

Abstract
Introduction:
Cardiovascular disease persists as the leading cause of death worldwide despite continued advances in diagnostics and therapeutics. Our current approach to patients with cardiovascular disease is rooted in reductionism, which presupposes that all patients share a similar phenotype and will respond the same to therapy; however, this is unlikely as cardiovascular diseases exhibit complex heterogeneous phenotypes.
Areas covered:
With the advent of high-throughput platforms for omics testing, phenotyping cardiovascular diseases has advanced to incorporate large-scale molecular data with classical history, physical examination, and laboratory results. Findings from genomics, proteomics, and metabolomics profiling have been used to define more precise cardiovascular phenotypes and predict adverse outcomes in population-based and disease-specific patient cohorts. These molecular data have also been utilized to inform drug efficacy based on a patient’s unique phenotype.
Expert opinion:
Multiscale phenotyping of cardiovascular disease has revealed diversity among patients that can be used to personalize pharmacotherapies and predict outcomes. Nonetheless, precision phenotyping for cardiovascular disease remains a nascent field that has not yet translated into widespread clinical practice despite its many potential advantages for patient care. Future endeavors that demonstrate improved pharmacotherapeutic responses and associated reduction in adverse events will facilitate mainstream adoption of precision cardiovascular phenotyping.
Keywords: cardiovascular disease, drug repurposing, genomics, metabolomics, phenotyping, precision medicine, proteomics
1. Introduction
Cardiovascular disease is highly prevalent worldwide and is associated with a high rate of morbidity, mortality, and increased healthcare costs. Cardiovascular disease remains the leading cause of death in the United States and worldwide with a 21.1% rise in cardiovascular deaths between 2007–2017 [2]. In 2017, the National Health Interview Survey reported that the age-adjusted prevalence of heart disease was 11.0% in whites, 9.7% in blacks, 7.4% in Hispanics, and 6.1% in Asians in the United States [3]. This survey also found that the prevalence of hypertension, stroke, and coronary artery disease was higher in men (26.0%, 3.3%, and 7.2%, respectively) than in women (23.1%, 2.5%, 4.2%, respectively) [3]. A large proportion of these cardiovascular diseases are attributable to modifiable risk factors, including diet, tobacco use, sedentary behavior, and obesity as well as treatable risk factors, such as hypertension, hypercholesterolemia, and hyperglycemia [4]. The resulting economic burden due to cardiovascular disease is substantial with an estimated cost of $351.3 billion in the United States alone between 2014–2015 [2].
Our current approach to the prevention, diagnosis, and treatment of cardiovascular disease is based on recommendations and guidelines supported by evidence from clinical trials conducted in community-based or patient-specific populations. This approach exemplifies reductionism in medicine and has been the mainstay of current medical practice (Figure 1). Reductionism is based on the assumption that all patients with similar symptoms, clinical exams, and test results have the same disease pathophenotype and will respond equally to medical therapies [5–7]. This, however, is unlikely considering that cardiovascular disease is a complex multifactorial systemic disorder [8]. Precision cardiovascular phenotyping has emerged as an alternative to reductionism by putting emphasis on the individual. This has been made possible, in part, by major advances in platforms that support omics (i.e., genomics, proteomics, metabolomics) profiling. These next generation technologies coupled with novel data analytics have facilitated in-depth biological and molecular phenotyping. When omics are merged with clinical information and exposure (i.e., natural, personal or social environments) history, a population’s or a patient’s individualized cardiovascular health and disease phenotypes can be determined [9]. The importance of considering a precision cardiovascular phenotyping approach was demonstrated in a study of 8,574 individuals referred for cardiac catheterization who had exome sequencing performed. Within this group, 149 individuals were found to have a heterogeneous group of monogenic cardiovascular diseases with only 35% of these individuals being diagnosed previously thereby illustrating the utility of omics to explain an individual’s phenotype [1]. Herein, the current state of omics to phenotype cardiovascular disease and select effective therapeutics is discussed.
Figure 1. Reductionism in cardiovascular medicine.
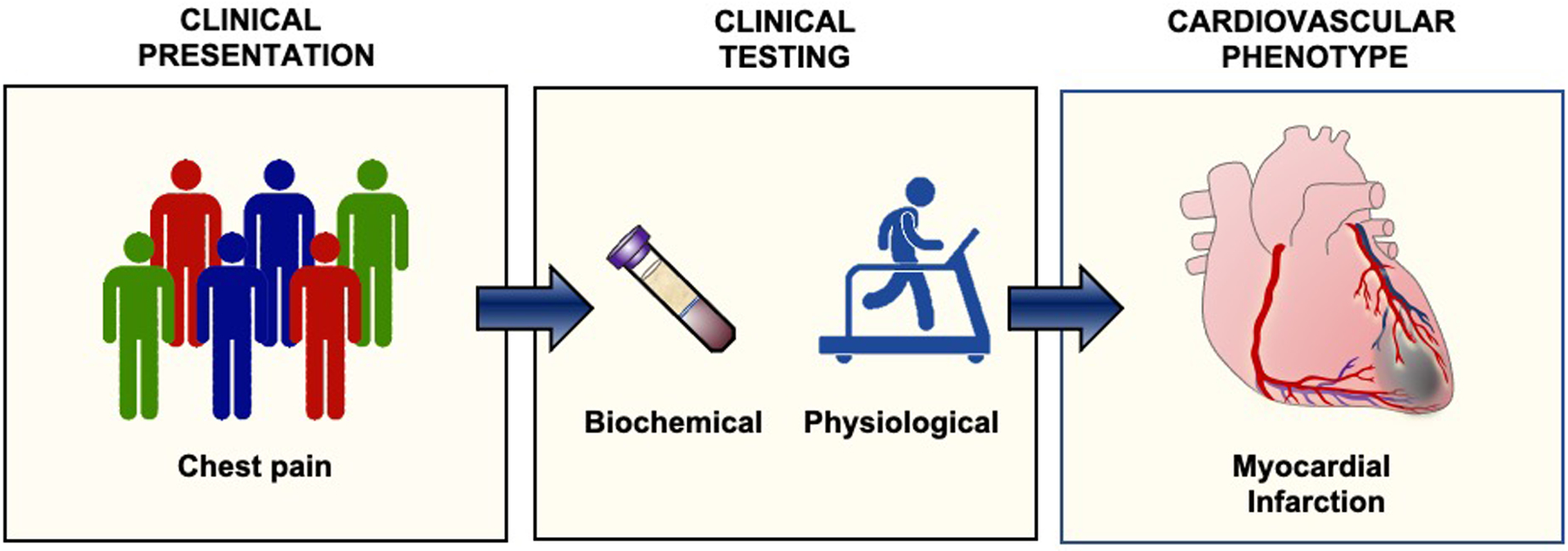
Reductionism in medicine assumes that all patients who have the same symptoms and exam findings are similar despite the fact that there is heterogeneity among individuals (left). These patients undergo clinical testing to phenotype their disease (center). The results of laboratory and functional testing identifies the cardiovascular phenotype and associated medical or interventional therapies (right).
2. Genomics and cardiovascular phenotyping
The value of genomics profiling to define cardiovascular phenotypes is underscored by the recognition that only a few cardiovascular diseases, such as familial hypercholesterolemia (mutation in the LDLR gene) or Marfan syndrome (mutation in the FBN1 gene) arise from a single causal gene mutation (see Table 1 for other monogenic cardiovascular disorders). As such, genome-wide association studies (GWAS) were undertaken to identify gene regions related to cardiovascular disease phenotypes. One of the earliest relationships identified was that between chromosome 9p21 and coronary artery atherosclerosis or myocardial infarction. When present, a single nucleotide polymorphism (SNP) in this region imparted a 29% increased risk of atherosclerotic cardiovascular disease [10–12].
Table 1.
Selected examples of monogenic cardiovascular disorders
| Cardiovascular Disease | Gene(s) | Clinical manifestations |
|---|---|---|
| LIPIDS | ||
| Familial hypercholesterolemia [97–100] | LDLR, PCSK9, | Elevated LDL and total cholesterol Xanthomas Early atherosclerosis Coronary artery disease and MI |
| Tangier disease [101–103] | ABCA1 | Low levels of HDL cholesterol Atherosclerosis Coronary artery disease and MI |
| CARDIOMYOPATHIES | ||
| Dilated cardiomyopathy [104–109] | TTN, LNMA, MYH7, BAG3, TNNT3, FLNC, RBM20, SCN5A, PLN, TNNC1, TNNI3, TPM1 | LV or biventricular dilatation Impaired contractility Congestive heart failure Arrhythmias |
| Hypertrophic cardiomyopathy [110–114] | MYH7, MYBPC3, TNNI3, TNNT2, MYL2, MYL3, ACTC1, TPM1, ACTA, MYO2, TNNC1 | Hypertrophy of the LV with stiffening Diastolic dysfunction Cardiac fibrosis Septal hypertrophy with outflow obstruction Congestive heart failure Syncope/sudden death |
| Arrhythmogenic right ventricular dysplasia [115–117] | PKP2, DSC2, DSG2, JUP, DSP, TMEM43, TGFB3 | RV cardiomyopathy Fibro-fatty replacement of RV cardiomyocytes Ventricular tachycardia Syncope/sudden death |
| AORTIC ANEURYSM | ||
| Marfans syndrome [118–121] | FBN1 | Pectus excavatum Aortic aneurysm and/or dissection Mitral valve prolapse Fatigue, dyspnea, palpitations |
| Type IV Ehlers-Danlos syndrome [122,123] | COL3A1 | Hypermobile joints Thoracic outlet syndrome Mitral valve prolapse Aortic aneurysm and/or dissection Fatigue, dyspnea, palpitations |
| Loeys-Dietz syndrome [124–128] | TGFβR1, TGFβR2, SMAD3, TGFβ2, TGFβ3 | Pectus excavatum Arterial tortuousity Aortic aneurysm and/or dissection Bicuspid aortic valve Patent ductus arteriosis or ASD Fatigue, dyspnea, palpitations |
| Familial aortic aneurysm [129–131] | ACTA2, MYH11, MYLK, PRKG1 | Aortic aneurysm and/or dissection Fatigue, dyspnea, palpitations |
| Bicuspid aortic valve and thoracic aneurysm [132] | NOTCH1 | Aortic aneurysm and/or dissection; Bicuspid aortic valve Fatigue, dyspnea, palpitations |
| ARRHYTHMIAS | ||
| Brugada syndrome [133–135] | SCN5A | Syncope/sudden death Ventricular fibrillation Polymorphic VT |
| Long QT syndrome [136,137] | KCNQ1/H2/E1/J2, SCN5A, CAV3, CALM1/2 | Syncope/sudden death Seizures VT/torsades de pointes |
| PULMONARY HYPERTENSION | ||
| Pulmonary arterial hypertension [138–145] | BMPR2, BMPR1B, CAV1, KCNK3, SMAD9, ACVRL1, ENG, ABCC8, TBX4, SOX17, GDF2, AQP1, FBLN2, PDGFD, KDR | Occlusive remodeling of pulmonary arteries Exercise intolerance, dyspnea, fatigue, syncope |
| Pulmonary veno-occlusive disease [146,147] | EIF2AK4 | Occlusive remodeling of pulmonary veins Exercise intolerance, dyspnea, fatigue, syncope |
LDL, low-density lipoprotein; HDL, high-density lipoprotein; MI, myocardial infarction; LV, left ventricle; RV, right ventricle; ASD, atrial septal defect; VT, ventricular tachycardia.
GWAS studies of cardiovascular disease have evolved over time to increase coverage of the genome, the number of variants identified, and the sample size included in the analysis [13]. The 1,000 Genomes Project reported a GWAS meta-analysis of 48 studies that included ~185,000 coronary artery disease cases and controls. This analysis identified 10 new loci with a frequency of >5% that were associated with coronary artery disease and led to the conclusion that genetic susceptibility to atherosclerotic coronary artery disease was most likely related to a panel of common SNPs with a small effect size [14]. A meta-analysis of GWAS studies was also done for subclinical atherosclerotic cardiovascular disease assessed by examining carotid artery intima-to-media thickness. This analysis included 71,128 patients and identified 8 new susceptibility loci. Linkage score disequilibrium regression further revealed genetic correlations between carotid intima-to-media thickness and coronary heart disease and stroke [15].
GWAS meta-analyses have also examined coding variants associated with hypertension and identified 242,296 rare and low-frequency common genetic variants in 192,763 individuals. Of the 30 novel variant-blood pressure associations identified, variants in four genes, OBFC1, CERS5, TBX2, and RGL3, were related to systolic and diastolic blood pressure as well as prevalent hypertension [16]. Similar to what was concluded for atherosclerosis, this analysis suggested that blood pressure and hypertension are associated with a panel of variants with small effect sizes. This, however, has been hard to confirm as results from GWAS studies of hypertension have not been replicated in subsequent studies. For example, while the Framingham Heart Study reported on 33 SNPs associated with blood pressure or hypertension, a contemporary GWAS from the United Kingdom (UK) Biobank found 107 loci related to blood pressure [17,18]. A more recent GWAS that included more than 1 million individuals of European ancestry identified 535 new loci that were linked to systolic and diastolic blood pressure and pulse pressure [18]. These differences achieve importance when one considers that SNPs are incorporated into genetic risk scores. For instance, a genetic risk score derived from SNPs identified in a UK Biobank study associated higher genetic burden with hypertension as well as an increased risk of myocardial infarction, stroke, and all-cause cardiovascular outcomes [19]. While this genetic risk score performed well and was validated in a second cohort, a risk score derived from SNPs identified by other studies may not yield the same results.
It is also worthwhile to note that there is a bidirectional relationship between genetic variants and exposures. GWAS have also been used to examine lifestyle choices and habits that are contributors to the cardiovascular phenotype and believed to have a genetic basis. A GWAS of physical activity and sleep duration that included 91,105 patients enrolled in the UK Biobank study identified 14 significant loci related to these behaviors. Common genetic variation at these loci were believed to account for up to 18% of the variation in physical activity and sleep duration among participants [20]. Exposures, in turn, can also modify genetic risk for cardiovascular phenotypes. A lifestyle score comprised of tobacco use, diet, obesity, and physical activity status was used to categorize individuals as having a favorable, intermediate, or unfavorable cardiovascular lifestyle. When patients were stratified by a polygenic risk comprised of 50 SNPs and lifestyle category, lifestyle was shown to change the 10-year coronary event rate among individuals with high genetic risk (Figure 2) [21].
Figure 2. The effect of exposures on genetic risk.
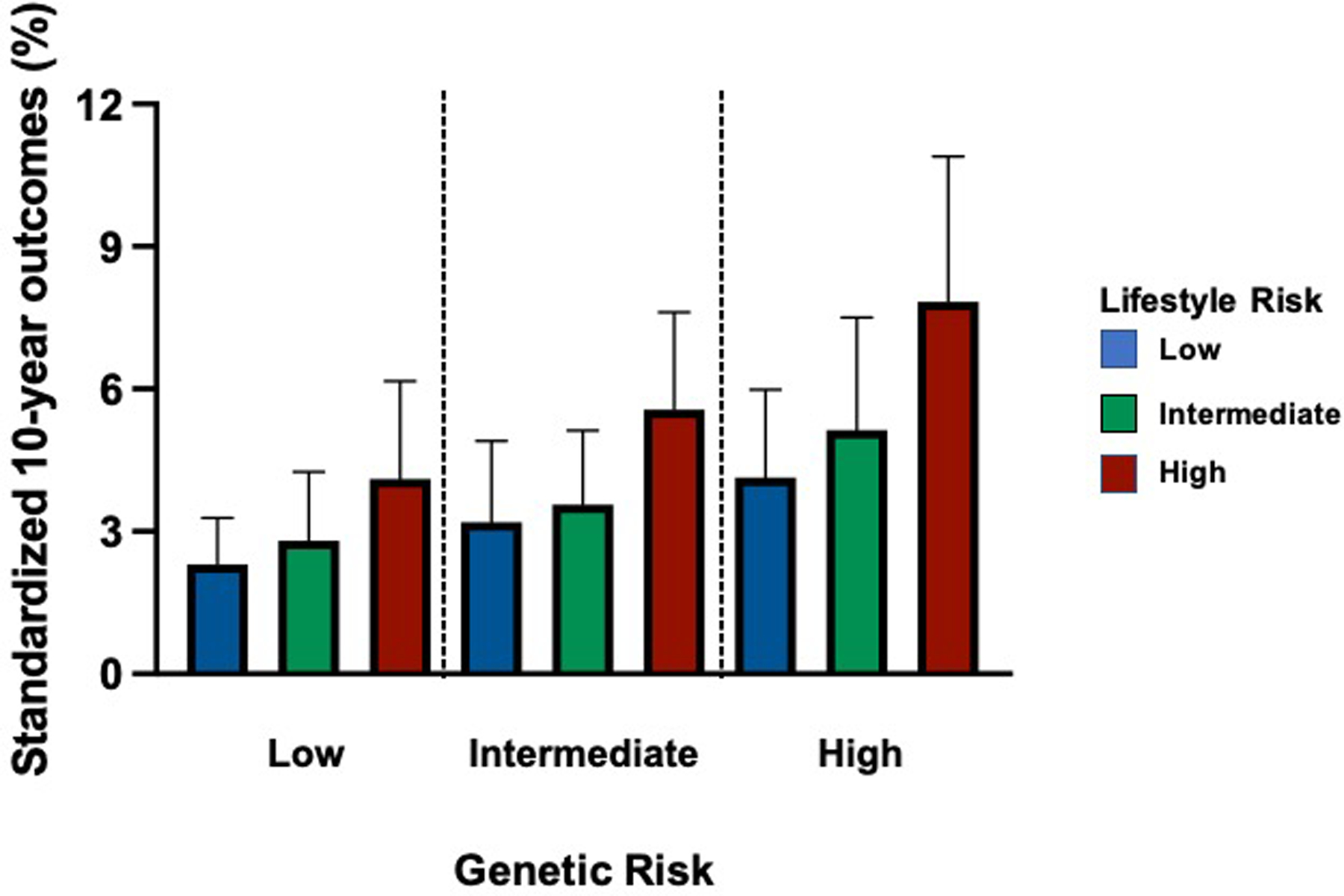
Low, intermediate, or high genetic risk was determined for individuals based on a risk score that included 50 variants and the effect of lifestyle behaviors (diet, weight, tobacco use, and exercise) on genetic risk was determined. Adapted from (20).
While GWAS studies have identified important associations between genes and cardiovascular disease phenotypes, a recent analysis of genetic data from the National Heart, Lung, and Blood Institute Trans-Omics for Precision Medicine (TOPMed) program highlights the relationship between rare variants and disease causality. This analysis of 53,831 diverse genomes identified >400 million single nucleotide, insertion, or deletion variants. Of these, 97% of the variants were found to have a frequency of <1% and 46% were present in only one individual [22]. These results confirm many of the findings from population-based GWAS studies: many low frequency variants drive the phenotype in complex diseases, like cardiovascular disease.
3. Cardiovascular phenotyping by the epigenome and transcriptome
Studies have examined the epigenome or the transcriptome to phenotype cardiovascular disease, although there are relatively few studies in this area. The relationship between genome-wide DNA methylation and blood pressure was evaluated in a study that included 17,010 individuals. Genome-wide analysis identified 13 significant cytosine-phosphate-guanine (CpG) dinucleotides (DNA methylation sites) that were independent of known SNPs related to blood pressure. Interestingly, only one of the CpG sites was found to have an effect on blood pressure while the other sites were affected by blood pressure [23]. A second genome-wide DNA methylation study in 4,820 individuals discovered 39 CpG sites related to blood pressure. When these sites were examined in twin cohorts, it was determined that the majority of methylation events could be explained by environmental factors [24].
Examination of the transcriptome revealed that 8 transcripts accounted for 13% of blood pressure variability. One of the transcripts, cysteine-rich protein 1(CRIP1) also correlated with cardiac hypertrophy, a marker of hypertension-mediated end-organ damage. When studied as a biomarker, CRIP1 levels were associated with incident stroke (HR=1.06, 95% CI: 1.03–1.09, p<5 × 10−5) [25]. Further studies found that 14–25% of the transcriptome related to the vascular endothelium was sex-based and may explain differences in vascular function between men and women [26]. In a search for other biomarkers of cardiovascular disease, circulating microRNAs and mRNAs associated with blood pressure, body mass index, fasting glucose, high-density lipoprotein cholesterol and triglycerides were determined in 2,812 individuals. Although many associations between microRNAs, mRNAs and the cardiometabolic phenotypes were identified, only four mRNAs that were linked to all of the traits and four microRNAs that associated with four of the traits. While interesting, the functional significance of these relationships wasn’t determined [27].
4. Proteomics and phenotyping cardiovascular disease
The human arterial proteome has been analyzed using 200 arterial specimens (coronary artery and aorta) obtained from 100 young adults at the time of autopsy. A comparison of these arterial proteomes revealed that there were differences in mitochondrial protein abundance between the vessels. When early atherosclerotic disease was present, compared to vessels without disease, differences in extracellular matrix protein expression as well as tumor necrosis factor-α, insulin receptor, peroxisome proliferator-activated receptor-α and peroxisome proliferator-activated receptor-γ signaling networks were observed [28]. Proteomics have also been employed to phenotype atherosclerotic plaques and identified subsets of CD4+ T cells and activated differentiated T cells in the atherosclerotic plaques [29]. These studies highlight the fact that there can be significant differences in the proteome within the same individual.
The majority of proteomic studies have examined the circulating proteome as a method to phenotype cardiovascular diseases because cardiac and/or vascular tissues are not readily available. Proteomic analyses from the Framingham Heart Study have found circulating proteins relevant for cardiovascular disease. One study that included 3,523 participants, identified 8 biomarkers associated with incident cardiovascular disease and 35 biomarkers associated with cardiovascular death. Several biomarkers were related to metabolism and adipocyte homeostasis, including insulin-like growth factor 1, leptin, and the insulin-like growth factor binding proteins 1 and 2 [30]. Within this study population, matching genomic data with the proteomic results found 120 genetic locus-circulating protein associations, which explained only ~66% of the interindividual variability in protein levels [31]. Sex differences in proteomic profiles have also been seen. Among 7,184 participants enrolled in the Framingham Heart Study, 37 of 71 circulating cardiovascular disease-related protein biomarkers were higher in women than men. In this predominantly Caucasian population, sex modified the relationship of specific biomarkers, such as CD14 and apolipoprotein B, with incident cardiovascular disease providing evidence that exposures can also modify the proteome [32].
Proteomic data has also been used to create protein-based risk scores to relate cardiovascular phenotypes with outcomes. Circulating proteomics from patients enrolled in the Heart and Soul study was used to create a 9-protein risk score that was found to perform better than the Framingham Risk Score at predicting cardiovascular events [33]. Other analyses found that the predictive ability of a proteomics protein panel for all-cause mortality was enhanced when machine learning and neural networks were employed to predict outcome [34]. A nested case-control study from the EPIC-Norfolk prospective cohort study also demonstrated the utility of a panel of 50 proteins to predict risk. This panel of proteins outperformed clinical risk predictors of acute myocardial infarction (area under the curve 0.754 vs., 0.730, p<0.001) during a 20-year follow-up period [35]. GWAS data has also been utilized to create a virtual proteome and protein risk score that was tested in 41,288 individuals. This analysis found that platelet-derived growth factor receptor-β and CC-type lectin domain family 1 member B were predictive of atherosclerotic carotid artery disease [36].
Proteomic profiling has also been used to identify circulating proteins related to myocardial injury. In patients undergoing alcohol septal ablation who have a predefined time course of myocardial injury, proteomic profiling of blood samples obtained prior to and 10 and 60 minutes after the ablation found 79 proteins that were differentially expressed between pre- and post-injury. This included the novel proteins Dickkopf-related protein 4 and crypto, which has been implicated in cardiac development. Interestingly, 23 of the identified proteins were also increased in patients who presented with acute myocardial infarction [37].
The circulating proteome associated with heart failure has been linked with echocardiographic measures of ventricular dysfunction and heart failure. After meta-analysis, 6 proteins were associated with incident heart failure, including N-terminal-proB-type natriuretic peptide (NT-pro-BNP), mannose-binding lectin, thrombospondin-2, growth differentiation factor-11/8, epidermal growth factor receptor, and hemojuvelin [38]. Circulating proteins predictive of early heart failure were related to coagulation, immunology, complement, matrix remodeling and natriuretic pathways. Interestingly, NT-pro-BNP, thrombospondin-2, interleukin-18 receptor, and gelsolin all appeared to have a cardiac origin while activated C5a and liver-specific C5 were enriched in coronary blood suggesting systemic origin [39]. Using network analysis to understand relationships between circulating proteins in patients with incident heart failure, 4 main clusters were identified that could be categorized broadly as related to inflammation and apoptosis, extracellular matrix remodeling and angiogenesis, blood pressure regulation, and metabolism, all of which are related to etiologies for heart failure [40]. Other studies have reported that there are differences in the proteome associated with different sub-types of heart failure, including heart failure with preserved ejection fraction, heart failure with mid-range ejection fraction and heart failure with reduced ejection fraction as well as between ischemic and non-ischemic cardiomyopathy [41].
5. Metabolomics as a snapshot of cardiovascular disease phenotypes
Changes in the metabolome (all metabolites, sugars, lipids, amino acids and small molecules) have been linked to cardiovascular disease. Untargeted metabolomic profiling of 3,867 individuals enrolled in the Multi-Ethnic Study of Atherosclerosis study found 30 metabolites that were associated with atherosclerosis. Pathway analysis identified branched chain amino acids and aromatic acid metabolism; lipid, fatty acid and carbohydrate metabolism; the tricarboxylic acid pathway and urea cycle; and muscle metabolism as being related to coronary and carotid atherosclerosis [42]. Another study identified 89 metabolites in plasma related to the progression of atherosclerotic coronary artery disease. This metabolic profile was characterized by decreases in metabolites related to phospholipid catabolism, the tricarboxylic acid cycle, bile acid biosynthesis and increases in amino acid metabolism and short chain acylcarnitines [43]. Metabolomic profiling of 7,256 individuals from the FINRISK study found that phenylalanine as well as monounsaturated and polyunsaturated fatty acids were biomarkers for a high-risk cardiovascular phenotype [44]. In a population-based study of individuals with familial coronary atherosclerosis and population-based controls, metabolomic profiling restricted to the lipidome revealed that 30 of 32 sphingolipid species were increased in patients with coronary artery disease [45].
Functional metabolomics has been utilized to link metabolites to disease mechanism. In a group of 2,324 patients that underwent coronary angiography, 36 metabolites (15 amino acids, 12 free fatty acids, 8 organic acids, and 1 sialic acid) were identified that were differentially expressed among patients with angiographically normal coronary arteries, nonobstructive disease, stable angina, unstable angina and myocardial infarction. Of these metabolites, Neu5Ac, a metabolite of the sialic acid family, was found to be related to the level of atherosclerotic coronary disease progression. When examined in vitro and in vivo, Neu5Ac promoted myocardial injury thereby confirming the functional role of this metabolite in myocardial injury [46].
Metabolomics profiling has identified metabolites that are predictive for developing a cardiovascular phenotype. Elevated levels of the metabolites isocitrate, aconitase, and malate have been associated with suboptimal cardiovascular health; however, only isocitrate was associated with incident cardiovascular disease [47]. Metabolites have also been incorporated into risk scores for vascular and myocardial disease. Metabolomic data from the Coronary Artery Risk Development in Young Adults (CARDIA) study found that metabolites related to microbial metabolism, nitic oxide regulation, oxidant stress, and collagen metabolism were associated with incident cardiovascular disease. Metabolite-based vascular risk scores were lower in men than women while myocardial risk scores were lower in blacks than other racial and ethnic groups [48].
Associations between circulating metabolites and hypertension or heart failure have also been examined in community-based populations. In the Bogalusa Heart Study, 24 metabolites were found that were associated with blood pressure. These metabolites included amino acids; nucleotide metabolites of tryptophan, pyrimidine, or histidine; several cofactor or xenobiotic metabolites; and lipid metabolites from sphingolipid pathways. Interestingly, many of these metabolites and pathways were also associated with atherosclerosis, indicating a shared metabolic profile [49]. Metabolomics are also perturbed in patients with an increased risk of heart failure. Among 2,199 black adults enrolled in the Jackson Heart study, metabolites related to incident heart failure included products of nitric oxide metabolism, polyamine metabolism, and posttranslational RNA modifications. Other metabolites related to pyrimidine metabolism and collagen turnover were identified as part of a metabolic signature that differentiated heart failure with preserved ejection fraction from reduced ejection fraction [50].
6. Multiomics testing and cardiovascular phenotype
While the aforementioned studies illustrate the relevance of phenotyping cardiovascular disease (either incident or prevalent) with individual omics platforms, there are few large-scale studies that have performed multiomics profiling with the notable exception of the Framingham Heart Study [51]. Other small studies have demonstrated the value of integrated multiomics analyses for cardiovascular phenotyping. Genomics, transcriptomics, proteomics, metabolomics, microbiomics and digital health data was collected from 109 individuals three times annually over the course of 8 years. Integrated analyses identified 67 clinically actionable health discoveries within the fields of cardiology, oncology, infectious disease, and hematology, as well as disease-related molecular pathways. These findings led to additional testing in 29% of participants while 82% reported that the results of the study led to a change in diet and exercise behaviors [52]. In another study, multiomics profiling of a well phenotyped group of 36 individuals was done to examine the response to exercise. A time-series analysis of the omics identified early (energy metabolism, oxidant stress, and immune responses) and late pathways (energy homeostasis, tissue repair, and remodeling) associated with exercise, including a post-exercise inflammatory signature. Many of these pathways were either less activated or inhibited in participants who were insulin resistant [53].
Recently, whole genome sequence analysis has been utilized to evaluate the plasma proteome in 1,852 Black adults that were part of the Jackson Heart Study. This analysis identified 114 novel locus-protein relationships, including previously unknown associations between the APOE gene locus and the proteins MMP-3 and ZAP70. The analysis also facilitated recognition of new associations between variants, diseases, and proteins. For example, the study reported a new association between ATTR amyloidosis and RBP4 protein levels as well as APOL1 chronic kidney disease and heart disease and CKAP2 [54]. Another study examined the utility of polygenic risk scores to identify proteins related to cardiovascular disease pathogenesis. The analysis included data from 3,087 individuals and found that 49 plasma proteins were associated with the polygenic risk scores. Over 7.7-years of follow-up, 28 proteins were found to be associated with future type 2 diabetes mellitus events or myocardial infarction [55].
7. Utilizing genomic profiling to predict drug efficacy
The overarching value of precision phenotyping at a molecular level is that it allows for the selection of therapeutics based on an individual’s genome, metabolome or proteome (Figure 3). A well-known example that illustrates this concept is patients who require dual-antiplatelet therapy with clopidogrel, a P2Y12 inhibitor, after stent implantation to prevent stent thrombosis. Some patients who experienced adverse events while adherent to their dual-antiplatelet therapy regimen were subsequently identified as “non-responders” to clopidogrel [56–59]. Non-responsiveness was linked to loss-of-function alleles in CYP2C19 (CYP2C19*2 and CYP2C19*3), which are known to have a higher prevalence in India, Pakistan and Japan as compared to the United States with the exception of individuals of African and Asian descent [60–63]. However, attempts to utilize this knowledge for point-of-care testing in patients undergoing percutaneous coronary interventions with drug-eluting stents did not lead to a decrease in adverse cardiovascular events [64]. A pooled analysis that included 15,949 patients with coronary artery disease compared outcomes in patients treated with ticagrelor or prasugrel compared to clopidogrel. This study reported that among individuals with a CYP2C19 loss-of-function allele, treatment with ticagrelor or prasugrel decreased the risk of cardiovascular death, stroke, myocardial infarction, severe recurrent ischemia, and stent thrombosis (RR: 0.70; 95% CI: 0.59–0.83). This beneficial effect was not observed in individuals who were noncarriers of the loss-of-function allele. Thus, among individuals with coronary artery disease, the clinical benefits associated with ticagrelor or prasugrel were attributable to CYP2C19 loss-of-function carrier status [65]. Similarly, a randomized double-blind, placebo-controlled clinical trial that examined the efficacy of ticagrelor as compared to clopidogrel for the secondary prevention of stroke among Chinese individuals who harbored CYP2C19 loss-of-function alleles found that ticagrelor was protective (HR = 0.77; 95% CI: 0.64–0.94, p=0.008) but associated with more bleeding events than clopidogrel [66].
Figure 3. Pharmaco-omics and precision cardiovascular phenotyping for pharmacotherapeutic selection.
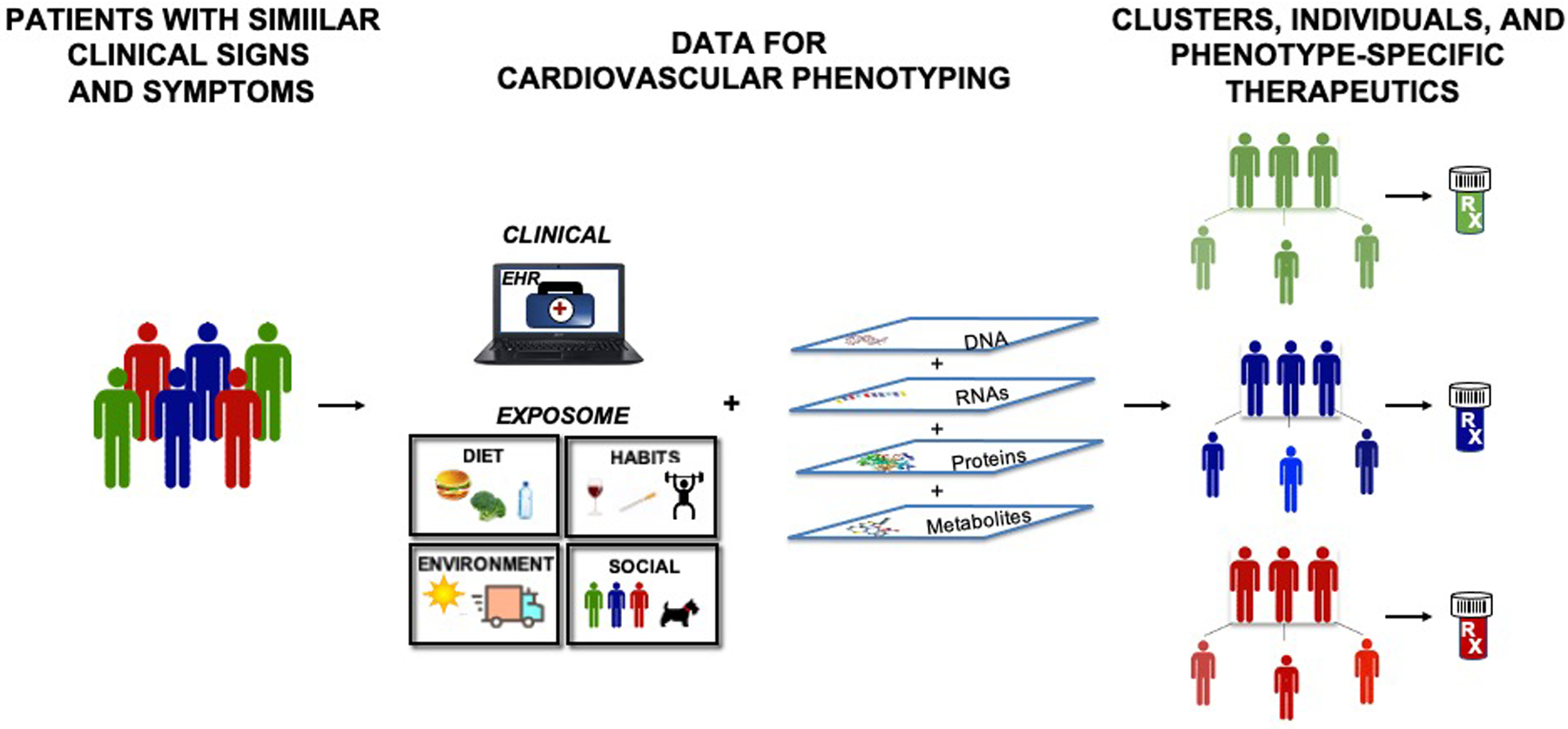
Precision cardiovascular phenotyping recognizes that patients with similar clinical signs and symptoms have significant heterogeneity. By utilizing molecular omics and in-depth clinical data to create a comprehensive cardiovascular phenotype, personalized therapeutics can be selected.
There is significant inter-individual variation in response to the anticoagulant warfarin with 10–50% of that variability attributed to genetic variants [67–69] and SNPs in CYP2C9 (CYP2C9*2, CYP2C9*3) and VKORC1 (rs9923231), which encode the enzymes that metabolize the S-isomer of warfarin and reduce vitamin K 2,3-epoxide to an active form, respectively, and thereby contribute to observed differences between patients [70–72]. Genotype-guided warfarin dosing was evaluated in two large randomized clinical trials [73,74]. While one trial, which enrolled 1,015 patients, found that genotyping did not affect the duration of time the International Normalized Ratio (INR) was in the therapeutic range, the other reported that genotype-based dosing resulted in fewer episodes of excess anticoagulation and patients were in the therapeutic range faster than those not guided by genotyping [73,74]. The reason for differences in the outcomes of these two trials was suggested to be related to differences in study design, study populations, and the prevalence of the SNPs in the study population [75].
Pharmacogenetic profiling has also identified a SNP related to statin intolerance in the solute carrier organic anion transporter 1B1 (SLCO1B1) gene, which encodes a membrane-bound sodium-independent organic anion transporter that transports statin drugs into the liver and has been associated with myopathy, the main reason why ~50% of patients discontinue the drug [76,77]. The relationship between SNPs in SLCO1B1 and statin discontinuation, however, remains controversial (68, [78].The noncoding rs4263657 polymorphism and the nonsynonymous rs4149056 polymorphism, which was in nearly complete linkage disequilibrium with rs4363657, have been associated with myopathy related to statin use [77,79]. A real-world population study that compared individuals with confirmed statin-induced myopathy with tolerant individuals who were genotyped found that only the rs4149056 polymorphism was associated with severe myopathy or rhabdomyolysis (OR: 5.15; 95% CI: 3.13–8.45, p=2.6 × 10−9) [80]. Genetic variants that modify responses to other cardiovascular disease-related drugs have also been identified. These drugs include angiotensin converting enzyme inhibitors (variants in ACE, AGTR1); beta-blockers (variants in ADRB1, ADRB2, GRK5, GRK4); and calcium channel blockers (variants in CACNB2, CACNA1C) (reviewed in [81]).
Genomic profiling has been utilized to identify optimal drug targets that would have both therapeutic efficacy with limited side effects. A GWAS of 69,479 Norwegian individuals identified 76 causal protein altering variants related to cardiovascular disease that did not affect liver function. Among these was ZNF529:pK405X, which was associated with low levels of low-density lipoprotein cholesterol with no relation to liver enzymes or blood glucose levels. In vitro studies confirmed that downregulation of ZNF529 in liver cells resulted in an increase in low-density lipoprotein cholesterol uptake indicating that this prevalent SNP may serve as candidate drug target to regulate cholesterol [82]. The value of this method will be determined only after it is compared to other highly effective lipid lowering agents that are currently available.
8. Drugs and pharmacometabolomics
Drugs have a broad impact on metabolism although in-depth knowledge of drug-metabolite interactions remains limited. Understanding drug-metabolite associations has implications for drug efficacy in patients with cardiovascular diseases. For instance, examining associations between 150 plasma metabolites and 87 commonly prescribed drugs revealed a total of 1,071 drug-metabolite associations. The top 15 drugs that were associated with the largest number of metabolites were broadly categorized as antihypertensive, glucose lowering and lipid modifying drugs; cardiovascular-related drugs; proton pump inhibitors; and selective serotonin reuptake inhibitors [83].
Other pharmacometabolomic studies have shown how clinical characteristics modulate the metabolome and their potential importance for cardiovascular disease-related therapeutics. For instance, race has been shown to have an effect on metabolomics. Studies have identified race-related differences in saturated free fatty acids (palmitic), monounsaturated free fatty acids (oleic, palmitoleic), and polyunsaturated free fatty acids (arachidonic, linoleic) with levels significantly lower in whites than blacks. The ketone body, 3-hydroxybutyrate, was also 33% lower in whites than blacks. Further investigation into the genetic causes for these findings revealed that there were differences in SNPs that regulate fatty acid metabolism with the rs9652472 SNP in the lipase C gene related to changes in whites while the SNP rs7250148 in the phospholipase A2 Group IV C gene was related to changes in oleic acid levels in blacks [84]. Thus, drugs that affect these targets are likely to exhibit differences in therapeutic efficacy for individuals that harbor these variants. Similarly, pharmacometabolomics identified race-based differences in the response to the antihypertensive diuretic hydrochlorothiazide with changes in metabolites related to purine metabolism, galactose metabolism, lactose synthesis, and gluconeogenesis seen only in white individuals treated with the drug [85]. Interestingly, other investigators found that elevated levels of N24:2 sphingomyelin had the most significant correlation with the blood pressure response to hydrochlorothiazide [86].
Pharmacometabolomics has also been utilized to determine the mechanism underlying side effects associated with cardiovascular medications. The beta-blocker atenolol, a commonly prescribed cardiovascular drug, is associated with hyperglycemia and incident diabetes as side effects. Metabolomics associated baseline levels of β-alanine with a change in plasma glucose after atenolol administration and this was related to a SNP in dihydropyrimidinase, an enzyme involved in β-alanine formation [87]. Other investigators who focused on acylcarnitines in patients treated with atenolol found that arachidonoyl-carnitine (C20:4) associated with a higher blood glucose level, lower plasma high-density lipoprotein cholesterol, and less of a therapeutic effect from atenolol than what was observed in individuals who didn’t have measurable levels of arachidonoyl-carnitine [88].
9. Network analysis, drug identification, and drug repurposing
Analysis of protein-protein interaction networks has been used to identify and repurpose drugs that may be effective in treating cardiovascular diseases [89]. A drug-disease proximity measure based on how close the target of a drug was located to a disease gene was used to quantify and predict the therapeutic effect of drugs. Examination of 78 diseases that had a minimum of 20 disease-related genes in the network found 238 drugs relevant for the diseases of interest. This analysis revealed that the drugs had an average of 3.5 targets in the network. The proximity measure was also used to investigate the relationship between drug targets and disease proteins. This identified 18,162 previously unknown drug-disease associations, which could represent new candidate drugs amenable for drug repurposing. This type of analysis can also give insight into drug mechanism of action as well as identify the mechanism underlying potential drug side effects. For example, two type 2 diabetes drugs are located proximal to cardiac arrhythmia in the disease network. Administration of these drugs is likely to affect disease genes related to arrhythmia and, therefore, explain some of the adverse cardiovascular events associated with these drugs [90].
More recently a drug-disease network was created that identified relationships between 22 types of cardiovascular diseases with 431 Food and Drug Administration approved drugs for non-cardiac diseases. The significance of these findings was tested using healthcare databases that contained information on more than 220 million patients, including their medication use history and clinical outcomes. Analyses revealed that the rheumatoid arthritis drug hydroxychloroquine was associated with a decreased risk of cardiovascular disease. In vitro studies provided mechanistic insight to explain this finding by demonstrating that hydroxychloroquine attenuated cytokine-mediated activation of human coronary artery endothelial cells [91,92].
10. Conclusion
Advances in genomics, proteomics, and metabolomics profiling have led to more precisely defined cardiovascular phenotypes that allow for prediction and prognostication. Whether selection of personalized therapeutics based on cardiovascular phenotyping will be broadly applicable to the population at-large or will be reserved for patients with select pathophenotypes remains to be determined. There are several hurdles in the pipeline that will have to be overcome, including widespread availability of user-friendly omics platforms that are cost-effective, acceptance by patients and the medical system at large, and commensurate growth in data analytics and data security (Figure 4). These factors are not insurmountable, and it is likely that in the future precision molecular and deep clinical phenotyping will be utilized for the prevention, diagnosis, and treatment of patients with cardiovascular disease.
Figure 4. Challenges to address to facilitate routine precision cardiovascular phenotyping.
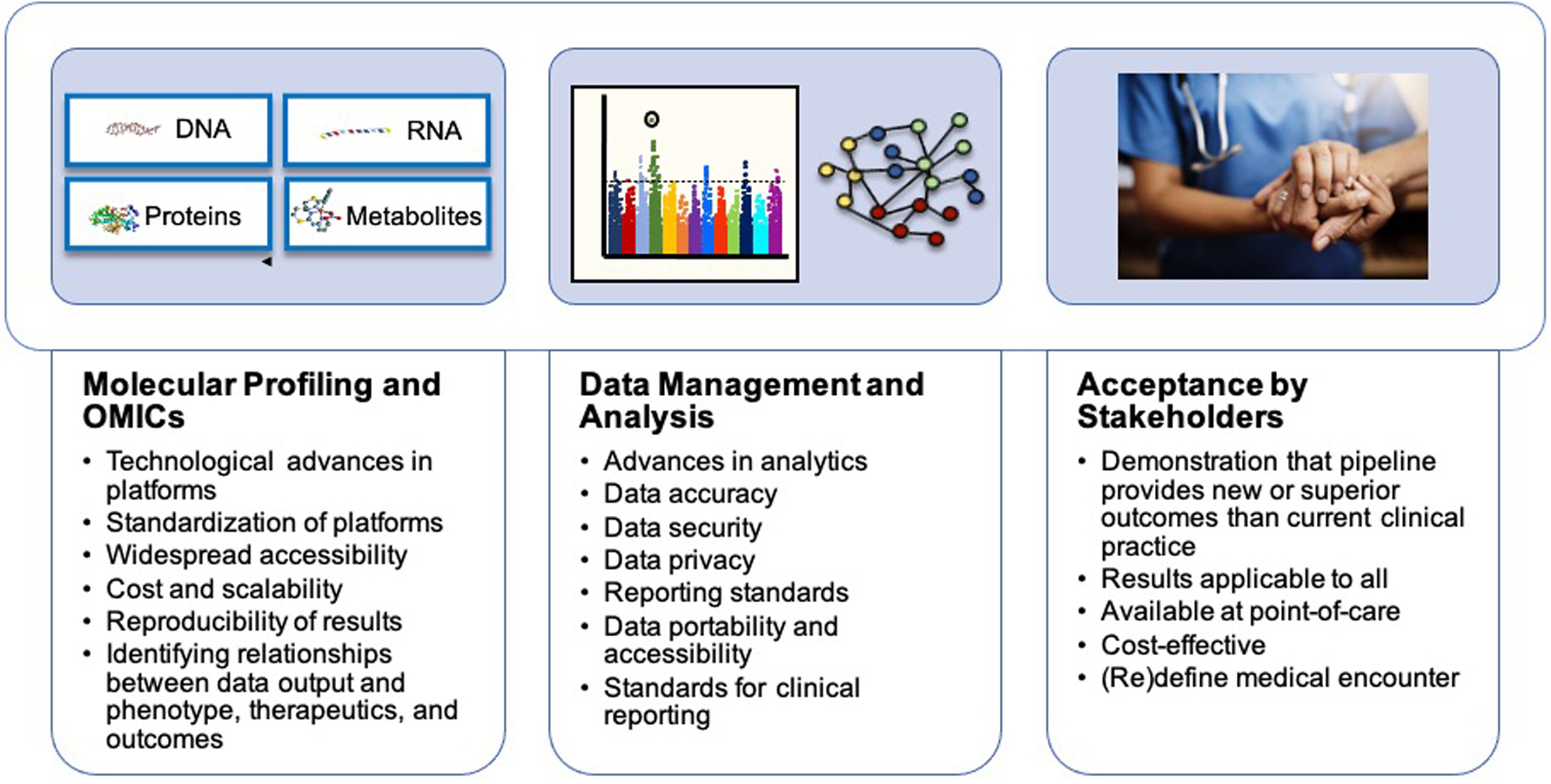
Several challenges to be addressed before precision cardiovascular phenotyping for personalized therapeutic selection becomes mainstream. These challenges are broadly related to molecular profiling and omics, data management and analysis, and acceptance by stakeholders.
11. Expert opinion
The current state-of-the-art in advanced phenotyping for cardiovascular disease remains firmly in the discovery phase as many studies have reported findings but have not provided a pathway to move towards the clinic. Two recent studies illustrate that the reception for precision cardiovascular phenotyping in current clinical practice is somewhat lukewarm. A pilot study of 110 patients was done to examine the impact of pharmacogenetic profiling and clinical decision-making support to decrease adverse drug reactions. Genomic analysis of CYP 450 genes to guide medication use was associated with a decrease in emergency department visits and hospitalizations. However, only 77% of the pharmacogenetic profiling and decision support recommendations were followed by clinicians [93]. The idea of using rapid genetic profiling to facilitate in-hospital treatment has also been studied. Rapid whole genome sequencing to guide drug selection for cardiovascular care was assessed in a small pilot study that included 50 patients with acute cardiovascular events or cardiac arrest. While rapid whole genome sequencing found that ~64% of the participants had one pharmacogenetic variant that would inform pharmacologic management of cardiovascular disease, only 14% were considered actionable [94]. Thus, in one case, results were not always followed by the managing physician and in the other the results were likely not superior to standard clinical practice.
While precision phenotyping of cardiovascular disease has the potential to change current practice, how the data is utilized clinically has not been optimized. Data from cardiovascular phenomic profiling can be used to create risk scores for prognostication, identify biomarkers for diagnosis, and determine effective pharmacotherapies. These goals have been accomplished and reported; however, there are limitations associated with the results from these studies that illustrate barriers to widespread adoption. Some clinical risk scores for prognostication, such as the Framingham Risk Score and the TIMI Risk Score, have performed remarkably well in practice, are validated, and are easy to use at the bedside [95,96]. In order for precision phenomic profiling to match the success of these scores, the pipeline will have to have the same ease of use, show superior results, or provide actionable information that changes clinical decision-making.
For true precision cardiovascular phenotyping to become mainstream, several factors that must be considered. The first is to identify the types of data that are required to construct the cardiovascular phenome. This typically includes data from the history and physical, laboratory tests, and imaging, but should be expanded to include a detailed history of exposures, social determinants of health, diet, and sleep as well as objective exercise data from wearable activity trackers [8]. These latter measures impact cardiovascular health and well-being yet are often not incorporated into the clinical record or assessed over time. The next decision is whether to perform molecular phenotyping using a singular or multiomics platform, if the tests will be performed as part of a cross-sectional or longitudinal evaluation, and whether analyses should focus on the population, the individual or both. Many of the studies published to date only partially address these important factors and this had likely contributed to the heterogeneous and often disparate published results.
Other issues that require consideration are related to standardization of testing, analysis, and reporting; data management; and acceptance of the practice by patients, physicians, and insurers. At present, there is no standardized template or minimal requirement for clinical data collection. There is also widespread variability in the platforms used for omics testing with results from one platform incompletely reproduced by others. In order to gain widespread adoption, these tests will need to be standardized and be as reliable as obtaining a simple routine blood test, such as a complete blood count that has the (relatively) same value when done at any testing site. Data management also remains a priority issue with strict rules needed for data privacy and data security owing to the large amount of data held in the electronic health record and generated by molecular testing. Acceptance of precision phenomics by relevant stakeholders will happen only when all these factors are resolved.
The future vision for precision cardiovascular phenotyping is that it will become routine for clinical care and pharmacotherapeutic selection. Whether this will require comprehensive molecular analyses along with deep clinical phenotyping or can be accomplished using a minimal dataset that includes genomics or proteomics only is unknown. Over time, molecular profiling platforms have become more user friendly, faster, and less expensive suggesting that this trend will continue. While it is unlikely that the pipeline from patient encounter to clinical and molecular phenotyping to therapeutic decision making will be mature in the next five years, it will be realized in the future.
Article highlights.
Cardiovascular diseases are associated with significant morbidity and are the leading cause of death around the world. Current medical practice based on clinical trial data and guidelines is an example of reductionism in medicine, which assumes that patients with the same outward phenotype will have the same response to interventions.
Studies utilizing high-throughput platforms for omics testing, including genomics, proteomics, and metabolomics, have identified molecular factors that characterize cardiovascular diseases and predict adverse outcomes. These studies have also revealed that patients with cardiovascular disease are heterogeneous and have complex pathophenotypes.
Clinical studies have confirmed that there are missed opportunities to utilize omics testing to phenotype cardiovascular disorders. Among patients being evaluated or treated for prevalent cardiovascular diseases, exome sequencing has diagnosed previously unrecognized monogenic cardiovascular disorders [1].
Cardiovascular phenotyping using omics methodology has been beneficial in defining the phenotype of aberrant lipid profiles, select arrhythmias, including Brugada syndrome and long QT syndrome; aortic diseases, such as Marfan’s syndrome and Ehlers Danlos syndrome; and hypertrophic cardiomyopathy among other cardiovascular diseases.
Precision phenotyping is capable of predicting drug efficacy and side effect profiles in patients and populations.
As more data become available from large studies, barriers to implementing precision cardiovascular phenotyping for personalized therapeutics will need to be addressed before it gains widespread acceptance in clinical practice. These barriers include standardizing testing platforms, addressing data privacy and security, and acceptance of the pipeline by patients and the medical community.
Funding
This work was supported by funding from the National Institutes of Health/National Heart, Lung, and Blood Institute U01 HL125215 and by the American Heart Association AIM 19AIML34980000. JA Leopold also receives research funding (to her institution) from Astellas.
Footnotes
Declaration of Interests
JA Leopold is a consultant for Abbott Vascular, speaker for United Therapeutics, and is a site principal investigator for a study sponsored by Aria CV. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
References:
- 1.Abdulrahim JW, Kwee LC, Alenezi F et al. Identification of Undetected Monogenic Cardiovascular Disorders. J Am Coll Cardiol, 76(7), 797–808 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virani SS, Alonso A, Benjamin EJ et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation, 141(9), e139–e596 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Blackwell DL, Villarroel MA. Tables of summary health statistics for US adults: 2017 National Health Interview Survey. (Centers for Disease Control and Prevention, http://www.cdc.gov/nchs/nhis/SHS/tables.htm., 2018) [Google Scholar]
- 4.Global Burden of Cardiovascular Diseases Collaboration, Roth GA, Johnson CO et al. The Burden of Cardiovascular Diseases Among US States, 1990–2016. JAMA Cardiol, 3(5), 375–389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snyderman R, Meade C, Drake C. Value of Personalized Medicine. JAMA, 315(6), 613 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Snyderman R, Dreke CD. Personalized Health Care: Unlocking the Potential of Genomic and Precision Medicine. Journal of Precision Medicine 1, 38–41 (2015). [Google Scholar]
- 7.Naylor S What’s In a Name? THe Evolution of P-Medicine. Journal of Precision Medicine, 1, 15–29 (2015). [Google Scholar]
- 8.Leopold JA, Loscalzo J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ Res, 122(9), 1302–1315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatnagar A Environmental Determinants of Cardiovascular Disease. Circ Res, 121(2), 162–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helgadottir A, Thorleifsson G, Manolescu A et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science, 316(5830), 1491–1493 (2007). [DOI] [PubMed] [Google Scholar]
- 11.McPherson R, Pertsemlidis A, Kavaslar N et al. A common allele on chromosome 9 associated with coronary heart disease. Science, 316(5830), 1488–1491 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samani NJ, Erdmann J, Hall AS et al. Genomewide association analysis of coronary artery disease. N Engl J Med, 357(5), 443–453 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leopold JA, Maron BA, Loscalzo J. The application of big data to cardiovascular disease: paths to precision medicine. J Clin Invest, 130(1), 29–38 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikpay M, Goel A, Won HH et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet, 47(10), 1121–1130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franceschini N, Giambartolomei C, de Vries PS et al. GWAS and colocalization analyses implicate carotid intima-media thickness and carotid plaque loci in cardiovascular outcomes. Nat Commun, 9(1), 5141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surendran P, Drenos F, Young R et al. Trans-ancestry meta-analyses identify rare and common variants associated with blood pressure and hypertension. Nat Genet, 48(10), 1151–1161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levy D, Ehret GB, Rice K et al. Genome-wide association study of blood pressure and hypertension. Nat Genet, 41(6), 677–687 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evangelou E, Warren HR, Mosen-Ansorena D et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet, 50(10), 1412–1425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warren HR, Evangelou E, Cabrera CP et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat Genet, 49(3), 403–415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doherty A, Smith-Byrne K, Ferreira T et al. GWAS identifies 14 loci for device-measured physical activity and sleep duration. Nat Commun, 9(1), 5257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khera AV, Emdin CA, Drake I et al. Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med, 375(24), 2349–2358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taliun D, Harris DN, Kessler MD et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature, 590(7845), 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richard MA, Huan T, Ligthart S et al. DNA Methylation Analysis Identifies Loci for Blood Pressure Regulation. Am J Hum Genet, 101(6), 888–902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Ollikainen M, Muniandy M et al. Identification, Heritability, and Relation With Gene Expression of Novel DNA Methylation Loci for Blood Pressure. Hypertension, 76(1), 195–205 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeller T, Schurmann C, Schramm K et al. Transcriptome-Wide Analysis Identifies Novel Associations With Blood Pressure. Hypertension, 70(4), 743–750 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartman RJG, Kapteijn DMC, Haitjema S et al. Intrinsic transcriptomic sex differences in human endothelial cells at birth and in adults are associated with coronary artery disease targets. Sci Rep, 10(1), 12367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McManus DD, Rong J, Huan T et al. Messenger RNA and MicroRNA transcriptomic signatures of cardiometabolic risk factors. BMC Genomics, 18(1), 139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrington DM, Mao C, Parker SJ et al. Proteomic Architecture of Human Coronary and Aortic Atherosclerosis. Circulation, 137(25), 2741–2756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez DM, Rahman AH, Fernandez NF et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med, 25(10), 1576–1588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho JE, Lyass A, Courchesne P et al. Protein Biomarkers of Cardiovascular Disease and Mortality in the Community. J Am Heart Assoc, 7(14) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benson MD, Yang Q, Ngo D et al. Genetic Architecture of the Cardiovascular Risk Proteome. Circulation, 137(11), 1158–1172 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lau ES, Paniagua SM, Guseh JS et al. Sex Differences in Circulating Biomarkers of Cardiovascular Disease. J Am Coll Cardiol, 74(12), 1543–1553 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ganz P, Heidecker B, Hveem K et al. Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients With Stable Coronary Heart Disease. JAMA, 315(23), 2532–2541 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Unterhuber M, Kresoja KP, Rommel KP et al. Proteomics-Enabled Deep Learning Machine Algorithms Can Enhance Prediction of Mortality. J Am Coll Cardiol, 78(16), 1621–1631 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Hoogeveen RM, Pereira JPB, Nurmohamed NS et al. Improved cardiovascular risk prediction using targeted plasma proteomics in primary prevention. Eur Heart J, 41(41), 3998–4007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mosley JD, Benson MD, Smith JG et al. Probing the Virtual Proteome to Identify Novel Disease Biomarkers. Circulation, 138(22), 2469–2481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ngo D, Sinha S, Shen D et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation, 134(4), 270–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nayor M, Short MI, Rasheed H et al. Aptamer-Based Proteomic Platform Identifies Novel Protein Predictors of Incident Heart Failure and Echocardiographic Traits. Circ Heart Fail, 13(5), e006749 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egerstedt A, Berntsson J, Smith ML et al. Profiling of the plasma proteome across different stages of human heart failure. Nat Commun, 10(1), 5830 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferreira JP, Verdonschot J, Collier T et al. Proteomic Bioprofiles and Mechanistic Pathways of Progression to Heart Failure. Circ Heart Fail, 12(5), e005897 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adamo L, Yu J, Rocha-Resende C, Javaheri A, Head RD, Mann DL. Proteomic Signatures of Heart Failure in Relation to Left Ventricular Ejection Fraction. J Am Coll Cardiol, 76(17), 1982–1994 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tzoulaki I, Castagne R, Boulange CL et al. Serum metabolic signatures of coronary and carotid atherosclerosis and subsequent cardiovascular disease. Eur Heart J, 40(34), 2883–2896 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fan Y, Li Y, Chen Y et al. Comprehensive Metabolomic Characterization of Coronary Artery Diseases. J Am Coll Cardiol, 68(12), 1281–1293 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Wurtz P, Havulinna AS, Soininen P et al. Metabolite profiling and cardiovascular event risk: a prospective study of 3 population-based cohorts. Circulation, 131(9), 774–785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poss AM, Maschek JA, Cox JE et al. Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. J Clin Invest, 130(3), 1363–1376 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, Wei TT, Li Y et al. Functional Metabolomics Characterizes a Key Role for N-Acetylneuraminic Acid in Coronary Artery Diseases. Circulation, 137(13), 1374–1390 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Cheng S, Larson MG, McCabe EL et al. Distinct metabolomic signatures are associated with longevity in humans. Nat Commun, 6, 6791 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murthy VL, Reis JP, Pico AR et al. Comprehensive Metabolic Phenotyping Refines Cardiovascular Risk in Young Adults. Circulation, 142(22), 2110–2127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He WJ, Li C, Mi X et al. An untargeted metabolomics study of blood pressure: findings from the Bogalusa Heart Study. J Hypertens, 38(7), 1302–1311 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tahir UA, Katz DH, Zhao T et al. Metabolomic Profiles and Heart Failure Risk in Black Adults: Insights From the Jackson Heart Study. Circ Heart Fail, 14(1), e007275 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andersson C, Johnson AD, Benjamin EJ, Levy D, Vasan RS. 70-year legacy of the Framingham Heart Study. Nat Rev Cardiol, 16(11), 687–698 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Schussler-Fiorenza Rose SM, Contrepois K, Moneghetti KJ et al. A longitudinal big data approach for precision health. Nat Med, 25(5), 792–804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Contrepois K, Wu S, Moneghetti KJ et al. Molecular Choreography of Acute Exercise. Cell, 181(5), 1112–1130 e1116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katz DH, Tahir UA, Bick AG et al. Whole Genome Sequence Analysis of the Plasma Proteome in Black Adults Provides Novel Insights into Cardiovascular Disease. Circulation, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ritchie SC, Lambert SA, Arnold M et al. Integrative analysis of the plasma proteome and polygenic risk of cardiometabolic diseases. Nat Metab, 3(11), 1476–1483 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oqueli E, Hiscock M, Dick R. Clopidogrel resistance. Heart Lung Circ, 16 Suppl 3, S17–28 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Ahmad T, Voora D, Becker RC. The pharmacogenetics of antiplatelet agents: towards personalized therapy? Nat Rev Cardiol, 8(10), 560–571 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Collet JP, Hulot JS, Pena A et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet, 373(9660), 309–317 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Mega JL, Close SL, Wiviott SD et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med, 360(4), 354–362 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Mega JL, Hochholzer W, Frelinger AL 3rd et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA, 306(20), 2221–2228 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Geisler T, Schaeffeler E, Dippon J et al. CYP2C19 and nongenetic factors predict poor responsiveness to clopidogrel loading dose after coronary stent implantation. Pharmacogenomics, 9(9), 1251–1259 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Sibbing D, Koch W, Gebhard D et al. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation, 121(4), 512–518 (2010). [DOI] [PubMed] [Google Scholar]
- 63.Koopmans AB, Braakman MH, Vinkers DJ, Hoek HW, van Harten PN. Meta-analysis of probability estimates of worldwide variation of CYP2D6 and CYP2C19. Transl Psychiatry, 11(1), 141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Price MJ, Berger PB, Teirstein PS et al. Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA, 305(11), 1097–1105 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Pereira NL, Rihal C, Lennon R et al. Effect of CYP2C19 Genotype on Ischemic Outcomes During Oral P2Y12 Inhibitor Therapy: A Meta-Analysis. JACC Cardiovasc Interv, 14(7), 739–750 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Meng X, Wang A et al. Ticagrelor versus Clopidogrel in CYP2C19 Loss-of-Function Carriers with Stroke or TIA. N Engl J Med, (2021). [DOI] [PubMed] [Google Scholar]
- 67.International Warfarin Pharmacogenetics Consortium, Klein TE, Altman RB et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med, 360(8), 753–764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zabalza M, Subirana I, Sala J et al. Meta-analyses of the association between cytochrome CYP2C19 loss- and gain-of-function polymorphisms and cardiovascular outcomes in patients with coronary artery disease treated with clopidogrel. Heart, 98(2), 100–108 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Cooper-DeHoff RM, Johnson JA. Hypertension pharmacogenomics: in search of personalized treatment approaches. Nat Rev Nephrol, 12(2), 110–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jorgensen AL, FitzGerald RJ, Oyee J, Pirmohamed M, Williamson PR. Influence of CYP2C9 and VKORC1 on patient response to warfarin: a systematic review and meta-analysis. PLoS One, 7(8), e44064 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cooper GM, Johnson JA, Langaee TY et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood, 112(4), 1022–1027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takeuchi F, McGinnis R, Bourgeois S et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet, 5(3), e1000433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kimmel SE, French B, Kasner SE et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med, 369(24), 2283–2293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pirmohamed M, Burnside G, Eriksson N et al. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med, 369(24), 2294–2303 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Ross S, Nejat S, Pare G. Use of genetic data to guide therapy in arterial disease. J Thromb Haemost, 13 Suppl 1, S281–289 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Wei MY, Ito MK, Cohen JD, Brinton EA, Jacobson TA. Predictors of statin adherence, switching, and discontinuation in the USAGE survey: understanding the use of statins in America and gaps in patient education. J Clin Lipidol, 7(5), 472–483 (2013). [DOI] [PubMed] [Google Scholar]
- 77.SEARCH Collaborative Group, Link E, Parish S et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med, 359(8), 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 78.Linskey DW, English JD, Perry DA et al. Association of SLCO1B1 c.521T>C (rs4149056) with discontinuation of atorvastatin due to statin-associated muscle symptoms. Pharmacogenet Genomics, 30(9), 208–211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hopewell JC, Offer A, Haynes R et al. Independent risk factors for simvastatin-related myopathy and relevance to different types of muscle symptom. Eur Heart J, 41(35), 3336–3342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carr DF, Francis B, Jorgensen AL et al. Genomewide Association Study of Statin-Induced Myopathy in Patients Recruited Using the UK Clinical Practice Research Datalink. Clin Pharmacol Ther, 106(6), 1353–1361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zaiou M, El Amri H. Cardiovascular pharmacogenetics: a promise for genomically-guided therapy and personalized medicine. Clin Genet, 91(3), 355–370 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Nielsen JB, Rom O, Surakka I et al. Loss-of-function genomic variants highlight potential therapeutic targets for cardiovascular disease. Nat Commun, 11(1), 6417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu J, Lahousse L, Nivard MG et al. Integration of epidemiologic, pharmacologic, genetic and gut microbiome data in a drug-metabolite atlas. Nat Med, 26(1), 110–117 (2020). [DOI] [PubMed] [Google Scholar]
- 84.Wikoff WR, Frye RF, Zhu H et al. Pharmacometabolomics reveals racial differences in response to atenolol treatment. PLoS One, 8(3), e57639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rotroff DM, Shahin MH, Gurley SB et al. Pharmacometabolomic Assessments of Atenolol and Hydrochlorothiazide Treatment Reveal Novel Drug Response Phenotypes. CPT Pharmacometrics Syst Pharmacol, 4(11), 669–679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shahin MH, Gong Y, Frye RF et al. Sphingolipid Metabolic Pathway Impacts Thiazide Diuretics Blood Pressure Response: Insights From Genomics, Metabolomics, and Lipidomics. J Am Heart Assoc, 7(1) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Oliveira FA, Shahin MH, Gong Y et al. Novel plasma biomarker of atenolol-induced hyperglycemia identified through a metabolomics-genomics integrative approach. Metabolomics, 12(8) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weng L, Gong Y, Culver J et al. Presence of arachidonoyl-carnitine is associated with adverse cardiometabolic responses in hypertensive patients treated with atenolol. Metabolomics, 12(10) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet, 12(1), 56–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guney E, Menche J, Vidal M, Barabasi AL. Network-based in silico drug efficacy screening. Nat Commun, 7, 10331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cheng F, Lu W, Liu C et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat Commun, 10(1), 3476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheng F, Desai RJ, Handy DE et al. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat Commun, 9(1), 2691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Elliott LS, Henderson JC, Neradilek MB, Moyer NA, Ashcraft KC, Thirumaran RK. Clinical impact of pharmacogenetic profiling with a clinical decision support tool in polypharmacy home health patients: A prospective pilot randomized controlled trial. PLoS One, 12(2), e0170905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aryan Z, Szanto A, Pantazi A et al. Moving Genomics to Routine Care: An Initial Pilot in Acute Cardiovascular Disease. Circ Genom Precis Med, 13(5), 406–416 (2020). [DOI] [PubMed] [Google Scholar]
- 95.Antman EM, Cohen M, Bernink PJ et al. The TIMI risk score for unstable angina/non-ST elevation MI: A method for prognostication and therapeutic decision making. JAMA, 284(7), 835–842 (2000). [DOI] [PubMed] [Google Scholar]
- 96.Lloyd-Jones DM, Wilson PW, Larson MG et al. Framingham risk score and prediction of lifetime risk for coronary heart disease. Am J Cardiol, 94(1), 20–24 (2004). [DOI] [PubMed] [Google Scholar]
- 97.Abifadel M, Varret M, Rabes JP et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet, 34(2), 154–156 (2003). [DOI] [PubMed] [Google Scholar]
- 98.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science, 232(4746), 34–47 (1986). [DOI] [PubMed] [Google Scholar]
- 99.Lehrman MA, Russell DW, Goldstein JL, Brown MS. Exon-Alu recombination deletes 5 kilobases from the low density lipoprotein receptor gene, producing a null phenotype in familial hypercholesterolemia. Proc Natl Acad Sci U S A, 83(11), 3679–3683 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lehrman MA, Schneider WJ, Sudhof TC, Brown MS, Goldstein JL, Russell DW. Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science, 227(4683), 140–146 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brooks-Wilson A, Marcil M, Clee SM et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet, 22(4), 336–345 (1999). [DOI] [PubMed] [Google Scholar]
- 102.Bodzioch M, Orso E, Klucken J et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet, 22(4), 347–351 (1999). [DOI] [PubMed] [Google Scholar]
- 103.Rust S, Rosier M, Funke H et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet, 22(4), 352–355 (1999). [DOI] [PubMed] [Google Scholar]
- 104.Schultheiss HP, Fairweather D, Caforio ALP et al. Dilated cardiomyopathy. Nat Rev Dis Primers, 5(1), 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res, 121(7), 731–748 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haas J, Frese KS, Peil B et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J, 36(18), 1123–1135a (2015). [DOI] [PubMed] [Google Scholar]
- 107.Fatkin D, MacRae C, Sasaki T et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med, 341(23), 1715–1724 (1999). [DOI] [PubMed] [Google Scholar]
- 108.Pugh TJ, Kelly MA, Gowrisankar S et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med, 16(8), 601–608 (2014). [DOI] [PubMed] [Google Scholar]
- 109.Tobita T, Nomura S, Fujita T et al. Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci Rep, 8(1), 1998 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Seeger T, Shrestha R, Lam CK et al. A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation, 139(6), 799–811 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Marian AJ. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ Res, 128(10), 1533–1553 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ho CY, Day SM, Ashley EA et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation, 138(14), 1387–1398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lopes LR, Syrris P, Guttmann OP et al. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart, 101(4), 294–301 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Coppini R, Ho CY, Ashley E et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J Am Coll Cardiol, 64(24), 2589–2600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.James CA, Syrris P, van Tintelen JP, Calkins H. The role of genetics in cardiovascular disease: arrhythmogenic cardiomyopathy. Eur Heart J, 41(14), 1393–1400 (2020). [DOI] [PubMed] [Google Scholar]
- 116.McKoy G, Protonotarios N, Crosby A et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet, 355(9221), 2119–2124 (2000). [DOI] [PubMed] [Google Scholar]
- 117.van Lint FHM, Murray B, Tichnell C et al. Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo. Circ Genom Precis Med, 12(8), e002467 (2019). [DOI] [PubMed] [Google Scholar]
- 118.Dietz HC, Cutting GR, Pyeritz RE et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature, 352(6333), 337–339 (1991). [DOI] [PubMed] [Google Scholar]
- 119.Arnaud P, Milleron O, Hanna N et al. Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet Med, 23(7), 1296–1304 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McKusick VA. The cardiovascular aspects of Marfan's syndrome: a heritable disorder of connective tissue. Circulation, 11(3), 321–342 (1955). [DOI] [PubMed] [Google Scholar]
- 121.Milewicz DM, Braverman AC, De Backer J et al. Marfan syndrome. Nat Rev Dis Primers, 7(1), 64 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med, 342(10), 673–680 (2000). [DOI] [PubMed] [Google Scholar]
- 123.Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med, 16(12), 881–888 (2014). [DOI] [PubMed] [Google Scholar]
- 124.Loeys BL, Chen J, Neptune ER et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet, 37(3), 275–281 (2005). [DOI] [PubMed] [Google Scholar]
- 125.van de Laar IM, Oldenburg RA, Pals G et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet, 43(2), 121–126 (2011). [DOI] [PubMed] [Google Scholar]
- 126.MacFarlane EG, Parker SJ, Shin JY et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J Clin Invest, 129(2), 659–675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lerner-Ellis JP, Aldubayan SH, Hernandez AL et al. The spectrum of FBN1, TGFbetaR1, TGFbetaR2 and ACTA2 variants in 594 individuals with suspected Marfan Syndrome, Loeys-Dietz Syndrome or Thoracic Aortic Aneurysms and Dissections (TAAD). Mol Genet Metab, 112(2), 171–176 (2014). [DOI] [PubMed] [Google Scholar]
- 128.Bertoli-Avella AM, Gillis E, Morisaki H et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol, 65(13), 1324–1336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Coady MA, Davies RR, Roberts M et al. Familial patterns of thoracic aortic aneurysms. Arch Surg, 134(4), 361–367 (1999). [DOI] [PubMed] [Google Scholar]
- 130.Guo DC, Papke CL, Tran-Fadulu V et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet, 84(5), 617–627 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pomianowski P, Elefteriades JA. The genetics and genomics of thoracic aortic disease. Ann Cardiothorac Surg, 2(3), 271–279 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic Bases of Bicuspid Aortic Valve: The Contribution of Traditional and High-Throughput Sequencing Approaches on Research and Diagnosis. Front Physiol, 8, 612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen Q, Kirsch GE, Zhang D et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature, 392(6673), 293–296 (1998). [DOI] [PubMed] [Google Scholar]
- 134.Brugada J, Campuzano O, Arbelo E, Sarquella-Brugada G, Brugada R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J Am Coll Cardiol, 72(9), 1046–1059 (2018). [DOI] [PubMed] [Google Scholar]
- 135.Sarquella-Brugada G, Campuzano O, Arbelo E, Brugada J, Brugada R. Brugada syndrome: clinical and genetic findings. Genet Med, 18(1), 3–12 (2016). [DOI] [PubMed] [Google Scholar]
- 136.Giudicessi JR, Wilde AAM, Ackerman MJ. The genetic architecture of long QT syndrome: A critical reappraisal. Trends Cardiovasc Med, 28(7), 453–464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Neira V, Enriquez A, Simpson C, Baranchuk A. Update on long QT syndrome. J Cardiovasc Electrophysiol, 30(12), 3068–3078 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Southgate L, Machado RD, Graf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol, 17(2), 85–95 (2020). [DOI] [PubMed] [Google Scholar]
- 139.Graf S, Haimel M, Bleda M et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun, 9(1), 1416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bohnen MS, Ma L, Zhu N et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ Genom Precis Med, 11(10), e002087 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Eyries M, Coulet F, Girerd B et al. ACVRL1 germinal mosaic with two mutant alleles in hereditary hemorrhagic telangiectasia associated with pulmonary arterial hypertension. Clin Genet, 82(2), 173–179 (2012). [DOI] [PubMed] [Google Scholar]
- 142.Eyries M, Montani D, Girerd B et al. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur Respir J, 55(4) (2020). [DOI] [PubMed] [Google Scholar]
- 143.Zhu N, Swietlik EM, Welch CL et al. Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH. Genome Med, 13(1), 80 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Swietlik EM, Greene D, Zhu N et al. Bayesian Inference Associates Rare KDR Variants with Specific Phenotypes in Pulmonary Arterial Hypertension. Circ Genom Precis Med, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Newman JH, Wheeler L, Lane KB et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med, 345(5), 319–324 (2001). [DOI] [PubMed] [Google Scholar]
- 146.Eyries M, Montani D, Girerd B et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet, 46(1), 65–69 (2014). [DOI] [PubMed] [Google Scholar]
- 147.Hadinnapola C, Bleda M, Haimel M et al. Phenotypic Characterization of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension. Circulation, 136(21), 2022–2033 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]