

Abstract
Metastatic Leydig cell tumors (LCT) are rare, difficult to treat malignancies without known underlying molecular-genetic events. We profiled 27 LCT cases using NGS and immunohistochemistry. Our study identified TERT gene fusions as a main genetic alteration and a potential therapeutic target in LCT. TOP1 and AR expressions may guide decisions on chemo- and/or hormone therapy for selected individual patients.
Objective:
Metastatic Leydig cell tumors (LCT) are rare, difficult-to-treat malignancies without known underlying molecular–genetic events. An index case of metastatic LCT showed an LDLR–TERT gene fusion upon routine genetic profiling for detection of therapeutic targets, which was then followed by an investigation into a cohort of additional LCTs.
Patients and Methods:
Twenty-nine LCT (27 male and 2 female patients) were profiled using next-generation sequencing and immunohistochemistry.
Results:
TERT gene fusions were detected only in testicular metastatic LCTs, in 3 of 7 successfully analyzed cases (RMST:TERT,LDLR:TERT, and B4GALT5:TERT). TOP1 and CCND3 amplifications were identified in the case with a B4GALT5:TERT fusion. A TP53 mutation was detected in 1 metastatic tumor without a TERT fusion. Five primary (4 testicular and 1 ovarian) LCTs showed multiple gene amplifications, without a consistent pattern. A single metastatic ovarian LCT showed BAP1 mutation and copy number amplifications affecting the NPM1, PCM1, and SS18 genes. At the protein level, 4 of 7 metastatic and 6 of 10 primary testicular LCTs overexpressed Topo1. Androgen receptor was overexpressed in 10 of 13 primary testicular tumors and 2 of 5 metastatic testicular LCTs (without detectable ARv7 messenger RNA or ARv7 protein). Only 1 metastatic testicular LCT exhibited a high tumor mutational burden; all tested cases were microsatellite instability stable and did not express programmed cell death ligand 1.
Conclusions:
Our study for the first time identified TERT gene fusions as a main genetic alteration and a potential therapeutic target in metastatic LCTs. Topo1 and androgen receptor may guide decisions on chemotherapy and/or hormone therapy for selected individual patients.
Keywords: Sex cord–stromal tumors, Molecular profiling, Sequencing, Targeted therapy, Leydig cell tumors
Introduction
Sex cord–stromal tumors are an uncommon group of neoplasms affecting gonads. In the testis, these tumors represent 4% of all neoplasms and are the second largest group of primary tumors after germ cell tumors.1 In ovary, sex cord–stromal tumors constitute 5% of all neoplasms, and 7% of malignant ovarian neoplasms belong to this group.2 Leydig cell tumors (LCT) are the most common pure form of sex cord–stromal tumor followed by Sertoli cell, granulosa cell, and pure stromal tumors.1 Little is known about the pathogenesis of these neoplasms beyond their rare association with germline fumarate hydratase mutations (hereditary leiomyomatosis and renal cell carcinoma syndrome, OMIM#150800) or the activating mutations that affect luteinizing hormone receptor in the pediatric population.1 In addition, DICER1 mutations have been reported in sporadic and hereditary ovarian sex cord stromal tumors.3–6 DICER1 gene mutations have been implicated in the dysregulation of the steroid hormone synthesis including androgen receptor (AR).7
Molecular profiling studies on these tumors are sparse owing to the overwhelmingly benign course of the disease and curative surgical resection.8 A recent whole exome sequencing study of Yuan et al8 revealed that LCTs frequently harbor somatic mutations of CDC27 (53%), DICER1 (21%), and MUC22 (21%) genes. Metastatic LCTs are clinically challenging and without a consensus treatment approach.
We have previously characterized multiple cancers with a comprehensive molecular profiling approach that uses various molecular techniques for the identification of potentially targetable biomarkers.9–13 Our initial case of metastatic LCT showed an LDLR:TERT gene fusion upon routine genetic profiling for the detection of therapeutic targets. This finding led us to investigate a cohort of additional LCTs.
Materials and Methods
Samples for the Study
Twenty-nine LCTs from 5 participating institutions (University Hospital Centre “Sestre milosrdnice,” Zagreb, Croatia; Caris Life Sciences, Phoenix, AZ; University of Oklahoma College of Medicine, Oklahoma City, OK; Charles University Hospital Plzen, Pilsen, Czech Republic; and National Cancer Institute, Bethesda, MD) were included in the current study.
Before molecular testing, each LCT case underwent confirmation of the histologic diagnosis and a review of the diagnostic immunohistochemical workup performed at the referring/participating pathology laboratories. For the study, all histopathologic reports and remnant LCT tissue samples provided by the referring laboratories and participating institutions were de-identified. Based on this, the study was compliant with 45 CFR 46.101(b) and was deemed exempt from institutional review board (IRB) approval and consent requirements were waived.
All molecular assays were performed at a CLIA/CAP/ISO15189/NYSDOH certified clinical laboratory (Caris Life Sciences, Phoenix, AZ).
Immunohistochemistry
PD-L1 expression was assessed in the tumor cells and immune cells using SP142 antibody (Ventana Medical Systems, Oro Valley, AZ). PD-L1 expression was considered positive if either tumor cell or immune cells exhibited any membranous or cytoplasmic staining.11,14 AR (clone 441, Leica Biosystems, Buffalo Grove, IL) was analyzed using a 10% or higher threshold for nuclear positivity.12–14 The ARv7 splice variant was explored at the protein level by immunohistochemistry (EPR15656; Abcam) and at the messenger RNA level using anchored multiplex polymerase chain reaction for targeted RNA sequencing (ArcherDX).9,11 Topoisomerase 1 (Topo1) expression (clone 1D6; Leica Biosystems, Nussloch, Germany) was scored as 0+, 1+, 2+, or 3+ depending on the staining intensity, and the percent tumor stained was also recorded. The threshold for Topo1 overexpression was a staining intensity of 2+ or higher in 30% or more of the cancer cells.15
Next-Generation Sequencing
The LCT samples were profiled using next-generation sequencing (NGS) of exons from 592 genes (SureSelect XT, Agilent, Santa Clara, CA; and the NextSeq instrument, Illumina, San Diego, CA). A full gene panel is available in the Supplemental Table 1.
For the study, the tumor mutational burden (TMB) was assessed by calculating the number of non-synonymous missense mutations, excluding common germline variants, per one megabase of DNA. TMB was considered high if 10 or more mutations/megabase (muts/Mb) were detected.16
Microsatellite instability (MSI) was calculated from the NGS data by direct analysis of short tandem repeat tracts in the target regions of sequenced genes. The count only included alterations that resulted in increases or decreases in the number of repeats; high MSI was defined as 46 or more altered microsatellite loci. This threshold was established by comparing NGS with the polymerase chain reaction–based microsatellite fragments analysis results from approximately 2100 samples.10
Copy number amplifications were assessed by comparing the depth of detected NGS sequence reads to calibrated control values. Genes having 6 or more copies were considered amplified.
The ArcherDx FusionPlex Assay (ArcherDX, Boulder, CO) was used for gene fusion assessment. The gene fusions panel (n = 54) is available in the Supplemental Table 2.
Results
Clinicopathologic Characteristics of the Cohort
Twenty-seven testicular (7 metastatic and 20 primary tumors) and 2 ovarian LCT (1 metastatic and 1 primary) were investigated. The mean patient age was 55.5 years (range, 23–94 years) for male patients; the 2 female patients with ovarian LCT were 45 and 69 years of age. The metastatic sites of testicular LCT included lung, liver, mediastinum, parasternal region, and retroperitoneum (×3). The only ovarian metastatic LCT site was peritoneum (4 years after the original ovarian tumor diagnosis).
Immunohistochemical Biomarkers
TOP1 was assessed in 17 testicular LCT: 6 of 10 primary (60%) and 4 of 7 metastatic (57%) LCT were positive (Table 1 and Fig. 1). Intriguingly, a single TOP1 amplified testicular LCT showed no Topo1 protein expression by immunohistochemistry. AR expression was more prevalent among the primary testicular LCT (10/13) compared with the metastatic cases (2/5). All cases were ARv7 negative (at either the messenger RNA or protein levels).
Table 1.
Molecular Findings in the Leydig Cell Tumors Cohort
| Biomarkers (number) | Testis (n = 27) |
|
|---|---|---|
| Primary ( n = 20) | Metastatic (n = 7) | |
| Topo1α protein* (IHC) (n = 17) | 6/10 (60%) | 4/7 (57%) |
| AR* (n = 18) | 10/13 (77%) | 2/5 (40%) |
| ARv7 (NGS and IHC) (n = 18) | All ARv7 negative (mRNA or protein) | All ARv7 negative (mRNA or protein) |
|
Genomic alterations
|
||
| TERT gene fusions (NGS)† (n = 19) | 0/12 (0%) | 3+/7 (43%): LDLR:TERT B4GALT5:TERT RMSTTERT |
| Mutational profile (NGS) (n = 15) | CTNNB1 (2/10), NBN (1/10), MTOR (1/10), FOXO4 (1/10), BAP1 (1/10), MEN1 (1/10), CREBBP (1/10) |
TP53 (1/5) FOXO4 (1/5) |
| Copy number amplifications (NGS) (n = 16) |
MDM2, TCF3, LRIG3, HMGA2, CYP2D6, ASPSCR1 (1 case) CDKN1B, DAXX, DDX5, PER1, VEGFB (1 case) MDM2, CDK4, CCND3, TFEB (1 case) GATA3, FGFR3, AKT2, TLX1, PIK3R2, MEF2B, JAK3, ERCC2, ELL, CIC, CD79A, CBFA2T3, BCL3 (1 case) |
TOP1, CCND3, MCL1 (1 case) FGFR4, FLT4 (1 case) |
|
I-O biomarkers
|
||
| PD-L1 expression* (n = 15) | 0/10 (0%) | 0/5 (0%) |
| TMB (n = 7) | 4–7/Mb (n = 4) | 4–11/Mb (n = 3) |
| MSI (n = 7) | Stable (n = 4) | Stable (n = 3) |
| Biomarkers |
Ovary (n = 2)
|
|
| Primary (n = 1) | Metastatic (n = 1) | |
|
|
||
| Topo1α protein* | Not available | Not available |
| AR* | Not available | Not available |
|
Genomic alterations
|
||
| TERT gene fusions (NGS)† | Absent | Absent |
| Mutational profile (NGS) | None | BAP1 |
| Copy number amplifications (NGS) | FGF3, FGFR3 | NPM1, PCM1, SS18 |
|
I-O biomarkers
|
||
| PD-L1 expression* | Not available | Not available |
| TMB | Not available | 3 mutations/Mb |
| MSI | Not available | Not available |
Assessed by IHC.
Archer FusionPlex assay; TERT promoter region was not covered by the analysis.AR, androgen receptor; CAN, copy number amplifications; IHC, immunohistochemistry; I-O, immuno-oncology; mRNA, messenger RNA; MSI, microsatellite instability; NGS, next-generation sequencing; PD-L1, programmed cell death ligand 1; TERT, telomerase reverse transcriptase; TMB, tumor mutational burden.
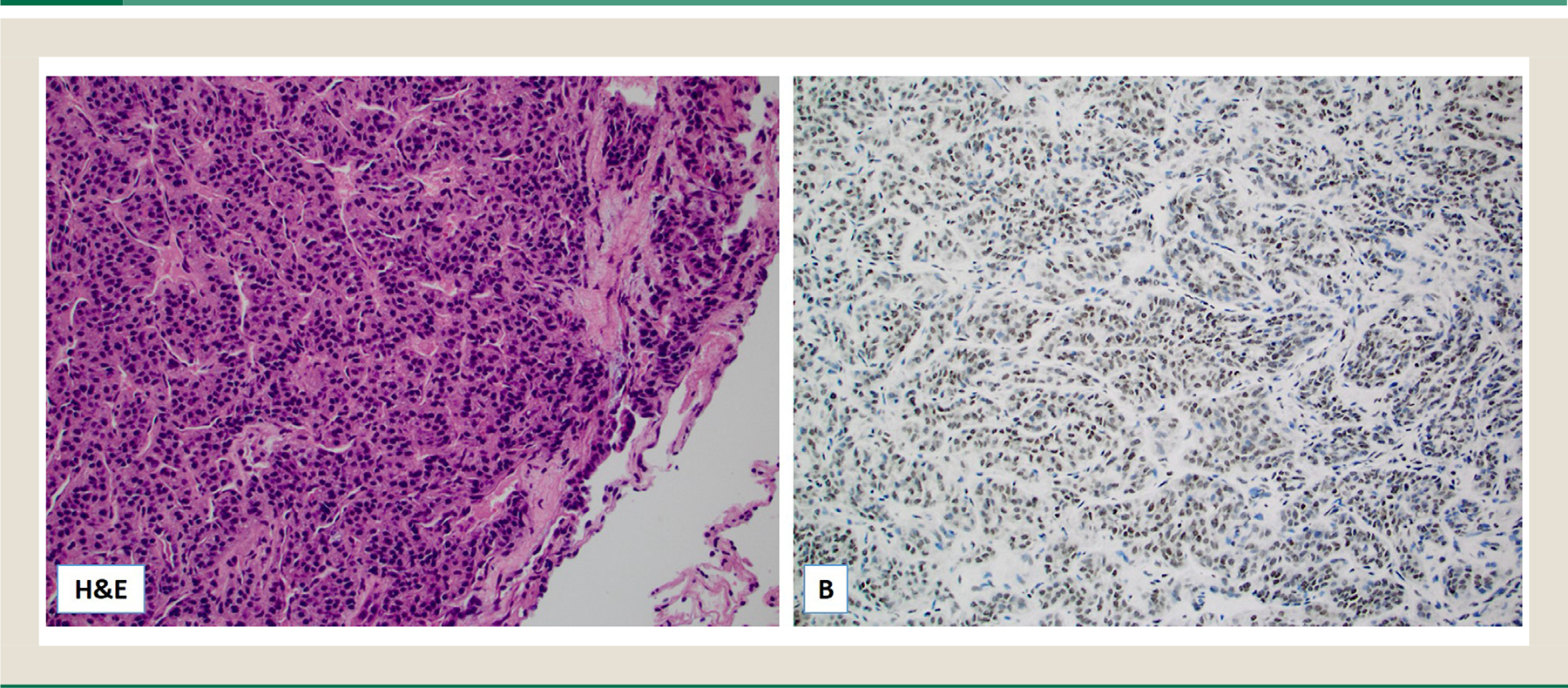
Fig. 1. Hematoxylin and eosin (H&E) slide of a metastatic Leydig cell tumor to the lung (A); the tumor cells were diffusely positive for Topo1 by immunohistochemistry (original magnification ×20).
Genomic Characteristics of LCT
Telomerase reverse transcriptase (TERT) gene fusions were exclusively seen in 3 of 7 successfully analyzed metastatic testicular LCT. The following fusions were detected: RMST:TERT, LDLR:TERT, and B4GALT5:TERT (Fig. 2). The specimen harboring the B4GALT5:TERT fusion also showed amplifications (>6 copies) of the TOP1 and CCND3 genes (Table 1). Neither of the 2 ovarian LCT harbored TERT-related fusions. TERT promoter mutations were not tested, because this region was not covered in the available commercial NGS panel at the time.
Fig. 2. Telomerase reverse transcriptase (TERT) gene fusions detected in 3 metastatic (malignant) Leydig cell tumors of the testis.

A NGS mutational profile was available for 15 testicular cases, which showed inconsistent and rare pathogenic mutations: 2 primary LCT harbored CTNNB1 gene mutations (encoding β-catenin protein); FOXO4 mutations were also observed in 2 cases (1 primary and 1 metastatic case), and a TP53 mutation was observed in 1 metastatic LCT. All other mutations were detected in single cases (NBN, MTOR, BAP1, MEN1, and CREBBP) (Table 1). A single metastatic ovarian LCT had a BAP1 mutation and copy number amplifications of the NPM1, PCM1, and SS18 genes.
Copy number amplifications were detected in 8 of 18 successfully tested cases (6 testicular and 2 ovarian LCTs). The more prevalent copy number amplifications included those affecting CCND3 (2 testicular) and genes in the fibroblast growth factor family: FGF3 (1 primary ovarian), FGFR3 (1 primary testicular and 1 primary ovarian), and FGFR4 (1 metastatic testicular) (Table 1).
Immuno-oncology Biomarkers
PD-L1 expression (threshold ≥1%) in the tumor cells or immune cells was not seen in any of the 15 tested testicular LCTs. All cases were MSI stable. A low tumor mutation burden (4–7 muts/Mb) characterized most of the testicular LCT except the peculiar metastatic case with a B4GALT5:TERT fusion and TOP1 and CCND3 amplifications that exhibited 11 muts/Mb (Table 1).
Discussion
Our study represents the first comprehensive molecular study to examine potentially targetable molecular alterations in LCT including its malignant variants. One of the key findings in our study was that TERT gene fusions were a major detected genetic alteration in malignant, metastatic LCTs. This finding is novel and had not been previously reported in sex cord–stromal tumors, including LCT.3,5,8 In addition, all 3 described gene fusions affecting TERT gene have not been previously reported in the literature (review of the literature covered PubMed/MEDLINE and COSMIC database). TERT activity plays a central role in the unlimited self-renewal potential of cancer cells via telomerase activity that maintains telomere ends through addition of telomere repeats TTAGGG).17 This mechanism is considered one of the hallmarks of cancer.18 Various genomic alterations including TERT promoter mutations, rearrangements, amplifications, fusions, and promoter methylation have been well-characterized across human cancers.19–21 Limited information on the therapeutic implications of TERT genomic alterations are currently available. One recent in vitro study conducted on acral melanoma cells revealed the cytotoxic effects of TERT inhibitors in melanoma cells harboring TERT genomic alterations.21
The family of topoisomerase enzymes (TOP1 and TOPO2) are the key players in unwinding coiled DNA to facilitate the cell replication and transcription.22 Given their active role in DNA replication and transcription, several classes of drugs targeting TOP1 and TOPO2 have been developed. One of these drugs is camptothecin against TOP1, whose derivatives irinotecan and topotecan have been widely used as cytotoxic drugs in a clinical setting. Topo1 protein overexpression has been described in various cancers,15 whereas TOP1 gene amplification is a much rarer event in cancers (the highest amplification rate [>10%] was reported in gallbladder, esophageal, and gastroesophageal carcinomas).15 Our study revealed a common (50%–60%) Topo1 protein expression in both primary and metastatic LCT, whereas TOP1 gene amplification was observed in 1 metastatic case. This finding may be clinically relevant for malignant LCTs and provide a rationale for the treatment with camptothecin derivatives alone or combined with novel anticancer treatments such as antibody–drug conjugates that contain irinotecan.
Hormone therapy with antiandrogens has been used therapeutically in prostate cancer patients.23 Some of the commonly used antiandrogens (eg, bicalutamide) competitively inhibit ligand binding to the active AR. Our study also confirmed AR activity in LCTs without the presence of splice variant ARv7. In prostate cancer cells, ARv7 stems from aberrant messenger RNA splicing of AR exons 1 to 3, loss of exons 4 to 8, and inclusion of cryptic exon 3 into the transcribed AR gene.24,25 Consequently, the affected protein is constitutively active in the absence of androgens and facilitates the growth of prostate cancer in the presence of antiandrogens.26,27 We found AR expression in 40% of metastatic LCT without the ARv7 splice variant, which indicates a potential for treatment with antiandrogens.
Immunotherapy with immune checkpoint inhibitors against PD-1/PD-L1 has markedly improved the treatment and outcome of multiple solid and hematological cancers (eg, non–small cell lung carcinoma, melanoma, renal cell carcinoma, urothelial bladder carcinoma, triple-negative breast carcinoma, and classical Hodgkin lymphoma). Several currently available predictive biomarkers (PD-L1 expression, high TMB, and high MSI status) with approved clinical usefulness have been explored in this study. In contrast with testicular germ cell tumors,28,29 we found no PD-L1 expression in LCTs. With the exception of 1 case with high TMB (11 muts/Mb), all cases exhibited a low TMB, and all cases were MSI stable. Based on these results, it is unlikely that these patients would benefit from targeted therapy from immune checkpoint inhibitors.
There are several limitations of our study. There was a lack of matched primary sample analysis for cases with TERT fusion-positive metastases to determine whether the fusions represent early events in more aggressive cancers or later events associated with metastasis. If the fusions are early events, patients with fusion-positive primary tumors could have increased surveillance. In addition, the TERT promoter mutations, commonly observed in other malignancies (eg, gliomas, bladder, thyroid cancers, and melanoma), were not possible to examine in this study owing to the lack of the gene promoter coverage in the NGS panel available at the time of study.30–33 Finally, there is lack of feedback information on the usefulness of molecular profiling in the treatment of metastatic LCT with potentially actionable findings detected in our cohort (eg, overexpression of Topo1 and AR).
In conclusion, we identified for the first time TERT gene fusions as a main genetic alteration and several potential therapeutic targets in malignant, metastatic LCTs, including Topo1 and AR, which may help guide decisions on chemotherapy and/or hormone therapy for selected individual patients.
Supplementary Material
Clinical Practice Points.
Metastatic Leydig cell tumors are rare and difficult to treat malignancies without known underlying molecular-genetic events.
We identified TERT gene fusions exclusively in malignant, metastatic Leydig cell tumors.
Additional predictive biomarkers (Topo1 and AR) may help guide decisions on chemo- and/or hormone therapy for selected individual patients.
Acknowledgments
The preliminary results from the current study were presented at the 44th European Society of Medical Oncology Congress (ESMO 2019) that was held in period 27 September through 1 October 2019 in Barcelona, Spain.
Footnotes
Disclosure
Elma Contreras, Joanne Xiu, Elena Florento, Michelle Ellis, and Jeffrey Swensen are employees of Caris Life Sciences. Zoran Gatalica reports Caris stock ownership. The other authors declare no conflict of interest.
CRediT authorship contribution statement
Bozo Kruslin: Formal analysis, resources, Writing – original draft. Zoran Gatalica: Conceptualization, Data curation, Writing – original draft, Supervision. Ondrej Hes: Formal analysis, resources. Faruk Skenderi: Formal analysis, resources. Markku Miettinen: Formal analysis, resources. Elma Contreras: Formal analysis, Validation. Joanne Xiu: Formal analysis, Validation. Michelle Ellis: Formal analysis, Validation. Elena Florento: Formal analysis, Validation. Semir Vranic: Data curation, Writing – original draft, Supervision. Jeffrey Swensen: Conceptualization, Data curation, Writing – original draft, Supervision.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.clgc.2021.02.002.
Data availability statement
The data presented in the current study are available from the corresponding authors upon reasonable request.
References
- 1.Idrees MT, Ulbright TM, Oliva E, et al. The World Health Organization 2016 classification of testicular non-germ cell tumours: a review and update from the International Society of Urological Pathology Testis Consultation Panel. Histopathology. 2017;70:513–521. [DOI] [PubMed] [Google Scholar]
- 2.Momenimovahed Z, Tiznobaik A, Taheri S, Salehiniya H. Ovarian cancer in the world: epidemiology and risk factors. Int J Womens Health. 2019;11:287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuller PJ, Leung D, Chu S. Genetics and genomics of ovarian sex cord-stromal tumors. Clin Genet. 2017;91:285–291. [DOI] [PubMed] [Google Scholar]
- 4.de Kock L, Terzic T, McCluggage WG, et al. DICER1 mutations are consistently present in moderately and poorly differentiated Sertoli-Leydig cell tumors. Am J Surg Pathol. 2017;41:1178–1187. [DOI] [PubMed] [Google Scholar]
- 5.Garg K, Karnezis AN, Rabban JT. Uncommon hereditary gynaecological tumour syndromes: pathological features in tumours that may predict risk for a germline mutation. Pathology. 2018;50:238–256. [DOI] [PubMed] [Google Scholar]
- 6.McCluggage WG, Chong AL, de Kock L, Foulkes WD. Somatic tumour testing establishes that bilateral DICER1-associated ovarian Sertoli-Leydig cell tumours represent independent primary neoplasms. Histopathology. 2020;77:223–230. [DOI] [PubMed] [Google Scholar]
- 7.Kato N, Kusumi T, Kamataki A, Tsunoda R, Fukase M, Kurose A. DICER1 hotspot mutations in ovarian Sertoli-Leydig cell tumors: a potential association with androgenic effects. Hum Pathol. 2017;59:41–47. [DOI] [PubMed] [Google Scholar]
- 8.Yuan Z, Huo X, Jiang D, et al. Clinical Characteristics and mutation analysis of ovarian Sertoli-Leydig cell tumors. Oncologist. 2020;25:e1396–e1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gargano SM, Senarathne W, Feldman R, et al. Novel therapeutic targets in salivary duct carcinoma uncovered by comprehensive molecular profiling. Cancer Med. 2019;8:7322–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11, 348 patients. Cancer Med. 2018;7:746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatalica Z, Vranic S, Kruslin B, et al. Comparison of the biomarkers for targeted therapies in primary extra-mammary and mammary Paget’s disease. Cancer Med. 2020;9:1441–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vranic S, Palazzo J, Sanati S, et al. Potential novel therapy targets in neuroendocrine carcinomas of the breast. Clin Breast Cancer. 2019;19:131–136. [DOI] [PubMed] [Google Scholar]
- 13.Vranic S, Senarathne W, Stafford P, Poorman K, Pockaj BA, Gatalica Z. Biomarkers of targeted therapy and immuno-oncology in cancers metastatic to the breast. Appl Immunohistochem Mol Morphol. 2020;28:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vranic S, Stafford P, Palazzo J, et al. Molecular profiling of the metaplastic spindle cell carcinoma of the breast reveals potentially targetable biomarkers. Clin Breast Cancer. 2020;20:326–331. [DOI] [PubMed] [Google Scholar]
- 15.Heestand GM, Schwaederle M, Gatalica Z, Arguello D, Kurzrock R. Topoisomerase expression and amplification in solid tumours: analysis of 24, 262 patients. Eur J Cancer. 2017;83:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marabelle A, Fakih MG, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with select advanced solid tumours treated with pembrolizumab in KEYNOTE-158. Ann Oncol. 2019;30:v477–v478. [DOI] [PubMed] [Google Scholar]
- 17.Telomeres Saretzki G., Telomerase and ageing. Subcell Biochem. 2018;90:221–308. [DOI] [PubMed] [Google Scholar]
- 18.Leao R, Apolonio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: clinical impacts in cancer. J Biomed Sci. 2018;25:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barthel FP, Wei W, Tang M, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49:349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiba K, Lorbeer FK, Shain AH, et al. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science. 2017;357:1416–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang WS, Hendricks W, Kiefer J, et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017;27:524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cummings J, Smyth JF. DNA topoisomerase I and II as targets for rational design of new anticancer drugs. Ann Oncol. 1993;4:533–543. [DOI] [PubMed] [Google Scholar]
- 23.Reid P, Kantoff P, Oh W. Antiandrogens in prostate cancer. Invest New Drugs. 1999;17:271–284. [DOI] [PubMed] [Google Scholar]
- 24.Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, Sharp A, Anderson CM, et al. Novel junction-specific and quantifiable in situ detection of AR-V7 and its clinical correlates in metastatic castration-resistant prostate cancer. Eur Urol. 2018;73:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu R, Lu C, Mostaghel EA, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cierna Z, Mego M, Miskovska V, et al. Prognostic value of programmed-death-1 receptor (PD-1) and its ligand 1 (PD-L1) in testicular germ cell tumors. Ann Oncol. 2016;27:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fankhauser CD, Curioni-Fontecedro A, Allmann V, et al. Frequent PD-L1 expression in testicular germ cell tumors. Br J Cancer. 2015;113:411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salgado C, Roelse C, Nell R, Gruis N, van Doorn R, van der Velden P. Interplay between TERT promoter mutations and methylation culminates in chromatin accessibility and TERT expression. PLoS One. 2020;15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee DD, Komosa M, Nunes NM, Tabori U. DNA methylation of the TERT promoter and its impact on human cancer. Curr Opin Genet Dev. 2020;60:17–24. [DOI] [PubMed] [Google Scholar]
- 32.Yuan X, Larsson C, Xu D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene. 2019;38:6172–6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vinagre J, Almeida A, Populo H, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in the current study are available from the corresponding authors upon reasonable request.