 anti-mouse CD94 Antibody | BioLegend | Cat#105515; RRID: AB_2819808 |
| TotalSeq™-A0847 anti-mouse CD278 (ICOS) Antibody | BioLegend | Cat#117409; RRID: AB_2800585 |
| TotalSeq™-A0563 anti-mouse CX3CR1 Antibody | BioLegend | Cat#149041; RRID: AB_2783121 |
| TotalSeq™-A0250 anti-mouse/human KLRG1 (MAFA) Antibody | BioLegend | Cat#138431; RRID: AB_2800648 |
| TotalSeq™-A0930 anti-mouse Ly108 Antibody | BioLegend | Cat#134611; RRID: AB_2888706 |
| TotalSeq™-A0824 anti-mouse P2X7R Antibody | BioLegend | Cat#148711; RRID: AB_200683 |
| TotalSeq™-A0848 anti-mouse TIGIT (Vstm3) Antibody | BioLegend | Cat#142115; RRID: AB_2800656 |
| TotalSeq™-A0905 anti-mouse CD107a (LAMP-1) Antibody | BioLegend | Cat#121635; RRID: AB_2810369 |
| TotalSeq™-A0839 anti-mouse Ly49H Antibody | BioLegend | Cat#144715; RRID: AB_2814049 |
| TotalSeq™-A0111 anti-mouse CD5 Antibody | BioLegend | Cat#100637; RRID: AB_2749985 |
| TotalSeq™-A1019 anti-mouse CD215 (IL-15Rα) Antibody | BioLegend | Cat#153507; RRID: AB_2832537 |
| TotalSeq™-A0557 anti-mouse CD38 Antibody | BioLegend | Cat#102733; RRID: AB_2750556 |
| TotalSeq™-A0835 anti-mouse CD314 (NKG2D) Antibody | BioLegend | Cat#130215; RRID: AB_2814023 |
| TotalSeq™-A0834 anti-mouse CD39 Antibody | BioLegend | Cat#143813; RRID: AB_2800669 |
| TotalSeq™-A0013 anti-mouse Ly-6C Antibody | BioLegend | Cat#128047; RRID: AB_2749961 |
| TotalSeq™-A0885 anti-mouse CD270 (HVEM) Antibody | BioLegend | Cat#136307; RRID: AB_2810403 |
| TotalSeq™-A0989 anti-mouse CD98 (4F2) Antibody | BioLegend | Cat#128223; RRID: AB_2876456 |
| TotalSeq™-A0388 anti-mouse CD152 Antibody | BioLegend | Cat#106325; RRID: AB_2876417 |
| TotalSeq™-A0883 anti-mouse CD26 (DPP-4) Antibody | BioLegend | Cat#137811; RRID: AB_2810405 |
| TotalSeq™-A0157 anti-mouse CD45.2 Antibody | BioLegend | Cat#109853; RRID: AB_2783051 |
| TotalSeq™-A0178 anti-mouse CD45.1 Antibody | BioLegend | Cat#110753; RRID: AB_2800573 |
| TotalSeq™-A0911 anti-phycoerythrin (PE) Antibody | BioLegend | Cat#408109; RRID: AB_2820078 |
| TotalSeq™-A0227 anti-mouse CD122 (IL-2Rb) | BioLegend | Custom |
| Bacterial and viral strains | ||
| LCMV-Armstrong | This paper | N/A |
| Bcl-2 overexpression retrovirus | This paper | N/A |
| Eomes overexpression retrovirus | This paper | N/A |
| TGFβRII overexpression retrovirus | This paper | N/A |
| Empty vector retrovirus | This paper | N/A |
| shRNA non-target retrovirus | This paper | N/A |
| shRNA P2RX7 retrovirus | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Murine IL-7 | PeproTech | Cat#217-17 |
| TransIT-LT1 Transfection Reagent | Mirus | Cat#MIR 23000 |
| eBioscience Fixable Viability Dye eFluor 780 | ThermoFisher | Cat#65-0865-18 |
| eBioscience Fixation/Perm Diluent | ThermoFisher | Cat#00-5223-56 |
| Fixation/Permeabilization concentrate | ThermoFIsher | Cat#00-5123-43 |
| Permeabilization Buffer 10× | ThermoFisher | Cat#00-8333-56 |
| Paraformaldehyde 16% Aqueous solution, EM grade | Fisher Scientific | Cat#50-980-487 |
| Fetal Bovine Serum | Genesee Scientific | Cat#25-525H |
| Ethylenediaminetetraacetic acid | Fisher Scientific | Cat#BP2482100 |
| Percoll | Sigma Aldrich | Cat#P1644 |
| L-glutamine 200 mM (100×) | ThermoFisher | Cat#25030-081 |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher | Cat#15140-122 |
| HEPES | Sigma Aldrich | Cat#H3375 |
| 4-hydroxytamoxifen | Sigma Aldrich | Cat#T176 |
| Sunflower seed oil | Sigma Aldrich | Cat#S5007 |
| Isoflurane | Vetone | Cat#502017 |
| Ethyl alcohol | Sigma Aldrich | Cat#E7023 |
| Dulbecco’s Modified Eagle Medium | ThermoFisher | Cat#11965-092 |
| Collagenase from Clostridium hystolyticum (Type IV) | Sigma Aldrich | Cat#C5138 |
| Dithiothretiol (DTT) | ThermoFisher | Cat#R0861 |
| DNase I, grade II, from bovine pancreas | Roche | Cat#10104159001 |
| Hank’s Balanced Salt Solution | Corning | Cat#21-021-CV |
| Dulbecco’s Phosphate-Buffered Saline | Corning | Cat#21-031-CM |
| Critical commercial assays | ||
| CD8+ T Cell Isolation Kit, mouse | Miltenyi | Cat#130-104-075 |
| Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1 | 10X Genomics | Cat#PN-1000121 |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10X Genomics | Cat#PN-1000175 |
| Chromium Next GEM Chip G Single Cell Kit, 16 rxns | 10X Genomics | Cat#PN-1000127 |
| Chromium Next GEM Chip H Single Cell Kit, 16 rxns | 10X Genomics | Cat#PN-1000162 |
| Deposited data | ||
| CITE-seq data | This paper | GEO: GSE205942 |
| scATAC-seq data | This paper | GEO: GSE205942 |
| Teichmann Gut Atlas - T/NK cells | Elmentaite et al., 2021 | ArrayExpress: E-MTAB-9532 |
| Experimental models: Cell lines | ||
| Platinum-E (Plat-E) cell line | Cell Biolabs, Inc. | Cat#RV-101; RRID: CVCL_B488 |
| BHK-21 (C13) | ATCC | Cat#CCL-10; RRID: CVCL_1915 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) | Jackson Laboratory | RRID: IMSR_JAX:002014 |
| Mouse: C57BL/6J (CD45.2) | Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: B6.Cg-Tcratm1Mom Tg(TcrLCMV)327Sdz (P14) | Jackson Laboratory | RRID: MMRRC_037394-MU |
| Mouse: B6.129S1(Cg)-Eomestm1.1Bflu/J (Eomes fl/fl) | Jackson Laboratory | RRID: IMSR_JAX:017293 |
| Mouse: B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ (CD4-Cre) | Jackson Laboratory | RRID: IMSR_JAX:022071 |
| Mouse: B6.Cg-Tg(CAG-cre/Esr1*)5Amc/J (ER-Cre) | Jackson Laboratory | RRID: IMSR_JAX:004682 |
| Mouse: B6;129-Tgfbr2tm1Karl/J (TGFβR2 fl/fl) | Jackson Laboratory | RRID: IMSR_JAX:012603 |
| Recombinant DNA | ||
| pCL-Eco Retrovirus packaging vector | Addgene | RRID: Addgene_12371 |
| pMIG Vector | Addgene | RRID: Addgene_9044 |
| pMIG Eomes Vector | Dr. Steven Reiner | N/A |
| pMIG Bcl-2 Vector | Dr. Michael Croft | RRID: Addgene_8793 |
| pMIG TGFβRII Vector | Dr. Wanjun Chen | N/A |
| pLMPd-Ametrine | Transomic | shRNA retroviral target gene set |
| shERWOOD UltramiR Lentiviral shRNA target gene set for gene P2RX7 | Transomic | Car#TLHSU1435 |
| Software and algorithms | ||
| FlowJo v10.8.1 | FlowJo | https://www.flowjo.com/ |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Cell Ranger 6.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat 4.1.0.9001 | Hao et al., 2021 | https://satijalab.org/seurat/ |
| Metascape | Zhou et al., 2019 | https://metascape.org/ |
| scVelo v0.2.4 | Bergen et al., 2020 | https://scvelo.readthedocs.io/ |
| Signac v1.6.0 | Stuart et al., 2021 | https://satijalab.org/signac/ |
| pheatmap v1.0.8 (R package) | Kolde, 2019 | https://cran.r-project.org/package=pheatmap |
| VennDiagram v1.7.3 (R package) | Chen, 2022 | https://cran.r-project.org/package=VennDiagram |
HIGHLIGHTS.
Small intestine (SI) and colon CD8+ TRM cells are molecularly and functionally distinct
Anatomically distinct SI TRM cells exhibit disparate degrees of developmental plasticity
Eomes supports maintenance of established TRM cells in the SI, but not in the colon
Eomes promotes expression of the anti-apoptotic regulator Bcl-2
ACKNOWLEDGMENTS
CITE-seq and scATAC-seq were performed at the UCSD IGM Genomics Center and supported by NIH grants P30KC063491, P30CA023100, and S10OD026929. M.S.T. was supported by NIH grant DK007202. P.H. was supported by NIH grant AR064194. This work was supported by the NIDDK-funded San Diego Digestive Diseases Research Center (P30DK120515) and grants from the NIH: AI129973, BX005106, and CX002396 (J.T.C.); AI123202 (G.W.Y. and J.T.C.); AI132122 (A.W.G., J.T.C., G.W.Y.); HG004659 (G.W.Y.).
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research.
Footnotes
DECLARATION OF INTERESTS
G.W.Y. is a co-founder, member of the Board of Directors, on the SAB, equity holder, and paid consultant for Locanabio and Eclipse BioInnovations. In addition, G.W.Y. is a visiting professor at the National University of Singapore. G.W.Y.’s interest(s) have been reviewed and approved by the University of California, San Diego in accordance with its conflict- of-interest policies. J.T.C. reported grants from Takeda and Eli Lilly outside the submitted work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Chang JT, Wherry EJ, and Goldrath AW (2014). Molecular regulation of effector and memory T cell differentiation. Nature immunology 15, 1104–1115. 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milner JJ, Nguyen H, Omilusik K, Reina-Campos M, Tsai M, Toma C, Delpoux A, Boland BS, Hedrick SM, Chang JT, and Goldrath AW (2020). Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc Natl Acad Sci U S A 117, 25667–25678. 10.1073/pnas.2008571117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, and von Andrian UH (2016). The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 45, 1270–1284. 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sallusto F, Lenig D, Forster R, Lipp M, and Lanzavecchia A (1999). Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712. 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 5.Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, Scott MC, Fares-Frederickson NJ, Wijeyesinghe S, Thompson EA, et al. (2020). Developmental plasticity allows outside-in immune responses by resident memory T cells. Nature immunology 21, 412–421. 10.1038/s41590-020-0607-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stolley JM, Johnston TS, Soerens AG, Beura LK, Rosato PC, Joag V, Wijeyesinghe SP, Langlois RA, Osum KC, Mitchell JS, and Masopust D (2020). Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J Exp Med 217. 10.1084/jem.20192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, and Carbone FR (2009). Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nature immunology 10, 524–530. 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 8.Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, et al. (2010). Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med 207, 553–564. 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masopust D, Vezys V, Wherry EJ, Barber DL, and Ahmed R (2006). Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol 176, 2079–2083. 10.4049/jimmunol.176.4.2079. [DOI] [PubMed] [Google Scholar]
- 10.Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, and Bevan MJ (2012). The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol 189, 3462–3471. 10.4049/jimmunol.1201305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. (2017). Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257. 10.1038/nature24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, McBain N, Wagner T, Edwards J, McConville R, et al. (2019). Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 565, 366–371. 10.1038/s41586-018-0812-9. [DOI] [PubMed] [Google Scholar]
- 13.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, et al. (2018). Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 24, 986–993. 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 14.Boland BS, He Z, Tsai MS, Olvera JG, Omilusik KD, Duong HG, Kim ES, Limary AE, Jin W, Milner JJ, et al. (2020). Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol 5. 10.1126/sciimmunol.abb4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, and Nestle FO (2004). Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med 199, 731–736. 10.1084/jem.20031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheuk S, Schlums H, Gallais Serezal I, Martini E, Chiang SC, Marquardt N, Gibbs A, Detlofsson E, Introini A, Forkel M, et al. (2017). CD49a Expression Defines Tissue-Resident CD8(+) T Cells Poised for Cytotoxic Function in Human Skin. Immunity 46, 287–300. 10.1016/j.immuni.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang JT (2020). Pathophysiology of Inflammatory Bowel Diseases. N Engl J Med 383, 2652–2664. 10.1056/NEJMra2002697. [DOI] [PubMed] [Google Scholar]
- 18.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. (2013). The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nature immunology 14, 1294–1301. 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 19.Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, and Jameson SC (2013). Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nature immunology 14, 1285–1293. 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, et al. (2016). Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463. 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 21.Behr FM, Parga-Vidal L, Kragten NAM, van Dam TJP, Wesselink TH, Sheridan BS, Arens R, van Lier RAW, Stark R, and van Gisbergen K (2020). Tissue-resident memory CD8(+) T cells shape local and systemic secondary T cell responses. Nature immunology 21, 1070–1081. 10.1038/s41590-020-0723-4. [DOI] [PubMed] [Google Scholar]
- 22.Christo SN, Evrard M, Park SL, Gandolfo LC, Burn TN, Fonseca R, Newman DM, Alexandre YO, Collins N, Zamudio NM, et al. (2021). Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nature immunology 22, 1140–1151. 10.1038/s41590-021-01004-1. [DOI] [PubMed] [Google Scholar]
- 23.Crowl JT, Heeg M, Ferry A, Milner JJ, Omilusik KD, Toma C, He Z, Chang JT, and Goldrath AW (2022). Tissue-resident memory CD8(+) T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments. Nature immunology 23, 1121–1131. 10.1038/s41590-022-01229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Behr FM, Kragten NAM, Wesselink TH, Nota B, van Lier RAW, Amsen D, Stark R, Hombrink P, and van Gisbergen K (2019). Blimp-1 Rather Than Hobit Drives the Formation of Tissue-Resident Memory CD8(+) T Cells in the Lungs. Front Immunol 10, 400. 10.3389/fimmu.2019.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, Lietzenmayer M, Kroehling L, Takumi A, Kometani K, et al. (2018). KLRG1(+) Effector CD8(+) T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity 48, 716–729 e718. 10.1016/j.immuni.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holz LE, Prier JE, Freestone D, Steiner TM, English K, Johnson DN, Mollard V, Cozijnsen A, Davey GM, Godfrey DI, et al. (2018). CD8(+) T Cell Activation Leads to Constitutive Formation of Liver Tissue-Resident Memory T Cells that Seed a Large and Flexible Niche in the Liver. Cell Rep 25, 68–79 e64. 10.1016/j.celrep.2018.08.094. [DOI] [PubMed] [Google Scholar]
- 27.Schenkel JM, Fraser KA, Casey KA, Beura LK, Pauken KE, Vezys V, and Masopust D (2016). IL-15-Independent Maintenance of Tissue-Resident and Boosted Effector Memory CD8 T Cells. J Immunol 196, 3920–3926. 10.4049/jimmunol.1502337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng C, Huggins MA, Wanhainen KM, Knutson TP, Lu H, Georgiev H, Mittelsteadt KL, Jarjour NN, Wang H, Hogquist KA, et al. (2022). Engagement of the costimulatory molecule ICOS in tissues promotes establishment of CD8(+) tissue-resident memory T cells. Immunity 55, 98–114 e115. 10.1016/j.immuni.2021.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, and Lefrancois L (2014). Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 40, 747–757. 10.1016/j.immuni.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang N, and Bevan MJ (2013). Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696. 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, and Carbone FR (2015). T-box Transcription Factors Combine with the Cytokines TGF-beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 43, 1101–1111. 10.1016/j.immuni.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Parga-Vidal L, Behr FM, Kragten NAM, Nota B, Wesselink TH, Kavazovic I, Covill LE, Schuller MBP, Bryceson YT, Wensveen FM, et al. (2021). Hobit identifies tissue-resident memory T cell precursors that are regulated by Eomes. Sci Immunol 6. 10.1126/sciimmunol.abg3533. [DOI] [PubMed] [Google Scholar]
- 33.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Southern PJ, and Masopust D (2015). Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749. 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, and Masopust D (2012). Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol 189, 2702–2706. 10.4049/jimmunol.1201682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865–868. 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borges da Silva H, Beura LK, Wang H, Hanse EA, Gore R, Scott MC, Walsh DA, Block KE, Fonseca R, Yan Y, et al. (2018). The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 559, 264–268. 10.1038/s41586-018-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stark R, Wesselink TH, Behr FM, Kragten NAM, Arens R, Koch-Nolte F, van Gisbergen K, and van Lier RAW (2018). T RM maintenance is regulated by tissue damage via P2RX7. Sci Immunol 3. 10.1126/sciimmunol.aau1022. [DOI] [PubMed] [Google Scholar]
- 39.Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, Manton JH, Heath WR, and Mueller SN (2014). Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci U S A 111, 5307–5312. 10.1073/pnas.1322292111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, and Held W (2010). Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci U S A 107, 9777–9782. 10.1073/pnas.0914127107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao DM, Yu S, Zhou X, Haring JS, Held W, Badovinac VP, Harty JT, and Xue HH (2010). Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J Immunol 184, 1191–1199. 10.4049/jimmunol.0901199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 38, 1408–1414. 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 43.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, et al. (2005). Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nature immunology 6, 1236–1244. 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 45.Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, Wherry EJ, and Reiner SL (2010). Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol 185, 4988–4992. 10.4049/jimmunol.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borges da Silva H, Peng C, Wang H, Wanhainen KM, Ma C, Lopez S, Khoruts A, Zhang N, and Jameson SC (2020). Sensing of ATP via the Purinergic Receptor P2RX7 Promotes CD8(+) Trm Cell Generation by Enhancing Their Sensitivity to the Cytokine TGF-beta. Immunity 53, 158–171 e156. 10.1016/j.immuni.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyman O, Ramsey C, Kim DM, Sprent J, and Surh CD (2008). IL-7/anti-IL-7 mAb complexes restore T cell development and induce homeostatic T Cell expansion without lymphopenia. J Immunol 180, 7265–7275. 10.4049/jimmunol.180.11.7265. [DOI] [PubMed] [Google Scholar]
- 48.Kavazovic I, Han H, Balzaretti G, Slinger E, Lemmermann NAW, Ten Brinke A, Merkler D, Koster J, Bryceson YT, de Vries N, et al. (2020). Eomes broadens the scope of CD8 T-cell memory by inhibiting apoptosis in cells of low affinity. PLoS Biol 18, e3000648. 10.1371/journal.pbio.3000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fung HY, Teryek M, Lemenze AD, and Bergsbaken T (2022). CD103 fate mapping reveals that intestinal CD103(−) tissue-resident memory T cells are the primary responders to secondary infection. Sci Immunol 7, eabl9925. 10.1126/sciimmunol.abl9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Hoesslin M, Kuhlmann M, de Almeida GP, Kanev K, Wurmser C, Gerullis AK, Roelli P, Berner J, and Zehn D (2022). Secondary infections rejuvenate the intestinal CD103(+) tissue-resident memory T cell pool. Sci Immunol 7, eabp9553. 10.1126/sciimmunol.abp9553. [DOI] [PubMed] [Google Scholar]
- 51.Kurd NS, He Z, Louis TL, Milner JJ, Omilusik KD, Jin W, Tsai MS, Widjaja CE, Kanbar JN, Olvera JG, et al. (2020). Early precursors and molecular determinants of tissue-resident memory CD8(+) T lymphocytes revealed by single-cell RNA sequencing. Sci Immunol 5. 10.1126/sciimmunol.aaz6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z, Beckermann KE, Wu Y, Jenkins JW, et al. (2019). The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8(+) T Cell Fitness and Functionality. Immunity 51, 491–507 e497. 10.1016/j.immuni.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boddupalli CS, Nair S, Gray SM, Nowyhed HN, Verma R, Gibson JA, Abraham C, Narayan D, Vasquez J, Hedrick CC, et al. (2016). ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J Clin Invest 126, 3905–3916. 10.1172/JCI85329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirai T, Yang Y, Zenke Y, Li H, Chaudhri VK, De La Cruz Diaz JS, Zhou PY, Nguyen BA, Bartholin L, Workman CJ, et al. (2021). Competition for Active TGFbeta Cytokine Allows for Selective Retention of Antigen-Specific Tissue- Resident Memory T Cells in the Epidermal Niche. Immunity 54, 84–98 e85. 10.1016/j.immuni.2020.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, et al. (2003). Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043. 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 56.Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, Fonseca R, Burbach BJ, Hickman HD, Vezys V, et al. (2018). Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nature immunology 19, 173–182. 10.1038/s41590-017-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, et al. (2018). Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nature immunology 19, 183–191. 10.1038/s41590-017-0027-5. [DOI] [PubMed] [Google Scholar]
- 58.Thompson EA, Mitchell JS, Beura LK, Torres DJ, Mrass P, Pierson MJ, Cannon JL, Masopust D, Fife BT, and Vezys V (2019). Interstitial Migration of CD8alphabeta T Cells in the Small Intestine Is Dynamic and Is Dictated by Environmental Cues. Cell Rep 26, 2859–2867 e2854. 10.1016/j.celrep.2019.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beura LK, Anderson KG, Schenkel JM, Locquiao JJ, Fraser KA, Vezys V, Pepper M, and Masopust D (2015). Lymphocytic choriomeningitis virus persistence promotes effector-like memory differentiation and enhances mucosal T cell distribution. J Leukoc Biol 97, 217–225. 10.1189/jlb.1HI0314-154R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labarta-Bajo L, Nilsen SP, Humphrey G, Schwartz T, Sanders K, Swafford A, Knight R, Turner JR, and Zuniga EI (2020). Type I IFNs and CD8 T cells increase intestinal barrier permeability after chronic viral infection. J Exp Med 217. 10.1084/jem.20192276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Macleod BL, Elsaesser HJ, Snell LM, Dickson RJ, Guo M, Hezaveh K, Xu W, Kothari A, McGaha TL, Guidos CJ, and Brooks DG (2020). A network of immune and microbial modifications underlies viral persistence in the gastrointestinal tract. J Exp Med 217. 10.1084/jem.20191473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khan TN, Mooster JL, Kilgore AM, Osborn JF, and Nolz JC (2016). Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med 213, 951–966. 10.1084/jem.20151855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maier T, Guell M, and Serrano L (2009). Correlation of mRNA and protein in complex biological samples. FEBS Lett 583, 3966–3973. 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 64.Myers SA, Rhoads A, Cocco AR, Peckner R, Haber AL, Schweitzer LD, Krug K, Mani DR, Clauser KR, Rozenblatt-Rosen O, et al. (2019). Streamlined Protocol for Deep Proteomic Profiling of FAC-sorted Cells and Its Application to Freshly Isolated Murine Immune Cells. Mol Cell Proteomics 18, 995–1009. 10.1074/mcp.RA118.001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reimegard J, Tarbier M, Danielsson M, Schuster J, Baskaran S, Panagiotou S, Dahl N, Friedlander MR, and Gallant CJ (2021). A combined approach for single-cell mRNA and intracellular protein expression analysis. Commun Biol 4, 624. 10.1038/s42003-021-02142-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stuart T, Srivastava A, Madad S, Lareau CA, and Satija R (2021). Single-cell chromatin state analysis with Signac. Nat Methods 18, 1333–1341. 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes for intestinal TRM cells separated by CITE-seq clusters, Related to Figures 3 and S2
Table S2. Gene and protein expression, and pathway analyses of intestinal TRM cells by CITE-seq clusters, Related to Figures 3 and S2
Table S3. Gene and protein expression of intestinal TRM cells, and differentially expressed genes of the same, Related to Figure 3
Table S4. Differentially accessible chromatin regions of intestinal TRM cells, Related to Figures 3 and S4
Table S5. Enriched transcription factor binding motifs within differentially accessible chromatin regions of intestinal TRM cells, Related to Figures 3 and S4
Table S6. Uniquely enriched or shared transcription factor binding motifs within differentially accessible chromatin regions of intestinal TRM cells, Related to Figures 3 and S4.
Table S7. Gene and protein expression, and pathway analysis of control vs. Eomes iKO intestinal TRM cells, Related to Figure 7.
Data Availability Statement
All data reported in this paper are available at Gene Expression Omnibus under accession number GSE205942.